

Abstract
Chimeric antigen receptor (CAR) T cells are engineered constructs composed of synthetic receptors that direct T cells to surface antigens for subsequent elimination. Many CAR constructs are also manufactured with elements that augment T-cell persistence and activity. To date, CAR T cells have demonstrated tremendous success in eradicating hematological malignancies (e.g., CD19 CARs in leukemias). This success is not yet extrapolated to solid tumors, and the reasons for this are being actively investigated. Here in this mini-review, we discuss some of the key hurdles encountered by CAR T cells in the solid tumor microenvironment.
Infusion of T cells directed against specific antigens has demonstrated promise in HIV and cancer therapy. Along with immune checkpoint blockade,1 this approach is triggering a paradigm shift in cancer immunotherapy. Perhaps the most exciting of these approaches has been the use of T cells that have been genetically modified to express chimeric antigen receptor (CAR) genes. CARs are comprised of an extracellular single-chain variable fragment (scFv), which serves as the targeting moiety, a transmembrane spacer, and intracellular signaling/activation domain(s) (Figure 1). The CAR constructs are transfected into T cells, using plasmid transfection, mRNA or via viral vector transduction, to direct them toward tumor-associated antigens (TAAs). CAR structure has evolved significantly from the initial composition involving only the CD3ζ signaling domain, dubbed a “first-generation CAR.” Since then, in an effort to augment T-cell persistence and proliferation, costimulatory endodomains were added, giving rise to second- (e.g., CD3ζ plus 41BB- or CD28-signaling domains) and third-generation (e.g., CD3ζ plus 41BB- and CD28-signaling domains) CARs. CARs have also been constructed in the context of human leukocyte antigen targeting intracellular molecules.2
Figure 1.

Building blocks of chimeric antigen receptor (CAR) T cell. The single chain (scFv) targeting moiety is taken from the antigen-binding domain of antibodies, fused to the CD3ζ transmembrane and intracellular signaling domains from the T-cell receptor complex; this is the first-generation CAR. Later, additional intracellular signaling domains were added for costimulatory signals, such as the CD28 and 41BB signaling domains, to yield second- and third-generation CARs.
The adoptive transfer of CAR T cells has demonstrated remarkable success in treating blood-borne tumors; prominently, the use of CD19 CARs in leukemias,3 and indications in patients with lymphoma and myeloma are being explored. A growing number of clinical trials have focused on solid tumors, targeting surface proteins including carcinoembryonic antigen (CEA), the diganglioside GD2, mesothelin, interleukin 13 receptor α (IL13Rα), human epidermal growth factor receptor 2 (HER2), fibroblast activation protein (FAP), and L1 cell adhesion molecule (L1CAM) (reviewed in Gill et al.3 and Fousek et al.4). Unfortunately, the clinical results have been much less encouraging. To date, the two most positive trials reported have used GD2 CARs to target neuroblastoma (3 of 11 patients with complete remissions),5 and HER2 CARs for sarcoma (4 of 17 patients showing stable disease).6
The reason for this is not yet known, but is likely multifactorial. The solid tumor landscape presents unique barriers that are absent in hematological malignancies, and these barriers, either by themselves or in combination with various tumor- and/or host cell-borne factors eventually neutralize CAR activity. Unlike the “liquid tumor” environment of blood malignancies, CAR T cells must successfully traffic to solid tumor sites in spite of potential T-cell chemokine receptor-/tumor-derived chemokine mismatches and successfully infiltrate the stromal elements of solid tumors in order to elicit TAA-specific cytotoxicity, regardless of antigen loss or heterogeneity. Even after successful trafficking and infiltration, T cells must surmount challenges conferred by: (i) an environment characterized by oxidative stress, nutritional depletion, acidic pH, and hypoxia; (ii) the presence of suppressive soluble factors and cytokines; (iii) suppressive immune cells (regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSC), tumor-associated macrophages (TAM) or neutrophils (TAN); and (iv) T-cell-intrinsic negative regulatory mechanisms (e.g., upregulation of cytoplasmic and surface inhibitory receptors) and overexpression of inhibitory molecules. Lastly, the CAR T cells, themselves, may be problematic given their potential immunogenicity and toxicity.
In this mini-review, we discuss some of the key immunosuppressive barriers and other negative elements within solid tumors that ultimately neutralize the function of antitumor T cells, and CAR T cells in particular (Figure 2).
Figure 2.
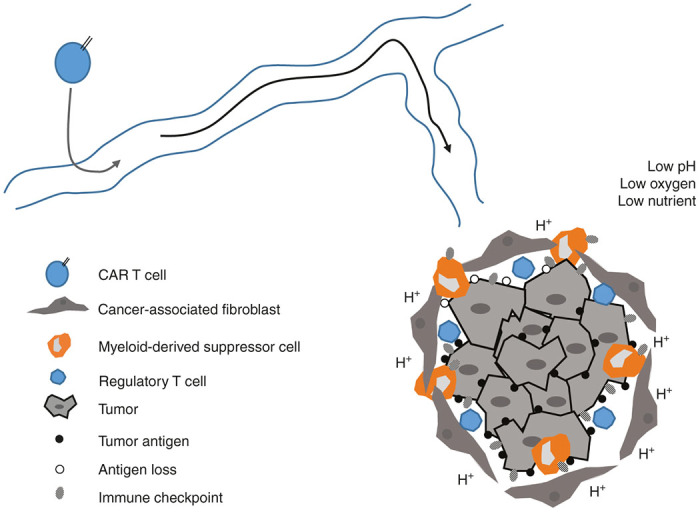
Immunosuppressive tumor microenvironment. This diagram represents a simplified schema of the negative elements that barrage activated chimeric antigen receptor T cells as they navigate through the tumor landscape, thereby inactivating them. These barriers in solid tumors rapidly neutralize the antitumor effect.
Antigen Specificity and Heterogeneity
The first step in adoptive T-cell therapy is choosing an optimal TAA for CAR T-cell targeting. Ideally, the TAA is highly expressed on the surface of all tumor cells but not on important normal tissues (or at least lowly expressed). However, unlike the success story of CD19 CARs in leukemias, where CD19 is consistently expressed on tumors and only on “dispensable” B cells, specific target antigens on solid tumors have been more difficult to identify. So far, roughly 30 solid tumor antigens are being evaluated for CAR T-cell therapy; these include neoantigens (for instance, mutated sequences), oncofetal or developmental antigens, tumor-selective antigens (i.e., enriched expression of antigens on neoplastic cells, but low basal expression on normal cells), and endogenous tumor-specific antigens; a recent list of CAR targets that are currently being evaluated in clinical trials is available.3 It should be noted that the scFv avidity to TAA may also be important, and immunoediting and subsequent removal of the most immunogenic epitopes may lead to tumor escape.7
Neoantigens on the surface of solid tumors represent an especially attractive target for CAR therapy as their expression is restricted to tumor cells. It is now recognized that most neoantigens are likely to due to tumor-specific mutations and are thus highly individualized (and hence not practical for CAR therapy). However, several neoepitopes that are more generalized have been identified. CAR T cells targeting the mutated EGF receptor is an illustration of this approach; EGFR variant 3 (EGFRvIII) is only expressed on malignant tumor cells (mostly glioblastomas). EGFRvIII CARs have shown promise in treating animal models of glioblastomas8,9 and clinical trials testing the efficacy of EGFRvIII CAR in patients with glioblastomas are underway (NCT02209376, NCT01454596). Abnormal glycosylation of extracellular glycoprotein MUC1 is also seen in a large variety of cancers; MUC-1-targeting CAR T cells against MUC1-overexpressing breast cancer xenografts were shown to significantly delay tumor progression.10 A similar success was reported for CAR T cells targeting MUC16, which is overexpressed in many ovarian carcinomas.11 CEA is an example of an antigen expressed during developmental growth, but restricted in normal adult tissues and in transformed cells. Evidence of tumor eradication by CEA-CAR T cells in mice has been reported.12 A recent phase 1 trial using CEA-redirected transgenic T-cell receptor (TCR) T cells, however, showed that while one out of three metastatic colon cancer patients demonstrated an objective response to the therapy as exhibited by regression of metastasis to the lungs and liver, all three patients had to endure transient colitis.13 Cancers arising from virus transformation may express viral products that are attractive targets for therapy since these products are not displayed on normal tissues; for instance, human papillomavirus (HPV)-transformed ovarian cancers.14
Tumor-selective (versus tumor-specific) antigens include targets that are overexpressed on transformed cells but expressed at low levels on normal tissues. This includes mesothelin, a glycoprotein whose overexpression in mesothelioma, ovarian, and pancreatic carcinomas, and low expression on peritoneal, pleural, and pericardial surfaces, has made it an attractive target for CAR therapy.15,16 Two mesothelin-specific CARs have been reported; one based on the SS1 antibody17—a mouse anti-human scFv which is currently being evaluated in a clinical trial at the University of Pennsylvania (NCT02159716), and another designated P4, a fully human scFv.18 A fully human scFv targeting mesothelin was recently described by another group, and is currently being tested in a clinical trial (NCT02414269).19 Treatment using T cells electroporated with the mRNA encoding SS1-CAR, while promising, raised concerns about potential immunogenicity-related toxicity (see below). The notion of targeting multiple antigens (for instance, the expression of 2 scFvs,20) or non-tumor cell–related antigens (i.e., a CAR-targeting the stromally-expressed FAP,21 or the VEGF receptor on tumor endothelium has also been explored.22
It should be noted that for any of the targets chosen, the scFv avidity to TAA may also be important. Another issue is that immunoediting and subsequent removal of the most immunogenic epitopes may lead to tumor escape.7
Trafficking
Once a CAR that targets appropriate tumor antigen is generated and infused into a patient, an immediate obstacle is the ability of these CAR T cells to successfully target and infiltrate the solid tumor. This process is dependent on the appropriate expression of adhesion receptors on both T cells and the tumor endothelium, and a “match” between the chemokine receptors on the CAR (primarily CXCR3 and CCR5) and the chemokines secreted by the tumors. Unfortunately, there is often a chemokine/chemokine receptor “mismatch”, with tumors producing very small amounts of, for instance, CXCR3 ligands, resulting in inefficient targeting of CXCR3high CD8+ effectors to tumor sites.23 One approach to overcome this problem is to design CAR T cells that coexpress “better-matched” chemokine receptors. For example, using mesothelioma tumors that make large amounts of CCL2, we and others demonstrated enhanced intratumoral migration of CAR T cells when they coexpressed the appropriate CCR2b transgene, thus leading to subsequent tumor eradication.24 Similarly, the use of GD2-CAR T cells coexpressing CCR2b exhibited improved trafficking and tumor control compared to GD2-CAR alone.25 We have also recently found that the genetic inhibition of protein kinase A activation in CAR T cells increased their ability to infiltrate tumors in vivo due to higher baseline expression of CXCR3.26
Due to poor trafficking after intravenous injection, local instillation of CARs is also being explored; there are a number of clinical trials that are evaluating the merits of site-specific (i.e., systemic versus regional versus intratumoral) administration of CAR T cells in solid tumors (NCT02498912, NCT02414269, NCT01818323). One potential limitation is that local instillation is often more technically challenging than simple intravenous administration. Another potential issue is that although site-specific injection of CAR T cells will likely result in higher T-cell levels locally, the ability of these CARs to exit the tumor, enter the blood and then traffic to other tumor sites (which presumably exist in advanced cancer patients) is uncertain. The ongoing studies will help to address these issues.
Several groups have also demonstrated the successful use of oncolytic viruses armed with chemotactic chemokines in attempts to attract CAR T cells to tumor sites. Oncolytic viruses have been shown to successfully and specifically infect tumor cells, and lyse them. The use of oncolytic adenoviral vector expressing CCL5 and GD2-CAR T cells robustly controlled neuroblastoma progression in mice and improved CAR T-cell influx,27 and similar observations were attained with the use of HER2-CAR T cells loaded with modified oncolytic viruses.28
The Hostile Tumor Microenvironment: Physical and Metabolic Barriers
The solid tumor microenvironment presents many problems for CAR T cells. There are purely physical/anatomical barriers, such as stroma that characterizes many types of cancers, and the associated high tissue pressure that prevents extravasation. Countering these barriers by reducing tumor fibroblast numbers using FAP-CAR T cells21 or by having the CARs secrete an enzyme that degrades matrix29 have both shown some success in augmenting CAR T-cell function in animal models.
The metabolic landscape within the tumor microenvironment is markedly stressful and inhospitable toward T cells. Prominent hallmarks of the tumor microenvironment include hypoxia and nutrient starvation; under these conditions, elevated lactate generation (leading to acidosis) and the lack of glucose and other metabolites inhibit T-cell proliferation and cytokine production.30,31 The lack of nutrients (specifically amino acids such as tryptophan, arginine, and lysine) may also activate the integrated stress response, causing protein translation shutdown or autophagy responses in effector T cells as a means of survival in order to generate an intracellular source of nutrients.32 For example, the amino acid tryptophan is essential for many biological functions though it cannot be synthesized, and hence must be obtained via dietary means. Tryptophan metabolism as catalyzed by tumor- and MDSC-expressed indolamine-2,3-dioxygenase leads to T-cell anergy and death, and Treg accumulation. In a solid tumor xenograft model of CD19-expressing tumor cells transduced with indolamine-2,3-dioxygenase, Ninomiya and colleagues showed the failure of adoptively transferred CD19 CAR T cells to control progression of indolamine-2,3-dioxygenase-expressing tumors.33 MDSC may also reduce the bioavailability of the key amino acid arginine (see below). Preliminary work has suggested that manipulation of key cellular regulators of protein synthesis (i.e., the mammalian target of rapamycin) might augment the efficacy of adoptively transferred cells.34 Sukumar and colleague also showed that inhibiting glycolysis promoted the formation of memory cells, and enhanced antitumor activity.35
The Hostile Tumor Microenvironment: Tumor-Derived Soluble Factors and Cytokines
Many studies have reported the presence of immunosuppressive soluble factors in the sera, ascites fluid, and tissue extracts from cancer patients that could inhibit CAR T cells. Prostaglandin E2 (PGE2), a small molecule derivative of arachidonic acid produced by the inducible cyclooxygenase 2 (COX2) enzyme, is generated by both tumor cells and macrophages; many studies have reported PGE2-mediated inhibition of T-cell proliferation, suppression of CD4 help, and subversion of CD8 differentiation.36 Adenosine, a purine nucleoside seen at high levels during hypoxia, is another potent inhibitor of T-cell proliferation and activity. Both PGE2 and adenosine illicit their immunosuppressive effects via signaling through their own G-coupled receptors which activate protein kinase A in a cyclic AMP-dependent manner.37 We recently demonstrated that genetic inhibition of protein kinase A activation in CAR T cells can enhance their antitumor efficacy.26
Cytokines, implicated in inflammatory responses at tumor sites, can be a double-edged sword, which may bolster or inhibit the antitumor response. One of the most important inhibitory tumor cytokines is TGFβ. In addition to its ability to promote epithelial-to-mesenchymal transition, enhance matrix production, promote metastasis, and skew the immune response toward a Th2 phenotype,38 TGFβ has direct negative effects on T-cell effector functions.39 A few approaches have been used to counteract this effect. We previously showed that systemic blockade of TGFβ was efficacious in augmenting adoptive T-cell therapy.40 To counteract TGFβ effects specifically in T cells, CAR T cells expressing a dominant negative TGFβ receptor have been created. These CAR T cells were resistant to TGFβ suppression and demonstrated augmented efficacy in animal models.41
Other inhibitory cytokines include IL10 and IL4; although these have not been directly targeted by alterations in T cells, two groups have constructed chimeric IL4 receptors so that IL4 engagement resulted in signaling that mimicked that of IL2.42,43 One of these groups combined this with the use of CAR T cells targeting the tumor-associated antigen MUC1 and showed enhanced efficacy.43
Finally, it has also been possible to use introduce activating cytokines to improve the tumor microenvironment milieu to augment CAR function. A few groups have designed CARs or T cells that release the stimulatory cytokine IL12 upon TCR engagement.44 Although the approach worked extremely well in animal models, a recent clinical trial in which the IL12 gene, driven by an NFAT promoter in adoptively transferred tumor-infiltrating lymphocytes (TILs) resulted in unacceptable toxicity.45 Finding ways to more tightly control IL12 release or the use of less toxic cytokines (i.e., type 1 interferons) might allow this strategy to proceed in the clinic.
The Hostile Tumor Microenvironment: Immunosuppressive Immune Cells
Within the tumor microenvironment, various suppressive surveilling immune cells, Tregs, MDSC, and TAM/TAN with the so called M2 and N2 phenotype are known to present a barrier against successful antitumor immunity. Although there is extensive literature describing the immunosuppressive nature of these cells, to date, their effects on CAR T-cell therapy has not been extensively examined. One technical factor to consider is that in order to study these cell-cell interactions, mouse CAR T cells must be injected into immunocompetent mice. Given the major differences between the behavior of mouse versus human CAR T cells (e.g., in our experience, mouse CAR T cells are much more sensitive to activation-induced cell death and have a very short persistence compared to human CAR T cells), the relevance of these studies to human CAR T cells is not certain.
MDSC, M2-TAM, and N2-TAN are well-known producers of TGFβ, PGE2, reactive oxygen/nitrogen species, and arginase.46,47 As discussed above, all these factors likely blunt the efficacy of CAR T cells. In addition, TAM can express high levels of PDL1, which can interact with PD1 on CAR T cells and inhibit them (see below). MDSC may also recruit Treg cells. On the other hand, TAM and TAN activated in the proper fashion (the so-called M1 or N1 phenotype) can work to eliminate tumor cells.
The role of myeloid cells in CAR therapy is not yet clear. Burga and colleagues found that depletion of GR1+ cells (targeting TAN and MDSC) augmented the ability CEA-CAR T cells to control colorectal cancer liver metastases.48 In contrast, Spear et al.49 found in an ovarian cancer model that CARs activated F4/80high TAMs, and enhanced production of nitric oxide by TAMs, leading to tumor lysis. Further studies are needed to more precisely define the role of myeloid cells in CAR efficacy.
CD4+/FOXP3+ Tregs are well-documented suppressors of T-cell activity acting through multiple mechanisms including cell-to-cell contact inhibition, and via soluble factors such as TGFβ, and interleukin 10 (IL10).50 It has been difficult to study the effects of Tregs on CAR therapy since it is difficult to selectively deplete Tregs. For example, depletion using anti-CD25 antibody will also affect CAR T cells. Nonetheless, some studies have been performed using genetic depletion approaches or adoptive transfer of Tregs with CAR T cells. Zhou et al.51 studied adoptively transferred cytotoxic T-lymphocytes in a mouse leukemia model and found that antibody blockade of PDL1, combined with genetic depletion (using a diphtheria toxin model) of Tregs, markedly increased efficacy of T-cell adoptive transfer, although depletion of Tregs alone had relatively minor effects. Our lab has recently conducted studies using a selective inhibitor of Tregs52 and shown augmentation of a mouse CAR T cells targeted to mesothelin (Wang et al., Submitted).
There is some data in humans to suggest an inhibitory effect of Tregs on adoptive T-cell transfer. Perna et al.53 describe a model in which the efficacy of human GD2-CARs is inhibited by coinjection of human Tregs with IL2. An analysis of four T-cell adoptive therapy clinical trials employing nonmyeloablative chemotherapy with/without total body irradiation before adoptive T-cell transfer revealed that the percentage and number of reconstituting CD4+/FOXP3+ Tregs observed in the peripheral blood was higher in nonresponders than in responders.54 In addition, the number of administered doses of IL2 was found to be positively associated with peripheral Treg reconstitution. These latter data highlight the complex role of IL2 in CAR therapy. Although IL2 can support CAR T cells in vivo and has been used pre-clinically and in many clinical trials,54 it also, and perhaps preferentially, activates and induces proliferation of Tregs.55 Thus, the use of alternative T cells homeostatic cytokines, such as IL7 and IL21, was explored, and shown to enhance CAR efficacy.56 High IL2 levels with subsequent Treg stimulation may also be an issue in CAR constructs containing the CD28 cytoplasmic domain which produce much higher levels of IL2 than do CARs with the 41BB cytoplasmic domain.57
T-Cell-Intrinsic Regulatory Mechanisms
In order to maintain tolerance, T cells express activation-induced surface molecules, such as CTLA4 and PD1, which can have antagonistic effects on the overall antitumor immune response, generally restricting the extent and strength of the immune response upon receptor ligation. The importance of these inhibitory receptors has now been established in multiple clinical trials.1 Since these receptors are upregulated on infused CAR T cells and even further increased on CAR TILs,58 a number of groups have shown that blockade of these receptors can augment therapy. For example, using mouse T cells, a combinatorial strategy of HER2-CAR T-cell adoptive transfer and PD1 blockade led to significant tumor regression.59 In experiments studying human CAR T cells in an immunodeficient animal tumor model, our group showed that PD1 blockade using anti-human antibodies enhanced antitumor effects of human mesothelin-directed CARs.58 We60 and Kobold and colleagues61 showed that it is also possible to reverse the inhibitory effects of PD1 by transducing T cells with a PD1 “switch receptor”; that is, the extracellular domain of PD1 fused to the cytoplasmic domain of an activating receptor like CD28. Antibodies against CTLA4 have also been shown to augment adoptive T-cell transfer.62
In addition to surface inhibitory receptors, T cells activate a range of intracellular negative feedback loops after TCR stimulation that work to shut down T-cell activity.63 Some examples include: (i) enzymes (such as diacylglycerol kinase; (ii) phosphatases (such as SHP1; (ii) ubiquitin ligases (such as Cbl-B); and (iv) transcription factors (such as Ikaros). Augmenting CAR T cells function by reducing the expression or function of these inhibitors is an active area of investigation; for example, CAR T cells lacking expression of diacylglycerol kinase showed markedly increased efficacy.64
Another process that can limit CAR function is receptor- or activation-induced cell death. In many cases, this is affected by activation of Fas (CD95) on the T cells through the engagement by Fas ligand (FasL) that is upregulated in most tumor cells, tumor vasculature, and on activated T cells. Engagement of Fas induces T-cell apoptosis, thereby dampening T-cell-mediated immunity. Along these lines, engineering T cells to express higher levels of antiapoptotic proteins was undertaken.65
Immunogenicity and Toxicity
Despite the lack of proven efficacy to date, there have been some safety concerns in solid tumor CAR T cells trials that will need to be kept in mind as clinical trials progress. The major toxicity seen in the CAR19 T cells trials has been attributed to severe “cytokine storm” seen in conjunction with rapid T-cell proliferation.66 It is thought that the infused CAR product causes a widespread, toxic release of proinflammatory cytokines, thus leading to clinical manifestations such as fever, rash, and potentially organ failure.67 Fortunately (or perhaps unfortunately), this has not yet been observed in trials for solid tumors, likely due to the fact that the degree of T-cell engraftment and proliferation seems to be quite low compared to the leukemia patients. However, as enhanced CARs are developed, and/or as stronger lymphodepletion regimens are employed, this potential toxicity may be observed.
The most feared complication of CAR therapy, a catastrophic and rapid “on target-off tumor” event, has been documented. A fatal event occurred rapidly after infusion with a high affinity HER2-CAR, which was attributed to low-level expression of the antigen on normal endothelium and epithelium.68 Approaches to avoid this type of event include extensive preclinical toxicology studies, use of “self-limited CARs” that use mRNA rather than lentivirus to transiently express the CAR receptor, and careful dose escalation trial designs. Some groups are also advocating the insertion of suicide genes which can be activated in case of adverse events. Success in preclinical models has been shown with use of the herpes simplex virus thymidine kinase (HSV-TK) gene or an inducible caspase 9 (iCasp9) gene. The activation of these suicide genes leads to the specific and permanent eradication of CAR T cells. Another approach could be to increase the specificity of CARs by requiring the CAR to recognize two antigens to promote activity.69,70
Finally, the potential immunogenicity of transduced genes must be considered. For example, since the viral gene HSV-TK is immunogenic, the use of iCasp9 seems more attractive as it manipulates the endogenous caspase pathway, and was shown to be very efficient in inducing apoptosis.71 Phase 1 trials utilizing GD2-CAR T cells with iCasp9 are ongoing (NCT01822652, NCT01953900). Another possible problem with immunogenicity relates to the fact that some of the scFvs incorporated in the CARs used in current clinical trials are of murine origin and can thus elicit a human anti-mouse protein immune response. This can be a cellular immune response that eliminates the transduced T cells after 4–6 weeks, but can also result in the generation of anti-murine scFv IgG or even IgE antibodies. In our recent mRNA mesothelin-CAR trial, we observed an anaphylactic reaction when CARs expressing a murine scFv were readministered to a patient after a period of 6 weeks.72 Because of this, most groups are now using human or humanized scFvs in their CAR constructs.
Conclusions and Future Perspectives
A better understanding of the multiples barriers seen in solid tumors will drive advances in CAR engineering and in clinical trial design. For example, it is currently unclear if aggressive lymphodepletion suggested for TIL therapy7 will also be needed for CAR T-cell infusion. A number of groups are currently exploring this issue. In preliminary studies from our institution, the use of cyclophosphamide appears to increase blood levels of CARs after infusion, suggesting that some sort of lymphodepletion may be needed in solid tumor therapy.
Some approaches to overcome solid tumor barriers were discussed above, however many other strategies are being tested. To mention just a few, the use of alternative cytoplasmic activation domains, such as ICOS, 41BB, OX40, or CD27 are being explored.73,74 Even more radical design changes are also being evaluated. Wang et al.75 have fused a scFv for antigen recognition to the transmembrane and cytoplasmic domains of KIR2DS2, a stimulatory killer immunoglobulin-like receptor (KIR). This KIR-based CAR (KIR-CAR), when fused to the adaptor DAP12, proliferated in an antigen-specific manner, and demonstrated enhanced effector function.
The compelling success of CAR therapy in hematologic malignancies is propelling the development of CARs that can show similar efficacy in solid tumors. The ability to genetically manipulate infused CAR T cells provides almost limitless opportunities for additional changes and improvements, and thus provides strong hope for future success.
The authors declare no conflict of interest.
References
- Topalian, SL, Drake, CG and Pardoll, DM (2015). Immune checkpoint blockade: a common denominator approach to cancer therapy. Cancer Cell 27: 450–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen, RA, Debets, R, Hart, E, Hoogenboom, HR, Bolhuis, RL and Chames, P (2001). A phage display selected fab fragment with MHC class I-restricted specificity for MAGE-A1 allows for retargeting of primary human T lymphocytes. Gene Ther 8: 1601–1608. [DOI] [PubMed] [Google Scholar]
- Gill, S, Maus, MV and Porter, DL (2015). Chimeric antigen receptor T cell therapy: 25 years in the making. Blood Rev (Epub ahead of print). [DOI] [PubMed]
- Fousek, K and Ahmed, N (2015). The evolution of T-cell therapies for solid malignancies. Clin Cancer Res 21: 3384–3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis, CU, Savoldo, B, Dotti, G, Pule, M, Yvon, E, Myers, GD et al. (2011). Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood 118: 6050–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed, N, Brawley, VS, Hegde, M, Robertson, C, Ghazi, A, Gerken, C et al. (2015). Human epidermal growth factor receptor 2 (HER2)-specific chimeric antigen receptor-modified T cells for the immunotherapy of HER2-positive sarcoma. J Clin Oncol 33: 1688–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restifo, NP, Dudley, ME and Rosenberg, SA (2012). Adoptive immunotherapy for cancer: harnessing the T cell response. Nat Rev Immunol 12: 269–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, LA, Scholler, J, Ohkuri, T, Kosaka, A, Patel, PR, McGettigan, SE et al. (2015). Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 7: 275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Johnson, LA, Davis, JL, Zheng, Z, Woolard, KD, Reap, EA et al. (2012). Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther 23: 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie, S, Picco, G, Foster, J, Davies, DM, Julien, S, Cooper, L et al. (2008). Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol 180: 4901–4909. [DOI] [PubMed] [Google Scholar]
- Chekmasova, AA, Rao, TD, Nikhamin, Y, Park, KJ, Levine, DA, Spriggs, DR et al. (2010). Successful eradication of established peritoneal ovarian tumors in SCID-Beige mice following adoptive transfer of T cells genetically targeted to the MUC16 antigen. Clin Cancer Res 16: 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski, M, Hahn, O, Rappl, G, Nowak, M, Schmidt-Wolf, IH, Hombach, AA et al. (2012). T cells that target carcinoembryonic antigen eradicate orthotopic pancreatic carcinomas without inducing autoimmune colitis in mice. Gastroenterology 143: 1095–107.e2. [DOI] [PubMed] [Google Scholar]
- Parkhurst, MR, Yang, JC, Langan, RC, Dudley, ME, Nathan, DA, Feldman, SA et al. (2011). T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther 19: 620–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenter, GG, Welters, MJ, Valentijn, AR, Lowik, MJ, Berends-van der Meer, DM, Vloon, AP et al. (2009). Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med 361: 1838–1847. [DOI] [PubMed] [Google Scholar]
- Morello, A, Sadelain, M and Adusumilli, PS (2016). Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov 6: 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastan, I and Hassan, R (2014). Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer Res 74: 2907–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury, PS, Viner, JL, Beers, R and Pastan, I (1998). Isolation of a high-affinity stable single-chain Fv specific for mesothelin from DNA-immunized mice by phage display and construction of a recombinant immunotoxin with anti-tumor activity. Proc Natl Acad Sci USA 95: 669–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanitis, E, Poussin, M, Hagemann, IS, Coukos, G, Sandaltzopoulos, R, Scholler, N et al. (2012). Redirected antitumor activity of primary human lymphocytes transduced with a fully human anti-mesothelin chimeric receptor. Mol Ther 20: 633–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adusumilli, PS, Cherkassky, L, Villena-Vargas, J, Colovos, C, Servais, E, Plotkin, J et al. (2014). Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med 6: 261ra151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie, S, van Schalkwyk, MC, Hobbs, S, Davies, DM, van der Stegen, SJ, Pereira, AC et al. (2012). Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J Clin Immunol 32: 1059–1070. [DOI] [PubMed] [Google Scholar]
- Wang, LC, Lo, A, Scholler, J, Sun, J, Majumdar, RS, Kapoor, V et al. (2014). Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol Res 2: 154–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnasamy, D, Tran, E, Yu, Z, Morgan, RA, Restifo, NP and Rosenberg, SA (2013). Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res 73: 3371–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlin, H, Meng, Y, Peterson, AC, Zha, Y, Tretiakova, M, Slingluff, C et al. (2009). Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 69: 3077–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon, EK, Carpenito, C, Sun, J, Wang, LC, Kapoor, V, Predina, J et al. (2011). Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res 17: 4719–4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craddock, JA, Lu, A, Bear, A, Pule, M, Brenner, MK, Rooney, CM et al. (2010). Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J Immunother 33: 780–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newick, K, O’Brien, S, Sun, J, Kapoor, V, Maceyko, S, Lo, A et al. (2016). Augmentation of CAR T cell trafficking and antitumor efficact by blocking protein kinase A (PKA) localization. Cancer Immunol Res. In press. [DOI] [PMC free article] [PubMed]
- Nishio, N, Diaconu, I, Liu, H, Cerullo, V, Caruana, I, Hoyos, V et al. (2014). Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res 74: 5195–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanSeggelen, H, Hammill, JA, Dvorkin-Gheva, A, Tantalo, DG, Kwiecien, JM, Denisova, GF et al. (2015). T cells engineered with chimeric antigen receptors targeting NKG2D ligands display lethal toxicity in mice. Mol Ther 23: 1600–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana, I, Savoldo, B, Hoyos, V, Weber, G, Liu, H, Kim, ES et al. (2015). Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat Med 21: 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs, SR, Herman, CE, Maciver, NJ, Wofford, JA, Wieman, HL, Hammen, JJ et al. (2008). Glucose uptake is limiting in T cell activation and requires CD28-mediated Akt-dependent and independent pathways. J Immunol 180: 4476–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, K, Hoffmann, P, Voelkl, S, Meidenbauer, N, Ammer, J, Edinger, M et al. (2007). Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109: 3812–3819. [DOI] [PubMed] [Google Scholar]
- Howie, D, Waldmann, H and Cobbold, S (2014). Nutrient sensing via mTOR in T cells maintains a tolerogenic microenvironment. Front Immunol 5: 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninomiya, S, Narala, N, Huye, L, Yagyu, S, Savoldo, B, Dotti, G et al. (2015). Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood 125: 3905–3916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veliça, P, Zech, M, Henson, S, Holler, A, Manzo, T, Pike, R et al. (2015). Genetic regulation of fate decisions in therapeutic T cells to enhance tumor protection and memory formation. Cancer Res 75: 2641–2652. [DOI] [PubMed] [Google Scholar]
- Sukumar, M, Liu, J, Ji, Y, Subramanian, M, Crompton, JG, Yu, Z et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123: 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, JS, Bankhurst, AD and Messner, RP (1977). Suppression of human T-cell mitogenesis by prostaglandin. Existence of a prostaglandin-producing suppressor cell. J Exp Med 146: 1719–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su, Y, Huang, X, Raskovalova, T, Zacharia, L, Lokshin, A, Jackson, E et al. (2008). Cooperation of adenosine and prostaglandin E2 (PGE2) in amplification of cAMP-PKA signaling and immunosuppression. Cancer Immunol Immunother 57: 1611–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh, SA and Li, MO (2013). TGF-ß: guardian of T cell function. J Immunol 191: 3973–3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué, J (2008). TGFbeta in Cancer. Cell 134: 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace, A, Kapoor, V, Sun, J, Mrass, P, Weninger, W, Heitjan, DF et al. (2008). Transforming growth factor-beta receptor blockade augments the effectiveness of adoptive T-cell therapy of established solid cancers. Clin Cancer Res 14: 3966–3974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollard, CM, Rössig, C, Calonge, MJ, Huls, MH, Wagner, HJ, Massague, J et al. (2002). Adapting a transforming growth factor beta-related tumor protection strategy to enhance antitumor immunity. Blood 99: 3179–3187. [DOI] [PubMed] [Google Scholar]
- Leen, AM, Sukumaran, S, Watanabe, N, Mohammed, S, Keirnan, J, Yanagisawa, R et al. (2014). Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther 22: 1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie, S, Burbridge, SE, Chiapero-Stanke, L, Pereira, AC, Cleary, S, van der Stegen, SJ et al. (2010). Selective expansion of chimeric antigen receptor-targeted T-cells with potent effector function using interleukin-4. J Biol Chem 285: 25538–25544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chmielewski, M, Hombach, AA and Abken, H (2014). Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunol Rev 257: 83–90. [DOI] [PubMed] [Google Scholar]
- Zhang, L, Morgan, RA, Beane, JD, Zheng, Z, Dudley, ME, Kassim, SH et al. (2015). Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res 21: 2278–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich, DI and Nagaraj, S (2009). Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9: 162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mussai, F, Egan, S, Hunter, S, Webber, H, Fisher, J, Wheat, R et al. (2015). Neuroblastoma Arginase Activity Creates an Immunosuppressive Microenvironment That Impairs Autologous and Engineered Immunity. Cancer Res 75: 3043–3053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burga, RA, Thorn, M, Point, GR, Guha, P, Nguyen, CT, Licata, LA et al. (2015). Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother 64: 817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear, P, Barber, A, Rynda-Apple, A and Sentman, CL (2012). Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-? and GM-CSF. J Immunol 188: 6389–6398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi, S, Miyara, M, Costantino, CM and Hafler, DA (2010). FOXP3+ regulatory T cells in the human immune system. Nat Rev Immunol 10: 490–500. [DOI] [PubMed] [Google Scholar]
- Zhou, Q, Munger, ME, Highfill, SL, Tolar, J, Weigel, BJ, Riddle, M et al. (2010). Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood 116: 2484–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y, Wang, L, Predina, J, Han, R, Beier, UH, Wang, LC et al. (2013). Inhibition of p300 impairs Foxp3? T regulatory cell function and promotes antitumor immunity. Nat Med 19: 1173–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perna, SK, Pagliara, D, Mahendravada, A, Liu, H, Brenner, MK, Savoldo, B et al. (2014). Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res 20: 131–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X, Ahmadzadeh, M, Lu, YC, Liewehr, DJ, Dudley, ME, Liu, F et al. (2012). Levels of peripheral CD4(+)FoxP3(+) regulatory T cells are negatively associated with clinical response to adoptive immunotherapy of human cancer. Blood 119: 5688–5696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler, DM, Chmielewski, M, Rappl, G, Hombach, A, Riet, T, Schmidt, A et al. (2011). CD28 costimulation Impairs the efficacy of a redirected t-cell antitumor attack in the presence of regulatory T cells which can be overcome by preventing Lck activation. Mol Ther 19: 760–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markley, JC and Sadelain, M (2010). IL-7 and IL-21 are superior to IL-2 and IL-15 in promoting human T cell-mediated rejection of systemic lymphoma in immunodeficient mice. Blood 115: 3508–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney, HM, Akbar, AN and Lawson, AD (2004). Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol 172: 104–113. [DOI] [PubMed] [Google Scholar]
- Moon, EK, Wang, LC, Dolfi, DV, Wilson, CB, Ranganathan, R, Sun, J et al. (2014). Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 20: 4262–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John, LB, Devaud, C, Duong, CP, Yong, CS, Beavis, PA, Haynes, NM et al. (2013). Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res 19: 5636–5646. [DOI] [PubMed] [Google Scholar]
- Moon, EK, Ranganathan, R, Eruslanov, E, Kim, S, Newick, K, O’Brien, S et al. (2016). Blockade of programmed death 1 augments the ability of human T cells engineered to target NY-ESO-1 to control tumor growth after adoptive transfer. Clin Cancer Res 22: 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobold, S, Grassmann, S, Chaloupka, M, Lampert, C, Wenk, S, Kraus, F et al. (2015). Impact of a new fusion receptor on PD-1-mediated immunosuppression in adoptive T cell therapy. J Natl Cancer Inst 107:djv146 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahvi, DA, Meyers, JV, Tatar, AJ, Contreras, A, Suresh, M, Leverson, GE et al. (2015). Ctla-4 blockade plus adoptive T-cell transfer promotes optimal melanoma immunity in mice. J Immunother 38: 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, LC, Riese, MJ, Moon, EK and Albelda, SM (2013). Overcoming intrinsic inhibitory pathways to augment the antineoplastic activity of adoptively transferred T cells: Re-tuning your CAR before hitting a rocky road. Oncoimmunology 2: e26492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riese, MJ, Wang, LC, Moon, EK, Joshi, RP, Ranganathan, A, June, CH et al. (2013). Enhanced effector responses in activated CD8+ T cells deficient in diacylglycerol kinases. Cancer Res 73: 3566–3577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charo, J, Finkelstein, SE, Grewal, N, Restifo, NP, Robbins, PF and Rosenberg, SA (2005). Bcl-2 overexpression enhances tumor-specific T-cell survival. Cancer Res 65: 2001–2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos, CA, Savoldo, B and Dotti, G (2014). CD19-CAR trials. Cancer J 20: 112–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, DW, Gardner, R, Porter, DL, Louis, CU, Ahmed, N, Jensen, M et al. (2014). Current concepts in the diagnosis and management of cytokine release syndrome. Blood 124: 188–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, RA, Yang, JC, Kitano, M, Dudley, ME, Laurencot, CM and Rosenberg, SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18: 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanitis, E, Poussin, M, Klattenhoff, AW, Song, D, Sandaltzopoulos, R, June, CH et al. (2013). Chimeric antigen receptor T cells with dissociated signaling domains exhibit focused antitumor activity with reduced potential for toxicity in vivo. Cancer Immunol Res 1: 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloss, CC, Condomines, M, Cartellieri, M, Bachmann, M and Sadelain, M (2013). Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol 31: 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tey, SK, Dotti, G, Rooney, CM, Heslop, HE and Brenner, MK (2007). Inducible caspase 9 suicide gene to improve the safety of allodepleted T cells after haploidentical stem cell transplantation. Biol Blood Marrow Transplant 13: 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty, GL, Haas, AR, Maus, MV, Torigian, DA, Soulen, MC, Plesa, G et al. (2014). Mesothelin-specific chimeric antigen receptor mRNA-engineered T cells induce anti-tumor activity in solid malignancies. Cancer Immunol Res 2: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedan, S, Chen, X, Madar, A, Carpenito, C, McGettigan, SE, Frigault, MJ et al. (2014). ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 124: 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, DG, Ye, Q, Poussin, M, Harms, GM, Figini, M and Powell, DJ Jr (2012). CD27 costimulation augments the survival and antitumor activity of redirected human T cells in vivo. Blood 119: 696–706. [DOI] [PubMed] [Google Scholar]
- Wang, E, Wang, LC, Tsai, CY, Bhoj, V, Gershenson, Z, Moon, E et al. (2015). Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol Res 3: 815–826. [DOI] [PMC free article] [PubMed] [Google Scholar]