

Abstract
Alcoholic liver disease is a significant contributor to global liver failure. In murine models, chronic ethanol consumption dysregulates PTEN/Akt signaling. Hepatospecific deletion of phosphatase and tensin homolog deleted on chromosome 10 (PTENLKO) mice possess constitutive activation of Akt(s) and increased de novo lipogenesis resulting in increased hepatocellular steatosis. This makes PTENLKO a viable model to examine the effects of ethanol in an environment of preexisting steatosis. The aim of this study was to determine the impact of chronic ethanol consumption and the absence of PTEN (PTENLKO) compared to Alb-Cre control mice (PTENf/f) on hepatocellular damage as evidenced by changes in lipid accumulation, protein carbonylation and alanine amino transferase (ALT). In the control PTENf/f animals, ethanol significantly increased ALT, liver triglycerides and steatosis. In contrast, chronic ethanol consumption in PTENLKO mice decreased hepatocellular damage when compared to PTENLKO pair-fed controls. Consumption of ethanol elevated protein carbonylation in PTENf/f animals but had no effect in PTENLKO animals. In PTENLKO mice, overall hepatic mRNA expression of genes that contribute to GSH homeostasis as well as reduced glutathione (GSH) and oxidized glutathione (GSSG) concentrations were significantly elevated compared to respective PTENf/f counterparts. These data indicate that during conditions of constitutive Akt activation and steatosis, increased GSH homeostasis assists in mitigation of ethanol-dependent induction of oxidative stress and hepatocellular damage. Furthermore, data herein suggest a divergence in EtOH-induced hepatocellular damage and increases in steatosis due to polyunsaturated fatty acids downstream of PTEN.
Introduction
Alcoholic liver disease (ALD) and non-alcoholic fatty liver disease (NAFLD) are two of the leading causes of liver disease in the United States today. Both ALD and NAFLD are characterized by progressive hepatocellular damage manifested in increased steatosis, steatohepatitis, fibrosis and ultimately progression to cirrhosis [1]. In the western world, the prevalence of diet-induced nonalcoholic steatohepatitis (NASH) has dramatically increased in the last decade. According to the latest data from the Centers of Disease Control, current estimates indicate 30–35% of all Americans are obese and 69% are overweight (http://www.cdc.gov/nchs/fastats/obesity-overweight.htm). Moderate alcohol consumption by human subjects with increased body mass index is strongly correlated with increased hepatic damage as determined by plasma alanine amino transferase (ALT) and Gamma-Glutamyl Transferase levels [2]. Given these statistics, obesity and its concomitant steatosis are predictable cofactors for ALD.
In the liver, insulin is a major regulator of lipid metabolism and alteration of insulin signaling can induce hepatocellular lipid accumulation evident in both ALD and NAFLD [3–7]. Downstream of the insulin receptor, the PTEN/Akt pathway regulates insulin signaling [8]. Recruitment and full activation of Akt requires interaction of N-terminal pleckstrin homology domains with phosphatidylinositol (3,4,5) trisphosphate (PIP3) on intracellular membranes [9, 10]. Through its ability to catalyze the hydrolysis of the 3’ position phosphate on PIP3, PTEN negatively regulates Akt activation and insulin signaling [11]. This is highlighted by bypassing the Akt arm of insulin receptor signaling using mice containing a hepatospecific deletion of PTEN (PTENLKO). PTENLKO mice possess constitutive Akt activation, hepatic insulin hypersensitivity and increased steatohepatitis [12–14]. In our previous study using the PTENLKO model, we determined that increased consumption of a diet high in polyunsaturated fatty acids potentiates hepatocellular damage and decreases hepatocellular redox capacity when compared to chow fed controls [15].
During chronic alcohol consumption, there is pronounced lipid accumulation and enhanced oxidative stress [16]. In murine models of ALD, increased de novo lipogenesis (DNL) has been proposed to be a contributing factor in lipid accumulation [17, 18]. Chronic consumption of EtOH combined with dietary polyunsaturated fatty acids decreases PTEN expression/activity increasing activation of Akt2 [19]. In other models, chronic EtOH consumption has also been demonstrated to enhance the Akt activated transcription factor SREBP1, increasing fatty acid synthesis [17]. Furthermore, using a short term model of EtOH toxicity, pre-administration of insulin reduced oxidative stress and hepatocellular damage but did not diminish steatosis further demonstrating the contribution of insulin signaling in EtOH toxicity [20]. Most recently, an environment of increased insulin hypersensitivity created by PTENLKO was demonstrated to provide protection against murine endotoxemia [21]. Thus, it would be reasonable to predict that if insulin signaling was completely bypassed by using mice possessing a constitutively activated form of Akt, then EtOH administration would not increase hepatocellular damage but still increase steatosis. In the present study, mice possessing constitutively activated Akt (PTENLKO) were used in a well characterized 6-week model of chronic EtOH consumption to further elucidate the contribution of PTEN/Akt signaling in EtOH-induced steatosis and hepatotoxicity. In PTENLKO mice, chronic EtOH consumption did not increase hepatocellular damage and corresponded with elevated glutathione metabolism. We hypothesize that the aforementioned elevation in glutathione metabolism assists in mitigation of hepatocellular damage induced by EtOH.
Materials and Methods
Animal Model
To generate liver-specific PTEN deletion mice (PTENLKO), mice carrying PTEN conditional knockout alleles (PTENloxP/loxP; Alb-Cre− (PTENf/f)) were bred with an Albumin (Alb)-Cre-recombinase transgenic mouse as previously described [12, 15]. At 5 weeks of age, tail snips were taken and mice were genotyped using PTENf/f and Cre specific primers (Transnetyx, Memphis TN). At 12 weeks of age, mice were isocalorically pair fed (PF) in groups of 6 either a modified Lieber-DeCarli diet (45% polyunsaturated fat derived calories mostly from corn oil) (Bio-Serv, Frenchtown, NJ) or EtOH-fed (EtOH-derived caloric content with an initial EtOH concentration of 10.8%, which was subsequently increased weekly to 16.2, 21.5, 26.9, 29.2, 31.8% ethanol derived calories for a total of 6-weeks. [19, 22]. Upon completion of the study, animals were anesthetized via intraperitoneal injection with sodium pentobarbital and euthanized by exsanguination. Blood was collected from the inferior vena cava and plasma was separated through centrifugation at 4°C and assayed for ALT activity (Sekisui Diagnostics, P.E.I., Canada). Excised livers were weighed, homogenized and subjected to differential centrifugation and subcellular fractionation (cytosolic, mitochondrial, microsomal and nuclear fractions) as previously described [23]. All procedures involving animals were approved by the Institutional Animal Care and Use Committee of the University of Colorado and were performed in accordance with published National Institutes of Health guidelines.
Western Blotting
Proteins from either whole liver extracts or subcellular fractions were subjected to standard SDS-PAGE and transferred to PVDF (GE Healthcare, Picataway, NJ). Membranes were processed using the following antibody dilutions: ALDH2, 1:2000, Cat #15310 (Protein Tech Rosemont, IL.), AHD1, 1:1000 Cat#GTX62515 (Genetex, Irvine, CA.), beta actin 1:5000, Cat#A5441 (SIGMA, Saint Louis, MO) as previously described [15, 19, 22, 24]. Chemiluminescence was visualized using either film or a Storm 860 scanner from Molecular Dynamics (Sunnyvale, CA).
Biochemical Analysis
Liver triglycerides were measured using a 2:1 chloroform:MEOH extract of liver homogenate using a kit from Diagnostic Research Inc. Protein concentrations were determined using a modified Lowry Protein Assay from Bio-Rad (Hercules, CA). Blood ethanol concentrations were determined from freshly isolated serum as previously described. [25, 26]
Microarray analysis: For microarrays, total RNA was extracted from fresh frozen pooled tissue isolated from triplicates of PF/EtOH-fed PTENf/f/PTENLKO mice. Following transcription, cDNA was processed and analyzed as previously described [22, 27].
Immunohistochemistry
Sections of freshly excised liver tissue were placed in 10% neutral buffered formalin (SIGMA) for 16 hours, followed by incubation in 70% EtOH overnight. Samples were then processed, embedded in paraffin, cut and sections were mounted on slides by the UC Denver Histology Core. Immunohistochemical characterization was performed using rabbit polyclonal antibodies directed against acrolein (Dilution 1:250, Cat#PA2049, Cell Sciences, Canton, MA), 4-HNE (Dilution 1:500), MDA (Dilution 1:100) and CYP2E1 (Dilution 1:750, Cat#1252, Millipore, Billerica, MA), Protein-SSG (Dilution 1:100, Cat#101-A-100, Virogen, Watertown, MA) as described [19, 24, 28]. Histologic images were captured on an Olympus BX51 microscope equipped with a four megapixel Macrofire digital camera (Optronics; Goleta, CA) using the PictureFrame Application 2.3 (Optronics).
Statistical Analysis
Relative densitometry of Western blots was quantified using ImageJ (http://rsb.info.nih.gov/ij/). All data and statistical analysis was performed by two-way Analysis of Variance or a students t-test using Prism 5 for Windows (GraphPad Software, San Diego, CA). All data are expressed as mean +/- S.E.M. and p values <0.05 were considered significant.
Results
Chronic EtOH consumption increases Akt phosphorylation in WT mice but has no further effect in PTENLKO mice
To verify deletion of PTEN in our model, hepatic cytosolic extracts were prepared from PF and EtOH-fed PTENf/f and PTENLKO mice. As shown in S1A and S1B Fig, EtOH consumption decreased overall PTEN expression and increased Akt phosphorylation in PTENf/f mice. In PF PTENLKO mice, PTEN expression was decreased by greater than 95% and cytoplasmic pSer473Akt levels significantly increased by 7-fold above normal. Addition of EtOH did not significantly affect total cytosolic levels of or phosphorylation of the cytosolic Akt in the PTENLKO mice. Combined these data support constitutive activation of Akt’s in PTENLKO mice irrespective of EtOH addition.
Deletion of PTEN confers significant protective effects against EtOH induced liver injury. Effects of PTENLKO and EtOH on hepatocellular function
We have previously demonstrated the consumption of a diet rich in polyunsaturated fatty acids exerts an additive effect with respect to hepatocellular damage in PTENLKO mice. The data presented in Table 1 presents the effects of either a PF diet or PTENLKO on EtOH induced hepatotoxicity. In the PTENLKO PF animals, serum ALT increased 10.7-fold when compared to PTENf/f animals. In the PTENf/f animals, chronic EtOH resulted in a 2.18-fold increase in serum ALT, a result comparable to values obtained using the C57BL6/J strain [22]. Interestingly, EtOH ingestion in the PTENLKO background resulted in a significant (p<0.05) 42% decrease in ALT relative to the PTENLKO pair fed group. Comparing liver to body weight ratios, EtOH consumption significantly increased liver/body weight in the PTENf/f group. Compared to PF PTENf/f mice, liver to body weight was 3.2-fold higher in the PTENLKO PF animals. The addition of EtOH did not have a further effect on liver to body weight ratio in PTENLKO animals. As expected, under conditions of sustained lipid synthesis, hepatic triglycerides were elevated by 6-fold in the PF PTENLKO animals compared PF PTENf/f groups [12, 29]. In the EtOH-fed groups, a significant increase in hepatic triglycerides in occurred in PTENf/f animals but triglycerides significantly decreased in EtOH-fed PTENLKO animals when compared to PF PTENLKO animals (p = 0.05). Overall, 2-way ANOVA indicated a significant interaction between PTENLKO and EtOH with respect to ALT, liver triglycerides and liver to body weight. In summary, when compared to the PTENLKO PF group, chronic EtOH consumption results in decreased hepatocellular damage as evidenced by decreased ALT and hepatic triglycerides in PTENLKO mice.
Table 1. Biochemical analysis of liver homogenates of PF and EtOH PTENf/f and PTENLKO mice.
| Parameter╪ | Pair-fed | EtOH | Pair-fed | EtOH | Genotype | EtOH | Interaction |
|---|---|---|---|---|---|---|---|
| ALT (U/L) | 16.70±3.235a | 36.44±5.604b | 178.47±24.04a,c | 103.98±11.62b,c | <0.0001 | 0.0490 | 0.0022 |
| Liver weight | 0.94±0.05a | 1.08±0.05b | 2.63±0.21a,c | 2.22±0.07b,c | <0.0001 | 0.0261 | 0.2675 |
| Body weight | 29.87±0.20a | 27.15±0.19b | 25.61±0.77a,c | 22.37±0.45b,c | <0.0001 | <0.0001 | 0.5786 |
| Change in body weight | 6.39±0.73a | 2.21±0.40b | 3.83±0.39a | 0.37±0.59b | 0.0004 | <0.0001 | 0.5125 |
| Liver/Body Weight | 3.20±0.11a | 4.03±0.18b | 11.16±0.31a,c | 10.23±0.23b,c | <0.0001 | 0.8182 | 0.0005 |
| Liver Triglycerides (mmol/mg tissue) | 0.003±0.001a | 0.008±0.001b | 0.020±0.001a,c | 0.017±0.001b,c | <0.0001 | 0.4005 | 0.0062 |
| Food Intake (mls/day) | 16.83±0.44 | 17.87±0.36 | 16.92±0.44 | 17.30±0.27 | 0.5462 | 0.0756 | 0.4031 |
Serum ALT, liver weight, body weight, change in body weight, liver to body weight, liver triglycerides and food intake were determined as described in methods. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (PTENf/f group compared to PTENLKO group). Means without a common superscript letter are significantly different (N = 7 pairs of PTENf/f PF/EtOH and 7 pairs of PTENLKO PF/EtOH mice/group (p<0.05)). Values in bold indicate significance by two-way ANOVA.
╪Letter superscripts (a,b,c) denote significant difference of P<0.05 by unpaired t-test, values in bold significant difference by Two-Way ANOVA.
Chronic EtOH consumption decreases periportal steatosis in PTENLKO mice
Given the finding that the addition of EtOH resulted in a decrease in ALT, the effects of EtOH in the PTENLKO background was further explored with respect to hepatocellular pathology using histology. Using hematoxylin and eosin staining, at low magnification (100X), mild steatosis occurred in EtOH-fed PTENf/f mice that was barely visible (Fig 1 panels A-D). Steatosis was much more significant in liver sections from PTENLKO PF mice but steatosis decreased with EtOH (Arrows Panel D). To better assess specific changes in hepatic pathology a higher magnification was employed. At higher magnification (400X) (Fig 1 panels E-H), formation of mild steatosis is much more visible following consumption of EtOH in PTENf/f mice. In the PTENLKO mice, consumption of the high fat diet in the PF group induced severe panlobular steatosis. In agreement with hepatic triglyceride accumulation, EtOH consumption by the PTENLKO mice decreased steatosis primarily in the periportal region which parallels the observed decrease in hepatic triglycerides presented in Table 1. As an additional method to support the change in periportal lipid accumulation, tissues sections were stained with adipophilin. As shown in Fig 1 Panels I-L, compared to the PTENLKO PF group, adipophilin staining decreased in the periportal region of PTENLKO EtOH mice. These data support decreased periportal lipid accumulation in EtOH-fed PTENLKO mice.
Fig 1. Effects of chronic EtOH on hepatic lipid accumulation and fibrosis in PTENf/f and PTENLKO mice.

Panels: (A-D), hematoxylin and eosin staining (100X) of liver sections isolated from PF/EtOH PTENf/f and PTENLKO mice (N = 7 pairs of PTENf/f PF/EtOH and 7 pairs of PTENLKO PF/EtOH mice/group), (E-H), (400X), (I-L), adipophilin staining (N = 3 pairs of PTENf/f PF/EtOH and 3 pairs of PTENLKO PF/EtOH mice/group), (M-P), picrosirius red staining of liver sections (N = 4 pairs of PTENf/f PF/EtOH and 3 pairs of PTENLKO PF/EtOH mice/group), (Q-T), polarized light exposure of picrosirius red staining. (CV, central vein, PT, portal triad). Original Magnification 400X, H&E staining, 100X, picrosirius red collagen staining.
To further explore differences in overall pathology in PTENLKO PF/EtOH fed mice, a modified Kleiner Scoring was performed on hematoxylin and eosin stained hepatic tissue sections [30]. As shown in Table 2, with the exception of a mild increase in steatosis, PTENf/f PF/EtOH groups did not exhibit significant differences in overall hepatic pathology. Not surprisingly, both PF and EtOH-fed PTENLKO groups exhibited increased biliary hyperplasia [12, 13]. Although there was an increasing trend, at least by pathology, steatosis was not significantly increased by EtOH in the PTENf/f control groups. Steatosis in the PTENLKO PF group was extensive and panlobular. Addition of EtOH resulted in a significant decrease in steatosis primarily in zone 1. Both PTENLKO groups exhibited macro and microsteatosis. Overall, the combined modified Kleiner score was significantly decreased by 6-weeks consumption of EtOH in PTENLKO mice. An examination by Two-Way analysis of variance revealed a significant interaction between the PTENLKO genotype and EtOH with respect to overall hepatic steatosis and overall modified Kleiner score.
Table 2. Modified Kleiner scoring of tissue sections isolated from PF and EtOH PTENf/f and PTENLKO mice.
| PTENf/f | PTENLKO | Two-Way ANOVA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Parameter╪ | PF | EtOH | PF | EtOH | Genotype | EtOH | Interaction | ||
| Steatosis (0–3) | 0.04±0.12a | 0.43±0.13b | 2.93±0.13a,c | 2.07±0.16b,c | <0.0001 | 0.0965 | 0.0206 | ||
| Ballooning/Degeneration (0–2) | 0.0±0.0 | 0.0±0.0 | 1.14±0.23c | 0.43±0.28c | 0.0002 | 0.0618 | 0.0618 | ||
| Lobular Inflammation (0–2) | 0.0±0.0 | 0.0±0.0 | 1.00±0.0c | 1.00±0.0c | <0.0001 | 1 | 1 | ||
| Microsteatosis (0–3) | 0.0±0.0 | 0.0±0.0 | 1.00±0.0c | 1.00±0.0c | 0.0001 | 1 | 1 | ||
| Score Modified Kleiner (0–10) | 0.04±0.12a | 0.43±0.13b | 6.07±0.25a,c | 4.50±0.30b,c | <0.0001 | 0.0099 | 0.0002 | ||
| Biliary Hyperplasia | 0.0±0.0 | 0.0±0.0 | 2.00±0.29c | 2.14±0.23c | <0.0001 | 0.7086 | 0.7068 |
Overall hepatic steatosis, hepatocyte ballooning, lobular inflammation, microsteatosis, biliary hyperplasia were quantified according to Brunt and Kleiner with minor modifications [30]. Data are means± SEM as analyzed by two-way ANOVA with a Bonferroni post hoc analysis (PTENf/f group compared to PTENLKO group). To determine specific effects of EtOH within each group, an unpaired student’s t-test was performed. Means without a common superscript letter are significantly different (N = 7 pairs of PTENf/f PF/EtOH and 7 pairs of PTENLKO PF/EtOH mice/group (p<0.05)). Values in bold indicate significance by two-way ANOVA.
╪Letter superscripts (a,b,c) denote significant difference of P<0.05 by unpaired t-test, values in bold significant difference by Two-Way ANOVA.
In a previous report, PTENLKO mice develop fibrosis by 40 weeks [12, 13]. Although the mice used in the present study were only 18 weeks old at study completion, we hypothesized that EtOH may also affect fibrosis. Therefore, we examined the effects of 6 weeks of EtOH consumption on fibrosis using Picrosirius red staining [31]. To eliminate showing “selected” fields of the liver, lower magnification are presented in Panels M-P (white light) and lower panels ((Panels Q-T) polarized light). Chronic EtOH consumption did not result in an increase in picrosirius red staining in the PTENf/f animals. In PF PTENLKO, only mild fibrosis was present compared to PF PTENf/f mice. Following 6-weeks consumption of EtOH, no significant differences were evident in PTENLKO mice (quantification not shown), indicating that fibrosis as evidenced by collagen deposition was not affected by EtOH consumption in PTENLKO mice.
Effects of PTENLKO and EtOH on ADH and ALDH2 expression
When ingested, EtOH is first metabolized by alcohol dehydrogenase 1 (ADH1) forming acetaldehyde which is then further metabolized by aldehyde dehydrogenase 2 (ALDH2) to produce acetate [32]. To determine the effects of PTENLKO on EtOH metabolism, expression of ADH1, and ALDH2 was examined. As shown in Fig 2A and 2B, EtOH ingestion significantly increased ALDH2 expression in PTENf/f and PTENLKO mice. Chronic EtOH consumption had no effect on ADH expression in the PTENf/f mice but in PTENLKO, a significant increase was present in both PF and EtOH mice. This suggested that metabolism of EtOH might be increased in PTENLKO mice. Therefore, overall blood ethanol concentrations (BEC) were examined using serum isolated from each group. In EtOH-fed PTENf/f and PTENLKO mice, BEC was increased (PTENf/f 137.38±66.69, PTENLKO 120.25±26.48) but no significant differences were evident between the two genotypes indicating that metabolism of EtOH is not significantly affected by hepatospecific deletion of PTEN.
Fig 2. Effects of PTENLKO and EtOH on expression of EtOH metabolizing enzymes.
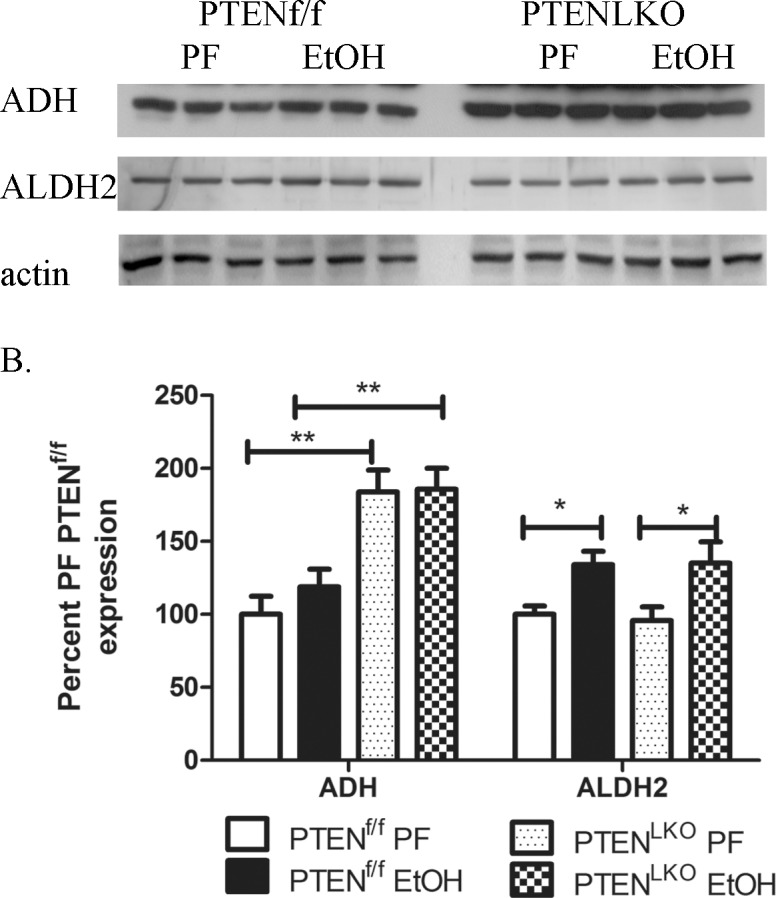
Whole cell extracts fractions from PF control and EtOH PTENf/f/PTENLKO groups were analyzed via SDS PAGE, Western blotted and probed for ADH and ALDH2. (A) Western blotting analysis of ALDH2 and ADH expression in PF and EtOH-fed PTENf/f and PTENLKO mice. (B) Quantification of the Western blots presented in Fig 2A (actin normalized). Data are means± SEM as analyzed by students t-test (PF/EtOH) and two-way ANOVA with a Bonferroni post hoc analysis (PTENf/f group compared to PTENLKO group) (N = 6 mice/group (*p<0.05, ***p<0.001)).
Consumption of EtOH does not increase protein carbonylation in PTENLKO mice
Chronic EtOH consumption results in increased accumulation of lipid aldehyde modified hepatic proteins (protein carbonylation) [19, 33]. To examine the effects of constitutive Akt activation on protein carbonylation, liver sections prepared from PTENf/f and PTENLKO pairs were examined for expression of Cyp2E1, acrolein, 4-HNE and MDA via immunohistochemistry (Fig 3). Expression of Cyp2E1 was clearly elevated in the centrilobular region of both genotypes following EtOH consumption. Examining protein carbonylation, in PTENf/f animals, EtOH-induced a periportal increase in post-translational modification of proteins by oxidative stress induced lipid peroxidation products acrolein, MDA and 4-HNE. Surprisingly, in the PTENLKO model, this increase was not evident. As interesting, strong acrolein staining occurred in the cholangiocytes of PTENLKO mice although the ramifications of increased cholangiocyte acrolein staining are not clear. Collectively, these data indicate chronic consumption of EtOH results in Cyp2E1 induction that is not associated with increased protein carbonylation in PTENLKO mice.
Fig 3. Effects of chronic EtOH on protein carbonylation in PTENLKO mice.

(A) Hepatic tissue sections isolated from PF and EtOH-fed PTENf/f/PTENLKO mice were analyzed for protein modification by acrolein, MDA and 4-HNE and Cyp2E1 expression using immunohistochemistry. Figures are representative of five PF/EtOH groups of PTENf/f and PTENLKO mice respectively.
GSH homeostasis is increased in PTENLKO mice
In mice, 6-weeks consumption of EtOH results in a 30% decrease in total hepatic GSH concentrations [34]. By its ability to conjugate GSH with reactive aldehydes, glutathione S-transferase A4 (GSTA4) represents a primary mechanism for regulation of hepatic protein carbonylation [35–38]. To determine if alterations in GSH metabolism are contributing factors in mitigation of oxidative stress occurring in PTENLKO mice, tissue was isolated and pooled from 4 PF/EtOH fed PTENf/f/PTENLKO mice respectively. Microarray analysis comparing PF/EtOH fed PTENLKO, PF PTENf/f /PF PTENLKO and PTENf/f PF/EtOH fed was then performed using cDNA transcribed from mRNA. From the heat map presented in Fig 4, chronic EtOH administration increased mRNA expression of GSTa2/4, GSTm1/2/4/6, glutathione synthetase (GSS) and glutathione cysteine ligase (GCLC). When compared to the PF PTENf/f group, PF PTENLKO mice exhibited increased mRNA expression of almost all GSTs as well as GSH synthesizing enzymes. Addition of EtOH decreased GSTa1/2/3/5 and GSTm6 in PTENLKO mice. Overall these data support increased GSH homeostasis in PTENLKO mice. KEGG pathways analysis was used to gain addition understanding of the ramifications of changes in the genes presented in Fig 4. From the data presented in S1 Table, hepatospecific deletion of PTEN significantly affected glutathione metabolic and xenobiotic detoxification pathways.
Fig 4. Effects of PTENLKO and EtOH on expression of GSH homeostatic genes.
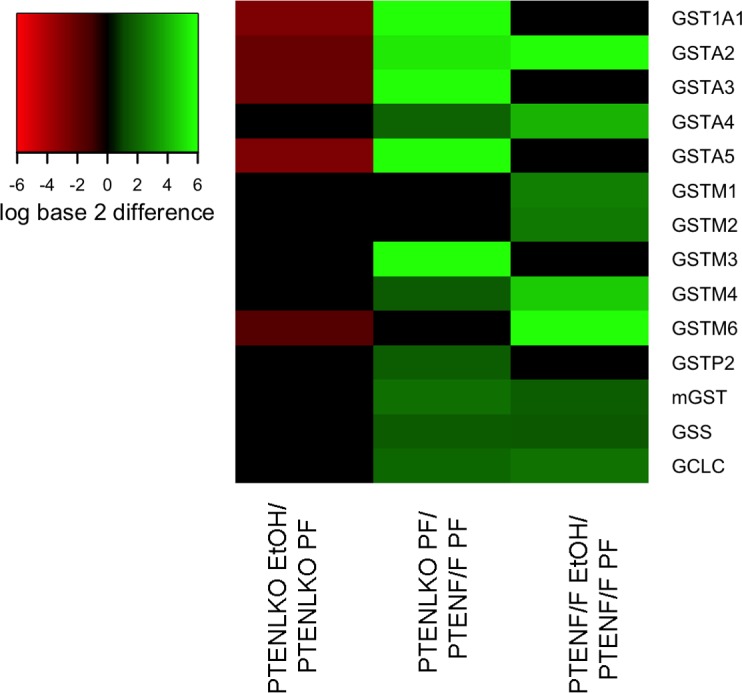
A limited microarray was performed using pooled tissue isolated from 3 pairs of PF/EtOH fed PTENLKO and PTENf/f respectively. The values represented in the heat map are log base 2 differences in gene expression compared to the respective control. The values are indicated by color with red representing lower expression in the following: PTENLKO EtOH (lane 1), PTENLKO PF (lane 2), PTENf/f EtOH (lane 3), compared to the control: PTENLKO PF (lane 1), PTENf/f PF (lane 2), PTENf/f PF (lane 3) group and with bright green representing higher expression in the treatment groups compared to the controls. Differences in expression that were between -1.5 and 1.5 are represented using the color black.
Effects of PTENLKO and EtOH on overall glutathione oxidation and reduction (redox) capacity
In hepatocytes, reactive aldehydes are removed via conjugation to GSH [39]. Oxidation of glutathione (GSSG) occurs under conditions of oxidative stress and a significant decrease in the ratio of GSH:GSSG is an accepted marker of increased oxidative stress [40, 41]. In wild type murine models, chronic addition of EtOH results in a decrease in total reduced GSH but no significant change in GSSG [34]. We hypothesized that in PTENLKO mice, an increase in GSH contributes to decreased carbonylation following chronic EtOH consumption. From Fig 5A, EtOH decreased GSH by 30% in the PTENf/f animals, a result similar to data previously obtained in WT C57BL6/J mice [16]. Comparing both genotypes, deletion of PTEN significantly increased GSH and GSSG. In the PTENLKO model, EtOH slightly increased hepatic GSH but did not affect GSSG. Examining redox status, the ratio of GSH:GSSG significantly decreased in PTENf/f mice following EtOH. This effect was reversed in PTENLKO mice. Compared to either group of PTENf/f mice, cellular redox ratios significantly decreased in the PTENLKO mice. Using 2-way ANOVA, genotype effects were evident for all three parameters and a significant interaction occurred with respect to GSH and redox ratio. Combined with data presented in Figs 1, 2 and 3, these data indicate that in PTENLKO mice there is increased GSH, enhanced mRNA expression of glutathione S-transferases correlating with mitigation of protein carbonylation, decreased ALT and decreased hepatic triglycerides.
Fig 5. Oxidative stress measurements in hepatic tissue isolated from PF and EtOH-fed PTENf/f and PTENLKO mice.
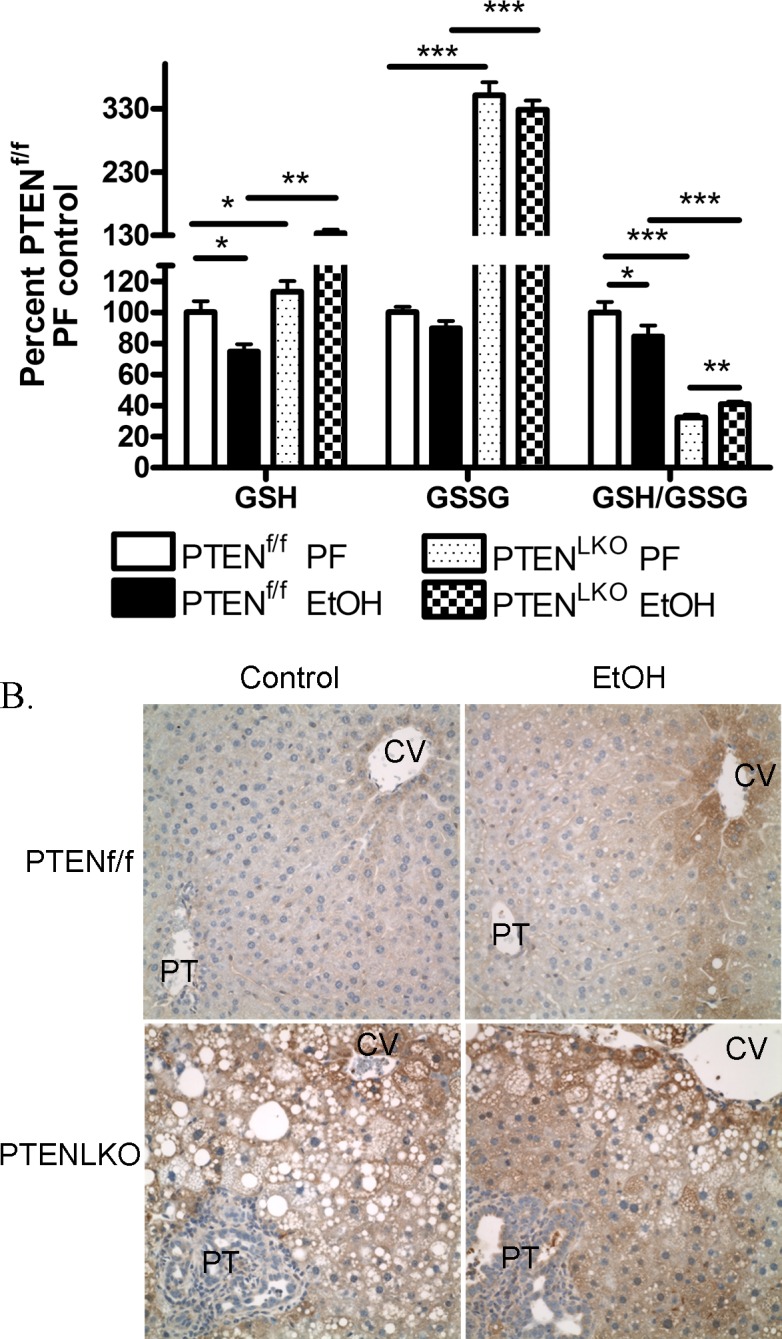
(A) GSH, GSSG and GSH/GSSG. Total hepatic concentrations of GSH and GSSG were determined from 6 pairs of PF/EtOH-fed PTENf/f and 7 pairs of PTENLKO respectively. Data are means± SEM as analyzed by students t-test (PF/EtOH) and two-way ANOVA with a Bonferroni post hoc analysis (PTENf/f group compared to PTENLKO group) (N = 6 mice/group (*p<0.05, ***p<0.001)) (B) Hepatic tissue sections isolated from PF and EtOH-fed PTENf/f/PTENLKO mice were analyzed for Protein-SSG using immunohistochemistry. Figures are representative of 3 PF/EtOH groups of PTENf/f and PTENLKO mice respectively.
Protein glutathionylation is increased in PTENLKO mice
In the PTENLKO mice, there is a significant increase in GSH and GSSG. We hypothesized that increased cellular GSH would also result in an increased in protein glutathionylation. As shown in Fig 5B, in the PTENf/f group, protein-SSG is increased around the central vein following consumption of EtOH indicating an increase in oxidative stress. Examining PF/EtOH fed PTENLKO mice, staining of protein SSG was increased when compared to respective PTENf/f controls but no effect was evident with respect to EtOH.
Discussion
Steatosis is an early consequence of ALD as well as NASH. Given that in the Western world, obesity and NASH are rapidly increasing it is critical to understand that combinatorial effects of alcohol and NASH. Recent research has clearly demonstrated that a major regulator of hepatocellular lipid accumulation is the PTEN/Akt pathway [12]. We previously demonstrated that in C57BL/6J WT mice, chronic consumption of EtOH increases PTEN phosphorylation, carbonylation and decreases PTEN expression [19]. This translated to an increase in Akt activation contributing to the formation of steatosis [19, 42]. To further elucidate the contribution of PTEN/Akt signaling in EtOH-induced hepatocellular toxicity, we utilized PTENLKO mice as a model of increased Akt activation and preexisting steatosis [11, 15, 19]. Our initial hypothesis was that hepatocellular damage would not increase following EtOH consumption and that concurrent steatosis in PTENLKO mice in conjunction with EtOH would exert an additive effect with respect to hepatic triglyceride accumulation. From the results obtained in this study, chronic consumption of EtOH reversed high fat diet induced increases in hepatocellular damage as evidenced by decreased ALT and also decreased hepatic triglycerides.
Compared to the pair-fed PTENf/f genotype, pair-fed PTENLKO mice displayed a dramatic 3-fold increase in overall liver weight, an increased liver:body weight ratios. Hepatic triglycerides and ALT both increased by 10-fold accompanied by a dramatic reduction of GSH/GSSG ratio. These data demonstrate that the high polyunsaturated fat PF diet produces significant liver injury by itself in the PTENLKO mice and is associated with increased oxidative stress and inflammation. In the PTENf/f animals, chronic EtOH consumption resulted in a mild but significant increase in hepatic triglycerides, steatosis and hepatocellular damage as shown by increased ALT as well as by increased protein carbonylation. In the PTENLKO model, surprisingly, chronic EtOH addition resulted in a significant decrease in ALT, periportal steatosis and hepatic triglycerides. Furthermore, compared to PF PTENLKO, an increase in protein modification by reactive aldehydes did not occur. This is in agreement with previous data that demonstrates that PTENLKO mice are resistant to additional hydrogen peroxide-induced oxidative stress [43]. Furthermore, in chow-fed PTENLKO mice, basal oxidative stress is increased when compared to WT controls [44].
In the PTENLKO PF group, expression of GSH metabolizing enzymes (GST’s, GCLC, GSS) are increased when compared to PTENf/f controls. This corresponds to increased GSH and GSSG and indicates a plausible mechanism for resistance to EtOH-induced oxidative stress. In a previous report, expression of GSTm6 was decreased in PTENLKO mice whereas we find no significant change [14]. In that report, array analysis was performed at 10 weeks of age and the authors hypothesize that downregulation of GSTm6 may contribute to an increase in inflammation that occurs after 10 weeks of age. Our data originated from 18 week old PTENLKO mice and demonstrate no significant change in GSTm6 expression in PTENLKO PF groups when compared to PTENf/f PF groups. In the previous study, analysis did not report differences in other GST isoforms. In this study, we find that GSTa1/2/3/4/5 and GSTm3 are all upregulated in PF PTENLKO mice when compared to PF PTENf/f mice indicating an enhanced response to diet induced inflammation. Chronic EtOH challenge however, decreases GSTm6 expression and decreased GSTA isoforms suggesting that inflammation may be decreased. In a recent publication, PTENLKO mice were protected against endotoxemia [21]. In that study the authors determined that heme oxygenase was upregulated by PPARγ. We do not see HO-1 upregulation in this study but we do see increased PPARγ in the PTENLKO model [15]. A difference between our study, is that Guenzl et al, used mice younger than 12-weeks for their studies and there are some reports that suggest that 12 weeks of age is necessary for the full Cre-recombination and PTEN deletion to take effect [12–14, 45].
Interestingly, using the same model, 6-weeks consumption of alcohol and WT SV and C57BL/6J mice, protein adducts are increased in the periportal region [19, 34, 46]. This also occurs in the PTENf/f controls. In this study, protein-SSG is only increased around the central venous region in PTENf/f mice but is increased panlobularly with EtOH in the PTENLKO model. This suggests that glutathionylation may also prevent carbonylation of proteins. Recent evidence suggests that GSTμ by its protein-SSG regulatory function, exerts a positive influence against ER stress [47]. In support of this mechanism, both GSTμ and protein-SSG are elevated in PF PTENLKO mice when compared to PF PTENf/f controls. Increased GST activity exerts a protective effect in NASH [48]. Post-translational modification of cysteine residues occurs in both glutathionylation and carbonylation [48–51]. Increased glutathionylation would protect critical cysteine residues by preventing carbonylation in a reversible mechanism. Future studies will be necessary to fully elucidate the impact of increased glutathionylation and inflammation in PTENLKO mice chronically fed EtOH and to determine clinical relevance.
In conclusion, this study examined the effects of chronic EtOH consumption in the background of constitutively activated de novo lipogenesis, increased steatosis and increased cellular respiration. The data obtained provide new insight into the hepatocellular outcomes of preexisting steatosis due to increased Akt activation during alcohol consumption [19]. In PTENLKO mice, EtOH consumption did not exacerbate hepatocellular damage suggesting that an environment of increased hepatocellular glutathione concentration may be protective. We hypothesize that reduced injury evident in this study is in part from enhancement of protective oxidative stress responses due to increased cellular respiration occurring during constitutive Akt activation (PTENLKO) (S1 Fig). This preexisting condition in PTENLKO mice results in an alteration of glutathione homeostasis creating a compensatory hepatocellular environment primed to mitigate increased oxidative stress and hepatocellular damage by EtOH. Alternatively, in the background of increased de novo lipogenesis, the addition of EtOH mitigates the effects of a high fat diet. Additional studies will be necessary to determine the sustainability these compensatory mechanisms under conditions of long-term ethanol ingestion and to determine proteins downstream of PTEN that contribute to the protective effect.
Supporting Information
(A) Cytosolic extracts from PF and EtOH-fed PTENf/f/PTENLKO groups were analyzed via SDS PAGE, Western blotted and probed for PTEN, pSer473 Akt, and total Akt. (B) Quantification of the Western blots presented in S1A Fig (actin normalized). Data are means± SEM as analyzed by students t-test (PF/EtOH) and two-way ANOVA with a Bonferroni post hoc analysis (PTENf/f group compared to PTENLKO group) (N = 3 mice/group (*p<0.05, ***p<0.001)).
(DOCX)
Proteins identified in Fig 4 were examined using KEGG pathway analysis as previously described [49].
(XLSX)
Acknowledgments
This research was supported by the following grants from the National Institutes of Health; F32 AA018613-03 CTS, R37AA009300-18 DRP. The authors also wish to thank E. Erin Smith, HTL(ASCP)CMQIHC of the University of Colorado Denver Cancer Center Research Histology Core for assistance in preparing histology slides. The UCDCCRHC is supported in part by NIH/NCRR Colorado CTSI Grant Number UL1 RR025780 and the University of Colorado Cancer Center Grant (P30 CA046934).
Abbreviations
- 4-HNE
4-Hydroxynonenal
- ALD
Alcoholic Liver Disease
- ALT
Alanine aminotransferase
- ANOVA
Analysis of variance
- BEC
Blood ethanol concentration
- Cyp2E1
Cytochrome P450 2E1
- DNL
De novo lipogenesis
- GCLC
Glutathione cysteine ligase
- GSH
Reduced glutathione
- GSS
Glutathione synthetase
- GSSG
Glutathione disulfide
- GSTA4
Glutathione S-transferase A4
- MDA
Malondialdehyde
- NAFLD
Nonalcoholic fatty liver disease
- PF
Pair-fed
- PTEN
Phosphatase and tensin homolog on chromosome 10
- PTENf/f
PTEN mice with floxed alleles but without Cre-Recombinase
- PTENLKO
Liver-specific PTEN deletion mice
- PtdIns
Phosphatidylinositol
- SEM
Standard error of the mean
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
Funding was provided by National Institutes of Health (NIH) Division of Alcohol Abuse and Alcoholism, www.niaaa.nih.gov: F32 AA018613-03 CTS, R37AA009300-18 DRP. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.De Minicis S, Brenner DA. Oxidative stress in alcoholic liver disease: role of NADPH oxidase complex. J Gastroenterol Hepatol. 2008;23 Suppl 1:S98–103. 10.1111/j.1440-1746.2007.05277.x [DOI] [PubMed] [Google Scholar]
- 2.Alatalo PI, Koivisto HM, Hietala JP, Puukka KS, Bloigu R, Niemela OJ. Effect of moderate alcohol consumption on liver enzymes increases with increasing body mass index. Am J Clin Nutr. 2008;88(4):1097–103. Epub 2008/10/10. doi: 88/4/1097 [pii]. . [DOI] [PubMed] [Google Scholar]
- 3.Matsuda S, Kobayashi M, Kitagishi Y. Roles for PI3K/AKT/PTEN Pathway in Cell Signaling of Nonalcoholic Fatty Liver Disease. ISRN Endocrinol. 2013;2013:472432 10.1155/2013/472432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yeon JE, Califano S, Xu J, Wands JR, De La Monte SM. Potential role of PTEN phosphatase in ethanol-impaired survival signaling in the liver. Hepatology. 2003;38(3):703–14. . [DOI] [PubMed] [Google Scholar]
- 5.de la Monte SM, Yeon JE, Tong M, Longato L, Chaudhry R, Pang MY, et al. Insulin resistance in experimental alcohol-induced liver disease. J Gastroenterol Hepatol. 2008;23(8 Pt 2):e477–86. 10.1111/j.1440-1746.2008.05339.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denucci SM, Tong M, Longato L, Lawton M, Setshedi M, Carlson RI, et al. Rat strain differences in susceptibility to alcohol-induced chronic liver injury and hepatic insulin resistance. Gastroenterol Res Pract. 2010;2010 Epub 2010/09/04. 10.1155/2010/312790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peyrou M, Bourgoin L, Foti M. PTEN in non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and cancer. Dig Dis. 2010;28(1):236–46. Epub 2010/05/13. doi: 000282095 [pii] 10.1159/000282095 . [DOI] [PubMed] [Google Scholar]
- 8.Leavens KF, Easton RM, Shulman GI, Previs SF, Birnbaum MJ. Akt2 is required for hepatic lipid accumulation in models of insulin resistance. Cell Metab. 2009;10(5):405–18. 10.1016/j.cmet.2009.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ross SH, Lindsay Y, Safrany ST, Lorenzo O, Villa F, Toth R, et al. Differential redox regulation within the PTP superfamily. Cell Signal. 2007. . [DOI] [PubMed] [Google Scholar]
- 10.Maehama T, Dixon JE. PTEN: a tumour suppressor that functions as a phospholipid phosphatase. Trends Cell Biol. 1999;9(4):125–8. . [DOI] [PubMed] [Google Scholar]
- 11.Shearn CT, Petersen DR. Understanding the tumor suppressor PTEN in chronic alcoholism and hepatocellular carcinoma. Adv Exp Med Biol. 2015;815:173–84. 10.1007/978-3-319-09614-8_10 . [DOI] [PubMed] [Google Scholar]
- 12.Stiles B, Wang Y, Stahl A, Bassilian S, Lee WP, Kim YJ, et al. Liver-specific deletion of negative regulator Pten results in fatty liver and insulin hypersensitivity [corrected]. Proc Natl Acad Sci U S A. 2004;101(7):2082–7. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horie Y, Suzuki A, Kataoka E, Sasaki T, Hamada K, Sasaki J, et al. Hepatocyte-specific Pten deficiency results in steatohepatitis and hepatocellular carcinomas. J Clin Invest. 2004;113(12):1774–83. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sato W, Horie Y, Kataoka E, Ohshima S, Dohmen T, Iizuka M, et al. Hepatic gene expression in hepatocyte-specific Pten deficient mice showing steatohepatitis without ethanol challenge. Hepatol Res. 2006;34(4):256–65. . [DOI] [PubMed] [Google Scholar]
- 15.Shearn CT, Mercer KE, Orlicky DJ, Hennings L, Smathers-McCullough RL, Stiles BL, et al. Short Term Feeding of a High Fat Diet Exerts an Additive Effect on Hepatocellular Damage and Steatosis in Liver-Specific PTEN Knockout Mice. PLoS One. 2014;9(5):e96553 Epub 2014/05/14. 10.1371/journal.pone.0096553 PONE-D-14-01221 [pii]. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galligan JJ, Smathers RL, Fritz KS, Epperson LE, Hunter LE, Petersen DR. Protein carbonylation in a murine model for early alcoholic liver disease. Chem Res Toxicol. 2012;25(5):1012–21. Epub 2012/04/17. 10.1021/tx300002q [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.You M, Crabb DW. Molecular mechanisms of alcoholic fatty liver: role of sterol regulatory element-binding proteins. Alcohol. 2004;34(1):39–43. Epub 2005/01/27. doi: S0741-8329(04)00152-1 [pii] 10.1016/j.alcohol.2004.07.004 . [DOI] [PubMed] [Google Scholar]
- 18.You M, Fischer M, Deeg MA, Crabb DW. Ethanol induces fatty acid synthesis pathways by activation of sterol regulatory element-binding protein (SREBP). J Biol Chem. 2002;277(32):29342–7. Epub 2002/05/31. 10.1074/jbc.M202411200 M202411200 [pii]. . [DOI] [PubMed] [Google Scholar]
- 19.Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, Petersen DR. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radic Biol Med. 2013;65:680–92. Epub 2013/07/23. doi: S0891-5849(13)00342-0 [pii] 10.1016/j.freeradbiomed.2013.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu J, Wang X, Peng Z, Zhang T, Wu H, Yu W, et al. The effects of Insulin Pre-Administration in Mice Exposed to Ethanol: Alleviating Hepatic Oxidative Injury through Anti-Oxidative, Anti-Apoptotic Activities and Deteriorating Hepatic Steatosis through SRBEP-1c Activation. Int J Biol Sci. 2015;11(5):569–86. 10.7150/ijbs.11039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guenzl PM, Raim R, Kral J, Brunner J, Sahin E, Schabbauer G. Insulin hypersensitivity induced by hepatic PTEN gene ablation protects from murine endotoxemia. PLoS One. 2013;8(6):e67013 10.1371/journal.pone.0067013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shearn CT, Smathers RL, Jiang H, Orlicky DJ, Maclean KN, Petersen DR. Increased dietary fat contributes to dysregulation of the LKB1/AMPK pathway and increased damage in a mouse model of early-stage ethanol-mediated steatosis. J Nutr Biochem. 2013;24(8):1436–45. Epub 2013/03/08. doi: S0955-2863(12)00300-2 [pii] 10.1016/j.jnutbio.2012.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galligan JJ, Fritz KS, Tipney H, Smathers RL, Roede JR, Shearn CT, et al. Profiling impaired hepatic endoplasmic reticulum glycosylation as a consequence of ethanol ingestion. J Proteome Res. 2011;10(4):1837–47. Epub 2011/02/16. 10.1021/pr101101s [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shearn CT, Backos DS, Orlicky DJ, Smathers-McCullough RL, Petersen DR. Identification of 5' AMP activated kinase as a target of reactive aldehydes during chronic ingestion of high concentrations of ethanol. J Biol Chem. 2014. Epub 2014/04/12. doi: M113.543942 [pii] 10.1074/jbc.M113.543942 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ronis MJ, Hennings L, Stewart B, Basnakian AG, Apostolov EO, Albano E, et al. Effects of long-term ethanol administration in a rat total enteral nutrition model of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2011;300(1):G109–19. Epub 2010/11/06. doi: ajpgi.00145.2010 [pii] 10.1152/ajpgi.00145.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Setshedi M, Longato L, Petersen DR, Ronis M, Chen WC, Wands JR, et al. Limited therapeutic effect of N-acetylcysteine on hepatic insulin resistance in an experimental model of alcohol-induced steatohepatitis. Alcohol Clin Exp Res. 2011;35(12):2139–51. Epub 2011/07/28. 10.1111/j.1530-0277.2011.01569.x . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Z, Gasser DL, Rappaport EF, Falk MJ. Cross-platform expression microarray performance in a mouse model of mitochondrial disease therapy. Mol Genet Metab. 2010;99(3):309–18. Epub 2009/12/01. doi: S1096-7192(09)00472-7 [pii] 10.1016/j.ymgme.2009.10.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roede JR, Orlicky DJ, Fisher AB, Petersen DR. Overexpression of peroxiredoxin 6 does not prevent ethanol-mediated oxidative stress and may play a role in hepatic lipid accumulation. J Pharmacol Exp Ther. 2009;330(1):79–88. 10.1124/jpet.109.152983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He L, Hou X, Kanel G, Zeng N, Galicia V, Wang Y, et al. The critical role of AKT2 in hepatic steatosis induced by PTEN loss. Am J Pathol. 2010;176(5):2302–8. 10.2353/ajpath.2010.090931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kleiner DE, Brunt EM. Nonalcoholic fatty liver disease: pathologic patterns and biopsy evaluation in clinical research. Semin Liver Dis. 2012;32(1):3–13. 10.1055/s-0032-1306421 . [DOI] [PubMed] [Google Scholar]
- 31.Junqueira LC, Bignolas G, Brentani RR. Picrosirius staining plus polarization microscopy, a specific method for collagen detection in tissue sections. Histochem J. 1979;11(4):447–55. Epub 1979/07/01. . [DOI] [PubMed] [Google Scholar]
- 32.Albano E, Clot P, Morimoto M, Tomasi A, Ingelman-Sundberg M, French SW. Role of cytochrome P4502E1-dependent formation of hydroxyethyl free radical in the development of liver damage in rats intragastrically fed with ethanol. Hepatology. 1996;23(1):155–63. . [DOI] [PubMed] [Google Scholar]
- 33.Galligan JJ, Smathers RL, Fritz KS, Epperson E, Hunter LE, Petersen DR. Protein Carbonylation in a Murine Model for Early Alcoholic Liver Disease. Chem Res Toxicol. 2012. Epub 2012/04/17. 10.1021/tx300002q . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Galligan JJ, Smathers RL, Shearn CT, Fritz KS, Backos DS, Jiang H, et al. Oxidative Stress and the ER Stress Response in a Murine Model for Early-Stage Alcoholic Liver Disease. J Toxicol. 2012;2012:207594 Epub 2012/07/26. 10.1155/2012/207594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nanduri B, Hayden JB, Awasthi YC, Zimniak P. Amino acid residue 104 in an alpha-class glutathione S-transferase is essential for the high selectivity and specificity of the enzyme for 4-hydroxynonenal. Arch Biochem Biophys. 1996;335(2):305–10. Epub 1996/11/15. doi: S0003-9861(96)90511-1 [pii] 10.1006/abbi.1996.0511 . [DOI] [PubMed] [Google Scholar]
- 36.Yang Y, Sharma R, Sharma A, Awasthi S, Awasthi YC. Lipid peroxidation and cell cycle signaling: 4-hydroxynonenal, a key molecule in stress mediated signaling. Acta Biochim Pol. 2003;50(2):319–36. Epub 2003/07/02. doi: 035002319. . [PubMed] [Google Scholar]
- 37.Engle MR, Singh SP, Czernik PJ, Gaddy D, Montague DC, Ceci JD, et al. Physiological role of mGSTA4-4, a glutathione S-transferase metabolizing 4-hydroxynonenal: generation and analysis of mGsta4 null mouse. Toxicol Appl Pharmacol. 2004;194(3):296–308. Epub 2004/02/06. 10.1016/j.taap.2003.10.001 S0041008X03004873 [pii]. . [DOI] [PubMed] [Google Scholar]
- 38.Vaillancourt F, Fahmi H, Shi Q, Lavigne P, Ranger P, Fernandes JC, et al. 4-Hydroxynonenal induces apoptosis in human osteoarthritic chondrocytes: the protective role of glutathione-S-transferase. Arthritis Res Ther. 2008;10(5):R107 10.1186/ar2503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hartley DP, Ruth JA, Petersen DR. The hepatocellular metabolism of 4-hydroxynonenal by alcohol dehydrogenase, aldehyde dehydrogenase, and glutathione S-transferase. Arch Biochem Biophys. 1995;316(1):197–205. . [DOI] [PubMed] [Google Scholar]
- 40.Jones DP. Radical-free biology of oxidative stress. Am J Physiol Cell Physiol. 2008;295(4):C849–68. 10.1152/ajpcell.00283.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Go YM, Pohl J, Jones DP. Quantification of redox conditions in the nucleus. Methods Mol Biol. 2009;464:303–17. 10.1007/978-1-60327-461-6_17 [DOI] [PubMed] [Google Scholar]
- 42.Shearn CT, Smathers RL, Stewart BJ, Fritz KS, Galligan JJ, Hail N Jr., et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) inhibition by 4-hydroxynonenal leads to increased Akt activation in hepatocytes. Mol Pharmacol. 2011;79(6):941–52. Epub 2011/03/19. doi: mol.110.069534 [pii] 10.1124/mol.110.069534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zeng N, Li Y, He L, Xu X, Galicia V, Deng C, et al. Adaptive basal phosphorylation of eIF2alpha is responsible for resistance to cellular stress-induced cell death in Pten-null hepatocytes. Mol Cancer Res. 2011;9(12):1708–17. Epub 2011/10/20. doi: 1541-7786.MCR-11-0299 [pii] 10.1158/1541-7786.MCR-11-0299 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, He L, Zeng N, Sahu D, Cadenas E, Shearn C, et al. Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) signaling regulates mitochondrial biogenesis and respiration via estrogen-related receptor alpha (ERRalpha). J Biol Chem. 2013;288(35):25007–24. Epub 2013/07/10. doi: M113.450353 [pii] 10.1074/jbc.M113.450353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Watanabe S, Horie Y, Suzuki A. Hepatocyte-specific Pten-deficient mice as a novel model for nonalcoholic steatohepatitis and hepatocellular carcinoma. Hepatol Res. 2005;33(2):161–6. . [DOI] [PubMed] [Google Scholar]
- 46.Ronis MJ, Mercer KE, Gannon B, Engi B, Zimniak P, Shearn CT, et al. Increased 4-hydroxynonenal protein adducts in male GSTA4-4/PPARalpha double knockout mice enhance injury during early stages of alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol. 2014:ajpgi 00154 2014. Epub 2014/12/17. doi: ajpgi.00154.2014 [pii] 10.1152/ajpgi.00154.2014 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ye Z, Zhang J, Ancrum T, Manevich Y, Townsend DM, Tew KD. S-Glutathionylation of Endoplasmic Reticulum Proteins Impacts Unfolded Protein Response Sensitivity. Antioxid Redox Signal. 2016. 10.1089/ars.2015.6486 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nobili V, Pastore A, Gaeta LM, Tozzi G, Comparcola D, Sartorelli MR, et al. Glutathione metabolism and antioxidant enzymes in patients affected by nonalcoholic steatohepatitis. Clin Chim Acta. 2005;355(1–2):105–11. Epub 2005/04/12. doi: S0009-8981(05)00058-6 [pii] 10.1016/j.cccn.2004.12.022 . [DOI] [PubMed] [Google Scholar]
- 49.Shearn CT, Fritz KS, Shearn AH, Saba LM, Mercer KE, Engi B, et al. Deletion of GSTA4-4 results in increased mitochondrial post-translational modification of proteins by reactive aldehydes following chronic ethanol consumption in mice. Redox Biol. 2015;7:68–77. 10.1016/j.redox.2015.11.013 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Codreanu SG, Zhang B, Sobecki SM, Billheimer DD, Liebler DC. Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol Cell Proteomics. 2009;8(4):670–80. Epub 2008/12/05. doi: M800070-MCP200 [pii] 10.1074/mcp.M800070-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fitzpatrick AM, Teague WG, Burwell L, Brown MS, Brown LA. Glutathione oxidation is associated with airway macrophage functional impairment in children with severe asthma. Pediatr Res. 2011;69(2):154–9. Epub 2010/10/27. 10.1203/PDR.0b013e3182026370 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(A) Cytosolic extracts from PF and EtOH-fed PTENf/f/PTENLKO groups were analyzed via SDS PAGE, Western blotted and probed for PTEN, pSer473 Akt, and total Akt. (B) Quantification of the Western blots presented in S1A Fig (actin normalized). Data are means± SEM as analyzed by students t-test (PF/EtOH) and two-way ANOVA with a Bonferroni post hoc analysis (PTENf/f group compared to PTENLKO group) (N = 3 mice/group (*p<0.05, ***p<0.001)).
(DOCX)
Proteins identified in Fig 4 were examined using KEGG pathway analysis as previously described [49].
(XLSX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.