

Abstract
The secreted peptide hormone hepcidin regulates systemic and local iron homeostasis through degradation of the iron exporter ferroportin. Dysregulation of hepcidin leads to altered iron homeostasis and development of pathological disorders including hemochromatosis, and iron loading and iron restrictive anemias. Therapeutic modulation of hepcidin is a promising method to ameliorate these conditions. Several approaches have been taken to enhance or reduce the effects of hepcidin in vitro and in vivo. Based on these approaches, hepcidin modulating drugs have been developed and are undergoing clinical evaluation. In this article we review the rationale for development of these drugs, the data concerning their safety and efficacy, their therapeutic uses, and potential future prospects.
Keywords: Hepcidin, ferroportin, anemia of chronic disease, iron overload, iron deficiency, clinical trials
Overview
Iron is an essential nutrient that functions as a cofactor in many proteins and enzymes necessary for cell growth and maintenance.[1] However, excess iron can be cytotoxic due to its ability to contribute to free radical-generating reactions.[2] Therefore, iron homeostasis is carefully maintained. Iron from the diet is absorbed by duodenal enterocytes and is effluxed to the circulation through ferroportin (FPN), the only known iron pump in mammalian cells.[3] Iron is then loaded onto a carrier protein, transferrin (TF), for circulation and delivery to peripheral tissues. [4] Transferrin receptor 1 (TFR1), which is displayed on most cells, binds diferric TF, and the complex of diferric TF and TFR1 is endocytosed. Following endocytosis, iron is released from the TF/TFR1 complex for intracellular use and the apo-TF/TFR1 complex recycles back to the cell surface to participate in multiple rounds of iron delivery. The bone marrow, in particular, requires a large amount of iron to maintain erythropoiesis (red blood cell (RBC) production). During erythropoiesis, iron is incorporated into hemoglobin, an oxygen transport metalloprotein that is the major constituent of RBCs. Senescent RBCs are catabolized by macrophages of the reticuloendothelial system, which release iron from hemoglobin. Iron is exported from macrophages by FPN, loaded onto TF, and reenters the circulation for further utilization. An overview of systemic iron trafficking and regulation is provided in Figure 1.
Figure 1.
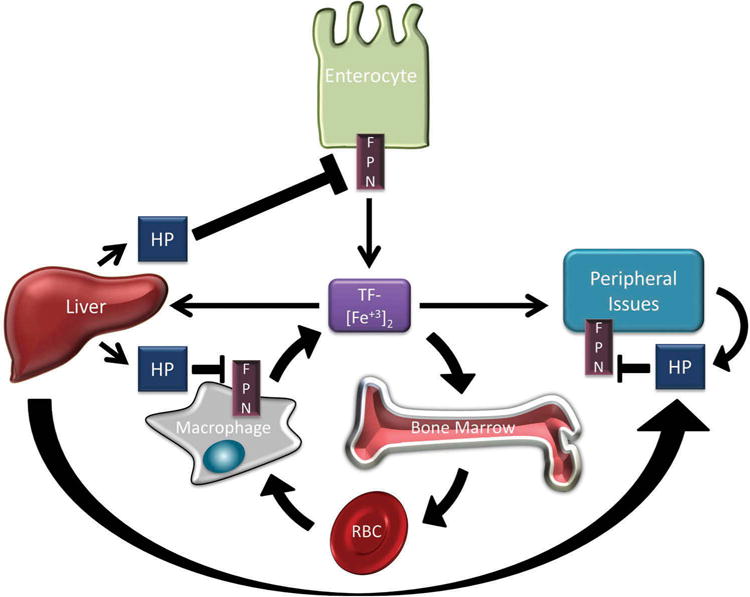
Overview of iron trafficking and regulation. Dietary iron is absorbed by duodenal enterocytes and exported into circulation by the iron export protein ferroportin (FPN). In circulation, iron is loaded onto transferrin (TF-[Fe+3]2) for delivery to peripheral tissues. Large amounts of iron are transported to the bone marrow for incorporation into hemoglobin during red blood cell (RBC) synthesis. Macrophages recycle iron from senescent RBCs and efflux iron back into circulation through FPN. Hepcidin (HP) is secreted peptide produced predominantly in the liver but also by cells in peripheral tissues. HP binds to FPN and targets it for degradation. HP secreted by the liver regulates systemic iron homeostasis by reducing iron absorption from the gut, impairing iron recycling by macrophages, and by reducing the release of iron from peripheral tissues. HP is secreted by peripheral tissues at lower concentrations and predominantly regulates local iron homeostasis.
As there is no iron excretory pathway, intake of iron must be tightly regulated. Hepcidin, a secreted peptide hormone produced predominantly by the liver, is the master regulator of iron intake and systemic iron homeostasis. It was first discovered by independent groups in 2000 and 2001 as a peptide with antimicrobial activity. Soon after, hepcidin was determined to play a central role in maintaining systemic iron levels and controlling iron storage in macrophages.[5–8] Encoded by the HAMP gene, human hepcidin is synthesized in the liver as an 84 amino acid prepropeptide that contains a typical N-terminal endoplasmic reticulum targeting sequence, as well as a C-terminal consensus furin cleavage site.[6] Upon cleavage at both sites, hepcidin is secreted into the circulation as a 25 amino acid bioactive peptide hormone.[6,7,9]
Hepcidin regulates body iron by binding to FPN, causing the internalization and subsequent degradation of hepcidin and FPN in the lysosome.[10,11] FPN expression is most prominent on the surface of enterocytes and macrophages due to their respective roles in uptake of dietary iron and iron recycling (Figure 1).[12] When systemic iron levels are high, hepcidin is produced and targets FPN for degradation, reducing the release of iron into the plasma. When systemic iron levels are low, hepcidin decreases, and more iron is released into the plasma through FPN. In both circumstances, manipulation of hepcidin restores normal serum iron levels. Thus, hepcidin and FPN constitute a regulatory axis that is essential for the maintenance of systemic iron homeostasis. In addition to its role in controlling systemic iron metabolism, the FPN/hepcidin regulatory axis is also operant in peripheral tissues, such as the breast, prostate, and brain [13–15] Hepcidin is secreted from cells within these tissues and acts locally to reduce iron export from cells, again by degrading FPN (Figure 1). Therefore, the FPN/hepcidin regulatory axis allows precise control of iron at both the systemic as well as the cellular level. Disrupted regulation of this axis can result in pathological elevation or reduction of iron levels.
Disordered hepcidin & disease
Hepcidin deficiency
Deficiency in hepcidin production is the main cause of iron overload. When hepcidin is deficient, intestinal absorption of iron is inappropriately high leading to elevated serum iron levels and iron overload or hemochromatosis. Most iron overload disorders are inherited, including hereditary hemochromatosis (HH). Mutations that lead to HH have been identified in many of the genes that regulate hepcidin, as well as the HAMP gene itself, and the severity of iron overload depends on the gene mutated.[16] Less common mutations, such as a mutation in FPN that renders it insensitive to hepcidin degradation, can also lead to HH.[17] Current treatment options for patients with HH are limited to phlebotomy to deplete excess iron from circulation. However, phlebotomy also removes hepcidin, increasing iron absorption and creating a cycle that demands careful monitoring and repeated treatment.[18] Stimulation of hepcidin production may prove to be an alternative or additional therapy for some patients with iron overload.
Deficiency in hepcidin is also associated with iron loading anemias, such as β-thalassemia and congenital dyserythropoietic anemia, in which iron overload and anemia can present within the same disease.[19,20] These complex diseases are characterized by ineffective erythropoiesis, due to premature apoptosis of erythroid precursors before maturation into erythrocytes.[21] A lack of mature erythrocytes leads to anemia and an increase in erythropoietin production in attempt to increase erythropoiesis. Under these conditions, hepcidin synthesis is repressed by factors secreted by erythroid precursors: likely erythreferrone and/or growth differentiation factor 15 (GDF15).[22,23] Similar to HH, low hepcidin levels in turn result in excessive iron absorption and an iron overload phenotype.[24] Treatments of these patients include transfusions and/or administration of iron chelators. In transfused patients, anemia is moderately corrected, and due to reduced erythropoiesis, hepcidin levels can be partially restored. Iron overload can be corrected by iron chelator treatment; however, many adverse side effects can arise. Thus, hepcidin agonists may also be beneficial in correcting these complex set of diseases.
Hepcidin excess
In contrast to iron loading anemias, iron restrictive anemias may result from excess hepcidin production. The most common cause of abnormally elevated hepcidin is inflammation associated with chronic disease, which can result in development of anemia of chronic disease (ACD). ACD is observed in a wide range of chronic diseases including infection, kidney disease, inflammatory disorders, trauma, malignancy, and rheumatologic diseases.[25] Treatment of the underlying disorder is the preferred treatment for ACD, but this is not always feasible. In addition, two rare causes of increased hepcidin production are hepcidin secreting hepatomas [26] and iron-refractory iron deficiency anemia (IRIDA). IRIDA is characterized by resistance to oral iron treatment and is caused by hereditary mutations in the hepcidin inhibitor TMPRSS6.[27]The prevalence and morbidity of anemia associated with hepcidin excess has driven the development of hepcidin antagonists.
Pharmacological considerations
Successful therapeutic manipulation of hepcidin is dependent on knowledge of its pharmacologic and pharmacokinetic properties and, most importantly, the ability to reproducibly and reliably measure hepcidin levels. Methods to detect hepcidin have been difficult to develop. Early studies detected urinary secreted hepcidin by cation-exchange chromatography and subsequent immunodots, a technique that was limited by uncertainty as to whether excreted hepcidin correlated with plasma hepcidin levels. [28] Mass spectrometry (MS) methods, including time-of-flight MS and high performance liquid chromatography-tandem MS, have also been used for the detection of bioactive hepcidin,[29–31] and recently, a liquid chromatography tandem–mass spectrometry (LC-MS/MS) method was validated as a reliable method for quantifying hepcidin-25 in human urine and serum.[32,33] In addition to MS methods, the first serum ELISA assay for hepcidin was developed and validated in 2008.[34] It is currently the most frequently used assay for the measurement of hepcidin in serum. Based on this assay, normal physiologic levels of serum hepcidin range from 29 to 254 ng/mL in men and 17 to 286 ng/mL in women (with median concentrations 112 ng/mL versus 65 ng/mL, respectively).[34] However, it is important to note that current hepcidin ELISA assays utilize antibodies that are unable to distinguish between the bioactive peptide hepcidin-25 and smaller isoforms of hepcidin whose significance is still uncertain (hepcidin-20 and −22).[35,36] Although dramatic progress has been achieved in hepcidin detection, improvements in detection limits and assay validation are still needed, as large discrepancies remain between detection methods.[37] In addition to detection limitations, studies in non-human primates have revealed that hepcidin has a short half-life of only several minutes.[38] Hepcidin is rapidly cleared in the kidney, as its small size allows it to pass through the glomerular membrane where it is subsequently degraded in the proximal tubule.[39]
Regulation of hepcidin
Regulation of hepcidin synthesis has been extensively studied, and both positive and negative regulators have been identified. A brief overview of pathways that regulate hepcidin, as well as therapeutics targeting these pathways, is provided in Figure 3.
Figure 3.
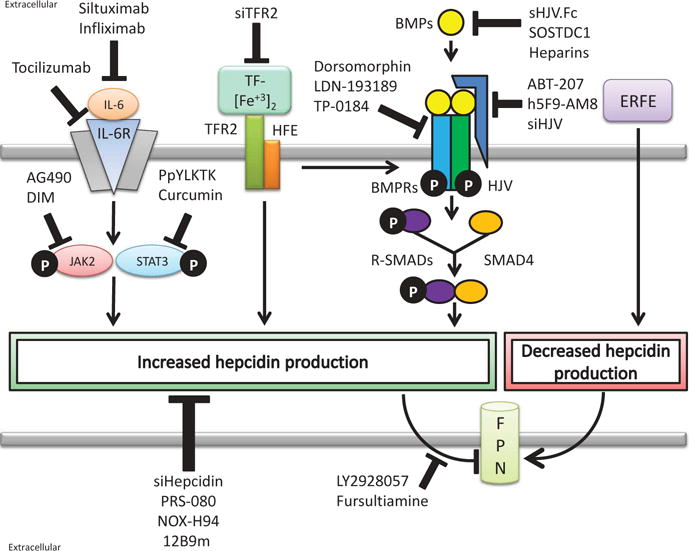
Overview of hepcidin antagonists. Hepcidin antagonists reduce hepcidin production or inhibit hepcidin function. The interleukin-6 (IL-6), iron sensing, and bone morphogenetic protein (BMP) pathways increase hepcidin production, while erythroferrone (ERFE) decreases hepcidin production. In the IL-6 pathway, secreted IL-6 binds the IL-6 receptor (IL-6R), activating janus kinase 2 (JAK2) and subsequently signal transducers and activators of transcription 3 (STAT3). STAT3 binds to a STAT3 response element in the hepcidin promoter to stimulate hepcidin synthesis. Targeting the IL-6 pathway, siltuximab and infliximab reduce IL-6 availability while tocilizumab interferes with IL-6R binding. Downstream, JAK2 inhibitors AG490 and 3,3′-Diindolylmethane (DIM), and STAT3 inhibitors PpYLKTK and curcumin, also interfere with IL-6-stimulated hepcidin synthesis. In the iron sensing pathway, diferric transferrin (TF-[Fe+3]2) binds to transferrin receptor 2 (TFR2) and associated human hemochromatosis protein (HFE) to increase hepcidin synthesis. Small interfering RNA targeting TFR2 (siTFR2) reduces hepcidin production by the iron sensing pathway. Downstream signaling of the iron sensing pathway is yet to be worked out but may involve stimulation of the BMP pathway. In the BMP signaling pathway, secreted BMPs bind to BMP receptors (BMPRs) activating intracellular receptor SMADs (R-SMADs) and SMAD4. SMADs bind to BMP-responsive elements in the hepcidin promoter stimulating hepcidin synthesis. BMP signaling can be enhanced by the hemojuvelin (HJV) coreceptor. A soluble HJV conjugate (sHJV.Fc), SOSTDC1, and heparins reduce hepcidin expression by sequestering BMPs. Dorsomophin, LDN-193189, and TP-0184 interfere with BMP receptors while small interfering RNA against HJV (siHJV) and HJV targeted antibodies (ABT-207 and h5F9-AM8) also reduce BMP signaling. Finally, several hepcidin antagonists interfere with hepcidin-FPN interactions. Small interfering RNA against hepcidin (siHepcidin), PRS-080, NOX-H94, and 12B9m specifically target hepcidin, while LY2928057 and fursultiamine prevent hepcidin binding to FPN.
Positive regulation
When plasma iron levels are high, hepatic hepcidin synthesis is elevated to reduce further iron absorption (Figure 1). To maintain this homeostatic loop, hepcidin is positively regulated by iron itself via a pathway that involves bone morphogenetic proteins (BMPs) and SMADs (verterbrate homologs of Sma and mothers against decapentaplegic) (Figure 3). BMPs are a subfamily of the transforming growth factor beta (TGF-β) superfamily of growth factors that control a wide range of biological functions including bone formation, embryonic neural crest organization, and post-natal tissue maintenance.[40] BMPs utilize the intracellular transducing SMAD molecules to activate their downstream target genes.[41] Aside from its function in controlling embryonic development and cell maintenance, BMP/SMAD signaling was recently identified to control iron homeostasis. A mouse hepatocyte-specific deletion of Smad4 resulted in a severe iron overload phenotype. [42] Interestingly, there was a 100-fold reduction in hepcidin mRNA in the livers of the same Smad4-deficient mice, revealing Smad4 as a potential transcriptional regulator of hepcidin. Subsequent studies confirmed this hypothesis. It is postulated that BMP6 is the main ligand that positively regulates hepcidin transcription in vivo, via a signaling pathway controlled by plasma iron.[43] BMP6-deficient mice exhibit extreme iron overload due to reduced hepcidin synthesis.[44] Others have shown that additional BMPs and other ligands of the TGFβ family can also positively regulate hepcidin in hepatocytes as well as other cell types.[14,45] However, BMP6 is believed to be the main ligand responsible for hepcidin production and maintaining normal body iron levels in vivo.
In recent years, other members of the BMP signaling pathway have been shown to further control hepcidin production, as inferred from mutational analysis in HH patients. Hemojuvelin (HJV) was found to be a critical BMP co-receptor that precisely controls hepcidin expression.[46] Mutations in HJV were identified in humans that develop juvenile hemochromatosis, one of the most severe forms of iron overload.[47,48] These patients were found to have extremely low levels of hepcidin, revealing HJV as an important regulator of hepcidin. Additionally, it has been postulated that interactions among hepatic TFR1, transferrin receptor 2 (TFR2), and human hemochromatosis protein (HFE), serve as the main sensors of plasma iron that result in BMP-SMAD activation and subsequent hepcidin production (Figure 3). Mutations in TFR2 or HFE result in partial hepcidin insufficiency, leading to a less severe form of HH.[49,50] It is thought that increasing concentrations of holo-transferrin results in the transfer of HFE from TFR1 to TFR2, stabilizing TFR2 and subsequently activating the downstream SMAD pathway.[51] However, the exact molecular pathways that connect HFE-TFR2 and BMP-SMAD signaling have yet to be understood.
Aside from control by iron itself, hepcidin is also regulated by inflammation. Interestingly, hepcidin was first described as an antimicrobial peptide, and it is thought that regulation of hepcidin by inflammation has evolved as a protective mechanism to reduce iron availability for microbes during host infection.[5,52] It has been widely observed that patients with chronic inflammation have reduced serum iron levels, which can result in ACD. A direct connection between inflammation and hepcidin was first shown in studies demonstrating that treatment of hepatocytes with lipopolysaccharide (LPS) resulted in an increase of hepcidin transcription.[7] It was later determined that inflammatory regulation of hepcidin was specific for type II acute phase cytokines, particularly Interleukin-6 (IL-6), and not type I cytokines.[53] Specifically, IL-6 activates Janus kinase 2 (JAK2) and downstream signal transducers and activators of transcription 3 (STAT3) (Figure 3). STAT3 translocates to the nucleus where it binds to a STAT3 response element on the hepcidin promoter, increasing hepcidin transcription.[54] IL-6 secreted during inflammation [55] is likely the major cause of elevated hepcidin in ACD [28] although BMPs seem to play a role as well.[43,56–59] Interestingly, cross-talk between the IL-6 and BMP signaling pathways may facilitate hepcidin induction.[60]
Negative regulation
In times when additional iron is needed in circulation, hepcidin production must be suppressed. One such instance is during stress erythropoiesis, such as that triggered by hemorrhage, hemolysis, and other stresses. Initially, it was postulated that erythropoietin (EPO), the main hormone that mediates erythropoiesis, directly suppressed hepcidin synthesis. Treatment of mouse and human hepatocytes as well as human subjects with EPO resulted in a striking reduction of hepcidin transcription and circulating levels, respectively.[61,62] However, additional studies investigating direct regulation of hepcidin by EPO yielded negative results, suggesting that EPO is not the “erythroid factor” that negatively regulates hepcidin. A recently discovered hormone, erythroferrone (ERFE), may represent a major contributor to hepcidin suppression.[23] ERFE is produced by erythroblasts in response to erythropoietin and was found to directly suppress hepcidin production, allowing release of iron into circulation to meet the demands of erythropoiesis. This negative regulation of hepcidin by ERFE seems to be induced at times when new red blood cell formation is crucial, such as hemorrhage, or during disease states, such as β-thalassemia, where hepcidin levels are extremely low.[23] Additional studies are needed to identify the molecular mechanisms of hepcidin repression by ERFE and whether additional erythroid regulators exist.
Parallel to erythropoietic control of hepcidin, low oxygen concentrations also lead to reduced hepcidin production. It is known that low oxygen conditions stimulate the production of RBC, which results in increased demand for iron.[63] Hepcidin expression must be repressed to achieve this increase in iron. The molecular connection between hepcidin and hypoxia was first observed when mice with hepatocyte deletion of Hif1α did not show a decrease in hepcidin after exposure to hypoxia or iron deficiency.[64] The same study went on to confirm that Hif1α binds to hypoxia responsive elements (HREs) on the HAMP promoter and, thus, directly represses hepcidin transcription in both human HEK293 cells as well as mouse liver tissue. However, additional studies have suggested that regulation of hepcidin by hypoxia involves solely Hif2α or that regulation of hepcidin by HIFs is indirect.[65,66] Others believe that hypoxia may increase levels of furin, the enzyme that can cleave HJV to soluble HJV (sHJV), which in turn can sequester BMPs rendering them unable to activate the BMP-SMAD pathway for induction of hepcidin.[67] Overall, the hypoxia-induced molecular players and whether they directly or indirectly regulate hepcidin still remains to be elucidated.
Another means to negatively regulate hepcidin is by inhibiting its positive regulators. Some of these regulators have been identified serendipitously through the discovery of mutations in genes that lead to an altered iron phenotype. One such instance is mutations in the TMPRSS6 gene which encodes a type II transmembrane serine protease, known as matriptase-2. TMPRSS6 mutations result in elevated hepcidin and severe anemia that is not responsive to therapy.[27] In vitro, matriptase-2 was observed to cleave HJV, the BMP co-receptor, reducing downstream signaling and induction of hepcidin.[68] Thus, matriptase-2 is believed to be a negative regulator of hepcidin (Figure 2). As mentioned above, soluble HJV is formed by furin cleavage of membrane HJV, and HJV can thus transition from a positive regulator of hepcidin to a negative regulator once cleaved.[69] This represents a sophisticated level of hepcidin regulation by utilization of either membrane or soluble HJV, depending on the amount of iron desired. Another BMP antagonist, known as Sclerostin domain containing 1 protein (SOSTDC1), has also been shown to negatively regulate hepcidin in prostate cancer cells.[14] SOSTDC1 antagonizes BMP signaling by sequestering BMP ligands, similar to sHJV (Figure 3).[70] Thus, by sequestering the BMPs, activation of downstream signaling for induction of hepcidin is compromised.
Figure 2.
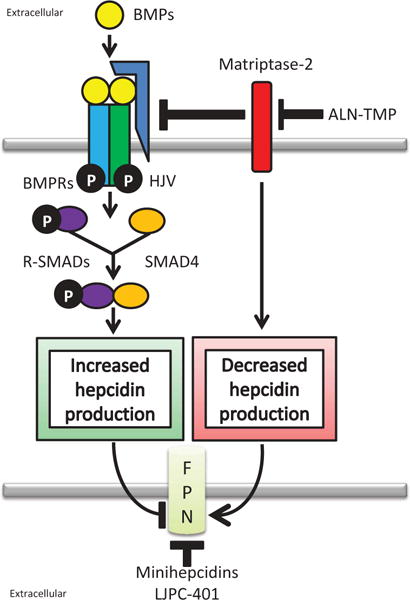
Overview of hepcidin agonists. The bone morphogenetic protein (BMP) pathway stimulates hepcidin production. Secreted BMPs bind to BMP receptors (BMPRs) activating intracellular receptor SMADs (R-SMADs) and SMAD4. SMADs bind to BMP-responsive elements in the hepcidin promoter stimulating hepcidin synthesis. BMP signaling is enhanced by the hemojuvelin (HJV) coreceptor or repressed by matriptase-2 which cleaves HJV and decreases hepcidin production. By regulating hepcidin production, modulation of BMP signaling can regulate the amount of ferroportin (FPN) degraded. Hepcidin agonists aim to increase hepcidin production and reduce FPN. Hepcidin mimetics, such as minihepcidins and LJPC-401, mimic hepcidin while other agonists target the BMP signaling pathways. ALN-TMP increases hepcidin synthesis by enhancing BMP signaling through inhibition of matripase-2. Administration of BMPs, particularly BMP6, also stimulate hepcidin production.
Additional regulators
In recent years, additional regulators of hepcidin have emerged, including hormones, growth factors, and other signaling molecules. Hormonal control of hepcidin was suspected based on the need for additional iron during menstruation. This was confirmed, as treatment of human liver cell lines with 17β-estradiol resulted in reduced hepcidin transcription.[71] Ultimately, a functional estrogen responsive element within the HAMP promoter was detected.[71] Thus, high levels of estrogen just before menstruation negatively regulate hepcidin, resulting in increased iron in circulation to compensate for elimination of iron through blood loss. However, additional studies contradicted negative regulation of hepcidin by estrogen and concluded that estrogen actually positively regulates hepcidin, via GPR30-BMP6-dependent mechanism.[72] Testosterone has also been shown to inhibit hepcidin transcription in mice.[73] Clarification of hormonal control of hepcidin is necessary to understand the pathways involved and rationalization of such regulation.
In addition to BMPs, other ligands, such as growth factors, have been shown to regulate hepcidin. GDF15 was identified by microarray as a regulator of erythroid maturation, and it was found that high levels of GDF15, comparable to those found in thalassemia patients, suppress hepcidin expression.[22] Although this suggests that GDF15 negatively regulates hepcidin, others have shown a positive correlation between levels of circulating GDF15 and hepcidin in a variety of cancer-related anemias.[74,75] Regulation of hepcidin by GDF15 seems to depend greatly on GDF15 concentration. However, more investigation into whether this relationship goes beyond a correlative one is necessary to understand the connection between GDF15 and hepcidin. Other growth factors, including epidermal growth factor (EGF) and hepatocyte growth factor (HGF), have been shown to decrease hepcidin transcription by inhibiting the BMP-SMAD pathway, specifically by inhibiting translocation of SMAD-1,5,8 to the nucleus for induction of hepcidin.[76]
Novel signaling pathways are also being revealed in regulation of hepcidin. Recently, our group demonstrated that hepcidin can be regulated by Wnt signaling in prostate cancer cells.[14] SOSTDC1, which antagonizes BMP signaling, can also antagonize Wnt signaling by binding to the Wnt receptor LRP5/6.[77] Treatment of prostate cancer cells with LiCl, an activator of Wnt signaling, increased hepcidin expression.[14] On the other hand, inhibition of Wnt signaling resulted in marked reduction of hepcidin production. Further investigation led to the discovery of a Wnt transcription factor responsive element, known as TCF/LEF in the hepcidin promoter that directly controls hepcidin transcription. It would be interesting to determine if this positive regulation of hepcidin by Wnt signaling is specific to cancer cells or if it is a more general regulatory mechanism. Other pathways that may regulate hepcidin include the mammalian target of rapamycin (mTOR), mitogen-activated protein kinase (MAPK) as well as the rapidly accelerated fibrosarcoma (Ras-RAF) pathways, all of which were identified by an RNAi screen that monitors hepcidin promoter activity after gene knockdown.[78] Verification of these pathways and their involvement in hepcidin regulation are ongoing.
Hepcidin therapeutic intervention
A number of hepcidin agonists and antagonists are currently being developed. Currently, hepcidin agonists are either hepcidin mimetics or stimulators of hepcidin production. Hepcidin production may be stimulated by enhancing positive regulators of hepcidin or by inhibiting negative regulators. In contrast, the antagonists take two main approaches: inhibiting hepcidin production and disrupting hepcidin function. Inhibiting hepcidin production is achieved by inhibiting positive regulators of hepcidin or enhancing negative regulators. Disruption of hepcidin function is accomplished by directly antagonizing hepcidin or interfering with the hepcidin-FPN interaction. An overview of targets for hepcidin agonists and antagonists is shown in Figures 2 and 3, and specific pharmacological entities are further described in Tables 1 and 2.
Table 1.
Hepcidin agonists.
| Mechanism of action | Drug | Target | Evidence | Clinical stage | Ref. | Implicated disorders |
|---|---|---|---|---|---|---|
| Hepcidin mimetics | Minihepcidins | FPN | Cell culture and animal models | Preclinical | [79–81] | Hereditary hemochromatosis; iron loading anemias |
| LJPC-401 | FPN | Animal models | Preclinical | [82,83] | ||
| Enhance positive regulators | BMP6 administration | BMP pathway | Cell culture and animal models | Preclinical | [84] | |
| Inhibit negative regulators | ALN-TMP | TMPRSS6 inhibitor | Cell culture and animal models | Preclinical | [85] |
Table 2.
Hepcidin antagonists.
| Mechanism of action | Drug | Target | Evidence | Clinical stage | Ref. | Implicated disorders |
|---|---|---|---|---|---|---|
| SOSTDC1 | BMP pathway | Cell culture | Preclinical | [14] | Anemia of chronic disease; iron refractory iron-deficient anemia | |
| Heparins | BMP pathway | Cell culture, animal models, patients | FDA-approved anti-coagulant | [59,86–90] | ||
| sHJV.Fc (FMX-8) | BMP pathway | Cell culture and animal models | Clinical trials terminated | [43,56,58,67,91,92] | ||
| Dorsomorphin\LDN-193189 | BMP pathway | Cell culture and animal models | Preclinical; orphan drug designation | [58,93–97] | ||
| TP-0184 | BMP pathway | Cell culture and animal models | Preclinical | [57,98] | ||
| ABT-207 | BMP pathway | Animal models including nonhuman primates | Preclinical | [99] | ||
| h5F9-AM8 | BMP pathway | Animal models | Preclinical | [99] | ||
| XEN701 | BMP pathway | None | Preclinical/discontinued | [100,101] | ||
| Inhibit positive regulators | siHJV | BMP pathway | Cell culture and animal models | Preclinical | [102] | |
| siTFR2 | Iron-sensing/BMP pathway | Cell culture and animal models | Preclinical | [102] | ||
| Siltuximab (Slyvant™) | IL-6 pathway | Patients | FDA approved for MCDa | [103–105] | ||
| Tocilizumab (ACTEMRA®) | IL-6 pathway | Patients | FDA approved for RA and JIAb | [106–108] | ||
| AG490 | IL-6 pathway | Cell culture and animal models | Preclinical | [109–112] | ||
| PpYLKTK | IL-6 pathway | Cell culture | Preclinical | [110,111] | ||
| Curcumin | IL-6 pathway | Cell culture and animal models | Phase 1/2/3/4 for several indications | [111,113–115] | ||
| 3,3′-Diindolyl-methane | IL-6 pathway | Rainbow trout model | Phase 1/2/3 for several indications | [116,117] | ||
| Infliximab (REMICADE®) | TNFα/IL-6 pathway | Cell culture and patients | FDA-approved | [107] | ||
| Enhance negative regulators | Erythroferrone | Erythroferrone | Cell culture and animal models | Preclinical | [23,118,119,120] | |
| siHepcidin | Hepcidin | Cell culture and animal models | Preclinical | [102] | ||
| 12B9m | Hepcidin | Cell culture and animal models including nonhuman primates | Preclinical | [121] | ||
| Disrupt hepcidin–ferroportin (FPN) interaction | PRS-080 | Hepcidin | Cell culture and animal models | Phase 1 | [122–124] | |
| NOX-H94 (Lexaptepid Pegol) | Hepcidin | Cell culture, nonhuman primates, patients | Phase 2 | [125–129] | ||
| LY2787106 | Hepcidin | None | Preclinical/discontinued | [130,131] | ||
| LY2928057 | FPN | Nonhuman primates | Phase 1 | [132,133] | ||
| Fursultiamine | FPN | Cell culture | Preclinical | [134] |
Multicentric Castleman’s Disease (MCD).
Rheumatoid (RA) and juvenile idiopathic arthritis (JIA).
Hepcidin agonists
Treatment options currently available for patients with iron overload are limited to phlebotomy to reduce the overall amount of iron from circulation and transfusion and iron chelation treatments for β-thalassemia and other iron loading anemias. An alternative approach to reducing body iron is to increase hepcidin production. Several classes of hepcidin agonists are beginning to emerge in hopes of successfully treating diseases of hepcidin deficiency.
The most direct approach to treat hepcidin deficiency is to utilize a hepcidin mimetic, including hepcidin itself (Figure 2). However, developing an active recombinant hepcidin has proven challenging. This is due to hepcidin-25’s small size, ability to aggregate, short half-life, as well as its cysteine rich and disulfide bond structure, making proper protein folding difficult. [135] To circumvent these issues, “minihepcidins” were developed.[79] These are small 7–9 amino acid peptides that mimic hepcidin function both in vitro and in vivo and were designed to retain the amino acids that were critical for Fpn binding.[79,80] Minihepcidins successfully prevented iron overload in mouse models of hemochromatosis and reduced basal iron levels in mice.[80] Currently, minihepcidins M009 and M012 are in preclinical development at Merganser Biotech.[81] In addition, La Jolla Pharmaceuticals Company has developed a novel formulation of hepcidin, LJPC-401, that has been accepted by the FDA as an Investigational New Drug.[82] LJPC-401 successfully reduced serum iron in rats,[83] and results from a Phase 1 clinical trial are expected by the end of 2015. The combination of synthetic hepcidins with existing therapies may improve treatment and quality of life for patients suffering from iron overload disorders.
The second approach to increase hepcidin production is to stimulate its positive regulators. For example, BMP6 is believed to be the main ligand responsible for induction of hepcidin in vivo. Twice daily administration of BMP6 increased hepcidin mRNA and corrected severe iron overload in HFE-deficient mice.[84] Although BMP2 and BMP7 are FDA approved for certain indications, the safety of BMP therapies is in question.[136] Therefore, it is unclear whether administration of BMPs will be clinically useful for modulation of hepcidin.
The third strategy that is being employed to increase hepcidin is to perturb negative regulators of hepcidin signaling, such as TMPRSS6. Homozygous deletion of TMPRSS6 in a mouse model of thalassemia increased hepcidin and improved anemia as well as erythropoiesis.[137] This study led to the development of novel therapies targeting TMPRSS6 for inactivation, and thus inducing hepcidin (Figure 2). Recently, researchers have successfully utilized RNA-based therapeutics for inhibiting TMPRSS6 in mice, resulting in increased hepcidin transcription and reduced iron levels in iron overload mice.[138–140] ALN-TMP is a TMPRSS6 inhibitor currently being developed for clinical use by Alnylam Pharmaceuticals, Inc.[85]
Hepcidin antagonists
Inhibiting BMP-mediated hepcidin production
BMPs are potent inducers of hepcidin production (Figure 3). BMP signaling inhibitors can directly sequester extracellular BMPs, prevent BMP receptor activation, or inhibit intracellular SMAD signaling. All of these approaches effectively interfere with hepcidin production.
BMP sequestration
Heparin is a glycosaminoglycan capable of directly binding to BMPs (Figure 3).[141,142] Repurposing heparin, an FDA-approved anticoagulant, is a promising approach to clinically reduce serum hepcidin. Commercially available heparin has been shown to inhibit hepcidin production in hepatocellular carcinoma cells in vitro.[59] Furthermore, heparin effectively reduced liver hepcidin in a mouse model and serum hepcidin in human patients by 15–20%.[59] While these findings are encouraging, the potency of heparin as an anticoagulant complicates its use to relieve hepcidin excess. Development of heparin as a hepcidin antagonist has therefore evolved to chemically modified heparins, termed “glycol-split” heparins (gs-heparin), which exhibit reduced anticoagulant ability.[86] Gs-heparins retain the ability to reduce hepcidin production in vitro and in vivo and ameliorated the anemia of inflammation in mice models induced by LPS and heat-killed Brucella abortus.[87] The main potential advantage of the reduced anticoagulant activity of gs-heparins is the ability to use higher doses in vivo. Dosing and safety of a gs-heparin, SST0001, is currently being assessed in a phase 1 clinical trial in multiple myeloma patients.[88–90] Positive data from this trial would be encouraging for further clinical development of gs-heparins for anemias associated with hepcidin overexpression.
Soluble hemojuvelin-Fc fusion protein (sHJV.Fc) is another BMP signaling inhibitor that directly binds BMPs (Figure 3).[56] sHJV.Fc is a fusion of the extracellular domain of the BMP coreceptor HJV and the Fc portion of human immunoglobulin G.[56] sHJV.Fc is a promising approach designed around sHJV which naturally modulates hepcidin levels in vivo (Figure 3).[67] In rodent models, sHJV.Fc was shown to significantly reduce hepcidin levels and correct anemia of inflammation.[43,56,58] Ferrumax Pharmaceuticals, Inc. initiated clinical trials for sHJV.Fc (FMX-8) in patients with renal disease-associated anemia; however, these studies were recently terminated due to an inability to recruit patients meeting the inclusion criteria.[91,92] Further clinical development of sHJV.Fc is unclear.
BMP receptor inhibitors
BMP-mediated hepcidin induction relies on a number of receptors and coreceptors (Figure 3). Dorsomorphin is a small molecule inhibitor that blocks SMAD activation by BMP type I receptors ALK2, ALK3, and ALK6.[93] LDN-193189, an optimized molecule derived from dorsomophin,[94] is a more potent inhibitor of BMP type I receptors (Figure 3). Both dorsomorphin and LDN-193189 reduced BMP and IL-6-mediated hepcidin transcription in vitro in either primary rat hepatocytes or human hepatoma cells.[58,95] in vivo, LDN-193189 reversed amemia of inflammation in rodent models induced by Group A Streptococcal Peptidoglycan-Polysaccharide,[58] IL-6, or turpentine.[95] LDN-193189 has not progressed to clinical trials, although La Jolla Pharmaceutical Company has received orphan drug designation for LDN-193189 (LJPC-6417) for the treatment of fibrodysplasia ossificans progressive.[96] Nonspecificity of LDN-193189 [97] may complicate its further development as a clinical modulator of hepcidin. Nevertheless, LDN-193189 provides proof-of-concept that BMP receptor inhibitors can modulate hepcidin and ameliorate anemia.
TP-0184 takes a similar approach as LDN-193189 to modulate hepcidin levels. TP-0184 is a small molecule inhibitor of the BMP type 1 receptor ALK2 (Figure 3).[57] TP-0184 is in preclinical development by Tolero Pharmaceuticals, Inc. and has been shown to downregulate hepcidin expression in BMP-induced cell culture models.[57] Furthermore, TP-0184 reduced hepcidin induction and elevated serum iron levels in turpentine and lung cancer mouse models of ACD. [57,98] According to Tolero Pharmaceuticals, Inc., TP-0184 demonstrates favorable pharmacokinetics and drug development potential.[98] The company intends to further develop TP-0184 for treatment of ACD.
BMP coreceptor inhibitors
BMP coreceptors, such as HJV, present an alternative approach to target BMP-mediated hepcidin production. Membrane-bound HJV enhances SMAD activation by BMP receptors and subsequent hepcidin transcription. [56]Two monoclonal antibodies targeting HJV, ABT-207 and h5F9-AM8, have been developed by AbbVie.[99] Both ABT-207 and h5F9-AM8 reduce hepcidin levels and increase serum iron in Sprague–Dawley (SD) rats. [99] In this study, single dose treatment with ABT-207 (200 mg/kg) and h5F9-AM8 (20 mg/kg) increased serum iron and reduced unsaturated iron binding capacity (UIBC) for 3 weeks and 6 weeks, respectively.[99] ABT-207 was further shown to increase serum iron and reduce UIBC in cynomologus monkeys.[99] In addition to efficacy, these antibodies exhibited favorable safety data and pharmacokinetics.[99] ABT-207 and h5F9-AM8 are therefore good candidates for further clinical development.
Xenon Pharmaceuticals Inc. initiated development of XEN701, a HJV-hepcidin pathway targeted antisense oligonucleotide, to antagonize hepcidin production. [100] However, development was discontinued due to discouraging preclinical data,[101] and the future of XEN701 is unclear. In a related approach, Alnylam Pharmaceuticals, Inc. is utilizing small interfering RNAs to inhibit HJV (siRNAs). Like antisense oligonucleotides, siRNAs aim to modulate gene expression by reducing HJV mRNA (Figure 3); the two methods have been compared previously.[143] In contrast to XEN701, siRNA targeting HJV (siHJV) has shown to be effective in reducing hepcidin expression and increasing serum TF saturation in a turpentine-induced anemia model. [102] Although toxicity and in vivo delivery are issues commonly associated with the siRNA approach, in vivo delivery is feasible to the liver,[144] the primary site of systemic hepcidin production.[7] In a related approach, Alnylam Pharmaceuticals, Inc. is utilizing a proprietary Enhanced Stabilization Chemistry (ESC)-GalNAc-conjugate system to deliver siRNA. (ESC)-GalNAc-conjugate delivery to the liver has been shown to be effective in preclinical [145] and clinical [146] studies. In addition to targeting HJV, Alnylam is developing several other siRNAs targeting hepcidin production: siRNAs targeting human hemochromatosis protein, BMP6, BMP receptor type 1, BMP receptor type 2, SMAD4, TFR2, hepcidin, and IL-6 receptor. Data suggest that siRNA directly targeting hepcidin and TfR2 are the most potent in reducing hecpidin levels and elevating serum iron.[102]
IL-6 signaling inhibitors
IL-6 signaling though JAK2 and STAT3 stimulates hepcidin production, particularly during inflammation. [54,55] Therapies targeting IL-6 have shown to be effective in reducing hepcidin levels and improving anemia. Siltuximab (Sylvant™) is a murine-human chimeric monoclonal antibody directed against IL-6 and is FDA-approved for use in multicentric Castleman’s Disease (MCD) (Figure 3). In a retrospective analysis of a phase 1 clinical trial, siltuximab reduced serum hepcidin in 97% of patients with Multiple Myeloma, or MCD.[147] Seventy-five percent of these patients showed an elevation in hemoglobin (hgb) of at least 1.5 g/dL.[147] In a randomized, double-blind study of siltuximab in patients with MCD, siltuximab reduced median hepcidin levels 47% from baseline.[103] The placebo group showed an 11% increase in hepcidin from baseline at the same timepoint.[103] Decreases in hepcidin correlated with an increase in hgb (≥15 g/L) in the siltuximab-treated group.[103] Siltuximab treatment in 4 cohorts of patients with advanced solid tumors showed 34% to 58% reduction in serum hepcidin levels from baseline.[104] Across cohorts, 29 of the 33 patients (88%) who showed an increase in hgb (≥1 g/dL) showed a decreased in hepcidin.[104] Furthermore, siltuximab treatment of renal cell carcinoma patients reduced median baseline serum hepcidin 61.1%.[105] Decreased serum hepcidin moderately correlated (r = −0.56, n = 19) with an increase in hgb (≥1 g/dL) in these patients.[105] These data support the use of siltuximab (Sylvant™) for treatment of anemia of chronic inflammation.
Tocilizumab (ACTEMRA®) is a humanized anti-IL-6 receptor antibody approved by the FDA for use in rheumatoid and juvenile idiopathic arthritis (Figure 3). Tocilizumab was first shown to reduce serum hepcidin in patients with MCD. In this study, tocilizumab prevented IL-6-mediated induction of hepcidin in hepatoma cells in vitro and reduced serum hepcidin in patients.[106] In 5 of 6 MCD patients (83%), tocilizumab reduced serum hepcidin within 24 hours.[106] A 12 month study of 9 MCD patients treated with tocilizumab showed a significant reduction in serum hepcidin and progressive improvement in iron status markers, including hgb, red blood count (RBC), mean corpuscular volume (MCV), and serum iron and ferritin levels.[106] Significant reduction of hepcidin and elevated hgb was also observed in a larger cohort of 46 rheumatoid arthritis patients treated with tocilizumab. [107] In the same study, a TNFα inhibitor (infliximab) also reduced serum hepcidin indirectly through reduction of IL-6.[107] Retrospective analysis of a phase 3B clinical trial for tocilizumab in rheumatoid arthritis patients (n = 70) further confirmed a reduction in hepcidin and increase in hgb.[108] Consistent in each of these studies was a rapid reduction in hepcidin followed by a prolonged, progressive increase in hgb.
Three JAK2-STAT3 signaling inhibitors have been shown to reduce hepcidin expression (Figure 3). Two synthetic inhibitors, AG490 and PpYLKTK, which target JAK2 [109] and STAT3 [110] respectively, reduced hepcidin expression in vitro.[111] AG490 was also shown to reduce hepatic hepcidin expression in vivo.[112] The third inhibitor, curcumin, is a naturally occurring anti-inflammatory compound that prevents STAT3 activation.[148] Curcumin treatment effectively reduced hepcidin expression in primary murine hepatocytes [111] and human hepatocellular carcinoma cells.[113] These findings indicate that inhibiting the JAK2-STAT3 pathway is a potential therapeutic approach for treatment of aberrant hepcidin expression. However, despite significantly reducing hepcidin expression, these inhibitors have not progressed to clinical development. PpYLKTK is a phosphorylated peptide fragment targeting the SH2 domain of STAT3.[110] Phosphopeptide mimetics, such as PpYLKTK and its derivatives, exhibit unfavorable pharmacokinetics due to poor in vivo stability and cell permeability. Attempts to improve pharmacokinetics for further clinical development of SH2 targeted phosphopeptides are ongoing.[149] AG490 clinical development was also hindered by poor pharmacokinetics. Safety and delivery of curcumin in vivo, however, is not an issue. Treatment with curcumin reduced hepatic hepcidin expression in vivo,[113,114] but conversely induced anemia through direct iron chelation.[113] Based on these findings, curcumin is also not the ideal candidate for treatment of anemia associated with excess hepcidin. In fact, curcumin may be more useful to treat iron overload conditions.[115]
Currently, several other JAK-STAT inhibitors are being developed for clinical use, primarily as anticancer agents. The clinical development of these inhibitors has been extensively reviewed.[150–152] One clinical JAK2 inhibitor,[116] 3,3′-Diindolylmethane (DIM), is of particular interest as it reduced hepatic hepcidin expression in rainbow trout (Figure 3).[117] Conversely, JAK-STAT inhibitor Sorafenib has been shown to increase hepcidin expression in human hepatocellular carcinoma cells and primary murine hepcatocytes, likely through Raf inhibition.[78] Furthermore, paclitaxel and vinorelbine are well described JAK-STAT inhibitors but induce anemia in some patients.[153] While JAK-STAT inhibitors are promising targets for therapeutic reduction of hepcidin, off-target effects may limit the efficacy of these drugs.
Erythroferrone
ERFE is a recently discovered hormone released from erythroblasts that mediates hepcidin repression during erythropoiesis (Figure 3). ERFE-deficient mice showed delayed and incomplete recovery from anemia induced by heat-killed Brucella abortus.[118] This finding suggested that administration of ERFE may be beneficial for relieving certain anemias with elevated hepcidin. Silarus Therapeutics is proceeding with further development of ERFE-targeted therapeutics for treatment of iron disorders.[119]
Hepcidin functional antagonists
A separate approach to alleviating hepcidin overload disorders is to disrupt hepcidin function. Several companies are developing therapies directly targeting hepcidin (Figure 3). Pieris Pharmaceuticals, Inc. is currently developing PRS-080, an anticalin targeted against hepcidin. Anticalins are small ligand-binding proteins based on a lipocalin scaffold. Anticalins offer several potential advantages over antibodies that have recently been summarized.[154] PRS-080 effectively neutralized hepcidin activity in vitro and in a murine model in vivo.[122] Another Anticalin developed by Pieris Pharmaceuticals, Inc. was well tolerated in a phase 1 trial.[123] Results are pending from a phase 1 clinical trial of PRS-080 which concluded this year.[124] Pending favorable results, Pieris Pharmaceuticals, Inc. intends to initiate phase 2 trials in patients with chronic kidney disease suffering from functional iron deficiency anemia.
In an analogous approach, NOXXON Pharma AG is developing a Spiegelmer hepcidin inhibitor called NOX-H94 (Lexaptepid Pegol). Spiegelmers are ligand-binding L-enantiomeric oligonucleotides which are designed to antagonize target function with high in vivo stability. [155] NOX-H94 inhibited hepcidin-induced FPN degradation in a murine macrophage cell line.[125] Furthermore, NOX-H94 prevented hypoferremia in an IL-6-induced nonhuman primate model.[125] NOX-H94 showed favorable pharmacokinetics, safety, and dose-dependent increases in iron parameters (serum iron, serum ferritin, and serum transferrin saturation).[126] In a separate phase 1 trial, hypoferremia induced by LPS injection in healthy subjects was prevented by NOX-H94.[127] A phase 2a pilot study was conducted in patents with anemia due to hematologic malignancies. In this study, NOX-H94 treatment showed an increase in hgb (≥1 g/dL) in 5 of 12 (43%) patients. [128] A separate phase 2 study is ongoing in dialysis patients with erythropoiesis stimulating agents-hyporesponsive anemia.[129]
A third approach to directly antagonize hepcidin has been taken by AMGEN and Eli Lilly and Company. Both companies developed humanized monoclonal antibodies targeting hepcidin. 12B9m, from AMGEN, was generated following the observation that mouse anti-hepcidin antibodies could rescue anemia of inflammation in combination with erythropoiesis-stimulating agent (ESA).[156] 12B9m was subsequently shown to increase serum iron and rescue ESA-refractory anemia when co-treated with ESA in a mouse model.[121] In this model, treatment with 12B9m increased mean corpuscular hgb with additional increases in hgb, and reticulocyte number, mean corpuscular volume, and mean corpuscular hgb upon co-treatment with ESA.[121] Furthermore, 12B9m increased serum iron levels in male cynomolgus monkeys.[121] Eli Lilly and Company completed a phase 1 clinical trial for their hepcidin-targeted antibody, LY2787106, in late 2014.[130] In January of 2015, Eli Lilly and Company announced that LY2787106 was no longer in development.[131] It appears the company is shifting focus to the development of LY2928057, an anti-FPN antibody that disrupts hepcidin binding to FPN but not FPN function (Figure 3). [132] Preclinical data in nonhuman primates showed that LY2928057 increased serum iron in a dose-dependent manner.[132] LY2928057 is currently in phase 1 clinical trials in anemic ESRD patients undergoing dialysis.[133]
Like LY2928057, fursultiamine interferes with hepcidin-FPN interactions.[134] Fursultiamine is a thymidine derivative used clinically for the treatment of thymidine deficiency. A high throughput screening identified fur-sultiamine as a hepcidin antagonist.[134] Subsequently, fursultiamine was shown to bind FPN and inhibit hepcidin-mediated degradation in vitro.[134] Fursultiamine was inconsistent in antagonizing hepcidin-mediated serum iron reduction in vivo, possibly due to its rapid conversion to thymidine.[134] Modifications to fursultiamine that improve in vivo effectiveness would be necessary for further consideration as a hepcidin-targeted therapeutic.
Expert commentary
The maintenance of iron homeostasis is critical for normal cellular function. Hepcidin, through its control of the iron efflux pump FPN, regulates dietary iron uptake and iron recycling. Dysregulation of hepcidin induces pathologic perturbations in iron levels. Examples of disorders associated with inadequate hepcidin synthesis include hereditary hemochromatosis and anemias. Treatment of these disorders remains suboptimal and has driven the clinical development of hepcidin modulators. One approach is the use of hepcidin agonists, such as minihepcidins, which act as hepcidin mimetics. Early results suggest that minihepcidins may represent an effective strategy for the treatment of iron overload disorders. These small peptides, which are easier to synthesize than full-length hepcidin, maintain functional degradation of FPN in animal models. There is also a substantial interest in development of hepcidin antagonists, some of which are already approved for clinical use in other indications. For example, siltuximab and tocilizumib are both FDA-approved IL-6 signaling inhibitors that reduce hepcidin and increase hgb in patients with Multicentric Castleman’s Disease and arthritis. Repurposing these medications could provide a faster path to more effective treatment of the ACD, particularly for patients in whom IL-6 inhibitors also treat the underlying disease. However, IL6 inhibitors have pleitrophic and possibly undesirable effects. Consequently, interfering more specifically with hepcidin function may offer therapeutic advantages. In this regard, the most clinically advanced hepcidin functional antagonist, NOX-H94, elevated hgb in 43% of patients in a pilot phase 2a trial. The development of this class of drugs may be particularly important for the long term treatment of ACD.
Five-year view
Modulation of hepcidin offers substantial clinical promise. Since the discovery of hepcidin in 2001, the development of strategies targeting both hepcidin excess and deficiency has progressed rapidly, with several entities in clinical trials and many more under preclinical investigation (Tables 1 and 2). It is not unreasonable to conjecture that modulation of hepcidin will become the standard for the treatment of a variety of disorders, including hereditary hemochromatosis, iron restrictive anemias, ACD and others, although the timeframe for delivery to market will likely extend beyond 5 years.
Nevertheless, questions remain. Although this review has focused on hepatic hepcidin and its interaction with ferroportin on the surface of enterocytes and macrophages, peripheral tissues also express ferroportin.[13–15] Ferroportin expressed on these cells is also subject to degradation following binding of hepcidin, resulting in an increase in intracellular iron.[13,14] In effect, hepatic hepcidin acts to reduce circulating iron, but also acts to increase cellular iron. This paradox may create a hurdle in the development of hepcidin modulators. For example, although minihepcidins may reduce iron absorption in HH patients due to their ability to bind and degrade FPN in the small intestine, minihepcidins may bind to and degrade FPN on cells in peripheral tissues as well. Such binding would result in increased intracellular iron in these tissues. Since excess iron can lead to DNA damage, mutagenesis, and possible malignant transformation, such an effect may have long-term undesirable consequences, particularly in patients with an increased risk of developing cancer. [157] The dichotomy between systemic and tumor iron may similarly pose a challenge for treatment of cancer patients with ACD. Cancer patients frequently suffer from the anemia of chronic disease as a result of increased inflammation and/or chemotherapy and subsequently high circulating hepcidin and are thus potential candidates for hepcidin antagonists. However, several groups have shown that increasing iron levels in tumors contributes to tumor growth.[13,158] Thus, it is possible that hepcidin antagonists administered in this setting may correct the anemia, but also augment growth of cancer cells.
Further, peripheral tissues not only express ferroportin, but synthesize their own hepcidin.[7,13,14] Hepcidin that is synthesized locally has the potential to act on adjacent cells, decreasing ferroportin and thereby increasing intracellular iron in these target cells.[13,14] The relative contributions of these two sources of hepcidin, and whether they can both be targeted with equal efficiency, has yet to be unraveled.
As discovery of pathways that regulate hepcidin continue to expand, the next 5 years may also witness the development of new hepcidin modulators that target elements of these pathways. An example is erythroferrone (EFRE), a very recently discovered regulator of hepcidin. Understanding how EFRE signals to suppress hepcidin synthesis, and whether other hepcidin suppressors complement or augment its activity, may reveal completely new signaling pathways that can be targeted to inhibit or stimulate hepcidin.
One of the most promising future uses of hepcidin modulators may be in cancer therapy. Although the role of hepcidin in promoting cancer growth is still under study, hepcidin appears to be elevated in a number of cancers.[159] Utilizing hepcidin antagonists to reduce ferroportin degradation in tumor cells, increasing iron efflux and potentially decreasing tumor growth, is an example of a potentially novel indication for hepcidin modulators. However, as discussed above, the potential of hepcidin antagonists to simultaneously increase systemic iron (thus possibly increasing the availability of iron for tumor growth and counteracting the effect of enhanced tumor iron efflux) may complicate this approach. Understanding the causes of dysregulated hepcidin production in cancer cells may prove fruitful in circumventing this problem. For example, if novel cancer-specific pathways of hepcidin regulation can be identified, hepcidin antagonists that specifically or preferentially block this pathway may prove most useful as anticancer agents.
Key issues.
Hepcidin is a secreted peptide that binds to ferroportin, the only known iron exporter in mammalian cells.
Binding of hepcidin to ferroportin triggers ferroportin degradation and thus reduces iron export.
Hepcidin regulates systemic iron homeostasis by regulating both dietary iron absorption in the gut and iron recycling by macrophages of the reticuloendothelial system.
Hepcidin dysregulation contributes to numerous disorders, ranging from hereditary hemochromatosis to the anemia of chronic disease.
Identification of signaling pathways that regulate hepcidin has revealed novel targets for restoring appropriate hepcidin production and normal iron homeostasis in disease states.
Hepcidin-targeted therapies are being actively developed and include both agonists and antagonists.
Hepcidin agonists under development include minihepcidins and agents that block TMPRSS6, a negative regulator of hepcidin. Hepcidin agonists may be useful in the treatment of hereditary hemochromatosis and iron-loading anemias.
Hepcidin antagonists include antibodies that directly bind hepcidin and inhibitors of the hepcidin biosynthetic pathway. These agents show promise in the treatment of the anemia of chronic disease.
Acknowledgments
Supported in part by grants R01 CA188025 (S Torti), R01 CA171101 (F Torti), and T90 DE021989 (D Manz) from the US National Institutes of Health. S Torti has received research support from AbbVie.
Biographies




Footnotes
Financial & competing interests disclosure
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
References
Papers of special note have been highlighted as:
• of interest
•• of considerable interest
- 1.Crichton RR. Iron metabolism: from molecular mechanisms to clinical consequences. Chichester (UK): John Wiley & Sons; 2009. [Google Scholar]
- 2.Dizdaroglu M, Rao G, Halliwell B, et al. Damage to the DNA bases in mammalian chromatin by hydrogen peroxide in the presence of ferric and cupric ions. Arch Biochem Biophys. 1991;285(2):317–324. doi: 10.1016/0003-9861(91)90366-q. [DOI] [PubMed] [Google Scholar]
- 3.Donovan A, Brownlie A, Zhou Y, et al. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403(6771):776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 4.Schade AL, Reinhart RW, Levy H. Carbon dioxide and oxygen in complex formation with iron and siderophilin, the iron-binding component of human plasma. Arch Biochem. 1949;20(1):170–172. [PubMed] [Google Scholar]
- 5.Krause A, Neitz S, Magert HJ, et al. LEAP-1, a novel highly disulfide-bonded human peptide, exhibits antimicrobial activity. FEBS Lett. 2000;480(2–3):147–150. doi: 10.1016/s0014-5793(00)01920-7. [DOI] [PubMed] [Google Scholar]
- 6.Park CH, Valore EV, Waring AJ, et al. Hepcidin, a urinary antimicrobial peptide synthesized in the liver. J Biol Chem. 2001;276(11):7806–7810. doi: 10.1074/jbc.M008922200. [DOI] [PubMed] [Google Scholar]
- 7.Pigeon C, Ilyin G, Courselaud B, et al. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276(11):7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 8•.Nicolas G, Bennoun M, Devaux I, et al. Lack of hepcidin gene expression and severe tissue iron overload in upstream stimulatory factor 2 (USF2) knockout mice. Proc Natl Acad Sci USA. 2001;98(15):8780–8785. doi: 10.1073/pnas.151179498. This study serendipitously identified hepcidin as a critical regulator of iron homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Valore EV, Ganz T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol Dis. 2008;40(1):132–138. doi: 10.1016/j.bcmd.2007.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10••.Nemeth E, Tuttle MS, Powelson J, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306(5704):2090–2093. doi: 10.1126/science.1104742. This study identified ferroportin as the target of hepcidin. [DOI] [PubMed] [Google Scholar]
- 11.Preza GC, Pinon R, Ganz T, et al. Cellular catabolism of the iron-regulatory peptide hormone hepcidin. Plos One. 2013;8(3):e58934. doi: 10.1371/journal.pone.0058934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12•.Donovan A, Lima CA, Pinkus JL, et al. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005;1(3):191–200. doi: 10.1016/j.cmet.2005.01.003. This article highlights the sites of ferroportin expression and its role in maintaining iron homeostasis in vivo. [DOI] [PubMed] [Google Scholar]
- 13.Pinnix ZK, Miller LD, Wang W, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med. 2010;2(43):43ra56. doi: 10.1126/scisignal.3001127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tesfay L, Clausen KA, Kim JW, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res. 2015;75(11):2254–2263. doi: 10.1158/0008-5472.CAN-14-2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Raha AA, Vaishnav RA, Friedland RP, et al. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun. 2013;1:55. doi: 10.1186/2051-5960-1-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olynyk JK, Trinder D, Ramm GA, et al. Hereditary hemochromatosis in the post-HFE era. Hepatology. 2008;48(3):991–1001. doi: 10.1002/hep.22507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sham RL, Phatak PD, West C, et al. Autosomal dominant hereditary hemochromatosis associated with a novel ferroportin mutation and unique clinical features. Blood Cells Mol Dis. 2005;34(2):157–161. doi: 10.1016/j.bcmd.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 18.Piperno A, Girelli D, Nemeth E, et al. Blunted hepcidin response to oral iron challenge in HFE-related hemochromatosis. Blood. 2007;110(12):4096–4100. doi: 10.1182/blood-2007-06-096503. [DOI] [PubMed] [Google Scholar]
- 19.Pippard MJ, Callender ST, Warner GT, et al. Iron absorption and loading in beta-thalassaemia intermedia. Lancet. 1979;2(8147):819–821. doi: 10.1016/s0140-6736(79)92175-5. [DOI] [PubMed] [Google Scholar]
- 20.Iolascon A, Esposito MR, Russo R. Clinical aspects and pathogenesis of congenital dyserythropoietic anemias: from morphology to molecular approach. Haematologica. 2012;97(12):1786–1794. doi: 10.3324/haematol.2012.072207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassemia. Blood. 2000;96(7):2606–2612. [PubMed] [Google Scholar]
- 22.Tanno T, Bhanu NV, Oneal PA, et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13(9):1096–1101. doi: 10.1038/nm1629. [DOI] [PubMed] [Google Scholar]
- 23••.Kautz L, Jung G, Valore EV, et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46(7):678–684. doi: 10.1038/ng.2996. This article identified erythroferrone as a key erythroid factor that controls iron homeostasis by suppressing hepcidin during stress erythropoiesis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Origa R, Galanello R, Ganz T, et al. Liver iron concentrations and urinary hepcidin in beta-thalassemia. Haematologica. 2007;92(5):583–588. doi: 10.3324/haematol.10842. [DOI] [PubMed] [Google Scholar]
- 25.Roy CN. Anemia of inflammation. Hematol Am Soc Hematol Educ Program. 2010;2010:276–280. doi: 10.1182/asheducation-2010.1.276. [DOI] [PubMed] [Google Scholar]
- 26.Chung A, Leo K, Wong G, et al. Giant hepatocellular adenoma presenting with chronic iron deficiency anemia. Am J Gastroenterol. 2006;101(9):2160–2162. doi: 10.1111/j.1572-0241.2006.00607.x. [DOI] [PubMed] [Google Scholar]
- 27•.Finberg KE, Heeney MM, Campagna DR, et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA) Nat Genet. 2008;40(5):569–571. doi: 10.1038/ng.130. This study revealed that TMPRSS6 is commonly mutated in patients suffering from iron-refractory iron deficiency anemia and demonstrated the importance of TMPRSS6 in iron homeostasis. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28•.Nemeth E, Rivera S, Gabayan V, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest. 2004;113(9):1271–1276. doi: 10.1172/JCI20945. This study demonstrated that IL-6 is responsible for induction of hepcidin by inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ward DG, Roberts K, Stonelake P, et al. SELDI-TOF-MS determination of hepcidin in clinical samples using stable isotope labelled hepcidin as an internal standard. Proteome Sci. 2008;6:28. doi: 10.1186/1477-5956-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Laarakkers CM, Wiegerinck ET, Klaver S, et al. Improved mass spectrometry assay for plasma hepcidin: detection and characterization of a novel hepcidin isoform. Plos One. 2013;8(10):e75518. doi: 10.1371/journal.pone.0075518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kobold U, Dulffer T, Dangl M, et al. Quantification of hepcidin-25 in human serum by isotope dilution micro-HPLC-tandem mass spectrometry. Clin Chem. 2008;54(9):1584–1586. doi: 10.1373/clinchem.2008.107029. [DOI] [PubMed] [Google Scholar]
- 32.Murao N, Ishigai M, Yasuno H, et al. Simple and sensitive quantification of bioactive peptides in biological matrices using liquid chromatography/selected reaction monitoring mass spectrometry coupled with trichloroacetic acid clean-up. Rapid Commun Mass Spectrom. 2007;21(24):4033–4038. doi: 10.1002/rcm.3319. [DOI] [PubMed] [Google Scholar]
- 33.Wolff F, Deleers M, Melot C, et al. Hepcidin-25: measurement by LC-MS/MS in serum and urine, reference ranges and urinary fractional excretion. Clin Chim Acta. 2013;423:99–104. doi: 10.1016/j.cca.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 34.Ganz T, Olbina G, Girelli D, et al. Immunoassay for human serum hepcidin. Blood. 2008;112(10):4292–4297. doi: 10.1182/blood-2008-02-139915. [DOI] [PubMed] [Google Scholar]
- 35.Delaby C, Vialaret J, Bros P, et al. Clinical measurement of Hepcidin-25 in human serum: is quantitative mass spectrometry up to the job? EuPA Open Proteomics. 2014;3:60–67. [Google Scholar]
- 36.Tomosugi N, Kawabata H, Wakatabe R, et al. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood. 2006;108(4):1381–1387. doi: 10.1182/blood-2005-10-4043. [DOI] [PubMed] [Google Scholar]
- 37.Kroot JJ, Van Herwaarden AE, Tjalsma H, et al. Second round robin for plasma hepcidin methods: first steps toward harmonization. Am J Hematol. 2012;87(10):977–983. doi: 10.1002/ajh.23289. [DOI] [PubMed] [Google Scholar]
- 38.Xiao JJ, Krzyzanski W, Wang YM, et al. Pharmacokinetics of anti-hepcidin monoclonal antibody Ab 12B9m and hepcidin in cynomolgus monkeys. Aaps J. 2010;12(4):646–657. doi: 10.1208/s12248-010-9222-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ganz T, Nemeth E. Hepcidin and iron homeostasis. Biochim Biophys Acta. 2012;1823(9):1434–1443. doi: 10.1016/j.bbamcr.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen D, Zhao M, Harris SE, et al. Signal transduction and biological functions of bone morphogenetic proteins. Front Biosci. 2004;9:349–358. doi: 10.2741/1090. [DOI] [PubMed] [Google Scholar]
- 41.Miyazono K, Kamiya Y, Morikawa M. Bone morphogenetic protein receptors and signal transduction. J Biochem. 2010;147(1):35–51. doi: 10.1093/jb/mvp148. [DOI] [PubMed] [Google Scholar]
- 42.Wang RH, Li C, Xu X, et al. A role of SMAD4 in iron metabolism through the positive regulation of hepcidin expression. Cell Metab. 2005;2(6):399–409. doi: 10.1016/j.cmet.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 43••.Andriopoulos B, Jr, Corradini E, Xia Y, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41(4):482–487. doi: 10.1038/ng.335. This study highlights a key role for BMP6 in regulation of hepcidin and iron metabolism in vivo. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Meynard D, Kautz L, Darnaud V, et al. Lack of the bone morphogenetic protein BMP6 induces massive iron overload. Nat Genet. 2009;41(4):478–481. doi: 10.1038/ng.320. [DOI] [PubMed] [Google Scholar]
- 45.Xia Y, Babitt JL, Sidis Y, et al. Hemojuvelin regulates hepcidin expression via a selective subset of BMP ligands and receptors independently of neogenin. Blood. 2008;111(10):5195–5204. doi: 10.1182/blood-2007-09-111567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46••.Babitt JL, Huang FW, Wrighting DM, et al. Bone morphogenetic protein signaling by hemojuvelin regulates hepcidin expression. Nat Genet. 2006;38(5):531–539. doi: 10.1038/ng1777. This study demonstrated that hemojuvelin, a gene mutated in juvenile hemochromatosis, encodes a BMP coreceptor and is involved in hepcidin regulation. [DOI] [PubMed] [Google Scholar]
- 47.Roetto A, Papanikolaou G, Politou M, et al. Mutant antimicrobial peptide hepcidin is associated with severe juvenile hemochromatosis. Nat Genet. 2003;33(1):21–22. doi: 10.1038/ng1053. [DOI] [PubMed] [Google Scholar]
- 48.Lee PL, Beutler E, Rao SV, et al. Genetic abnormalities and juvenile hemochromatosis: mutations of the HJV gene encoding hemojuvelin. Blood. 2004;103(12):4669–4671. doi: 10.1182/blood-2004-01-0072. [DOI] [PubMed] [Google Scholar]
- 49.Feder JN, Gnirke A, Thomas W, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 50.Camaschella C, Roetto A, Cali A, et al. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat Genet. 2000;25(1):14–15. doi: 10.1038/75534. [DOI] [PubMed] [Google Scholar]
- 51.Goswami T, Andrews NC. Hereditary hemochromatosis protein, HFE, interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J Biol Chem. 2006;281(39):28494–28498. doi: 10.1074/jbc.C600197200. [DOI] [PubMed] [Google Scholar]
- 52.Ganz T. The role of hepcidin in iron sequestration during infections and in the pathogenesis of anemia of chronic disease. Isr Med Assoc J. 2002;4(11):1043–1045. [PubMed] [Google Scholar]
- 53.Nemeth E, Valore EV, Territo M, et al. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood. 2003;101(7):2461–2463. doi: 10.1182/blood-2002-10-3235. [DOI] [PubMed] [Google Scholar]
- 54.Verga Falzacappa MV, Vujic Spasic M, Kessler R, et al. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood. 2007;109(1):353–358. doi: 10.1182/blood-2006-07-033969. [DOI] [PubMed] [Google Scholar]
- 55.Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. 2006;8(Suppl 2):S3. doi: 10.1186/ar1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Babitt JL, Huang FW, Xia Y, et al. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117(7):1933–1939. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Peterson PW. Targeting cancer-induced anemia with hepcidin lowering ALK2 inhibitors. Cancer Res. 2015;75(15 Supplement):3647. [Google Scholar]
- 58.Theurl I, Schroll A, Sonnweber T, et al. Pharmacologic inhibition of hepcidin expression reverses anemia of chronic inflammation in rats. Blood. 2011;118(18):4977–4984. doi: 10.1182/blood-2011-03-345066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Poli M, Girelli D, Campostrini N, et al. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011;117(3):997–1004. doi: 10.1182/blood-2010-06-289082. [DOI] [PubMed] [Google Scholar]
- 60.Mayeur C, Lohmeyer LK, Leyton P, et al. The type I BMP receptor Alk3 is required for the induction of hepatic hepcidin gene expression by interleukin-6. Blood. 2014;123(14):2261–2268. doi: 10.1182/blood-2013-02-480095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pinto JP, Ribeiro S, Pontes H, et al. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood. 2008;111(12):5727–5733. doi: 10.1182/blood-2007-08-106195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ashby DR, Gale DP, Busbridge M, et al. Erythropoietin administration in humans causes a marked and prolonged reduction in circulating hepcidin. Haematologica. 2010;95(3):505–508. doi: 10.3324/haematol.2009.013136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haase VH. Hypoxic regulation of erythropoiesis and iron metabolism. Am J Physiol Renal Physiol. 2010;299(1):F1–F13. doi: 10.1152/ajprenal.00174.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peyssonnaux C, Zinkernagel AS, Schuepbach RA, et al. Regulation of iron homeostasis by the hypoxia-inducible transcription factors (HIFs) J Clin Invest. 2007;117(7):1926–1932. doi: 10.1172/JCI31370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mastrogiannaki M, Matak P, Keith B, et al. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119(5):1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Volke M, Gale DP, Maegdefrau U, et al. Evidence for a lack of a direct transcriptional suppression of the iron regulatory peptide hepcidin by hypoxia-inducible factors. Plos One. 2009;4(11):e7875. doi: 10.1371/journal.pone.0007875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Silvestri L, Pagani A, Camaschella C. Furin-mediated release of soluble hemojuvelin: a new link between hypoxia and iron homeostasis. Blood. 2008;111(2):924–931. doi: 10.1182/blood-2007-07-100677. [DOI] [PubMed] [Google Scholar]
- 68.Silvestri L, Pagani A, Nai A, et al. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008;8(6):502–511. doi: 10.1016/j.cmet.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lin L, Goldberg YP, Ganz T. Competitive regulation of hepcidin mRNA by soluble and cell-associated hemojuvelin. Blood. 2005;106(8):2884–2889. doi: 10.1182/blood-2005-05-1845. [DOI] [PubMed] [Google Scholar]
- 70.Yanagita M, Oka M, Watabe T, et al. USAG-1: a bone morphogenetic protein antagonist abundantly expressed in the kidney. Biochem Biophys Res Commun. 2004;316(2):490–500. doi: 10.1016/j.bbrc.2004.02.075. [DOI] [PubMed] [Google Scholar]
- 71.Yang Q, Jian J, Katz S, et al. 17beta-Estradiol inhibits iron hormone hepcidin through an estrogen responsive element half-site. Endocrinology. 2012;153(7):3170–3178. doi: 10.1210/en.2011-2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ikeda Y, Tajima S, Izawa-Ishizawa Y, et al. Estrogen regulates hepcidin expression via GPR30-BMP6-dependent signaling in hepatocytes. Plos One. 2012;7(7):e40465. doi: 10.1371/journal.pone.0040465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Guo W, Bachman E, Li M, et al. Testosterone administration inhibits hepcidin transcription and is associated with increased iron incorporation into red blood cells. Aging Cell. 2013;12(2):280–291. doi: 10.1111/acel.12052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mei S, Wang H, Fu R, et al. Hepcidin and GDF15 in anemia of multiple myeloma. Int J Hematol. 2014;100(3):266–273. doi: 10.1007/s12185-014-1626-7. [DOI] [PubMed] [Google Scholar]
- 75.Jiang F, Yu WJ, Wang XH, et al. Regulation of hepcidin through GDF-15 in cancer-related anemia. Clin Chim Acta. 2014;428:14–19. doi: 10.1016/j.cca.2013.10.015. [DOI] [PubMed] [Google Scholar]
- 76.Goodnough JB, Ramos E, Nemeth E, et al. Inhibition of hepcidin transcription by growth factors. Hepatology. 2012;56(1):291–299. doi: 10.1002/hep.25615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280(20):19883–19887. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 78.Mleczko-Sanecka K, Roche F, da Silva AR, et al. Unbiased RNAi screen for hepcidin regulators links hepcidin suppression to proliferative Ras/RAF and nutrient-dependent mTOR signaling. Blood. 2014;123(10):1574–1585. doi: 10.1182/blood-2013-07-515957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Preza GC, Ruchala P, Pinon R, et al. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J Clin Invest. 2011;121(12):4880–4888. doi: 10.1172/JCI57693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ramos E, Ruchala P, Goodnough JB, et al. Minihepcidins prevent iron overload in a hepcidin-deficient mouse model of severe hemochromatosis. Blood. 2012;120(18):3829–3836. doi: 10.1182/blood-2012-07-440743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Merganser biotech. Development compound: M012 [Internet] 2015 [cited 2015 Sep 10]. Available from: http://merganserbiotech.com/hepcidin-mimetic-peptides/development-compound-m012/
- 82.La Jolla Pharmaceutical Company. LJPC-401 [Internet] 2015 [cited 2015 Sep 10]. Available from: http://lajollapharmaceutical.com/product-pipeline/ljpc-401/
- 83.La Jolla Pharmaceutical Company. Corportate presentation: developing innovative therapies for patients suffering from life-threatening diseases [Internet] 2015 [cited 2015 Sep 10]. Available from: http://lajollapharmaceutical.com/wp-content/uploads/2015/09/Sept-2015-Corporate-Presentation_9-17-2015-vJL.pdf.
- 84.Corradini E, Schmidt PJ, Meynard D, et al. BMP6 treatment compensates for the molecular defect and ameliorates hemochromatosis in Hfe knockout mice. Gastroenterology. 2010;139(5):1721–1729. doi: 10.1053/j.gastro.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Butler J, Fishman S, Racie T, et al. Aln-TMP: a subcutaneously administered RNAi therapeutic targeting TMPRSS6 for the treatment of β-thalassemia. Blood. 2013;122(21):2260. [Google Scholar]
- 86.Casu B, Diamantini G, Fedeli G, et al. Retention of antilipemic activity by periodate-oxidized non-anticoagulant heparins. Arzneimittelforschung. 1986;36(4):637–642. [PubMed] [Google Scholar]
- 87.Poli M, Asperti M, Naggi A, et al. Glycol-split nonanticoagulant heparins are inhibitors of hepcidin expression in vitro and in vivo. Blood. 2014;123(10):1564–1573. doi: 10.1182/blood-2013-07-515221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ritchie JP, Ramani VC, Ren Y, et al. SST0001, a chemically modified heparin, inhibits myeloma growth and angiogenesis via disruption of the heparanase/syndecan-1 axis. Clin Cancer Res. 2011;17(6):1382–1393. doi: 10.1158/1078-0432.CCR-10-2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sigma Tau Research Switzerland SA. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Phase I dose finding study assessing safety and tolerability of SST0001 in advanced multiple myeloma. [cited 2015 Sep 10]. Available from: https://clinicaltrials.gov/ct2/show/NCT01764880:NCT01764880. [Google Scholar]
- 90.Naggi A, Casu B, Perez M, et al. Modulation of the heparanase-inhibiting activity of heparin through selective desulfation, graded N-acetylation, and glycol splitting. J Biol Chem. 2005;280(13):12103–12113. doi: 10.1074/jbc.M414217200. [DOI] [PubMed] [Google Scholar]
- 91.FerruMax Pharmaceuticals I. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. An exploratory, uncontrolled, open-labeled trial to evaluate the effect of FMX-8 treatment for anemia in patients with chronic kidney disease (CKD), stage 4 or 5. [cited 2015 Sep 10]. Available from: https://clinicaltrials.gov/ct2/show/NCT02228655:NCT02228655. [Google Scholar]
- 92.FerruMax Pharmaceuticals I. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. A phase 2A, uncontrolled, open-labeled trial to evaluate the effect of FMX-8 treatment for anemia in patients with end stage renal disease (ESRD) on hemodialysis (HD) [cited 2015. Sep 10]. Available from: https://clinicaltrials.gov/ct2/show/NCT01873534:NCT01873534. [Google Scholar]
- 93.Yu PB, Hong CC, Sachidanandan C, et al. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4(1):33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu PB, Deng DY, Lai CS, et al. BMP type I receptor inhibition reduces heterotopic [corrected] ossification. Nat Med. 2008;14(12):1363–1369. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steinbicker AU, Sachidanandan C, Vonner AJ, et al. Inhibition of bone morphogenetic protein signaling attenuates anemia associated with inflammation. Blood. 2011;117(18):4915–4923. doi: 10.1182/blood-2010-10-313064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.La Jolla Pharmaceutical Company. La Jolla pharmaceutical company announces second-quarter 2013 financial results and highlights recent corporate progress [Internet] 2013 [cited 2015 Sep 10]. Available from: http://www.sec.gov/Archives/edgar/data/920465/000119312513301480/d573357dex991.htm.
- 97.Vogt J, Traynor R, Sapkota GP. The specificities of small molecule inhibitors of the TGFss and BMP pathways. Cell Signal. 2011;23(11):1831–1842. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 98.Tolero Pharmaceuticals, Inc. Tolero pharmaceuticals presents positive preclinical data on hepcidin inhibition for the treatment of anemia associated with chronic disease [Internet] 2015 [cited 2015 Sep 10]. Available from: http://www.businesswire.com/news/home/20150616006060/en/Tolero-Pharmaceuticals-Presents-Positive-Preclinical-Data-Hepcidin#.VgLnb8tVikp.
- 99.Boser P, Seemann D, Liguori MJ, et al. Anti-repulsive guidance molecule C (RGMc) antibodies increases serum iron in rats and cynomolgus monkeys by hepcidin downregulation. Aaps J. 2015;17(4):930–938. doi: 10.1208/s12248-015-9770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xenon Pharmaceuticals Inc. Xenon licenses antisense drug XEN701 from Isis and initiates preclinical toxicology studies. 2013 [cited 2015 Sep 10]. Available from: http://www.xenon-pharma.com/2013/06/isis/
- 101.Xenon Pharmaceuticals, Inc. FORM S-1 [Internet] 2014 [cited 2015 Sep 10]. Available from: http://www.sec.gov/Archives/edgar/data/1582313/000119312514337038/d582812ds1.htm.
- 102.Akinc A, Chan-Daniels A, Sehgal A, et al. Targeting the hepcidin pathway with RNAi therapeutics for the treatment of anemia [abstract]. ASH Annual Meeting and Exposition; 2011 Dec 10–13; San Diego, CA. Washington (DC): ASH; 2011. Abstract nr 688. [Google Scholar]
- 103.Casper C, Chaturvedi S, Munshi N, et al. Analysis of inflammatory and anemia-related biomarkers in a randomized, double-blind, placebo-controlled study of siltuximab (anti-IL6 monoclonal antibody) in patients with multicentric castleman disease. Clin Cancer Res. 2015 Oct 1;21(19):4294–4304. doi: 10.1158/1078-0432.CCR-15-0134. [DOI] [PubMed] [Google Scholar]
- 104.Angevin E, Tabernero J, Elez E, et al. A phase I/II, multiple-dose, dose-escalation study of siltuximab, an anti-interleukin-6 monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2014;20(8):2192–2204. doi: 10.1158/1078-0432.CCR-13-2200. [DOI] [PubMed] [Google Scholar]
- 105.Schipperus M, Rijnbeek B, Reddy M, et al. CNTO328 (anti-IL-6 mAb) treatment is associated with an increase in hemoglobin (Hb) and decrease in hepcidin levels in renal cell carcinoma (RCC) Blood. 2009;114(22):4045. [Google Scholar]
- 106.Song SN, Tomosugi N, Kawabata H, et al. Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease. Blood. 2010;116(18):3627–3634. doi: 10.1182/blood-2010-03-271791. [DOI] [PubMed] [Google Scholar]
- 107.Song SN, Iwahashi M, Tomosugi N, et al. Comparative evaluation of the effects of treatment with tocilizumab and TNF-alpha inhibitors on serum hepcidin, anemia response and disease activity in rheumatoid arthritis patients. Arthritis Res Ther. 2013;15(5):R141. doi: 10.1186/ar4323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Isaacs JD, Harari O, Kobold U, et al. Effect of tocilizumab on haematological markers implicates interleukin-6 signalling in the anaemia of rheumatoid arthritis. Arthritis Res Ther. 2013;15(6):R204. doi: 10.1186/ar4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.De Vos J, Jourdan M, Tarte K, et al. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br J Haematol. 2000;109(4):823–828. doi: 10.1046/j.1365-2141.2000.02127.x. [DOI] [PubMed] [Google Scholar]
- 110.Turkson J, Kim JS, Zhang S, et al. Novel peptidomimetic inhibitors of signal transducer and activator of transcription 3 dimerization and biological activity. Mol Cancer Ther. 2004;3(3):261–269. [PubMed] [Google Scholar]
- 111.Fatih N, Camberlein E, Island ML, et al. Natural and synthetic STAT3 inhibitors reduce hepcidin expression in differentiated mouse hepatocytes expressing the active phosphorylated STAT3 form. J Mol Med (Berl) 2010;88(5):477–486. doi: 10.1007/s00109-009-0588-3. [DOI] [PubMed] [Google Scholar]
- 112.Zhang SP, Wang Z, Wang LX, et al. AG490: an inhibitor of hepcidin expression in vivo. World J Gastroenterol. 2011;17(45):5032–5034. doi: 10.3748/wjg.v17.i45.5032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jiao Y, Wilkinson J, 4th, Di X, et al. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood. 2009;113(2):462–469. doi: 10.1182/blood-2008-05-155952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chin D, Huebbe P, Frank J, et al. Curcumin may impair iron status when fed to mice for six months. Redox Biol. 2014;2:563–569. doi: 10.1016/j.redox.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Badria FA, Ibrahim AS, Badria AF, et al. Curcumin attenuates iron accumulation and oxidative stress in the liver and spleen of chronic iron-overloaded rats. PLoS One. 2015;10(7):e0134156. doi: 10.1371/journal.pone.0134156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kandala PK, Srivastava SK. Regulation of Janus-activated kinase-2 (JAK2) by diindolylmethane in ovarian cancer in vitro and in vivo. Drug Discov Ther. 2012;6(2):94–101. [PubMed] [Google Scholar]
- 117.Tilton SC, Hendricks JD, Orner GA, et al. Gene expression analysis during tumor enhancement by the dietary phytochemical, 3,3′-diindolylmethane, in rainbow trout. Carcinogenesis. 2007;28(7):1589–1598. doi: 10.1093/carcin/bgm017. [DOI] [PubMed] [Google Scholar]
- 118.Kautz L, Jung G, Nemeth E, et al. Erythroferrone contributes to recovery from anemia of inflammation. Blood. 2014;124(16):2569–2574. doi: 10.1182/blood-2014-06-584607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Avalon Ventures. Avalon ventures and GlaxoSmithKline launch two new companies with $10 million each in series A financing and R&D support [Internet] 2014 [cited 2015 Sep 10]. Available from: http://www.prnewswire.com/news-releases/avalon-ventures-and-glaxosmithkline-launch-two-new-companies-with-10-million-each-in-series-a-financing-and-rd-support-275982981.html.
- 120.Kautz L. Erythroferrone, an erythroid regulator of iron metabolism. Med Sci (Paris) 2014;30(10):834–836. doi: 10.1051/medsci/20143010005. [DOI] [PubMed] [Google Scholar]
- 121.Cooke KS, Hinkle B, Salimi-Moosavi H, et al. A fully human anti-hepcidin antibody modulates iron metabolism in both mice and nonhuman primates. Blood. 2013;122(17):3054–3061. doi: 10.1182/blood-2013-06-505792. [DOI] [PubMed] [Google Scholar]
- 122.Hohlbaum AM, Trentman S, Gille H, et al. Discovery and preclinical characterization of a novel hepcidin antagonist with tunable PK/PD properties for the treatment of anemia in different patient populations [abstract]. ASH Annual Meeting and Exposition; 2011 Dec 10–13; San Diego, CA. Washington (DC): ASH; 2011. Abstract nr 687. [Google Scholar]
- 123.Mross K, Richly H, Fischer R, et al. First-in-human phase I study of PRS-050 (Angiocal), an Anticalin targeting and antagonizing VEGF-A, in patients with advanced solid tumors. PLoS One. 2013;8(12):e83232. doi: 10.1371/journal.pone.0083232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Pieris AG. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. A first-in-human, randomized, dose-escalation, double-blind, placebo-controlled single ascending dose study to establish safety, lack of immunogenicity, tolerability, pharmacokinetic parameters, target engagement and pharmacodynamic effects. [cited 2015 Sep 10]. Available from: https://clinicaltrials.gov/ct2/show/NCT02340572:NCT02340572. [Google Scholar]
- 125.Schwoebel F, Van Eijk LT, Zboralski D, et al. The effects of the anti-hepcidin Spiegelmer NOX-H94 on inflammation-induced anemia in cynomolgus monkeys. Blood. 2013;121(12):2311–2315. doi: 10.1182/blood-2012-09-456756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Riecke K, Zollner S, Boyce M, et al. Single and repeated dose first-in-human study with the anti-hepcidin spiegelmer Nox-H94 [abstract]. ASH Annual Meeting and Exposition; 2012 Dec 8–12; Atlanta, GA. Washington (DC): ASH; 2012. Abstract nr 2342. [Google Scholar]
- 127.Van Eijk L, Swinkels DW, John A, et al. Randomized double-blind placebo-controlled PK/PD study on the effects of a single intravenous dose of the anti-hepcidin Spiegelmer NOX-H94 on serum iron during experimental human endotoxemia. Crit Care. 2013;17(2):1–200. [Google Scholar]
- 128.Georgiev P, Lazaroiu M, Ocroteala L, et al. The anti-hepcidin Spiegelmer® Lexaptepid Pegol (NOX-H94) as treatment of anemia of chronic disease in patients with multiple myeloma, low grade lymphoma, and CLL: a phase II pilot study. Cancer Res. 2014;74(19 Supplement):3847. [Google Scholar]
- 129.NOXXON Pharma AG. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. Safety, PK/PD, and efficacy of NOX-H94 in dialysis patients with ESA-hyporesponsive anemia: a randomized, double blind, placebo controlled parallel group study with a single blind cross-over group. [cited 2015 Sep 10]. Available from. https://www.clinicaltrials.gov/ct2/show/NCT02079896:NCT02079896. [Google Scholar]
- 130.Eli Lilly and Company. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. A Phase 1 safety study of LY2787106 in patients with cancer and anemia. [cited 2015 Sep 10]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT01340976:NCT01340976) [Google Scholar]
- 131.Eli Lilly and Company. Q4 2014 financial review [Internet] 2015 Available from: http://files.shareholder.com/downloads/LLY/0x0x806041/8456ABB4-DE7C-4193-95EA-64400D334C44/Q4_2014_Slides.pdf.
- 132.Leung D, Hill KA, De Rosa DC, et al. LY2928057, an antibody targeting ferroportin, is a potent inhibitor of hepcidin activity and increases iron mobilization in normal cynomolgus monkeys. Blood. 2013;122(21):3433. [Google Scholar]
- 133.Eli Lilly and Company. ClinicalTrials.gov [Internet] Bethesda (MD): National Library of Medicine (US); 2000. A multiple-dose, dose-escalation study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of LY2928057 in hemodialysis patients. [cited. 2015 Sep 10]. Available from: https://www.clinicaltrials.gov/ct2/show/NCT01991483:NCT01991483. [Google Scholar]
- 134.Fung E, Sugianto P, Hsu J, et al. High-throughput screening of small molecules identifies hepcidin antagonists. Mol Pharmacol. 2013;83(3):681–690. doi: 10.1124/mol.112.083428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Gagliardo B, Faye A, Jaouen M, et al. Production of biologically active forms of recombinant hepcidin, the iron-regulatory hormone. Febs J. 2008;275(15):3793–3803. doi: 10.1111/j.1742-4658.2008.06525.x. [DOI] [PubMed] [Google Scholar]
- 136.Carragee EJ, Hurwitz EL, Weiner BK. A critical review of recombinant human bone morphogenetic protein-2 trials in spinal surgery: emerging safety concerns and lessons learned. Spine J. 2011;11(6):471–491. doi: 10.1016/j.spinee.2011.04.023. [DOI] [PubMed] [Google Scholar]
- 137.Nai A, Pagani A, Mandelli G, et al. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of beta-thalassemia. Blood. 2012;119(21):5021–5029. doi: 10.1182/blood-2012-01-401885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Guo S, Casu C, Gardenghi S, et al. Reducing TMPRSS6 ameliorates hemochromatosis and beta-thalassemia in mice. J Clin Invest. 2013;123(4):1531–1541. doi: 10.1172/JCI66969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schmidt PJ, Toudjarska I, Sendamarai AK, et al. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe(−/−) mice and ameliorates anemia and iron overload in murine beta-thalassemia intermedia. Blood. 2013;121(7):1200–1208. doi: 10.1182/blood-2012-09-453977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Schmidt PJ, Racie T, Westerman M, et al. Combination therapy with a Tmprss6 RNAi-therapeutic and the oral iron chelator deferiprone additively diminishes secondary iron overload in a mouse model of beta-thalassemia intermedia. Am J Hematol. 2015;90(4):310–313. doi: 10.1002/ajh.23934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wozney JM, Rosen V, Celeste AJ, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242(4885):1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 142.Rider CC. Heparin/heparan sulphate binding in the TGF-beta cytokine superfamily. Biochem Soc Trans. 2006;34(Pt 3):458–460. doi: 10.1042/BST0340458. [DOI] [PubMed] [Google Scholar]
- 143.Watts JK, Corey DR. Silencing disease genes in the laboratory and the clinic. J Pathol. 2012;226(2):365–379. doi: 10.1002/path.2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wittrup A, Lieberman J. Knocking down disease: a progress report on siRNA therapeutics. Nat Rev Genet. 2015;16(9):543–552. doi: 10.1038/nrg3978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nair JK, Willoughby JL, Chan A, et al. Multivalent N-acetylgalactosamine-conjugated siRNA localizes in hepatocytes and elicits robust RNAi-mediated gene silencing. J Am Chem Soc. 2014;136(49):16958–16961. doi: 10.1021/ja505986a. [DOI] [PubMed] [Google Scholar]
- 146.Sorensen B, Mant T, Akinc A, et al. A subcutaneously administered RNAi therapeutic (ALN-AT3) targeting antithrombin for treatment of hemophilia: interim Phase 1 study results in healthy volunteers and patients with hemophilia A or B. Blood. 2014;124(21):693. [Google Scholar]
- 147.Kurzrock R, Voorhees PM, Casper C, et al. A phase I, open-label study of siltuximab, an anti-IL-6 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma, multiple myeloma, or Castleman disease. Clin Cancer Res. 2013;19(13):3659–3670. doi: 10.1158/1078-0432.CCR-12-3349. [DOI] [PubMed] [Google Scholar]
- 148.Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloyl-methane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol. 2003;171(7):3863–3871. doi: 10.4049/jimmunol.171.7.3863. [DOI] [PubMed] [Google Scholar]
- 149.McMurray JS, Mandal PK, Liao WS, et al. The consequences of selective inhibition of signal transducer and activator of transcription 3 (STAT3) tyrosine705 phosphorylation by phosphopeptide mimetic prodrugs targeting the Src homology 2 (SH2) domain. Jakstat. 2012;1(4):263–347. doi: 10.4161/jkst.22682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Miklossy G, Hilliard TS, Turkson J. Therapeutic modulators of STAT signalling for human diseases. Nat Rev Drug Discov. 2013;12(8):611–629. doi: 10.1038/nrd4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lavecchia A, Di Giovanni C, Cerchia C. Novel inhibitors of signal transducer and activator of transcription 3 signaling pathway: an update on the recent patent literature. Expert Opin Ther Pat. 2014;24(4):383–400. doi: 10.1517/13543776.2014.877443. [DOI] [PubMed] [Google Scholar]
- 152.Sonbol MB, Firwana B, Zarzour A, et al. Comprehensive review of JAK inhibitors in myeloproliferative neoplasms. Ther Adv Hematol. 2013;4(1):15–35. doi: 10.1177/2040620712461047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Groopman JE, Itri LM. Chemotherapy-induced anemia in adults: incidence and treatment. J Natl Cancer Inst. 1999;91(19):1616–1634. doi: 10.1093/jnci/91.19.1616. [DOI] [PubMed] [Google Scholar]
- 154.Gebauer M, Skerra A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012;503:157–188. doi: 10.1016/B978-0-12-396962-0.00007-0. [DOI] [PubMed] [Google Scholar]
- 155.Vater A, Klussmann S. Turning mirror-image oligonucleotides into drugs: the evolution of Spiegelmer((R)) therapeutics. Drug Discov Today. 2015;20(1):147–155. doi: 10.1016/j.drudis.2014.09.004. [DOI] [PubMed] [Google Scholar]
- 156.Sasu BJ, Cooke KS, Arvedson TL, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010;115(17):3616–3624. doi: 10.1182/blood-2009-09-245977. [DOI] [PubMed] [Google Scholar]
- 157.Bradbear RA, Bain C, Siskind V, et al. Cohort study of internal malignancy in genetic hemochromatosis and other chronic nonalcoholic liver diseases. J Natl Cancer Inst. 1985;75(1):81–84. [PubMed] [Google Scholar]
- 158.Zhang S, Chen Y, Guo W, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal. 2014;26(11):2539–2550. doi: 10.1016/j.cellsig.2014.07.029. [DOI] [PubMed] [Google Scholar]
- 159.Torti SV, Torti FM. Iron and cancer: more ore to be mined. Nat Rev Cancer. 2013;13(5):342–355. doi: 10.1038/nrc3495. [DOI] [PMC free article] [PubMed] [Google Scholar]