

Abstract
Mechanistic target of rapamycin (mTOR) is a conserved threonine and serine protein kinase that was identified more than two decades ago as the target of immunosuppressive drug rapamycin. Since then considerable amount of information has been learned about the function of this kinase. It is now well-established that mTOR plays a pivotal role in governing cell growth and proliferation, hence making mTOR a therapeutic target for disease conditions caused by deregulated cell proliferation, such as cancer. In the past decade, numerous mTOR inhibitors have been developed and many are currently in clinical trials for cancer treatment. This commentary is to provide a brief summary of these mTOR inhibitors.
Keywords: mTOR, Phosphatidylinositol-3-kinase, Akt, Rapamycin, Kinase inhibitors
Introduction
mTOR belongs to the family of phosphatidylinositol-3-kinase-related kinases (PIKKs). Members in this family are large in size (>2,500 amino acids) and harbor a kinase domain at their C-terminals that shares sequence similarity to phosphatidylinositol-3-kinase (PI3K) (1). Despite having the sequence signature of a lipid kinase, mTOR is a protein kinase that phosphorylates threonine and serine residues in its substrates. In cells mTOR serves as the catalytic subunits of two multi-protein complexes termed as the mTOR complex 1 (mTORC1) and complex 2 (mTORC2) (2–4). TORC1 is a major downstream component of the PI3K/AKT pathway that relays the signals from tumor suppressors PTEN, LKB1 and TSC1/2, and oncoproteins PI3K and AKT (Figure 1) (5). Downstream mTORC1 controls cellular biogenesis through regulation of protein synthesis and turnover. It phosphorylates eIF4E binding protein 1 (4EBP1) and ribosomal protein S6 kinase (S6K), two factors involved in translation initiation (6). Its activity controls protein turnover through repressing autophagy (7). mTORC2 is also involved in the PI3K/AKT pathway but its function is independent of mTORC1. It phosphorylates and stimulates AKT activation, and hence plays a critical role in AKT mediated cell survival (8).
Rapamycin is the prototype of the first generation of mTOR inhibitors (9). It is a macrocyclic lactone that contains two binding moieties that are essential for its action (Figure 2). One moiety binds to FKBP12, a small cytosolic protein that displays peptidylprolyl isomerase activity. At pharmacological relevant concentration, rapamycin imposes no significant effect on the function of FKBP12. But binding with FKBP12 allows it to interact with its mechanistic target, mTOR, to form a ternary complex (10). The rapamycin-FKBP12 dimer binds to mTOR in a region (FKBP12-rapamycin binding domain) that is outside of the kinase domain. As such, the binding itself does not inhibit the kinase activity of mTOR. It is believed that the binding interferes with the association of the kinase with its substrates, although the precise mechanism remains to be elucidated (11). Despite the presence of mTOR in both mTORC1 and mTORC2, rapamycin only inhibits mTORC1 activity. The unique components of mTORC2 may exclude the drug from binding with the kinase. However, in some cells, prolonged incubation with the drug also affect mTORC2, presumably by binding the newly synthesized mTOR, hence preventing it from assembly into the complex (8).
Figure 2.
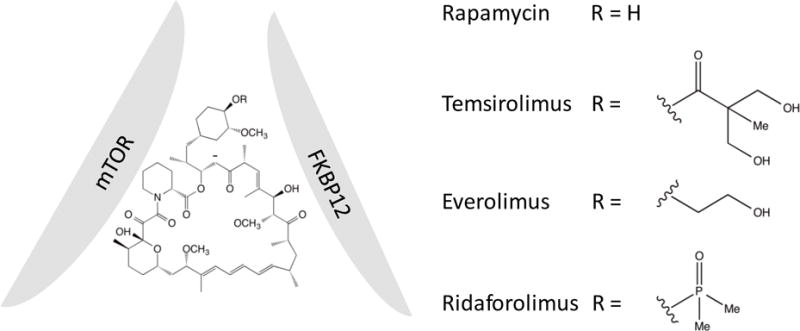
Structures of rapalogs.
Rapalogs, the first generation of mTOR inhibitors
Rapamycin was first developed into an immunosuppressive drug for its potent action in blocking T-cell activation. It was approved by the FDA in 1997 for use in transplantation to prevent allograft rejection, and in 2003 for use in coronary-artery stents to prevent restenosis (9). Although the anti-cancer activity of rapamycin was documented in early 1980s, its application in cancer therapy was not exploited until late 1990s, when several analogs of the drug, which often termed as rapalogs, were developed. Rapamycin is poorly water soluble, which affects its bioavailability. The development focused mainly on improving its pharmacokinetics and stability. However, because the drug requires two sides for binding with FKBP12 and mTOR, there is not much room for further modification. All rapalogs are created by replacing the hydrogen at C-40-O position with different moieties (Figure 2). For Temsirolimus (CCI-779), it is a dihydroxylmethyl propionic acid ester (12), Everolimus (RAD001), a hydroxylethyl group (13), and Ridaforolimus (AP23573), a dimethyl phosphate group (14) (Figure 2). In 2007 Temsirolimus became the first rapalog approved by FDA for cancer treatment (15).
Given the specificity of the drug and its potency in anticancer activity in many preclinical models, it was hard to image the need for new types of mTOR inhibitors. However, the clinical application of rapalogs in cancer treatment has so far met with limited success. Rapalogs are effective in treating a few cancers, including renal cell carcinoma and mantle cell lymphoma, but not in the majority of solid tumors (16). The mechanisms underlying the rapalog resistance are complex. Genetic variations of the factors associated with the mTOR signaling pathway in cancer cells contribute to de novo resistance to drug (17), however, the main reason is likely due to the fact that the drug does not directly cause cell death. Inhibition of mTORC1 by rapamycin actually induces stress responses, including reduction in protein synthesis and induction of autophagy, which are protective mechanisms for the cells to survive under stress conditions (18). Consequently, the direct effect of the drug is cytostatic rather than cytotoxic. In addition, in many types of cancer cells, inhibition of mTORC1 turns off a S6K-dependent negative feedback loop that downregulates upstream signaling of PI3K/AKT (Figure 1), resulting in enhanced PI3K/AKT activity that promotes cell survival (19).
Figure 1.
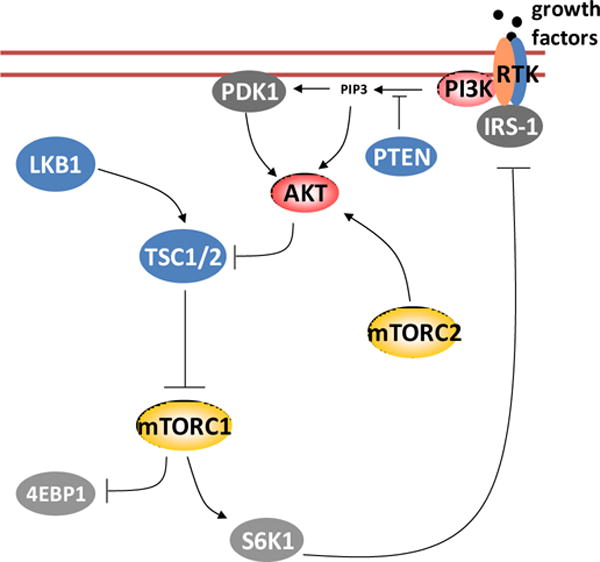
The mTOR pathway.
mTOR kinase inhibitors, the second generation of mTOR inhibitors
Two strategies have been explored to circumvent the limitation of rapalogs in cancer therapy (20). The first involves pairing the drugs with cytotoxic agents. For instance, rapalogs in combination with chemotherapeutic drugs such as paclitaxel and carboplatin are currently used in trials for treating advanced ovarian cancer and metastatic melanoma (NCT01196429 and NCT00976573). In addition, the drugs are also used in conjunction with hormonal therapy in endocrine cancers as a way to sensitize the cancer cells to the treatment (21–22). The second strategy involves developing inhibitors that target both PI3K and mTOR or selectively, mTOR (23–25). In the latter case, because mTOR serves as the catalytic subunits for both mTORC1 and mTORC2, a drug inhibiting the kinase activity is expected to affect both complexes, and consequently, block the mTORC2 dependent activation of AKT. In the second half of the last decade, many pharmaceutical companies and academic laboratories were actively engaged in development of this new generation of mTOR inhibitors and numerous compounds were discovered that possess potent inhibitory effect on PI3K and/or mTOR.
The development of PI3K and mTOR kinase inhibitors exploits the structure of the ATP binding pocket of the kinases with small molecules that compete for the binding pocket with ATP. Hence, these inhibitors are collectively called ATP competitive inhibitors. Because the sequence similarity of mTOR with PI3K, many ATP competitive PI3K inhibitors were found to display various degrees of mTOR inhibitory activity. These inhibitors were often used as prototype compounds for the PI3K/mTOR dual inhibitors. One of such compounds is PI-103, a pyrimidine derivative, which was originally developed as a pan-PI3K inhibitor but was subsequently found to inhibit PI3K-related kinases, including mTOR. PI-103 inhibits several isoforms of PI3K with an IC50 of 2–3 nM but is less selective for mTORC1 and mTORC2 with IC50 of 20 nM and 83 nM, respectively (26). Several other inhibitors were developed based on the structure of PI-103, including GDC-0980, which possess a similar IC50 for both PI3K and mTOR (27), and GNE-493, GNE-477 and PF-04691502, which are more selective for PI3K than for mTOR (28–30). BEZ-235, an imidazoquinoline derivative, represents another group of the inhibitors that also include BGT226 and GKS2126458. BEZ-235 inhibits several isoforms of PI3K and mTOR with IC50 of ~5 nM (31). GKS2126458 is one of the most potent PI3K/mTOR dual inhibitors. It exhibits an IC50 of 0.04 nM for PI3K and 0.18 and 0.3 nM for mTORC1 and mTORC2, respectively (32). XL765, a quinoxaline derivative, is another non-pyrimidine derivative that exhibits high potency for PI3K and mTOR (33) (Figure 3).
Figure 3.
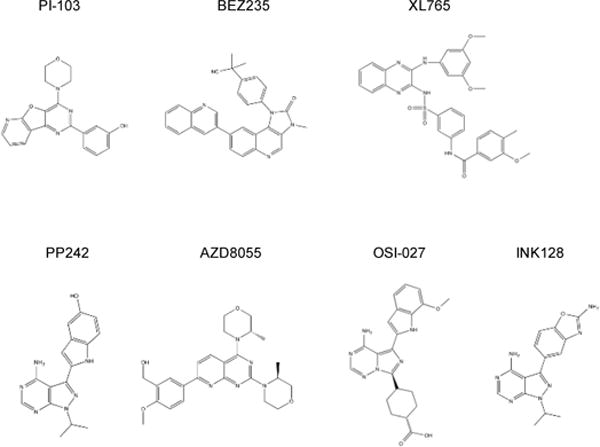
Structures of mTOR kinase inhibitors.
While simultaneously targeting PI3K and mTOR circumvents the limitation of rapalogs in blocking PI3K/AKT signaling, the potential toxicity associated with this type of inhibitors presents a big concern, owing to the diverse functions of different isoforms of PI3K. It is generally believed that inhibitors more selective for mTOR may be better tolerated than the dual inhibitors (34). In addition, because mTORC2 is required for activation of AKT, inhibiting this complex by mTOR kinase inhibitors is also expected to dampen the pro-survival function of AKT (35). These considerations fueled the development of inhibitors that are more specific to mTOR than to PI3K. In 2009, a series of selective mTOR inhibitors were reported. These selective mTOR inhibitors include pyrimidine derivatives PP242 and PP30 (36), morpholino-linked pyrimidine derivatives, WAY-600, WYE-687 and WYE354 (37), and KU0063794 and AZD8055 (38–39) (Figure 3). In comparison with many PI3K/mTOR dual inhibitors, these selective mTOR inhibitors exhibit 10–100 fold selectivity toward mTOR than to isoforms of PI3K. Additional mTOR inhibitors were also developed subsequently that are in general more potent and selective for mTOR inhibition. For instance, OSI-027, a triazine derivative (40), and AZD2014, a pyrimidine derivative similar to AZD8055 (41), both have IC50 below 4 nM and exhibit >300 fold selectivity for mTOR than for PI3K. INK128, a pyrimidine derivative similar to PP242 and PP30, has an IC50 of 1 nM and is >200 fold more selective for mTOR (42–43). Torin 1, a quinoline derivative developed based on BEZ-235, has an IC50 less than 10 nM and is >1,000 fold more specific (44). The high potency and selectivity of these new inhibitors ensure effective and specific downregulation of mTORC1 and mTORC2, while leave PI3K unaffected.
As expected, in both cell based and animal studies, the dual inhibitors, such as BEZ-235 and GSK2126458, are effective in inhibiting PI3K, mTORC1 processes and mTORC2-dependent processes (31–32, 45), while the mTOR selective inhibitors, such as OSI-027 and INK128, specifically reduce mTORC1 and mTORC2 activity (40, 46–47). In comparison with rapalogs, PI3K/mTOR and mTOR selective inhibitors are more potent in blocking cell proliferation and induction of apoptosis in many cultured cancer cells and in tumor xenograft models, including some of rapamycin resistant tumors (33, 40, 47–49). Several of this new generation of inhibitors have successfully made into clinical trials, among which BEZ235, INK128, AZD2014 and XL765 are currently in phase II trials for efficacy studies in treatment of various cancers. In phase I studies, these inhibitors exhibited toxicity that was tolerable and manageable (34), which, to certain degree, alleviates the safety concern association with the new generation drugs.
Conclusion
The past decade has seen an influx of many new mTOR inhibitors which have been proven to be valuable biological and pharmacological tools. The recent clinical success of some of these inhibitors validates the initial expectation for this new generation of mTOR inhibitors. However, a glance at NCI clinical trial website reveals that despite their superior potency, the new generation drugs are still not as favored as rapalogs, presumably over safety concern. As for many other drugs targeting intracellular signaling pathways, the challenges for the new mTOR inhibitors remain the identification of cancers that are sensitive to these inhibitors and optimization of the treatment strategy to obtain the maximal benefit of the drugs.
Acknowledgments
This study was supported by an NIH grant CA169186 to YJ.
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to declare.
References
- 1.Lempiainen H, Halazonetis TD. Emerging common themes in regulation of PIKKs and PI3Ks. EMBO J. 2009;28(20):3067–73. doi: 10.1038/emboj.2009.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–89. doi: 10.1016/s0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- 3.Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–75. doi: 10.1016/s0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- 4.Sarbassov DD, Ali SM, Kim DH, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–302. doi: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 5.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93. doi: 10.1016/j.cell.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nat Rev Mol Cell Biol. 2009;10(5):307–18. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 7.Jewell JL, Guan KL. Nutrient signaling to mTOR and cell growth. Trends Biochem Sci. 2013;38(5):233–42. doi: 10.1016/j.tibs.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huang J, Manning BD. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem Soc Trans. 2009;37(Pt 1):217–22. doi: 10.1042/BST0370217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sehgal SN. Sirolimus: its discovery, biological properties, and mechanism of action. Transplant Proc. 2003;35(3 Suppl):7S–14S. doi: 10.1016/s0041-1345(03)00211-2. [DOI] [PubMed] [Google Scholar]
- 10.Abraham RT, Wiederrecht GJ. Immunopharmacology of rapamycin. Annu Rev Immunol. 1996;14:483–510. doi: 10.1146/annurev.immunol.14.1.483. [DOI] [PubMed] [Google Scholar]
- 11.Lawrence JC, Lin TA, McMahon LP, et al. Modulation of the protein kinase activity of mTOR. Curr Top Microbiol Immunol. 2004;279:199–213. doi: 10.1007/978-3-642-18930-2_12. [DOI] [PubMed] [Google Scholar]
- 12.Elit L. CCI-779 Wyeth. Curr Opin Investig Drugs. 2002;3(8):1249–53. [PubMed] [Google Scholar]
- 13.Dumont FJ. Everolimus. Novartis. Curr Opin Investig Drugs. 2001;2(9):1220–34. [PubMed] [Google Scholar]
- 14.Mita M, Sankhala K, Abdel-Karim I, et al. Deforolimus (AP23573) a novel mTOR inhibitor in clinical development. Expert Opin Investig Drugs. 2008;17(12):1947–54. doi: 10.1517/13543780802556485. [DOI] [PubMed] [Google Scholar]
- 15.Kwitkowski VE, Prowell TM, Ibrahim A, et al. FDA approval summary: temsirolimus as treatment for advanced renal cell carcinoma. Oncologist. 2010;15(4):428–35. doi: 10.1634/theoncologist.2009-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carew JS, Kelly KR, Nawrocki ST. Mechanisms of mTOR inhibitor resistance in cancer therapy. Target Oncol. 2011;6(1):17–27. doi: 10.1007/s11523-011-0167-8. [DOI] [PubMed] [Google Scholar]
- 17.Huang S, Bjornsti MA, Houghton PJ. Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther. 2003;2(3):222–32. doi: 10.4161/cbt.2.3.360. [DOI] [PubMed] [Google Scholar]
- 18.Hung CM, Garcia-Haro L, Sparks CA, et al. mTOR-dependent cell survival mechanisms. Cold Spring Harb Perspect Biol. 2012;4(12):1–17. doi: 10.1101/cshperspect.a008771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Manning BD. Balancing Akt with S6K: implications for both metabolic diseases and tumorigenesis. J Cell Biol. 2004;167(3):399–403. doi: 10.1083/jcb.200408161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santoni M, Pantano F, Amantini C, et al. Emerging strategies to overcome the resistance to current mTOR inhibitors in renal cell carcinoma. Biochim Biophys Acta. 2014;1845(2):221–31. doi: 10.1016/j.bbcan.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Bitting RL, Armstrong AJ. Targeting the PI3K/Akt/mTOR pathway in castration-resistant prostate cancer. Endocr Relat Cancer. 2013;20(3):R83–99. doi: 10.1530/ERC-12-0394. [DOI] [PubMed] [Google Scholar]
- 22.Mohamed A, Krajewski K, Cakar B, et al. Targeted therapy for breast cancer. Am J Pathol. 2013;183(4):1096–112. doi: 10.1016/j.ajpath.2013.07.005. [DOI] [PubMed] [Google Scholar]
- 23.Yea SS, Fruman DA. Achieving cancer cell death with PI3K/mTOR-targeted therapies. Ann N Y Acad Sci. 2013;1280:15–8. doi: 10.1111/nyas.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dancey J. mTOR signaling and drug development in cancer. Nat Rev Clin Oncol. 2010;7(4):209–19. doi: 10.1038/nrclinonc.2010.21. [DOI] [PubMed] [Google Scholar]
- 25.Zaytseva YY, Valentino JD, Gulhati P, et al. mTOR inhibitors in cancer therapy. Cancer Lett. 2012;319(1):1–7. doi: 10.1016/j.canlet.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 26.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9(5):341–9. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutherlin DP, Bao L, Berry M, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54(21):7579–87. doi: 10.1021/jm2009327. [DOI] [PubMed] [Google Scholar]
- 28.Sutherlin DP, Sampath D, Berry M, et al. Discovery of (thienopyrimidin-2-yl)aminopyrimidines as potent, selective, and orally available pan-PI3-kinase and dual pan-PI3-kinase/mTOR inhibitors for the treatment of cancer. J Med Chem. 2010;53(3):1086–97. doi: 10.1021/jm901284w. [DOI] [PubMed] [Google Scholar]
- 29.Heffron TP, Berry M, Castanedo G, et al. Identification of GNE-477, a potent and efficacious dual PI3K/mTOR inhibitor. Bioorg Med Chem Lett. 2010;20(8):2408–11. doi: 10.1016/j.bmcl.2010.03.046. [DOI] [PubMed] [Google Scholar]
- 30.Yuan J, Mehta PP, Yin MJ, et al. PF-04691502, a potent and selective oral inhibitor of PI3K and mTOR kinases with antitumor activity. Mol Cancer Ther. 2011;10(11):2189–99. doi: 10.1158/1535-7163.MCT-11-0185. [DOI] [PubMed] [Google Scholar]
- 31.Maira SM, Stauffer F, Brueggen J, et al. Identification and characterization of NVP-BEZ235, a new orally available dual phosphatidylinositol 3-kinase/mammalian target of rapamycin inhibitor with potent in vivo antitumor activity. Mol Cancer Ther. 2008;7(7):1851–63. doi: 10.1158/1535-7163.MCT-08-0017. [DOI] [PubMed] [Google Scholar]
- 32.Knight SD, Adams ND, Burgess JL, et al. Discovery of GSK2126458, a Highly Potent Inhibitor of PI3K and the Mammalian Target of Rapamycin. ACS Med Chem Lett. 2010;1(1):39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu P, Laird AD, Du X, et al. Characterization of the activity of the PI3K/mTOR inhibitor XL765 (SAR245409) in tumor models with diverse genetic alterations affecting the PI3K pathway. Mol Cancer Ther. 2014;13(5):1078–91. doi: 10.1158/1535-7163.MCT-13-0709. [DOI] [PubMed] [Google Scholar]
- 34.Pallet N, Legendre C. Adverse events associated with mTOR inhibitors. Expert Opin Drug Saf. 2013;12(2):177–86. doi: 10.1517/14740338.2013.752814. [DOI] [PubMed] [Google Scholar]
- 35.Shor B, Gibbons JJ, Abraham RT, et al. Targeting mTOR globally in cancer: thinking beyond rapamycin. Cell Cycle. 2009;8(23):3831–7. doi: 10.4161/cc.8.23.10070. [DOI] [PubMed] [Google Scholar]
- 36.Feldman ME, Apsel B, Uotila A, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. PLoS Biol. 2009;7(2):e38. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Verheijen JC, Richard DJ, Curran K, et al. Discovery of 4-morpholino-6-aryl-1H-pyrazolo(3,4-d)pyrimidines as highly potent and selective ATP-competitive inhibitors of the mammalian target of rapamycin (mTOR): optimization of the 6-aryl substituent. J Med Chem. 2009;52(24):8010–24. doi: 10.1021/jm9013828. [DOI] [PubMed] [Google Scholar]
- 38.Malagu K, Duggan H, Menear K, et al. The discovery and optimisation of pyrido(2,3-d)pyrimidine-2,4-diamines as potent and selective inhibitors of mTOR kinase. Bioorg Med Chem Lett. 2009;19(20):5950–3. doi: 10.1016/j.bmcl.2009.08.038. [DOI] [PubMed] [Google Scholar]
- 39.Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70(1):288–98. doi: 10.1158/0008-5472.CAN-09-1751. [DOI] [PubMed] [Google Scholar]
- 40.Bhagwat SV, Gokhale PC, Crew AP, et al. Preclinical characterization of OSI-027, a potent and selective inhibitor of mTORC1 and mTORC2: distinct from rapamycin. Mol Cancer Ther. 2011;10(8):1394–406. doi: 10.1158/1535-7163.MCT-10-1099. [DOI] [PubMed] [Google Scholar]
- 41.Pike KG, Malagu K, Hummersone MG, et al. Optimization of potent and selective dual mTORC1 and mTORC2 inhibitors: the discovery of AZD8055 and AZD2014. Bioorg Med Chem Lett. 2013;23(5):1212–6. doi: 10.1016/j.bmcl.2013.01.019. [DOI] [PubMed] [Google Scholar]
- 42.Maiso P, Liu Y, Morgan B, et al. Defining the role of TORC1/2 in multiple myeloma. Blood. 2011;118(26):6860–70. doi: 10.1182/blood-2011-03-342394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schenone S, Brullo C, Musumeci F, et al. ATP-competitive inhibitors of mTOR: an update. Curr Med Chem. 2011;18(20):2995–3014. doi: 10.2174/092986711796391651. [DOI] [PubMed] [Google Scholar]
- 44.Thoreen CC, Kang SA, Chang JW, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–32. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Leung E, Kim JE, Rewcastle GW, et al. Comparison of the effects of the PI3K/mTOR inhibitors NVP-BEZ235 and GSK2126458 on tamoxifen-resistant breast cancer cells. Cancer Biol Ther. 2011;11(11):938–46. doi: 10.4161/cbt.11.11.15527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cassell A, Freilino ML, Lee J, et al. Targeting TORC1/2 enhances sensitivity to EGFR inhibitors in head and neck cancer preclinical models. Neoplasia. 2012;14(11):1005–14. doi: 10.1593/neo.121212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lou HZ, Weng XC, Pan HM, et al. The novel mTORC1/2 dual inhibitor INK-128 suppresses survival and proliferation of primary and transformed human pancreatic cancer cells. Biochem Biophys Res Commun. 2014;450(2):973–8. doi: 10.1016/j.bbrc.2014.06.081. [DOI] [PubMed] [Google Scholar]
- 48.Serova M, de Gramont A, Tijeras-Raballand A, et al. Benchmarking effects of mTOR, PI3K, and dual PI3K/mTOR inhibitors in hepatocellular and renal cell carcinoma models developing resistance to sunitinib and sorafenib. Cancer Chemother Pharmacol. 2013;71(5):1297–307. doi: 10.1007/s00280-013-2129-6. [DOI] [PubMed] [Google Scholar]
- 49.Gupta M, Hendrickson AE, Yun SS, et al. Dual mTORC1/mTORC2 inhibition diminishes Akt activation and induces Puma-dependent apoptosis in lymphoid malignancies. Blood. 2012;119(2):476–87. doi: 10.1182/blood-2011-04-346601. [DOI] [PMC free article] [PubMed] [Google Scholar]