

Abstract
Due to the significance of protein phosphorylation in various biological processes and signaling events, new analytical techniques for enhanced phosphoproteomics have been rapidly introduced in recent years. The combinatorial use of the phospho-specific enrichment techniques and prefractionation methods prior to MS analysis enables comprehensive profiling of the phosphoproteome and facilitates deciphering the critical roles that phosphorylation plays in signaling pathways in various biological systems. This review places special emphasis on the recent five-year (2009–2013) advances for enrichment and separation techniques that have been utilized for phosphopeptides prior to MS analysis.
Keywords: Enrichment, separation, phosphopeptides, phosphoproteomics, mass spectrometry
1. Introduction
Protein phosphorylation is one of the most common post-translational modifications (PTMs) in various organisms. One-third of proteins in eukaryotic cells are estimated to be phosphorylated at any time [1]. The reversible phosphorylation of proteins plays critical roles in the regulation of intracellular biological processes, such as signal transduction, transcription and translation regulation, and metabolism [2]. Aberrant phosphorylation can result in diseases including cancer [3] and cardiovascular disease [4]. Due to the significance of protein phosphorylation in biological processes, tremendous efforts have been made to investigate protein phosphorylation for decades [5]. Mass spectrometry (MS) as a promising tool to analyze protein phosphorylation has gained a great deal of attention due to its ability to profile thousands of proteins in a single analysis [1, 6]. However, investigation of protein phosphorylation is not straightforward. The low stoichiometry, wide dynamic range, and various isoforms of phosphorylated proteins present in the biological systems pose significant challenges to current analytical techniques. Fortunately, these challenges can be alleviated to some extent by enriching phosphoproteins or phosphopeptides prior to MS analysis [7, 8].
To increase the number of identified proteins from a biological sample, multidimensional separation strategies are commonly utilized in current MS analysis in order to reduce the complexity of samples [9]. In a typical MS-based phosphoproteomic analysis, prefractionation and enrichment steps are often employed before MS analysis [10]. Although some prefractionation methods can partially enrich phosphopeptides or phosphoproteins from biological samples, such as ion-exchange based chromatography, we categorize those techniques as separation strategies. Herein, we will focus on the advances in enrichment and separation techniques for MS-based phosphoproteomics that have been published in recent years during 2009–2013. The citations are not comprehensive due to vast amount of literature and explosive rate of growth in the field. Rather, we attempt to highlight some representative examples from recent work and advances.
2. General workflow for MS-based phosphoproteomics
In a general “bottom-up” phosphoproteomic workflow (Figure 1), protein mixtures are usually extracted from cells or tissues and digested. The generated peptides are separated into tens of fractions. Phosphopeptides in each fraction are isolated, and subjected to LC-MS/MS (tandem mass spectrometry) analysis. Finally, phosphopeptides are identified by a database search, and phosphorylation sites are confidently assigned by a site localization algorithm.
Figure 1.
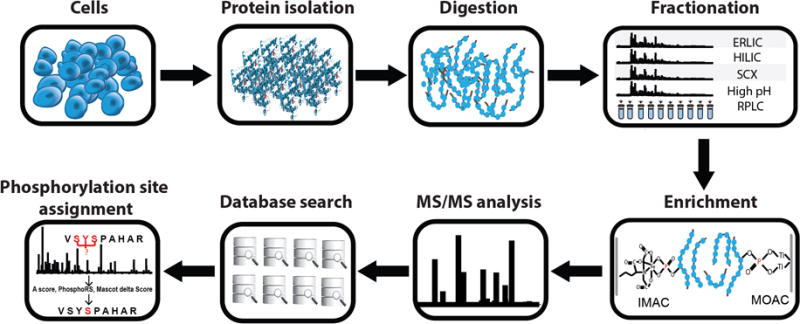
A general “bottom-up” phosphoproteomic workflow depicting the major steps, which consists of protein extraction from cells, enzymatic digestion, and fractionation of the resulting peptide mixtures. Phosphopeptides in each fraction are then enriched and subjected to LC-MS/MS analysis. Finally, phosphopeptides are identified by a database search and phosphorylation sites are confidently assigned by a site localization algorithm.
In a biological system, signaling pathways are usually activated or inactivated by phosphorylation or dephosphorylation of major proteins [5]. It is, therefore, very important to maintain the original biological state during sample preparation for a phosphoproteomic analysis. These phosphorylation dynamics are achieved by the interplay between protein kinases and protein phosphatases [11, 12]. Thus, adding protease and phosphatase inhibitors into cell lysis is required to avoid dephosphorylation of proteins during sample preparation. The choice of phosphatase inhibitors is also important as each has its unique specificity for inhibition [13]. Furthermore, storing cell pellets at −80°C before protein extraction and performing cell lysis at 4°C are also necessary since low temperatures reduce the activities of those enzymes. Trypsin is the most widely used enzyme in phosphoproteomic workflow. Glu-C, Lys-C, Asp-N or other enzymes are also used alone or with trypsin to achieve in-depth phosphoproteomic analysis [14, 15]. Different enrichment and LC methods are performed after digestion to facilitate the detection of phosphopeptides in subsequent MS analysis. Details on enrichment and separation techniques for phosphoproteomics will be covered in later sections.
During MS analysis, ions will be fragmented into small pieces in order to obtain sequence information. Although collision-induced dissociation (CID) is the most widely used fragmentation methods for peptide sequencing, phosphopeptides are often preferentially fragmented at the phosphate group. This gives rise to nonsequence neutral and charged losses from the precursor and sequence-specific product ions [16], limiting the identification of phosphopeptides and phosphorylation site localization. One way to deal with the loss of phosphate groups in CID is by performing MS3 or multi-stage activation in an ion trap [17–19]. In MS3, a neutral loss from phosphopeptides will trigger another stage of fragmentation for the neutral loss ions, providing more sequence information with the cost of reduced scan speed [17, 18]. Multi-stage activation, also called pseudo-MS3, operates in the same principle as MS3 except that it combines the precursor and the neutral loss reaction products in one spectrum without additional isolation of the neutral loss ions as in MS3 [19]. In comparison to CID, high-energy collision dissociation (HCD) was shown to identify more phosphopeptides and phosphorylation sites as it produces less abundant neutral loss and more interpretable sequence informative product ions [20, 21]. Electron capture dissociation (ECD) and electron transfer dissociation (ETD), unlike CID or HCD, fragment peptide ions independently of the sequence of peptides. As a result, labile PTMs, such as phosphate groups, will stay intact using these two approaches [22], however, these electron-based fragmentation techniques require the presence of multiply charged peptide ions [23]. Furthermore, ECD requires the use of FT-ICR instrumentation. These limitations prevent their broader usage in large-scale phosphoproteomics.
With thousands of tandem MS spectra identified by database searches, phosphorylation sites on each identified peptide need to be verified. Unfortunately, it is impossible to validate the assignments of phosphorylation sites by manual inspection of thousands of MS/MS spectra. To address this challenge, site localization algorithms, such as A score [24], PhosphoRS [25], and mascot delta score [26], are introduced in the field of phosphoproteomics to facilitate the validation process. Because of the biological significance of phosphorylation dynamics in signaling events, quantitative phosphoproteomics utilizing label-free or labeling approaches has gained increasing attention. Labeling approaches include SILAC (stable isotope labeling by metabolic incorporation of amino acids), and chemical labeling (such as isobaric tag and dimethylation), whereas label-free approaches include spectral counting and ion-intensity based quantitation [27]. Extreme caution is required when interpreting quantitative data in phosphoproteomics as the majority of dynamic variations can be caused by the abundance changes of the corresponding proteins. Thus, it is necessary to normalize the abundances of phosphopeptides to their corresponding protein abundances in order to extract phosphorylation-regulated dynamic information [28].
3. Advances in enrichment techniques
A variety of enrichment techniques, including immunoprecipitation (IP), chemical modification, immobilized metal affinity chromatography (IMAC), and metal oxide affinity chromatography (MOAC), have been developed and applied to study the phosphoproteome in different biological samples [3, 29]. Among these techniques, chemical modifications, such as β-elimination coupled with a Michael addition [30], often suffer from incomplete reactions resulting in significant sample loss and increased sample complexity. Despite being an effective enrichment strategy, IP is mainly used for enrichment of phosphotyrosine-containing proteins due to a lack of highly specific phosphoserine or phosphothreonine antibodies [31]. Thus, we will focus our discussion primarily on IMAC and MOAC in the following sections.
3.1. Immobilized metal affinity chromatography
Currently, IMAC is one of the most widely used enrichment techniques for phosphopeptides prior to MS analysis. It enriches phosphopeptides by utilizing the affinity of the positively charged metal ions for negatively charged phosphate. In IMAC, metal ions, such as Fe3+, Al3+, Ga3+, Zr4+ and Ti4+ [32–35], are chelated to a support, such as magnetic beads, stationary phase in a chromatographic column or MALDI plate, via a chelating group. To minimize the interference from the non-phosphorylated peptides with acidic residues, the pH of the loading and washing buffers should be in a range that keeps the carboxyl groups on the peptides protonated and phosphate groups on the phosphopeptides deprotonated. Thus, negatively-charged phosphopeptides will be retained by IMAC resins while non-phosphorylated peptides will be washed off [36, 37]. But complete protonation of all acidic residues can only be achieved in highly acidic pH (pH=1 to 1.5) and such low pH conditions could result in metal ions leaching from supports and contaminating phosphopeptides because the functional groups in IMAC materials frequently contain carboxyl groups [38]. One way to overcome this weakness is esterification of the phosphopeptides, which converts the carboxyl groups of amino acid residues into methyl esters [39]. However, sample loss and increased sample complexity caused by incomplete reactions and side reactions prevent the wide spread usage of esterification [40]. Significant efforts have been made to address these problems by optimization of the IMAC materials and enrichment protocols. Herein, we will highlight recent advances in IMAC in both of these aspects.
3.1.1 Advances in materials for immobilized metal affinity chromatography
Improvements in IMAC materials include selection of different metal ions, chelating groups and supports. Stationary phases of LC columns are the common supports for IMAC. Recently, various metal ions have been immobilized onto monolithic columns [41–43]. Because of the porous structure, a monolith provides a larger surface area to bind more metal ions with high specificity and enable rapid mass transport for phosphopeptides. A Ti4+-based IMAC monolith was used to perform a phosphoproteomic study of rat liver mitochondrion [41]. With tetraethoxysilane and 3-aminopropyltriethoxysilane as precursors, the sol-gel method was used to prepare the monolithic support, onto which Ti4+ ions were chelated by further functionalizing amine groups, in a 250 μm i.d. capillary. A total of 224 phosphopeptides were identified in comparison with 28 and 72 phosphopeptides that were identified by commercial Fe3+-IMAC and TiO2, respectively.
Enriching phosphopeptides in an LC column usually requires multiple sample-handling steps and a LC system to achieve sample loading, washing, and eluting. These limitations motivated researchers to design new supports to simplify the procedure of phosphopeptide enrichment. Magnetic beads have attracted a great deal of interest due to the simplicity for isolating phosphopeptides from a sample solution by an external magnetic field and provide a high throughput platform for automated enrichment processes [44]. Although magnetic non-porous beads with different metal ions immobilized have been successfully used for various biological samples and are commercially available [45], magnetic mesoporous beads with high density pores and large surface areas have gained increasing attention in the past five years [46, 47]. Zirconium-containing magnetic mesoporous beads have been synthesized and applied for enriching phosphopeptides by directly coating mesoporous silica on to Fe3O4 magnetic microspheres following addition of phosphate to chelate with Zr4+ ions [47]. The Zr4+-functionalized materials facilitated identification of 218 phosphopeptides in 100 μg rat brain which showed excellent potential for selective enrichment of phosphopeptides.
Directly enriching phosphopeptides on a modified MALDI plate has also been developed and successfully applied to enable rapid enrichment of a small amount of sample [34, 48–50]. Phosphopeptides in 20 fmol purified β-casein digests were successfully enriched by a functionalized MALDI plate with a phosphonate self-assembled monolayer immobilized to Zr4+ ions [50]. The sensitivity is significantly higher than TiO2 or Fe-IMAC microcolumns or magnetic beads due to reduced sample loss caused by additional sample transfer steps. Recently, Si wafers with a micro-array of functionalized microspots were fabricated as a MALDI plate [48]. In each microspot, Fe3+ ions were immobilized onto the Si wafers via nitrilotriacetate (NTA), which provides a potential platform for high throughput parallel enrichment of phosphopeptides.
Furthermore, to immobilize metal ions onto a solid support, a polymer-based metal ion affinity capture (PolyMAC), which immobilizes metal ions to soluble nanopolymers, was recently introduced [51]. This technique allows for enrichment of limited amounts of phosphopeptides in a homogeneous, aqueous solution, which overcomes the inconsistent enrichment resulting from the heterogeneity of solid phase extraction-based isolation methods. Phosphopeptides are typically bound to PolyMAC-Ti agents and isolated from solution through coupling to hydrazide-agarose gels. The entire enrichment process, including capture of phosphopeptides in solution, recovery of PolyMAC-phosphopeptide complexes on agarose beads, washing off non-phosphorylated peptides, and elution of phosphopeptides, requires less than 30 min and provides higher selectivity, reproducibility and greater yields than the commonly used TiO2 methods.
In addition to the improvements in IMAC supports, an increasing number of studies have focused on the improvements of chelating groups [41, 47, 50, 52–55]. The phosphate group has recently been employed in IMAC as a promising metal ion chelator. Unlike in NTA-IMAC or imidodiacetic acid-IMAC method where each metal ion only coordinates to one ligand, each metal ion binds to more than one phosphate group, producing strong binding between metal ions and ligands [56]. Ti or Zr ions immobilized on the phosphate materials have been reported to be coated onto various supports, including MALDI plates, magnetic beads, celluloses, and monolithic columns [41, 47, 50, 52].Moreover, many new types of complexes have also been utilized in IMAC, such as dinuclear zinc (II) complex Phos-tag, and polydopamine [53–55].
3.1.2 Advances in protocol for immobilized metal affinity chromatography
A recent study discovered that IMAC not only can enrich phosphopeptides, but also can bind to non-phosphorylated peptides with deamidation or carbamylation [57]. Therefore, it is recommended to avoid using urea, a commonly used denaturant, in phosphoprotemic studies due to its tendency to induce these modifications (deamidation and carbamylation) in peptides. Additionally, because interfering nucleic acids contain abundant phosphate groups that have strong affinities for IMAC beads, acetonitrile (ACN) precipitation has been employed for protein extraction to efficiently remove nucleic acids before the IMAC enrichment step [58]. To improve phosphopeptide enrichment, the loading, washing and elution buffers all need to be carefully adjusted. This topic has been extensively reviewed [37], and therefore is not covered here in detail.
To comprehensively map the heterogeneous types of phosphopeptides, different strategies have been developed for IMAC enrichment [59–61]. These strategies have included the improvements on enrichment steps and combinatorial use of different metal ion-based IMAC. For example, a tandem IMAC-IMAC strategy (Figure 2) was utilized to address the problem of the relatively low number of monophosphorylated peptides identified in IMAC enrichment, which is known for weak interactions with monophosphorylated peptides [59]. To recover the monophosphorylated peptides in the washing fractions from the first IMAC enrichment, a second IMAC enrichment step was performed to capture these unbound phosphopeptides. This tandem IMAC strategy yielded about a 60% increase in the number of phosphopeptides identified in comparison to one-step IMAC enrichment. Furthermore, more than 90% of the identified phosphopeptides in the second IMAC fractions were monophosphorylated peptides. Another strategy, which combined use of different metal ions for enriching phosphopeptides in a single experiment, has also been demonstrated [60, 61]. The Fe3+-IMAC and Ti4+-IMAC methods could complementarily enrich phosphopeptides in Raji cell lysate with only 10% overlapping [60]. Furthermore, sequential enrichment of phosphopeptides from Raji cell lysate by Ga3+-Fe3+IMAC also resulted in more than 1.5-fold greater coverage of phosphoproteome compared to that of single IMAC (Fe3+, Ti4+, Ga3+ and Al3+) [61].
Figure 2.
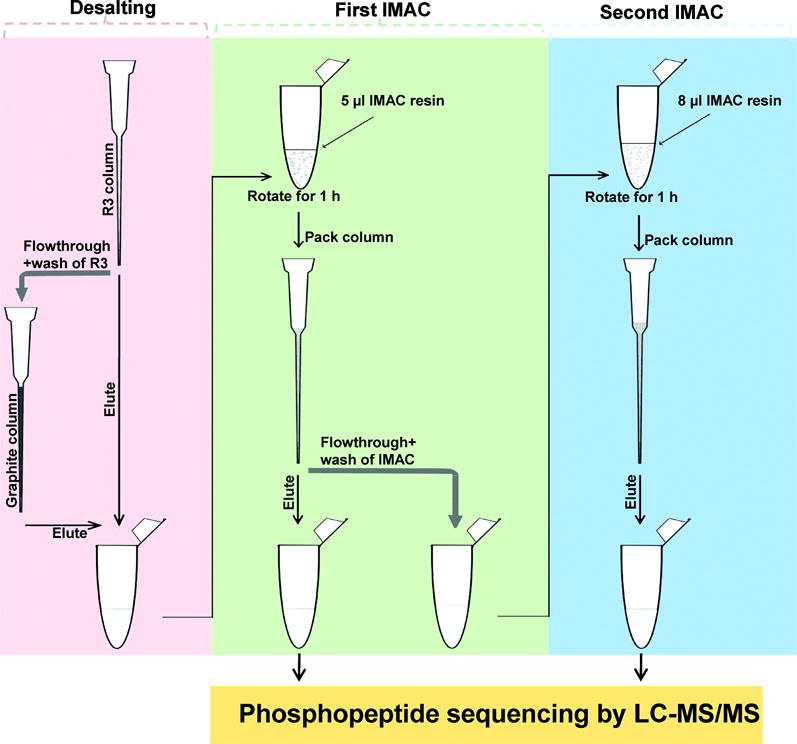
IMAC-IMAC strategy for enrichment of phosphopeptides from complex biological samples. Reprinted with permission from Ref [59]. Copyright American Chemical Society 2010.
3.2. Metal oxide affinity chromatography
In comparison with IMAC, which often involves chelation by proximal carboxyl groups, MOAC has a better tolerance for low pH loading and washing buffers that efficiently protonate carboxyl group while keeping the negative charges on phosphorylated residues[38]. This technique is based on the affinity of oxygen in the phosphate groups for metal atoms in the MOAC resin. Peptide mixtures are usually loaded onto the column under acidic conditions and the phosphopeptides are eluted by addition of basic solutions. Various metal oxides including TiO2, ZrO2, Fe3O4, SnO2, HfO2 and CeO2 have been used for enrichment and exhibit different affinity and specificity for phosphopeptides [48, 62–67]. The Turecek and Ge groups independently demonstrated that ZrO2 outperforms TiO2 [68, 69], however, the variations in phosphopeptide enrichment capabilities are highly dependent on the preparation techniques of metal oxide as the morphological properties of the solid coating, in addition to the differences in metal centers, can also significantly contribute to the enrichment capability of metal oxide. Enrichment of phosphopeptides is also influenced by the physical characteristics of the MOAC resin. For example, nanoparticles generate superior enrichment compared with microparticles due to larger surface area with high specificity to provide more ligand binding sites [70]. Herein, we will also cover recent advances in MOAC in terms of materials and enrichment protocols.
3.2.1 Advances in materials for metal oxide affinity chromatography
In the last five years, substrates with various structures and compositions were synthesized to improve the enrichment performance of MOAC [71, 72]. For example, TiO2 and iron oxide were coated onto a monolithic capillary column to enrich phosphopeptides from casein or human serum [73, 74]. Additionally, mesoporous TiO2 and ZrO2 aerogels were recently designed for selective enrichment of phosphopeptides in rat liver mitochondrion [75, 76], where the binding capacity of mesoporous TiO2 aerogel was shown to be six times higher than that of conventional TiO2 microparticles for a single phosphopeptide standard. A total of 216 phosphoprotein groups were identified by the mesoporous TiO2 aerogel from rat liver mitochondrion compared to less than 150 phosphoprotein groups identified by TiO2 nanoparticles or microparticles [76]. Magnetic particles decorated with metal oxide have also been widely applied for enriching phosphopeptides [77–79]. Carbon-encapsulated magnetic iron nanoparticles were functionalized with poly (acrylic acid) to produce a carboxyl-rich polymer surface which greatly facilitated the immobilization of TiO2 [79]. These nanoparticles enabled identification of 1415 unique phosphopeptides and 1093 phosphorylation sites from 200 μg of HeLa cell lysates, a performance significantly superior to that of commercial magnetic TiO2 beads.
Although metallic MALDI plates are the main supports used for on-plate enrichment of phosphopeptides, the high cost of these metallic plates make alternative substrates attractive, such as glass slides. A mesoporous TiO2 stripe coated microscope glass slide not only provided a platform for on-target enrichment but also enabled separation of phosphopeptides based on the number of phosphate groups on the peptides [80], for which ammonium dihydrogen phosphate solution was used to elute the phosphopeptides. In a similar way to thin layer chromatography, the stripe was placed in a chamber containing elution buffer, and the mono- and multiphosphorylated peptides of β-casein were clearly separated. TiO2 arrays were also fabricated on a gold-covered glass slide via photocatalysis and provided the capability for high-throughput processing [81]. Furthermore, single-use TiO2-coated aluminum foil was introduced for on-plate enrichment of phosphopeptides as an economical disposable layer attached to MALDI plates [82]. This platform avoided the time-consuming polishing and washing steps to regenerate MALDI plates and was used for matrix-free laser desorption/ionization of peptides with sample amounts down to several hundred femtomoles.
3.2.2 Advances in protocol for metal oxide affinity chromatography
To improve the enrichment performance of MOAC, considerable efforts have focused on the optimization of sample loading and elution buffers, which have been covered by other reviews [33]. In addition to conventional additives, including 2,5-dihydroxybenzoic acid (DHB) [83], glutamic acid [84] and lactic acid [85], which contain carboxyl groups, glycerol was demonstrated to improve the phosphopeptide selectivity of TiO2 [86]. It is speculated that glycerol added into loading and washing buffers could reduce the binding of acidic peptides to TiO2 by disruption of the carboxy group-based bidentate chelation. It was also demonstrated that NH4OH, a common elution solution, tended to elute shorter phosphopeptides (1000–1500 Da), while bis-Tris propane was suitable for elution of longer phosphopeptides (1000–4000 Da), which in some cases contained more hydrophilic and/or more acidic residues. Accordingly, an optimized method, which combined the glycerol additive and two-step elution (NH4OH and bis-Tris propane) was applied to enrich phosphopeptides in PC2 cell lysate. In comparison to the conventional method, a 1.4-fold increase in the number of identified phosphopeptides was obtained (Figure 3). In addition to these results, the glycerol additive enabled recovery of more monophosphorylated peptides than commonly used lactic acid [86]. Recently, asparagine (N) or glutamine (Q)-rich peptides, which did not show a propensity to binding to IMAC beads, were proven to exhibit non-specific binding to TiO2 beads [87]. Furthermore, phosphopeptide enrichment was particularly low for yeast cells compared to flies and human due to the relative higher abundance of N/Q-rich proteins in yeast (2.7%) than that in flies (1.5%) and human (1.3%). A mixture of asparagine and glutamine amino acids as decoy amino acids were added into washing buffers to reduce the extent of non-specific peptide binding and improve the recovery and detection of low abundance phosphopeptides. A 30% increase in the enrichment of phosphopeptides was observed compared to that of washing buffers without decoy amino acids.
Figure 3.
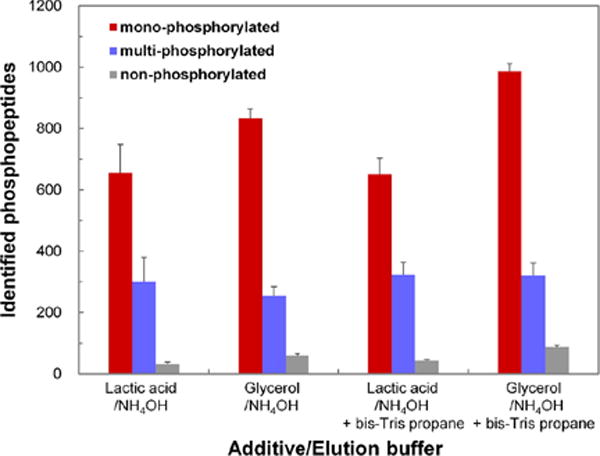
Comparison of the number of identified phosphopeptides obtained from PC3 cell lysate digest using different elution conditions for the TiO2 enrichment. LC-MS/MS measurement was performed in duplicate. Reprinted with permission from Ref [86]. Copyright American Chemical Society 2013.
The peptide-to-metal oxide beads ratio is also a significant factor for phosphopeptide enrichment (Figure 4) [88]. Insufficient or excessive TiO2 beads could decrease the selectivity. While excessive TiO2 beads could increase non-specific binding, insufficient TiO2 beads favor enrichment of multiphosphorylated peptides due to higher affinity compared to monophosphorylated peptides. Therefore, optimization of peptide-to-beads ratio is recommended when handling different biological samples. It is also suggested that incubation of samples with insufficient beads could be utilized as a strategy to separate multiphosphorylated peptides from monophosphorylated peptides, which can enhance the detection of multiphosphorylated peptides as monophosphorylated peptides can suppress the signal of the multiphosphorylated peptides. A pre-separation of mono- and multiphosphorylated peptides could also be achieved by eluting phosphopeptides at different high pH conditions [89]. Moreover, citric acid could be used for stepwise separation of phosphopeptides since the concentration of citric acid can greatly affect the binding of mono- and multiphosphorylated peptides with TiO2 [90]. In one example, 1 mg of HeLa cell digest was loaded onto TiO2 beads in loading buffer containing 1 M citric acid. The flow-through fraction was then diluted to a final concentration of 50 mM citric acid and enriched by another aliquot of TiO2 beads. The results showed that 69% of phosphopeptides in the first enrichment were multiphosphorylated and 92% of them in the second enrichment were monophosphorylated. In comparison to the commonly used DHB/TiO2 enrichment strategy, a 37% increase in the total number of identified phosphopeptides and 2.6-fold increase in the number of identified multiphosphorylated peptides was observed.
Figure 4.
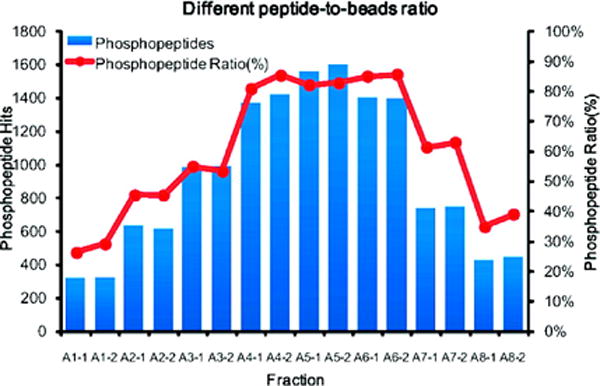
Profiles of phosphopeptides identified from 500 μg of HeLa cell lysate digest when different peptide-to-TiO2 bead ratios were used during the enrichment. The phosphopeptide ratio for each fraction is calculated by the number of phosphopeptides identified divided by the total identified peptides. A1 to A8 represent 125, 250, 500, 1000, 2000, 4000, 10 000, and 20 000 μg TiO2 beads respectively, and “−1” and “−2” correspond to duplicates of each experiment. Reprinted with permission from Ref [88]. Copyright American Chemical Society 2009.
4. Advances in separation techniques
Although IMAC or MOAC can effectively enrich thousands of phosphopeptides from tissue extract or cell lysate samples, single step affinity purification is often not adequate for a successful in-depth analysis of the phosphoproteome. Thus, prefractionation with different LC methods before routine RPLC-MS/MS analysis is essential to reduce sample complexity. In these prefractionation methods, strong cation exchange chromatography (SCX) is the most frequently used strategy prior to affinity purification. Hydrophilic interaction chromatography (HILIC), electrostatic repulsion-hydrophilic interaction chromatography (ERLIC), and high pH RPLC have also been used for fractionation of phosphopeptides before routine RPLC-MS/MS. Furthermore, capillary electrophoresis (CE) is also applied to separation of phosphopeptides or phosphoproteins prior to MS analysis. Each method has its own merits to provide optimal fractionation for different physicochemical properties of phosphopeptides. Herein, we will highlight advances from the past five years in separation methods for phosphoproteomics including ion exchange based chromatography, HILIC, ERLIC, RP and CE.
4.1. Ion exchange based chromatography
SCX separates phosphopeptides from non-phosphorylated peptides based on their net positive charge under acidic conditions. In an acidic condition (pH=3), phosphopeptides usually possess a lower net charge compared to non-phosphopeptides because of the protonation of acidic residues and deprotonation of phosphate groups. Thus, phosphopeptides have poor retention on the SCX column leading to multiphosphorylated peptides eluting first, followed by N-acetylated peptides, monophosphorylated peptides and finally regular peptides with a differing number of basic residues. However, some phosphopeptides with multiple basic residues will coelute with non-phosphorylated peptides because the net positive charge is similar. Instead of subsequent affinity enrichments, a successive SCX separation at a more acidic condition (pH=1) was recently used to address this problem by protonation of the phosphate groups [91]. The neutralization of phosphate groups results in increasing the net charge for phosphopeptides, whereas the net charge of regular peptides remains unchanged, causing the unaffected non-phosphopeptides to elute first followed by the “basic” phosphopeptides. This tandem SCX at different pH conditions approach enabled identification of over 10,000 unique “basic” phosphopeptides in HeLa cells. Unfortunately, this approach requires LC equipment and SCX columns to operate at pH=1, which is not compatible with the majority of chromatographic systems and columns.
Unlike the poor retention in SCX, phosphopeptides are eluted later compared to non-phosphorylated peptides during an anion exchange chromatography separation. It has been demonstrated that strong anion exchange chromatography (SAX)-TiO2 enabled identification of more acidic and multiphosphorylated peptides in comparison with SCX-TiO2 [92]. This difference in performance can be attributed to the fact that phosphopeptides with or without multiple acidic residues elute simultaneously from the SCX column due to their similar net charge at low pH conditions. Thus, the acidic phosphopeptides are not effectively separated by SCX. However, acidic phosphopeptides can be separated according to the number of acidic amino acid residues they have by SAX due to deprotonation of acidic residues under high pH condition. In contrast, phosphopeptides with multiple basic residues, which can be separated by SCX, cannot be distinguished from phosphopeptides without multiple basic residues in SAX. To take advantage of their varying properties, Dong et al. further separated basic phosphopeptides from the early-eluted fractions of SAX by SCX to improve the coverage for detection of basic phosphopeptides [93]. Based on similar principles, weak anion exchange chromatography, using a low concentration of salt to elute phosphopeptides compared to SAX, was applied to further separate a SCX fraction that was dominated by phosphopeptides with one basic amino acid and a free N-terminus [94].
4.2. Hydrophilic interaction chromatography
HILIC is chosen as a fractionation method for phosphopeptides due to its orthogonality to the subsequent routinely used low pH RPLC-MS/MS analysis [95]. In HILIC, samples are loaded at high organic solvent concentration and eluted by increasing the polarity of the mobile phase, which is opposite to the operation mode for RPLC [96]. Because of their considerable hydrophilicity, multiphosphorylated peptides and peptides with multiple acidic residues are strongly retained in HILIC and are more likely to elute in the same fractions. An increased selectivity for subsequent IMAC enrichment is obtained since multiphosphorylated peptides will compete more effectively for IMAC binding sites compared to monophosphorylated peptides in the presence of acidic peptides of high abundance [97]. Recently, HILIC was implemented into the workflow of sequential elution from IMAC (SIMAC) to improve the selectivity of downstream TiO2 enrichment [98]. A total of 400 μg protein digests from HeLa cells were first subjected to IMAC enrichment, and three fractions were generated: a flow-through fraction, an acidic elution and a basic elution. The flow-through fraction and acidic elution, which mainly contained non- and monophosphorylated peptides, were loaded onto the HILIC column. Phosphopeptides in each HILIC fraction were further enriched by TiO2. In most of the HILIC fractions, more than 90% of the identified peptides were phosphorylated.
4.3. Electrostatic repulsion-hydrophilic interaction chromatography
Recently, ERLIC, which involves both hydrophilic interaction and anion-exchange, has been introduced as a novel separation method for phosphopeptides [99]. In the low pH and high organic content mobile phase, the majority of peptides with acidic residues and carboxyl groups at the C terminus are largely protonated, thus poorly retained on an ERLIC column, whereas phosphopeptides will be retained due to electrostatic attraction by the deprotonated phosphate groups and their considerable hydrophilic interaction. Because of the characteristics of ERLIC, multiphosphorylated peptides possessing more negative charges are eluted later and separated with a relatively high resolution. As mentioned above, SCX usually generates more complex fractions for multiphosphorylated peptides but produces better resolution for monophosphorylated peptides. Therefore, combinatorial use of SCX and ERLIC to further separate their respective complex fractions has been investigated [100]. In this study, a peptide mixture was fractionated by SCX, and the flow-through fraction from SCX was further separated by ERLIC. Afterwards, phosphopeptides in each fraction were enriched by IMAC. In parallel experiments, ERLIC was used to fractionate peptide mixture first and the ERLIC-generated flow-through fractions were further separated by SCX. The number of identified nonredundant phosphopeptides from HeLa cell was increased by about 48% with the use of combinatorial strategies (SCX-ERLIC-TiO2 and ERLIC-SCX-TiO2) as compared to single step fractionation by SCX. A significant increase in the number of multiphosphorylated peptides was also noted when using combinatorial strategies that overcome the weakness of single SCX fractionation methods (Figure 5). These combination approaches have been further simplified by performing solid-phase extraction for both SCX and ERLIC [101]. In this simplified workflow, a total of 9952 unique phosphopeptides was identified for HeLa cells from 13 fractions. Of 9952 unique phosphopeptides, 2137 were multiphosphorylated.
Figure 5.
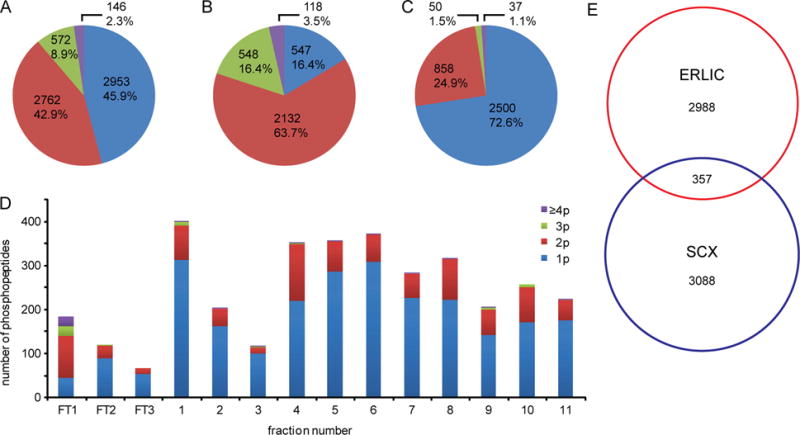
Distributions of identified phosphopeptides from HeLa cell lysate by combinatorial ERLIC–SCX fractionation. The number and percentage of nonredundant identified mono- and multiphosphorylated peptides by ERLIC-SCX-TiO2 (A). (B) The number and percentage of nonredundant identified mono- and multiphosphorylated peptides by ERLIC. (C) The number and percentage of nonredundant identified mono- and multiphosphorylated peptides from further separation of flow-through fractions of ERLIC by SCX. (D) Distribution of identified mono- and multiphosphorylated peptides in each SCX fraction. (E) Venn diagram for nonredundant identified phosphopeptides in both methods. Different colors represent the number of phosphate groups (1p–4,5p) carried by each phosphopeptide. Reprinted with permission from Ref [100]. Copyright American Chemical Society 2012.
4.4. Reversed phase liquid chromatography
RPLC is also widely used for fractionation of phosphopeptides due to its high resolution of peptides in general and capability to provide orthogonal separation in different pH conditions. A high pH RPLC separation is sometimes utilized as the first dimension separation followed by routine on-line low pH RPLC in a phosphoproteomic analysis [95, 102]. Complex peptide mixtures are usually fractionated into tens of fractions, but, to reduce the instrument analysis time, the convention is to pool adjacent fractions together, thus reducing the total number of fractions required to be analyzed. Recently, a new pooling strategy was applied to 2D RPLC in phosphoproteomic analysis resulting in a significantly increased number of phosphopeptides identified [103]. Phosphopeptides were firstly enriched by IMAC and loaded onto the high pH RPLC. The fractions were collected every minute over a 90 min gradient of the high pH RPLC separation. The fractions eluted in the first 45 min were labeled as early group and the last 45 min eluted fractions were called later eluted group. Next, every two fractions from each group with equal time intervals were mixed for the subsequent on-line low pH RPLC separation. This new strategy of pooling nonadjacent fractions yielded a 33% increase in the number of uniquely identified phosphopeptides for mouse liver lysate compared with that obtained by the conventional approach. This increase was likely due to a more random distribution of phosphopeptides in one pooled fraction. It reduced the probability of coelution of phosphopeptides in the second separation dimension since phosphopeptides with similar physiochemical properties were more likely to be pooled together in the conventional pooling approach. A combinatorial use of multistep IMAC and high pH reversed phase fractionation from solid-phase extraction was also introduced to simplify the RP-RP workflow for phosphoproteomics [104]. Phosphopeptides from MCF-10A cell lysate were firstly enriched by IMAC, and then fractionated by a hydrophilic−lipophilic-balanced reversed-phase cartridge using a serial mixed ratio of ACN/NH4HCO3 buffers. 3 mg of starting materials was used compared to 15 mg in a typical SCX-IMAC workflow and enabled the identification of 8969 unique phosphopeptides [104, 105].
4.5. Capillary electrophoresis
As the phosphate group introduces negative charges and modifies the pIs of peptides or proteins, capillary electrophoresis, which resolves analytes based on their different charge-to-size ratio, offers distinct advantages for separation of phosphorylated peptides or proteins. Both ESI and MALDI MS have been frequently employed in the CE-MS coupling for proteomic analysis [106]. Heemskerk et al. have explored the potential of sheathless ultra-low flow CE-ESI-MS in phosphoproteomics [107]. By using the high sensitivity porous sprayer (HSPS) for direct infusion of multiphosphorylated peptides, the authors demonstrated that a near equimolar ESI response could be approached when the flow rate was reduced below 15 nL/min, which might overcome the ionization bias against the phosphorylated peptides at conventional flow rates (> 50 nL/min). Although achieving this ultra-low flow by conventional nanoLC is difficult, it is possible to operate sheathless CE-ESI-MS at this low flow rate with the HSPS and a neutrally coated capillary that eliminates electroosmotic flow (EOF). Combined with an in-line preconcentration technique, transient isotachophoresis (t-ITP) was demonstrated to improve sensitivity of phosphopeptides from milk digest by CE-ESI-MS in comparison with RP nanoLC-MS under conditions of equal sample loading and equal sample concentrations. This sheathless nanospray CE-ESI-MS setup was also implemented for the characterization of post-translationally modified H1 histones isolated from rat testis by Sarg et al. [108]. With IMAC enrichment prior to MS analysis, a total of 55 phosphopeptides were identified by combining the results of RP nanoLC-MS and CE-MS, 22 of which were uniquely identified by CE-MS and 19 were uniquely identified by RPLC-MS, showing the complementarity of the two techniques. A major drawback of CE analysis is the small sample volume that could be injected onto the column. In addition to the t-ITP mentioned above, pH-mediated stacking strategies were also adopted to increase the loading amount of phosphopeptides [109, 110]. Dong et al. applied this method in CE-ESI-MS analysis of phosphopeptide isomers which have the same amino acid sequence but with phosphate group on different residues [110].
For MALDI-MS detection, the selection of background electrolytes (BGE) for CE analysis is more versatile compared to CE-ESI-MS. Bachmann et al. employed a latex-coated capillary and a BGE containing 80 mM of phosphoric acid, 40 mM of triethylamine and 20% ACN for CE-MALDI-TOF analysis of tryptic digests of phosphoproteins [109]. With pH-mediated stacking and pressure assisted sample deposition, even tetra- and penta-phosphorylated peptides (pI<2) from tryptic digests of α-casein and β-casein could be detected by MALDI-TOF MS.
5. Selected applications
Recently, Zhou et al. demonstrated that extensive fractionation in phosphoproteomics can boost the number of identified phosphopeptides in human cancer cells from 3700 to 22,000 by a three-dimensional LC-MS setup [111]. The increasing depth in the coverage of phosphoproteomes has led to advances in biology [3, 29, 112]. Herein, we highlight a few recent examples of phosphoproteomic applications with improved enrichment and separation strategies that enabled better understanding of biological processes. A recent study employed SCX and TiO2 to fractionate and enrich phosphopeptides from AT1R (angiotensin II type 1 receptor) stable transfected HEK293 (AT1R-HEK293) cells as AT1R is an important drug target in cardiovascular diseases [113]. A comprehensive understanding of the AT1R signaling pathways is critical to the development of effective treatments for cardiovascular diseases. A total of 10,965 unique phosphopeptides were identified and led to the discovery of protein kinase D as a critical mediator in the AT1R signaling pathways by comparing differential phosphoproteomes in AT1R-HEK293 cells treated with two different agonists. IMAC or MOAC combined with HILIC have also been implemented in a phosphoproteomic workflow to elucidate molecular pathways involved in biological processes [114, 115]. TiO2 and HILIC were utilized to facilitate monitoring cell signaling changes during mouse brain development [114], during which a total of 7682 unique phosphopeptides and 3246 unique formerly sialylated glycopeptides was identified. IMAC-HILIC was also used in conjunction to study 17β-Estradiol (E2)-modulated phosphorylation in order to investigate transcriptional activity regulated by E2 [115]. A collection of 2857 unique phosphorylation sites were quantified and E2 was found to modulate gene transcription through a HSP90 phosphorylation-mediated chaperoning process. Another work used ERLIC to enrich and separate phosphopeptides in partner of PIX 2 (POPX2) over-expressing cells in order to reveal the regulatory mechanism of cancer cell motility and invasiveness [116]. POPX2 is a serine/threonine phosphatase and is found in many cancer types, regulating cancer cell motility and invasiveness. After ERLIC enrichment and LC-MS/MS analysis, 3700 phosphopeptides were identified, from which POPX2 was found to exert its cellular function through the regulation of the activity of glycogen synthase kinase-3.
6. Concluding Remarks and Future Prospects
As we presented in this review, new materials and improved sample processing protocols have been rapidly developed for IMAC and MOAC strategies that enable phosphopeptide enrichment with high sensitivity and specificity. Furthermore, different separation methods with high resolution and efficiency have been utilized for enhanced separation of different types of phosphopeptides. Integrated use of enrichment techniques and multiple separation methods provides a powerful platform, enabling in-depth coverage of the phosphoproteome. Although MS-based phosphoproteomics have attained significant success in revealing the roles of phosphorylation in various biological systems [3, 29], several limitations still hinder the broader applications of MS-based phosphoproteomics. Non-specific binding of non-phosphorylated peptides and preferential enrichment of certain types of phosphopeptides still challenge the current enrichment techniques. Furthermore, although incorporating multidimensional separation strategies into phosphoproteomic workflow increases the phosphoproteome coverage, the workflow with increased complexity results in more sample-handling steps and greater sample losses. Therefore, the continuing development of simplified and integrated phosphoproteomic workflow that offer high selectivity for phosphopeptides and provide improved coverage and sensitivity would be in great demand. To better decipher biological processes under diseased and healthy conditions, quantitative approaches with multiplexing capability and reduced experimental cost are also highly attractive with great promise for systems phosphoproteomics.
Acknowledgments
The authors wish to thank Amanda Buchberger from the Li Research Group for critical reading of the manuscript. Preparation of this manuscript was supported in part by National Institutes of Health through grants (R01 NS071513 and P20 DK097826). L.L. acknowledges an H. I. Romnes Faculty Fellowship.
References
- 1.Mann M, Ong SE, Gronborg M, Steen H, Jensen ON, Pandey A. Trends in biotechnology. 2002;20:261–268. doi: 10.1016/s0167-7799(02)01944-3. [DOI] [PubMed] [Google Scholar]
- 2.Hunter T. Cell. 2000;100:113–127. doi: 10.1016/s0092-8674(00)81688-8. [DOI] [PubMed] [Google Scholar]
- 3.Harsha HC, Pandey A. Molecular oncology. 2010;4:482–495. doi: 10.1016/j.molonc.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun Z, Hamilton KL, Reardon KF. Journal of molecular and cellular cardiology. 2012;53:354–368. doi: 10.1016/j.yjmcc.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 5.Pawson T, Scott JD. Trends in biochemical sciences. 2005;30:286–290. doi: 10.1016/j.tibs.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 6.Piggee C. Analytical chemistry. 2009;81:2418–2420. doi: 10.1021/ac802740t. [DOI] [PubMed] [Google Scholar]
- 7.McLachlin DT, Chait BT. Current opinion in chemical biology. 2001;5:591–602. doi: 10.1016/s1367-5931(00)00250-7. [DOI] [PubMed] [Google Scholar]
- 8.Mann M, Jensen ON. Nature biotechnology. 2003;21:255–261. doi: 10.1038/nbt0303-255. [DOI] [PubMed] [Google Scholar]
- 9.Washburn MP, Wolters D, Yates JR., 3rd Nature biotechnology. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 10.Thingholm TE, Jensen ON, Larsen MR. Proteomics. 2009;9:1451–1468. doi: 10.1002/pmic.200800454. [DOI] [PubMed] [Google Scholar]
- 11.Hunter T. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 12.Schlessinger J. Cell. 2000;103:211–225. doi: 10.1016/s0092-8674(00)00114-8. [DOI] [PubMed] [Google Scholar]
- 13.Thingholm TE, Larsen MR, Ingrell CR, Kassem M, Jensen ON. Journal of proteome research. 2008;7:3304–3313. doi: 10.1021/pr800099y. [DOI] [PubMed] [Google Scholar]
- 14.Wisniewski JR, Mann M. Analytical chemistry. 2012;84:2631–2637. doi: 10.1021/ac300006b. [DOI] [PubMed] [Google Scholar]
- 15.Bian Y, Ye M, Song C, Cheng K, Wang C, Wei X, Zhu J, Chen R, Wang F, Zou H. Journal of proteome research. 2012;11:2828–2837. doi: 10.1021/pr300242w. [DOI] [PubMed] [Google Scholar]
- 16.Palumbo AM, Smith SA, Kalcic CL, Dantus M, Stemmer PM, Reid GE. Mass spectrometry reviews. 2011;30:600–625. doi: 10.1002/mas.20310. [DOI] [PubMed] [Google Scholar]
- 17.Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villen J, Li J, Cohn MA, Cantley LC, Gygi SP. Proceedings of the National Academy of Sciences of the USA. 2004;101:12130–12135. doi: 10.1073/pnas.0404720101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ulintz PJ, Yocum AK, Bodenmiller B, Aebersold R, Andrews PC, Nesvizhskii AI. Journal of proteome research. 2009;8:887–899. doi: 10.1021/pr800535h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schroeder MJ, Shabanowitz J, Schwartz JC, Hunt DF, Coon JJ. Analytical chemistry. 2004;76:3590–3598. doi: 10.1021/ac0497104. [DOI] [PubMed] [Google Scholar]
- 20.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Nature methods. 2007;4:709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 21.Nagaraj N, D’Souza RC, Cox J, Olsen JV, Mann M. Journal of proteome research. 2010;9:6786–6794. doi: 10.1021/pr100637q. [DOI] [PubMed] [Google Scholar]
- 22.Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Proceedings of the National Academy of Sciences of the USA. 2004;101:9528–9533. doi: 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Swaney DL, McAlister GC, Coon JJ. Nature methods. 2008;5:959–964. doi: 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. Nature biotechnology. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 25.Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, Mechtler K. Journal of proteome research. 2011;10:5354–5362. doi: 10.1021/pr200611n. [DOI] [PubMed] [Google Scholar]
- 26.Savitski MM, Lemeer S, Boesche M, Lang M, Mathieson T, Bantscheff M, Kuster B. Molecular & cellular proteomics. 2011;10:M110 003830. doi: 10.1074/mcp.M110.003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nilsson CL. Analytical chemistry. 2012;84:735–746. doi: 10.1021/ac202877y. [DOI] [PubMed] [Google Scholar]
- 28.Wu R, Dephoure N, Haas W, Huttlin EL, Zhai B, Sowa ME, Gygi SP. Molecular & cellular proteomics. 2011;10:M111 009654. doi: 10.1074/mcp.M111.009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nakagami H, Sugiyama N, Ishihama Y, Shirasu K. Plant & cell physiology. 2012;53:118–124. doi: 10.1093/pcp/pcr148. [DOI] [PubMed] [Google Scholar]
- 30.Nika H, Lee J, Willis IM, Angeletti RH, Hawke DH. Journal of biomolecular techniques. 2012;23:51–68. doi: 10.7171/jbt.2012-2302-002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leitner A, Sturm M, Lindner W. Analytica chimica acta. 2011;703:19–30. doi: 10.1016/j.aca.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 32.Zou XJ, Liu D, Zhong LJ, Yang B, Lou YX, Hu BH, Yin YX. Anal Bioanal Chem. 2011;401:1251–1261. doi: 10.1007/s00216-011-5186-x. [DOI] [PubMed] [Google Scholar]
- 33.Hu L, Zhou H, Li Y, Sun S, Guo L, Ye M, Tian X, Gu J, Yang S, Zou H. Analytical chemistry. 2009;81:94–104. doi: 10.1021/ac801974f. [DOI] [PubMed] [Google Scholar]
- 34.Hu Y, Peng Y, Lin K, Shen H, Brousseau LC, 3rd, Sakamoto J, Sun T, Ferrari M. Nanoscale. 2011;3:421–428. doi: 10.1039/c0nr00720j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qian K, Wan J, Liu F, Girault HH, Liu B, Yu C. ACS nano. 2009;3:3656–3662. doi: 10.1021/nn900739z. [DOI] [PubMed] [Google Scholar]
- 36.Dunn JD, Reid GE, Bruening ML. Mass spectrometry reviews. 2010;29:29–54. doi: 10.1002/mas.20219. [DOI] [PubMed] [Google Scholar]
- 37.Fila J, Honys D. Amino acids. 2012;43:1025–1047. doi: 10.1007/s00726-011-1111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Negroni L, Claverol S, Rosenbaum J, Chevet E, Bonneu M, Schmitter JM. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2012;891–892:109–112. doi: 10.1016/j.jchromb.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 39.Ficarro SB, McCleland ML, Stukenberg PT, Burke DJ, Ross MM, Shabanowitz J, Hunt DF, White FM. Nature biotechnology. 2002;20:301–305. doi: 10.1038/nbt0302-301. [DOI] [PubMed] [Google Scholar]
- 40.Stewart II, Thomson T, Figeys D. Rapid communications in mass spectrometry. 2001;15:2456–2465. doi: 10.1002/rcm.525. [DOI] [PubMed] [Google Scholar]
- 41.Hou C, Ma J, Tao D, Shan Y, Liang Z, Zhang L, Zhang Y. Journal of proteome research. 2010;9:4093–4101. doi: 10.1021/pr100294z. [DOI] [PubMed] [Google Scholar]
- 42.Krenkova J, Lacher NA, Svec F. Analytical chemistry. 2010;82:8335–8341. doi: 10.1021/ac1018815. [DOI] [PubMed] [Google Scholar]
- 43.Wang H, Duan J, Xu H, Zhao L, Liang Y, Shan Y, Zhang L, Liang Z, Zhang Y. Journal of separation science. 2011 doi: 10.1002/jssc.201100168. [DOI] [PubMed] [Google Scholar]
- 44.Ficarro SB, Adelmant G, Tomar MN, Zhang Y, Cheng VJ, Marto JA. Analytical chemistry. 2009;81:4566–4575. doi: 10.1021/ac9004452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Batalha IL, Lowe CR, Roque AC. Trends in biotechnology. 2012;30:100–110. doi: 10.1016/j.tibtech.2011.07.004. [DOI] [PubMed] [Google Scholar]
- 46.Li XS, Su X, Zhu GT, Zhao Y, Yuan BF, Guo L, Feng YQ. Journal of separation science. 2012;35:1506–1513. doi: 10.1002/jssc.201101067. [DOI] [PubMed] [Google Scholar]
- 47.Lu J, Li Y, Deng C. Nanoscale. 2011;3:1225–1233. doi: 10.1039/c0nr00896f. [DOI] [PubMed] [Google Scholar]
- 48.Wang WH, Bruening ML. The Analyst. 2009;134:512–518. doi: 10.1039/b815598d. [DOI] [PubMed] [Google Scholar]
- 49.Lu J, Liu S, Deng C. Chemical communications (Cambridge, England) 2011;47:5334–5336. doi: 10.1039/c0cc05524g. [DOI] [PubMed] [Google Scholar]
- 50.Hoang T, Roth U, Kowalewski K, Belisle C, Steinert K, Karas M. Analytical chemistry. 2010;82:219–228. doi: 10.1021/ac9017583. [DOI] [PubMed] [Google Scholar]
- 51.Iliuk AB, Martin VA, Alicie BM, Geahlen RL, Tao WA. Molecular & cellular proteomics. 2010;9:2162–2172. doi: 10.1074/mcp.M110.000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shen F, Hu Y, Guan P, Ren X. Journal of chromatography B, Analytical technologies in the biomedical and life sciences. 2012;902:108–115. doi: 10.1016/j.jchromb.2012.06.033. [DOI] [PubMed] [Google Scholar]
- 53.Yan Y, Zheng Z, Deng C, Li Y, Zhang X, Yang P. Analytical chemistry. 2013;85:8483–8487. doi: 10.1021/ac401668e. [DOI] [PubMed] [Google Scholar]
- 54.Nabetani T, Kim YJ, Watanabe M, Ohashi Y, Kamiguchi H, Hirabayashi Y. Proteomics. 2009;9:5525–5533. doi: 10.1002/pmic.200900341. [DOI] [PubMed] [Google Scholar]
- 55.Kinoshita E, Kinoshita-Kikuta E, Sugiyama Y, Fukada Y, Ozeki T, Koike T. Proteomics. 2012;12:932–937. doi: 10.1002/pmic.201100639. [DOI] [PubMed] [Google Scholar]
- 56.Han G, Ye M, Zou H. The Analyst. 2008;133:1128–1138. doi: 10.1039/b806775a. [DOI] [PubMed] [Google Scholar]
- 57.Worthington J, Cutillas PR, Timms JF. Proteomics. 2011;11:4583–4587. doi: 10.1002/pmic.201100143. [DOI] [PubMed] [Google Scholar]
- 58.Li Y, Luo Y, Wu S, Gao Y, Liu Y, Zheng D. Molecular biotechnology. 2009;43:59–66. doi: 10.1007/s12033-009-9176-6. [DOI] [PubMed] [Google Scholar]
- 59.Ye J, Zhang X, Young C, Zhao X, Hao Q, Cheng L, Jensen ON. Journal of proteome research. 2010;9:3561–3573. doi: 10.1021/pr100075x. [DOI] [PubMed] [Google Scholar]
- 60.Lai AC, Tsai CF, Hsu CC, Sun YN, Chen YJ. Rapid communications in mass spectrometry. 2012;26:2186–2194. doi: 10.1002/rcm.6327. [DOI] [PubMed] [Google Scholar]
- 61.Tsai CF, Hsu CC, Hung JN, Wang YT, Choong WK, Zeng MY, Lin PY, Hong RW, Sung TY, Chen YJ. Analytical chemistry. 2014;86:685–693. doi: 10.1021/ac4031175. [DOI] [PubMed] [Google Scholar]
- 62.Sun S, Ma H, Han G, Wu R, Zou H, Liu Y. Rapid communications in mass spectrometry. 2011;25:1862–1868. doi: 10.1002/rcm.5055. [DOI] [PubMed] [Google Scholar]
- 63.Lu Z, Duan J, He L, Hu Y, Yin Y. Analytical chemistry. 2010;82:7249–7258. doi: 10.1021/ac1011206. [DOI] [PubMed] [Google Scholar]
- 64.Wan H, Yan J, Yu L, Zhang X, Xue X, Li X, Liang X. Talanta. 2010;82:1701–1707. doi: 10.1016/j.talanta.2010.07.050. [DOI] [PubMed] [Google Scholar]
- 65.Chen SY, Juang YM, Chien MW, Li KI, Yu CS, Lai CC. The Analyst. 2011;136:4454–4459. doi: 10.1039/c1an15334j. [DOI] [PubMed] [Google Scholar]
- 66.Lu J, Qi D, Deng C, Zhang X, Yang P. Nanoscale. 2010;2:1892–1900. doi: 10.1039/c0nr00060d. [DOI] [PubMed] [Google Scholar]
- 67.Cheng G, Zhang JL, Liu YL, Sun DH, Ni JZ. Chemical communications (Cambridge, England) 2011;47:5732–5734. doi: 10.1039/c1cc10533g. [DOI] [PubMed] [Google Scholar]
- 68.Blacken GR, Volny M, Diener M, Jackson KE, Ranjitkar P, Maly DJ, Turecek F. Journal of the American Society for Mass Spectrometry. 2009;20:915–926. doi: 10.1016/j.jasms.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 69.Nelson CA, Szczech JR, Dooley CJ, Xu Q, Lawrence MJ, Zhu H, Jin S, Ge Y. Analytical chemistry. 2010;82:7193–7201. doi: 10.1021/ac100877a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vilasi A, Fiume I, Pace P, Rossi M, Pocsfalvi G. Journal of mass spectrometry. 2013;48:1188–1198. doi: 10.1002/jms.3254. [DOI] [PubMed] [Google Scholar]
- 71.Tang LA, Wang J, Lim TK, Bi X, Lee WC, Lin Q, Chang YT, Lim CT, Loh KP. Analytical chemistry. 2012;84:6693–6700. doi: 10.1021/ac301119r. [DOI] [PubMed] [Google Scholar]
- 72.Tan YJ, Sui D, Wang WH, Kuo MH, Reid GE, Bruening ML. Analytical chemistry. 2013;85:5699–5706. doi: 10.1021/ac400198n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang ST, Wang MY, Su X, Yuan BF, Feng YQ. Analytical chemistry. 2012;84:7763–7770. doi: 10.1021/ac301258q. [DOI] [PubMed] [Google Scholar]
- 74.Krenkova J, Foret F. Journal of separation science. 2011;34:2106–2112. doi: 10.1002/jssc.201100256. [DOI] [PubMed] [Google Scholar]
- 75.Zhang L, Xu J, Sun L, Ma J, Yang K, Liang Z, Zhang L, Zhang Y. Anal Bioanal Chem. 2011;399:3399–3405. doi: 10.1007/s00216-011-4657-4. [DOI] [PubMed] [Google Scholar]
- 76.Zhang L, Liang Z, Yang K, Xia S, Wu Q, Zhang L, Zhang Y. Analytica chimica acta. 2012;729:26–35. doi: 10.1016/j.aca.2012.04.005. [DOI] [PubMed] [Google Scholar]
- 77.Ji L, Wu JH, Luo Q, Li X, Zheng W, Zhai G, Wang F, Lu S, Feng YQ, Liu J, Xiong S. Analytical chemistry. 2012;84:2284–2291. doi: 10.1021/ac202897u. [DOI] [PubMed] [Google Scholar]
- 78.Lu J, Deng C, Zhang X, Yang P. ACS applied materials & interfaces. 2013;5:7330–7334. doi: 10.1021/am401662b. [DOI] [PubMed] [Google Scholar]
- 79.Zeng YY, Chen HJ, Shiau KJ, Hung SU, Wang YS, Wu CC. Proteomics. 2012;12:380–390. doi: 10.1002/pmic.201000726. [DOI] [PubMed] [Google Scholar]
- 80.Eriksson A, Bergquist J, Edwards K, Hagfeldt A, Malmstrom D, Hernandez VA. Analytical chemistry. 2011;83:761–766. doi: 10.1021/ac1027879. [DOI] [PubMed] [Google Scholar]
- 81.Wang H, Duan J, Cheng Q. Analytical chemistry. 2011;83:1624–1631. doi: 10.1021/ac1024232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bi H, Qiao L, Busnel JM, Devaud V, Liu B, Girault HH. Analytical chemistry. 2009;81:1177–1183. doi: 10.1021/ac8024448. [DOI] [PubMed] [Google Scholar]
- 83.Larsen MR, Thingholm TE, Jensen ON, Roepstorff P, Jorgensen TJ. Molecular & cellular proteomics. 2005;4:873–886. doi: 10.1074/mcp.T500007-MCP200. [DOI] [PubMed] [Google Scholar]
- 84.Wu J, Shakey Q, Liu W, Schuller A, Follettie MT. Journal of proteome research. 2007;6:4684–4689. doi: 10.1021/pr070481m. [DOI] [PubMed] [Google Scholar]
- 85.Sugiyama N, Masuda T, Shinoda K, Nakamura A, Tomita M, Ishihama Y. Molecular & cellular proteomics. 2007;6:1103–1109. doi: 10.1074/mcp.T600060-MCP200. [DOI] [PubMed] [Google Scholar]
- 86.Fukuda I, Hirabayashi-Ishioka Y, Sakikawa I, Ota T, Yokoyama M, Uchiumi T, Morita A. Journal of proteome research. 2013;12:5587–5597. doi: 10.1021/pr400546u. [DOI] [PubMed] [Google Scholar]
- 87.Kanshin E, Michnick SW, Thibault P. Journal of proteome research. 2013;12:2905–2913. doi: 10.1021/pr400198e. [DOI] [PubMed] [Google Scholar]
- 88.Li QR, Ning ZB, Tang JS, Nie S, Zeng R. Journal of proteome research. 2009;8:5375–5381. doi: 10.1021/pr900659n. [DOI] [PubMed] [Google Scholar]
- 89.Park SS, Maudsley S. Analytical biochemistry. 2011;409:81–88. doi: 10.1016/j.ab.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao X, Wang Q, Wang S, Zou X, An M, Zhang X, Ji J. Journal of proteome research. 2013;12:2467–2476. doi: 10.1021/pr301061q. [DOI] [PubMed] [Google Scholar]
- 91.Hennrich ML, van den Toorn HW, Groenewold V, Heck AJ, Mohammed S. Analytical chemistry. 2012;84:1804–1808. doi: 10.1021/ac203303t. [DOI] [PubMed] [Google Scholar]
- 92.Dai J, Wang LS, Wu YB, Sheng QH, Wu JR, Shieh CH, Zeng R. Journal of proteome research. 2009;8:133–141. doi: 10.1021/pr800381w. [DOI] [PubMed] [Google Scholar]
- 93.Dong M, Ye M, Cheng K, Song C, Pan Y, Wang C, Bian Y, Zou H. Journal of proteome research. 2012;11:4673–4681. doi: 10.1021/pr300503z. [DOI] [PubMed] [Google Scholar]
- 94.Hennrich ML, Groenewold V, Kops GJ, Heck AJ, Mohammed S. Analytical chemistry. 2011;83:7137–7143. doi: 10.1021/ac2015068. [DOI] [PubMed] [Google Scholar]
- 95.Gilar M, Olivova P, Daly AE, Gebler JC. Analytical chemistry. 2005;77:6426–6434. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- 96.Alpert AJ. Journal of chromatography. 1990;499:177–196. doi: 10.1016/s0021-9673(00)96972-3. [DOI] [PubMed] [Google Scholar]
- 97.McNulty DE, Annan RS. Molecular & cellular proteomics. 2008;7:971–980. doi: 10.1074/mcp.M700543-MCP200. [DOI] [PubMed] [Google Scholar]
- 98.Engholm-Keller K, Hansen TA, Palmisano G, Larsen MR. Journal of proteome research. 2011;10:5383–5397. doi: 10.1021/pr200641x. [DOI] [PubMed] [Google Scholar]
- 99.Alpert AJ. Analytical chemistry. 2008;80:62–76. doi: 10.1021/ac070997p. [DOI] [PubMed] [Google Scholar]
- 100.Zarei M, Sprenger A, Gretzmeier C, Dengjel J. Journal of proteome research. 2012;11:4269–4276. doi: 10.1021/pr300375d. [DOI] [PubMed] [Google Scholar]
- 101.Zarei M, Sprenger A, Gretzmeier C, Dengjel J. Journal of proteome research. 2013;12:5989–5995. doi: 10.1021/pr4007969. [DOI] [PubMed] [Google Scholar]
- 102.Delmotte N, Lasaosa M, Tholey A, Heinzle E, Huber CG. Journal of proteome research. 2007;6:4363–4373. doi: 10.1021/pr070424t. [DOI] [PubMed] [Google Scholar]
- 103.Song C, Ye M, Han G, Jiang X, Wang F, Yu Z, Chen R, Zou H. Analytical chemistry. 2010;82:53–56. doi: 10.1021/ac9023044. [DOI] [PubMed] [Google Scholar]
- 104.Yue XS, Hummon AB. Journal of proteome research. 2013;12:4176–4186. doi: 10.1021/pr4005234. [DOI] [PubMed] [Google Scholar]
- 105.Villen J, Gygi SP. Nature protocols. 2008;3:1630–1638. doi: 10.1038/nprot.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zhong X, Zhang Z, Jiang S, Li L. Electrophoresis. 2013 doi: 10.1002/elps.201300451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Heemskerk AA, Busnel JM, Schoenmaker B, Derks RJ, Klychnikov O, Hensbergen PJ, Deelder AM, Mayboroda OA. Analytical chemistry. 2012;84:4552–4559. doi: 10.1021/ac300641x. [DOI] [PubMed] [Google Scholar]
- 108.Sarg B, Faserl K, Kremser L, Halfinger B, Sebastiano R, Lindner HH. Molecular & cellular proteomics. 2013;12:2640–2656. doi: 10.1074/mcp.M112.024109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bachmann S, Bakry R, Huck CW, Polato F, Corradini D, Bonn GK. Electrophoresis. 2011;32:2830–2839. doi: 10.1002/elps.201000653. [DOI] [PubMed] [Google Scholar]
- 110.Dong YM, Chien KY, Chen JT, Lin SJ, Wang TC, Yu JS. Journal of separation science. 2013;36:1582–1589. doi: 10.1002/jssc.201300054. [DOI] [PubMed] [Google Scholar]
- 111.Zhou H, Di Palma S, Preisinger C, Peng M, Polat AN, Heck AJ, Mohammed S. Journal of proteome research. 2013;12:260–271. doi: 10.1021/pr300630k. [DOI] [PubMed] [Google Scholar]
- 112.Tobe BT, Hou J, Crain AM, Singec I, Snyder EY, Brill LM. Stem cell reviews. 2012;8:16–31. doi: 10.1007/s12015-011-9317-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Christensen GL, Kelstrup CD, Lyngso C, Sarwar U, Bogebo R, Sheikh SP, Gammeltoft S, Olsen JV, Hansen JL. Molecular & cellular proteomics. 2010;9:1540–1553. doi: 10.1074/mcp.M900550-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Palmisano G, Parker BL, Engholm-Keller K, Lendal SE, Kulej K, Schulz M, Schwammle V, Graham ME, Saxtorph H, Cordwell SJ, Larsen MR. Molecular & cellular proteomics. 2012;11:1191–1202. doi: 10.1074/mcp.M112.017509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Marcilla M, Alpizar A, Lombardia M, Ramos-Fernandez A, Ramos M, Albar JP. Molecular & cellular proteomics. 2013 doi: 10.1074/mcp.M113.034314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Singh P, Gan CS, Guo T, Phang HQ, Sze SK, Koh CG. Proteomics. 2011;11:2891–2900. doi: 10.1002/pmic.201100044. [DOI] [PubMed] [Google Scholar]