

Abstract
Pseudomonas aeruginosa is an opportunistic bacterial pathogen capable of causing a wide range of disease manifestations, including severe bacterial pneumonia. Recently, clinics have reported a rise in nosocomial infections with multidrug resistant (MDR) species, including MDR strains of P. aeruginosa. In order to quickly evaluate the efficacy of new therapeutics for MDR infections, highly reproducible and validated animal models need to be developed for pre-clinical testing. Here, we describe the characterization of two murine models to study MDR P. aeruginosa respiratory disease. We evaluated and compared these models using a non-invasive intratracheal instillation method and established the 50% lethal dose, course of infection, biometric parameters of disease and degree of pneumonia development for each model. Further, we tested meropenem as a proof-of-concept therapeutic and report efficacy data that suggests that the leukopenic model could serve a robust pre-clinical model to test novel therapeutics.
Keywords: MDR Pseudomonas aeruginosa, intubation-mediated intratracheal (IMIT) inoculation, murine respiratory disease, biometric endpoint criteria, therapeutic model
This article describes the development of a respiratory disease model for the testing of therapeutics against multidrug resistant Pseudomonas aeruginosa.
Graphical Abstract Figure.

This article describes the development of a respiratory disease model for the testing of therapeutics against multidrug resistant Pseudomonas aeruginosa.
INTRODUCTION
The Gram-negative bacterium Pseudomonas aeruginosa causes opportunistic infections of the skin, eyes, urinary tract, lungs and blood of humans. A frequent commensal inhabitant of skin, this species is known to readily infect immunocompromised individuals, the severely burned and those with the genetic disorder cystic fibrosis (CF). Up to 70% of CF patients are colonized at one time by P. aeruginosa which can cause acute or chronic infections in the lower respiratory tract. It is also responsible for 10–20% of nosocomial infections in hospital ICUs (Carmeli et al. 1999; Sader et al. 2014). P. aeruginosa is readily isolated from many soils, freshwaters and other moist environments making it difficult to eradicate. To date, there is no vaccine for this species.
P. aeruginosa is well-known for its ability to form biofilms in the environment, the lab, and clinically. The biofilms consist of matrix proteins, polysaccharides and extracellular DNA which serve to bind and protect individual cells from predation and environmental stresses. These biofilms are often difficult to treat clinically as they repel many antibiotics (Abdi-Ali, Mohammadi-Mehr and Agha Alaei 2006; Tseng et al. 2013) and immune cells (Jesaitis et al. 2003). This species encodes numerous Type III effectors for injecting toxins into host cells (Yahr et al. 1998; Shaver and Hauser 2004) and quorum sensing systems which modulate virulence properties (Gambello and Iglewski 1991; Ochsner et al. 1994). P. aeruginosa also has a large genome containing many genes responsible for the high levels of intrinsic antibacterial resistance observed in this species (Stover et al. 2000; Wei et al. 2011). In fact, many Pseudomonas strains are capable of metabolizing antibiotics as a sole carbon source (Dantas et al. 2008).
Recently, multidrug resistant (MDR) strains of P. aeruginosa have been observed both clinically and in the environment at rates of up to 36% of isolates tested being resistant to multiple antibiotics (Biswal et al. 2014). The development of multidrug resistance amongst P. aeruginosa isolates is associated with poor clinical outcome, an increased likelihood of inappropriate antibiotic therapy, as well as significant increases in mortality, particularly in cases with pneumonia-associated bacteremia (Tam et al. 2010). Thus, the increasing advent of MDR P. aeruginosa isolates represents a growing concern in healthcare settings for nosocomial disease, and particularly for susceptible populations, including patients with CF.
Bacterial pathogens are rapidly adapting to the modern use of antibiotics in clinical settings to become resistant to such therapies, leaving critical gaps in our ability to maintain efficacious treatment options. As new potential antibiotics are being discovered, it is imperative that standardized and well-characterized animal models are in place to rapidly perform pre-clinical testing of new therapeutics. Toward this goal, we tested two different animal models for susceptibility to P. aeruginosa lung infection, development of bacterial pneumonia, and biometric measurements that could be used as indicators of disease outcome. Subsequently, we determined the potential utility of these models to test the efficacy of antibiotic treatment through developing a platform system based on carbapenem treatment of a clinical P. aeruginosa isolate.
MATERIALS AND METHODS
Bacterial strains and media
P. aeruginosa UNC-D is a sputum isolate from a CF patient that is resistant to ceftazidime [32 μg/ml], meropenem [8 μg/ml], imipenem [16 μg/ml] and tobramycin [32 μg/ml], and sensitive to piperacillin [16 μg/ml], aztreonam [4 μg/ml] and colistin [1 μg/ml] (National Committee for Clinical Laboratory Standards 2014; kind gift from Dr Peter Gilligan, University of North Carolina). Bacteria were routinely cultured on trypticase soy broth (TSB) and trypticase soy agar (TSA) plates and in Lennox broth (LB) at 37°C with shaking of broth cultures. Stocks of UNC-D were prepared by single colony isolation of bacteria on TSA plates and subsequent broth culture in TSB at 37°C with shaking before –80°C cryopreservation in 25% glycerol (final concentration). Bacteria were prepared for animal infection studies by culturing TSA plate-grown bacteria in LB overnight and washing the bacteria into PBS. OD600-based estimates of bacterial concentration allowed accurate dilutions of bacterial inoculum to be prepared in a 50 μl delivery dose. All bacterial concentrations were accurately identified by plate counting on LB agar plates.
Mouse husbandry and induction of leukopenia
All animal studies were approved by the University of Louisville Institutional Animal Care and Use Committee (IACUC no. 13064). Six week old female BALB/cJ mice (Jackson Laboratories, Bar Harbor, ME) were acclimatized on site for eight days prior to bacterial challenge. Upon arrival, an identification/temperature transponder (Biomedic Data Systems; Seaford, DE) was implanted subcutaneously (s.c.) at the scruff of the neck. For the leukopenic model, mice were injected with cyclophosphamide (Cy) (Sigma) into the intraperitoneal (i.p.) cavity at 150 mg/kg on days –5 and –3 prior to bacterial challenge. Cy-treated mice were provided supportive care in the form of Napa nectar and 1 ml injections of saline administered s.c. on days –3, –2 and –1 prior to bacterial challenge. Blood collection was conducted on days –5, –1 and +3 by submandibular bleed to characterize leukopenia by complete blood counts (CBC) analysis (Hemavet 950, Drew Scientific).
Respiratory challenge with P. aeruginosa
Mice were challenged with saline suspensions of P. aeruginosa by intubation-mediated intratracheal inoculation (IMIT) as described (Lawrenz et al. 2014). Briefly, mice were anesthetized with isoflurane and 10 μl of a 2% lidocaine solution was applied to the back of their throat to suppress laryngeal spasm during intubation. The bacterial inoculum was loaded in to a 250 μl Hamilton Gastight syringe fit with a 22 G blunt needle, loading 150 μl of dead volume air, followed by 50 μl of bacterial saline suspension. Mice were intubated with a 20 G catheter before inserting a 22 G blunt needle into the lumen of the catheter to dispense the 50 μl bacterial suspension followed by 150 μl of air to aid distribution through the lung.
During the course of infection, temperature, heart rate and SpO2 were monitored every 8 h (q8h). Temperature was monitored via implanted transponders while heart rate and SpO2 were monitored using a MouseOx/collar sensor (Starr Life Sciences; Oakmont, PA). Studies were concluded at day 7 post-infection. Immunocompetent mice were provided supplemental heating during the first 24 h of infection by placing cages on 37°C water mats in response to the transient hypothermia observed in this model.
Euthanasia and necropsy
Mice were euthanized at moribund disease, or at day 7 post-infection, using carbon dioxide asphyxia. Immediately thereafter, exsanguination was performed by cardiac puncture, and blood was aliquoted into Li heparin tubes for clinical chemistry analysis (Comprehensive Metabolic Panel, Piccolo Xpress, Abaxis). A representative section of lung was excised, fixed in 10% neutral buffered formalin for 24 h, and submitted to Colorado Histoprep in 70% ethanol for hematoxylin and eosin (H&E) staining. Stained sections were scored by a Board Certified Veterinary Pathologist and compared to naïve control lungs. Pathology scoring was made on a four point, four criteria system with a maximum score of 16 points; tissues were scored in the areas of inflammation, infiltrate, necrosis and other (including hemorrhage). Points were assigned based on: no significant finding (0), minimal (1), mild (2), moderate (3) and severe (4) pathology. Representative H&E pathology images were captured using a 10X objective on a Zeiss Imager A1 with an AxioCam ICc 5. The remaining lung and the spleen were homogenized in PBS, treated with 1% Triton X-100 (final concentration) and serially diluted in PBS to enumerate bacterial burdens in tissue (Lawrenz et al. 2014).
Therapeutic intervention
Meropenem (Hospira) solutions were prepared fresh daily and diluted appropriately to administer the target dose in a 100 μl delivery volume in PBS q8h, and alternatively mock treated groups were injected with PBS vehicle. Therapeutics were administered s.c. to mice beginning 3 h post-infection using a 29 G needle/insulin syringe. Therapy continued for 5 days (15 total treatments).
Statistical analyses
Survival analyses, bacterial tissue burden and lung pathology statistics were conducted in Graphpad Prism using Log-Rank, One-way ANOVA (Tukey post-test) or unpaired student t-test as indicated. The 50% lethal dose (LD50) and 50% effective dose (ED50) were calculated by Probit (log normal distribution) analysis in StatPlus.
RESULTS
IMIT installation of BALB/c mice with MDR P. aeruginosa establishes a fatal pneumonia
To determine if IMIT instillation of P. aeruginosa could establish a lethal pneumonia in mice, seven week old female BALB/c mice were inoculated via IMIT and monitored for signs of infection (changes in temperature, heart rate and O2 levels) and survival over a 7 day period. Preliminary studies indicated that a dose >107 CFU was required to establish lethal infection with UNC-D via IMIT. Therefore, mice were inoculated with doses between 107 to 108 CFU. All mice, regardless of dose, demonstrated significant hypothermia 3 h post-instillation, but recovered to normal temperature ranges by 11 h, with the exception of three animals receiving the highest dose, whose temperatures increased but did not completely return to the normal range (Fig. 1A). A similar drop in temperature was not observed in animals that were instilled with PBS only. A second drop in temperature was observed in infected mice at ∼24 h post-instillation. Temperatures of mice receiving the highest dose (107.84 CFU) continued to decrease until they eventually reached euthanasia criteria (mean time to death = 43 h; Fig. 2A). This was in contrast to the mice receiving the lowest dose (107.09 CFU), which all survived the infection. In this group, temperatures began to rebound within 32 h post-instillation and remained within the normal range until the end of the observation period. Temperatures of animals receiving intermediate doses (107.59 and 107.34 CFU) followed the same trend, with temperatures returning to normal ranges in animals that eventually survived the infection or continuing to drop in animals that succumbed to infection. While more variable, changes in heart rate and blood O2 levels of infected animals also directly correlated to survival, with decreasing tends in both biometrics in animals that would eventually succumb to infection (Fig. 1B and C). For this model, the LD50 for UNC-D was calculated to be 107.44 CFU (Fig. 2A). UNC-D shares an exoS+/exoU− genotype with the commonly used lab strain of P. aeruginosa, PA01, and we therefore performed parallel LD50 assessment of the PA01 strain in this BALB/c respiratory model (Fig. 3A), which was found to be 106.59 CFU. Thus, the MDR UNC-D clinical isolate possesses approximately 7-fold less virulence potential than the PA01 lab strain.
Figure 1.

Biometrics of immunocompetent mice during P. aeruginosa UNC-D infection. BALB/c mice (n = 8) were inoculated with 107.84 (blue), 107.59 (green), 107.34 (red) or 107.09 (black) CFU of P. aeruginosa UNC-D via IMIT, and (A) temperature, (B) heart rate (BPM) and (C) concentration of blood oxygen (% O2) were monitored for 144 h. Each line represents an individual animal. Solid color symbols represent animals that survived the entire course of infection. Open symbols represent animals that succumbed to infection. The gray shaded area represents the mean ±3 standard deviations of the baseline values for each biometric. Data are representative of two independent experiments.
Figure 2.

Susceptibility of immunocompetent mice to P. aeruginosa UNC-D respiratory challenge. BALB/c mice (n = 8) were challenged with 107.84 (blue), 107.59 (green), 107.34 (red) or 107.09 (black) CFU of P. aeruginosa UNC-D via IMIT and (A) monitored for survival. MTD indicated in parenthesis; U = undefined. (B) At the time of euthanasia, lungs were harvested and severity of lung pathology was scored (ANOVA with Tukey: * = P < 0.05; *** = P < 0.001). Solid circles represent animals that survived the entire course of infection. Open circles represent moribund animals that were euthanized. Bar represents the mean. (C) Comparison of lung pathology between animals that met euthanasia criteria [E, open circles] or survived [S, solid circles] to the end of the study (Unpaired student's t-test: ***P < 0.001). (D) Representative lungs (H&E stain) from each group. Data are representative of two independent experiments.
Figure 3.

Susceptibility of BALB/c mice to P. aeruginosa PA01 respiratory challenge. BALB/c mice (n = 8) were infected with 107.08 (red) or 106.09 (black) CFU of P. aeruginosa PA01 and (A) monitored for survival. MTD indicated in parenthesis; U = undefined. The LD50 was calculated by the method of Reed and Muench to be 106.59 CFU. (B) At the time of euthanasia, lungs were harvest and pathology was determined. Solid circles represent animals that survived the entire course of infection and open cirlces represent moribund animals that were euthanized. Bar represents the mean.
To define the development of pneumonia in these animals, lungs were harvested at the time of euthanasia (either moribund animals or at 165 h post-instillation for animals that survived infection), and tissue sections were blindly analyzed and scored for inflammation and pathology (Fig. 2B–D). Lungs from animals instilled with 107.09 CFU did not demonstrate signs of inflammation and did not appear significantly different from uninfected PBS control animals. Increasing the dose to 107.34 resulted in the development of pronounced lung inflammation, and the severity of inflammation continued to increase as the dose of UNC-D increased (Fig. 2B). Independent of dose, the severity of lung pathology directly correlated with disease outcome, with significantly higher pathology in animals that succumbed to infection compared to those that survived to the end of the experiment (Fig. 2C, P<0.0001). Together these data demonstrate that IMIT instillation of UNC-D into healthy BALB/c mice can establish a lethal infection that is highlighted by hypothermia prior to death and the development of pneumonia. These findings are consistent with the description of human pneumonia caused by P. aeruginosa, including microabscesses with focal hemorrhage and periodic evidence of necrosis (Bonifacio, Kitterman and Ursell 2003).
Treatment with meropenem limits MDR P. aeruginosa growth in the lungs but does not protect immunocompetent mice from lethal infection
To determine the utility of the BALB/c model in testing potential therapeutics against MDR P. aeruginosa, mice were instilled via IMIT with a lethal dose of UNC-D (108.4 CFU) and treated s.c. with the antibiotic meropenem q8h beginning 3 h post-instillation. Meropenem was selected as a clinically important carbapenem used for treating P. aeruginosa infections, to which the UNC-D test strain is more sensitive (MIC 8 μg/ml) than another commonly used carbapenem, imipenem (MIC 16 μg/ml). Meropenem was administered at doses equivalent to human doses of 5.5, 7.0, 8.5 and 10 g/day (1000, 1250, 1500 and 1720 mg/kg/day, respectively) (Center for Drug Evaluation and Research (CDER) 2005; Reagan-Shaw, Nihal and Ahmad 2008). Mice were monitored for signs of infection and survival for 7 days, and tissue samples were harvested at euthanasia for bacterial enumeration and pathological analysis. Mock treated animals (PBS) succumbed to infection as expected with a mean time to death of 47 h (Fig. 4A). Treatment with meropenem, regardless of dose, did not significantly alter the outcome of the infection or the mean time to death (Fig. 4A). However, in spite of no change in survival, meropenem treatment >1,000 mg/kg/day significantly decreased the bacterial load in the lungs (Fig. 4B) to a similar level as bacterial numbers recovered from mice infected with a non-lethal dose of UNC-D (107.09 CFU challenge resulted in lung burden of 104.50 ± 101.83 CFU). Dissemination to the spleen was not a prominent feature of the immunocompetent model, as even mock treated mice splenic burdens were at or close to the limit of detection (Fig. 4C), thus we were unable to define a role for meropenem in protection against dissemination from the lung. Interestingly, while bacterial loads were lower in the lungs, we observed significantly more severe lung pathology in animals that received meropenem treatment than the mock treated animals (Fig. 4D), in spite of similar mean time to death (MTD) values amongst the groups. Together, these data indicate that in spite of limiting bacterial numbers in the lung, meropenem was unable to protect mice from mortality associated with bacterial-induced pneumonia.
Figure 4.

Efficacy of meropenem in immunocompetent mice challenged with P. aeruginosa UNC-D. BALB/c mice (n = 8) were challenged with 108.4 CFU of UNC-D and mock treated (PBS, black) or treated with meropenem beginning 3 h post-infection with 1750 (violet), 1500 (blue), 1250 (green) or 1000 (red) mg/kg/day and (A) monitored for survival. MTD indicated in parenthesis. At the time of euthanasia, tissues were harvest and bacteria were enumerated from the (B) lungs and (C) spleen (One-way ANOVA with Tukey: * = P < 0.05; *** = P < 0.001). (D) Severity of lung pathology at the time of euthanasia (Unpaired Student's t-test: * = P < 0.05). (B–D) Solid circles represent animals that survived the entire course of infection. Open circles represent moribund animals that were euthanized. X represents animals that succumbed to infection and tissues were not harvested. Bar represents the mean. Dotted line represents the limit of detection. Data are representative of two independent experiments.
Leukopenia decreases the LD50 for MDR P. aeruginosa
While meropenem treatment appeared to limit bacterial numbers in BALB/c mice, we did not observe a correlation between meropenem efficacy and improved survival. Another common model used to study P. aeruginosa and other opportunistic infectious diseases is the leukopenic mouse model (Scarff and Goldberg 2008; Louie et al. 2013). Therefore, we sought to characterize a chemically-induced leukopenic model for IMIT instillation of MDR P. aeruginosa. To induce leukopenia, BALB/c mice were administered Cy i.p. on days –5 and –3 prior to instillation with UNC-D. Blood samples were harvested on day –1, and CBCs were performed (Table 1). As expected, white blood cell (WBC) counts were significantly depleted after this treatment, with ≥80% depletion for all WBC populations. Treatment did not result in anemia, as red blood cell counts were minimally impacted (Table 1). However, platelet populations were marginally impacted with ∼19% decrease in total platelet numbers.
Table 1.
Impact of Cy on CBC.
| Pre-treatment | Post-treatment | Percent decrease | |
|---|---|---|---|
| White blood cells (per μL) | |||
| WBC (×103) | 6.41 (±1.85) | 0.78 (±0.41) | 88% |
| NE (×103) | 1.33 (±0.54) | 0.07 (±0.09) | 94% |
| LY (103) | 5.02 (±1.78) | 0.63 (±0.28) | 88% |
| MO (×103) | 0.29 (±0.16) | 0.06 (±0.03) | 80% |
| Red blood cells (per μL) | |||
| RBC (×106) | 9.31 (±1.08) | 8.75 (±0.61) | 6% |
| HCT% | 53.10 (±8.07) | 52.40 (±3.54) | 1% |
| PLT (×103) | 798.72 (±228.55) | 643.00 (±149.88) | 19% |
White blood cells (WBC), neutrophils (NE), lymphocytes (LY), monocytes (MO), red blood cells (RBC), hematocrit (HTC) and platelets (PLT).
One day after CBCs were performed, mice were inoculated via IMIT with UNC-D and monitored for signs of disease and survival over a 7 day period. Doses of UNC-D ranged from 103.34 to 105.58 CFU in order to establish an accurate LD50. Unlike the immunocompetent model, mice did not demonstrate hypothermia 3 h post-inoculation in this model using these infectious doses (Fig. 5A), suggesting that the early hypothermia seen in the previous model may be a result of the high inoculation dose. Temperatures of mice that eventually succumbed to infection began to decrease ∼27 h post-inoculation, which was a similar time frame observed for the second temperature decrease observed in the immunocompetent model (Fig. 1A). However, significant changes in temperature for leukopenic animals that survived infection were not observed. Similar to the immunocompetent model, decreases in heart rate and blood O2 levels correlated with disease outcome, with the exception of the 105.58 dose, but were also more variable than temperature (Fig. 5B and C). The LD50 in the neutropenic model was calculated to be 104.56 CFU (Fig. 6A). As a control, PA01 was examined in the leukopenic model, and the LD50 was found to be <102.84 CFU (Fig. 7A), thus the virulence potential of UNC-D is less than that of PA01 in the leukopenic model, consistent with our finding in the immunocompetent BALB/c model.
Figure 5.
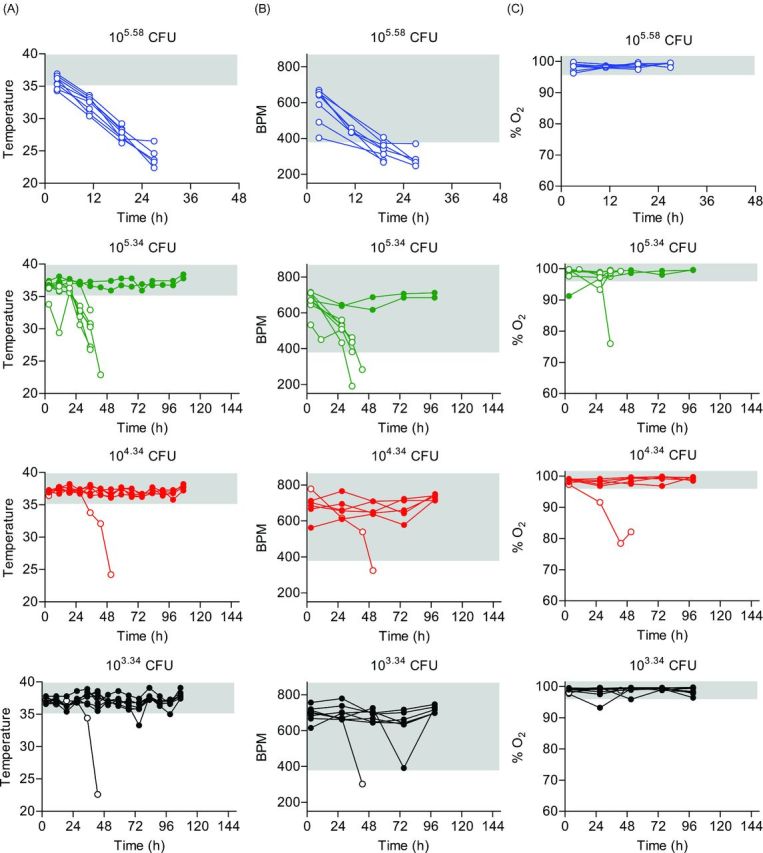
Biometrics of leukopenic mice during P. aeruginosa UNC-D infection. Cy-treated BALB/c mice (n = 8) were inoculated with 105.58 (blue), 105.34 (green), 104.34 (red) or 103.34 (black) CFU of P. aeruginosa UNC-D via IMIT and (A) temperature, (B) heart rate (BPM) and (C) concentration of blood oxygen (% O2) were monitored for 144 h. Each line represents an individual animal. Solid circles represent animals that survived the entire course of infection. Open circles represent animals that succumbed to infection. The gray shaded area represents the mean ±3 standard deviations of the baseline values for each biometric. Data are representative of two independent experiments.
Figure 6.

Susceptibility of leukopenic mice to P. aeruginosa UNC-D respiratory challenge. Cy-treated BALB/c mice (n = 8) were inoculated with 105.58 (blue), 105.34 (green), 104.34 (red) or 103.34 (black) CFU of P. aeruginosa UNC-D via IMIT and (A) monitored for survival. MTD indicated in parenthesis; U = undefined. (B) At the time of euthanasia, lungs were harvested and severity of lung pathology was scored (ANOVA with Tukey: * = P < 0.05; ** = P < 0.01). Solid circles represent animals that survived the entire course of infection. Open circles represent moribund animals that were euthanized. X represents animals that succumbed to infection and tissues were not harvested. Bar represents the mean. (C) Representative lungs (H&E stain) from each group. (D) Comparison of endpoint lung bacterial burden of leukopenic (Leuko) and immunocompetent (BALB/c) receiving an ∼LD100 of UNC-D (105.58 and 107.84 CFU, respectively). Data are representative of two independent experiments.
Figure 7.

Susceptibility of BALB/c mice to P. aeruginosa PA01 respiratory challenge. Cy-treated BALB/c mice (n = 8) were challenged with 102.84 CFU of P. aeruginosa PA01 and (A) monitored for survival. MTD indicated in parenthesis. (B) At the time of euthanasia, lungs were harvest and pathology was determined. Open circles represent moribund animals that were euthanized. Bar represents the mean.
Histopathological analysis of the lungs revealed dramatically less pathology compared to the immunocompetent BALB/c model (Fig. 6B and C). Furthermore, there were no significant differences between lungs from mice instilled with 103.34, 104.34 or 105.34 CFU compared to mock infected controls (Fig. 6B and C), even for animals that eventually succumbed to infection. However, pathology was observed in animals receiving 105.58 CFU, where all animals succumbed to infection, but pathology was still significantly lower than animals that succumbed to infection in the immunocompetent model (Fig. 6B compared to Fig. 2B; P<0.05, student's t-test). To determine if the decreased pathology was correlated with lower bacterial numbers in the lungs of leukopenic mice, bacterial numbers were enumerated from the lungs of moribund leukopenic mice instilled with 105.58 CFU and compared to moribund immunocompetent mice instilled with 107.84 bacteria (Fig. 6D). Tissues from both models had equivalent bacterial loads indicating that bacterial loads were not responsible for differences in pathology. Together these data demonstrate that IMIT instillation of MDR P. aeruginosa into leukopenic mice induces a lethal infection with an LD50 ∼1 000-fold lower than that for the immunocompetent BALB/c model, but infection appeared to result in less severe lung pathology.
Treatment with meropenem protects leukopenic mice from lethal MDR P. aeruginosa infection
To determine if the leukopenia model had a better correlation between meropenem efficacy and disease outcome, leukopenic mice were infected with a lethal dose (105.5 CFU) of UNC-D via IMIT and monitored for signs of disease and survival for 7 days. Three hours after infection, mice were administered meropenem q8h via s.c. injection. PBS mock-treated leukopenic animals succumbed to infection with a MTD of 43 h (Fig. 8A). Treatment with meropenem up to 1500 mg/kg/day slightly increased the MTD of the mice by delaying moribund disease by 16–28 h, but ultimately did not alter the outcome of the infection, as all mice in these groups succumbed to infection by 72 h post-infection. However, treatment with 1750 mg/kg/day significantly altered the course of the infection and protected animals from lethal infection (P = 0.0002). Increased survival directly correlated with bacterial loads in the lungs and spleen and was significantly lower in the 1750 mg/kg/day group (Fig. 8B and C). Furthermore, 1750 mg/kg/day treatment reduced the bacterial burden below the limit of detection in 80% of animals that survived to 7 days post-infection, suggesting that sterile clearance was achieved. The ED50 of meropenem was calculated to be 1736 mg/kg/day. Surprisingly, treatment with meropenem, regardless of dose, significantly increased lung pathology in leukopenic mice compared to mock treated animals (Fig. 8D). This trend was also observed in the 1750 mg/kg/day group, but this group showed less pathology than the groups receiving lower doses of meropenem. While bacteria were not enumerated from liver, we used clinical chemistries to measure tissue damage in the liver by changes in alanine transaminase (ALT) and aspartate transaminase (AST) activity. Mice protected with 1750 mg/kg/day meropenem had baseline ALT (Fig. 8E) and AST (Fig. 8F) levels while mock and sub-efficacious meropenem treatment groups exhibited increased evidence of liver damage. Thus, consistent with the spleen data demonstrating lack of disseminated disease, reduced tissue damage in the liver was evident when animals are protected by meropenem.
Figure 8.

Efficacy of meropenem in leukopenic mice challenged with P. aeruginosa UNC-D. Cy-treated BALB/c mice (n = 8) were challenged with 105.4 CFU of UNC-D and mock treated (PBS, black) or treated with meropenem beginning 3 h post-infection with 1750 (violet), 1500 (blue), 1250 (green) or 1000 (red) mg/kg/day and (A) monitored for survival. MTD indicated in parenthesis; U = undefined. At the time of euthanasia, tissues were harvest and bacteria were enumerated from the (B) lungs and (C) spleen. Dotted line represents the limit of detection. (D) Severity of lung pathology at the time of euthanasia. Clinical chemistries were performed on blood to evaluate (E) ALT or (F) AST markers of liver damage, with baseline enzyme values indicated by a horizontal dashed line. (B–F) Solid circles represent animals that survived the entire course of infection. Open circles represent moribund animals that were euthanized. X represents animals that succumbed to infection and tissues were not harvested. Bar represents the mean. (One-way ANOVA with Tukey: * = P < 0.05; ** = P < 0.01; *** = P < 0.001). Data are representative of two independent experiments.
DISCUSSION
The prevalence of MDR infections seen in hospitals has been on the rise, and new therapeutics is needed to fight these infections. Our goal in these studies was to develop a small animal model that could be used to reproducibly screen novel antimicrobial compounds, or combination therapeutics, against MDR P. aeruginosa respiratory disease. Our selected MDR strain exhibits moderate resistance to meropenem, thus our selection of meropenem as a reference antibiotic in these studies allows for an ideal benchmark for future testing of novel compounds, or potentially novel combination therapies which may enhance the efficacy of meropenem. We characterized both an immunocompetent and a leukopenic model of lung infection with an MDR clinical isolate of P. aeruginosa using a non-invasive intratracheal delivery method. We chose to use the IMIT method because it provides a highly reproducible and accurate instillation of bacteria directly to the lungs (>98% of inoculum is delivered to the lungs; Lawrenz et al. 2014) while avoiding potential complications associated with surgical intratracheal techniques. Furthermore, IMIT instillation can be rapidly performed (under 3 min per mouse) allowing for studies with large groups of mice to be conducted rapidly. These properties of IMIT delivery lend themselves to GLP-type studies requiring highly reproducible model systems, which is an important consideration for pre-clinical therapeutic models.
An important step in development of these models was defining appropriate and non-subjective endpoint criteria for the models. We monitored several biometrics, including animal temperature, heart rate and blood oxygen levels, to define those indicators that accurately predicted outcome of infection. In all cases, temperature was the earliest indicator of infection. All mice receiving a lethal dose of P. aeruginosa demonstrated hypothermia within the first 36 h post-infection. We established for both models that animals that reached a temperature of 26.6°C (80°F) would succumb to infection within the next 8 h and set this temperature as our primary endpoint criteria. Heart rate and oxygen levels were less consistent and did not begin to change until later during the course of infection, but we also observed a trend that if the heart rate reached 300 bpm or oxygen levels reached 70%, the animals would succumb to infection within the next 8 h. Based on these observations, we established the following health monitoring schedule and end point criteria. Temperature should be monitored q8h beginning 3 h after bacterial instillation, and heart rate and oxygen levels should be initially monitored every 24 h. However, once an animal's temperature drops below 32.2 °C (90°F) then heart rate and oxygen levels should be monitored q8h. Animals should be humanely euthanized if any one of four endpoint criteria is observed: (1) temperature ≤26.6°C, (2) heart rate ≤300 bpm, (3) oxygen levels ≤70%, or (4) animal has lost its righting reflex. The only exception to these criteria was within the first 8 h after infection in the immunocompetent model where we observed short term hypothermia that all mice recovered from (Fig. 1A). While the precise mechanism of this early hypothermia is not clear, it is likely a direct response to the high doses of bacteria required to establish infection, as we did not observe this hypothermia when using lower doses in preliminary studies. We found that using this criteria we could avoid unnecessary discomfort to moribund animals without euthanizing any animals that would eventually recover from infection.
We observed key differences between the immunocompetent and leukopenic models with respect to their response to therapeutic intervention. While meropenem treatment was able to limit bacterial growth and dissemination in both models, we only observed protection from lethal infection in leukopenic mice. We also observed much more severe pathology in the lungs of the immunocompetent mice, in spite of similar bacterial loads. We demonstrated that the large challenge dose of P. aeruginosa required to establish lethal infection in the immunocompetent model was successfully controlled by meropenem, but that the result of this treatment ultimately resulted in host morbidity. Thus, we caution the use of the immunocompetent model for therapeutic studies with P. aeruginosa due to the apparent inability of therapies to improve disease outcome against large challenge doses. Conversely, the leukopenic model requires a reduced inoculum to establish disease and we were able to demonstrate full clearance of UNC-D from the majority of surviving mice in the lung and spleen in the leukopenic model, in spite of the moderate resistance of this MDR clinical isolate to meropenem.
Carbapenem antibiotics represent an important class of antibiotics for treating P. aeruginosa infections, and meropenem was selected for use in these studies given that our MDR P. aeruginosa strain UNC-D is more sensitive to meropenem than another clinically important carbapenem, imipenem. Meropenem is used clinically with a maximum recommended daily dose of 6 g in an average human weighing 60 kg (100 mg/kg/day). Translating human dosing to surrogate animal models involves use of conversion factors which account for differences in metabolism, drug clearance rates and blood volume (Center for Drug Evaluation and Research (CDER) 2005; Reagan-Shaw, Nihal and Ahmad 2008). For mice, the 100 mg/kg/day human equivalent dose equates to a 1233 mg/kg/day maximum recommended dose. Given that meropenem is not recommended for use in humans with P. aeruginosa strains possessing an MIC >4 μg/ml, our identification of an ED50 of 1736 mg/kg/day for UNC-D is consistent with the recommendations that meropenem alone would not appropriate for clinical treatment of UNC-D whose MIC is 8 μg/ml. Our model therefore provides an excellent platform for investigation of novel therapeutics which would, in combination therapy with meropenem, increase meropenem efficacy to where it could be dosed at clinically relevant levels (1233 mg/kg/day equivalent in mice) for the treatment of MDR pathogens.
In conclusion, we have developed and validated two lung-specific murine disease models for MDR P. aeruginosa infections. Furthermore, head–head comparison of these two models using meropenem as a proof-of-concept suggests that the leukopenic model can serve a robust pre-clinical system to facilitate rapid testing of novel investigational drugs to combat MDR P. aeruginosa lung infections.
Acknowledgments
We thank Dr Peter Gilligan, PhD from the University of North Carolina School of Medicine for generously providing the UNC-D clinical isolate.
FUNDING
This work was supported by National Institutes of Health [contract HHSN272201000033I-HHSN27200001-Task A52].
Conflict of interest. None declared.
REFERENCES
- Abdi-Ali A, Mohammadi-Mehr M, Agha Alaei Y. Bactericidal activity of various antibiotics against biofilm-producing Pseudomonas aeruginosa. Int J Antimicrob Ag. 2006;27:196–200. doi: 10.1016/j.ijantimicag.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Biswal I, Arora BS, Kasana D, et al. Incidence of multidrug resistant Pseudomonas aeruginosa isolated from burn patients and environment of teaching institution. J Clin Diagn Res. 2014;8:DC26–9. doi: 10.7860/JCDR/2014/7483.4383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifacio SL, Kitterman JA, Ursell PC. Pseudomonas pneumonia in infants: an autopsy study. Hum Pathol. 2003;34:929–38. doi: 10.1016/s0046-8177(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Carmeli Y, Troillet N, Eliopoulos GM, et al. Emergence of antibiotic-resistant Pseudomonas aeruginosa: comparison of risks associated with different antipseudomonal agents. Antimicrob Agents Ch. 1999;43:1379–82. doi: 10.1128/aac.43.6.1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Center for Drug Evaluation and Research (CDER) Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers. Rockville, MD: Food and Drug Administration; 2005. [Google Scholar]
- Dantas G, Sommer MO, Oluwasegun RD, et al. Bacteria subsisting on antibiotics. Science. 2008;320:100–3. doi: 10.1126/science.1155157. [DOI] [PubMed] [Google Scholar]
- Gambello MJ, Iglewski BH. Cloning and characterization of the Pseudomonas aeruginosa lasR gene, a transcriptional activator of elastase expression. J Bacteriol. 1991;173:3000–9. doi: 10.1128/jb.173.9.3000-3009.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesaitis AJ, Franklin MJ, Berglund D, et al. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol. 2003;171:4329–39. doi: 10.4049/jimmunol.171.8.4329. [DOI] [PubMed] [Google Scholar]
- Lawrenz MB, Fodah RA, Gutierrez MG, et al. Intubation-mediated intratracheal (IMIT) instillation: a noninvasive, lung-specific delivery system. J Visualized Exp. 2014;17:e52261. doi: 10.3791/52261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louie A, Liu W, Fikes S, et al. Impact of meropenem in combination with tobramycin in a murine model of Pseudomonas aeruginosa pneumonia. Antimicrob Agents Ch. 2013;57:2788–92. doi: 10.1128/AAC.02624-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Committee for Clinical Laboratory Standards. Performance Standards for Antimicrobial Susceptibility Testing. M100-S24. Wayne, PA: Clinical Laboratory Standards Institute; 2014. [Google Scholar]
- Ochsner UA, Koch AK, Fiechter A, et al. Isolation and characterization of a regulatory gene affecting rhamnolipid biosurfactant synthesis in Pseudomonas aeruginosa. J Bacteriol. 1994;176:2044–54. doi: 10.1128/jb.176.7.2044-2054.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. Fed Am Soc Exp Biol J. 2008;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Sader HS, Farrell DJ, Flamm RK, et al. Antimicrobial susceptibility of Gram-negative organisms isolated from patients hospitalized in intensive care units in United States and European hospitals (2009–2011) Diagn Micr Infec Dis. 2014;78:443–8. doi: 10.1016/j.diagmicrobio.2013.11.025. [DOI] [PubMed] [Google Scholar]
- Scarff JM, Goldberg JB. Vaccination against Pseudomonas aeruginosa pneumonia in immunocompromised mice. Clin Vaccine Immunol. 2008;15:367–75. doi: 10.1128/CVI.00419-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaver CM, Hauser AR. Relative contributions of Pseudomonas aeruginosa ExoU, ExoS, and ExoT to virulence in the lung. Infect Immun. 2004;72:6969–77. doi: 10.1128/IAI.72.12.6969-6977.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature. 2000;406:959–64. doi: 10.1038/35023079. [DOI] [PubMed] [Google Scholar]
- Tam VH, Rogers CA, Chang KT, et al. Impact of multidrug-resistant Pseudomonas aeruginosa bacteremia on patient outcomes. Antimicrob Agents Ch. 2010;54:3717–22. doi: 10.1128/AAC.00207-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng BS, Zhang W, Harrison JJ, et al. The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycin. Environ Microbiol. 2013;15:2865–78. doi: 10.1111/1462-2920.12155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Q, Tarighi S, Dötsch A, et al. Phenotypic and genome-wide analysis of an antibiotic-resistant small colony variant (SCV) of Pseudomonas aeruginosa. PLoS ONE. 2011;6:e29276. doi: 10.1371/journal.pone.0029276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahr TL, Vallis AJ, Hancock MK, et al. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. P Natl Acad Sci USA. 1998;95:13899–904. doi: 10.1073/pnas.95.23.13899. [DOI] [PMC free article] [PubMed] [Google Scholar]