

Abstract
Purpose
Age-related macular degeneration is a common form of vision loss affecting older adults. The etiology of AMD is multifactorial and is influenced by environmental and genetic risk factors. In this study, we examine how 19 common risk variants contribute to drusen progression, a hallmark of AMD pathogenesis.
Methods
Exome chip data was made available through the International AMD Genomics Consortium (IAMDGC). Drusen quantification was carried out with color fundus photographs using an automated drusen detection and quantification algorithm. A genetic risk score (GRS) was calculated per subject by summing risk allele counts at 19 common genetic risk variants weighted by their respective effect sizes. Pathway analysis of drusen progression was carried out with the software package Pathway Analysis by Randomization Incorporating Structure.
Results
We observed significant correlation with drusen baseline area and the GRS in the age-related eye disease study (AREDS) dataset (ρ = 0.175, P = 0.006). Measures of association were not statistically significant between drusen progression and the GRS (P = 0.54). Pathway analysis revealed the cell adhesion molecules pathway as the most highly significant pathway associated with drusen progression (corrected P = 0.02).
Conclusions
In this study, we explored the potential influence of known common AMD genetic risk factors on drusen progression. Our results from the GRS analysis showed association of increasing genetic burden (from 19 AMD associated loci) to baseline drusen load but not drusen progression in the AREDS dataset while pathway analysis suggests additional genetic contributors to AMD risk.
Keywords: age-related macular degeneration, drusen, genetic risk score analysis, image analysis
Age-related macular degeneration is the leading cause of blindness in older adults in the developed world. Current prevalence estimates for AMD in the United States are 1.52% and 16.39% for ages 70 to 74 and 80 years.1 Age-related macular degeneration is a disease that results specifically in central vision loss as it affects the macula. Vision loss due to AMD is divided into “neovascular” and “dry” AMD.2 Neovascular AMD is the result of new blood vessels arising underneath the macula, which then begin to leak fluid into the space between Bruch's membrane and the RPE. Dry AMD is characterized by the presence of a broad range of abnormalities in the RPE and is the more common form of AMD.3 Dry AMD abnormalities include drusen, hyperpigmentation, or hypopigmentation in the RPE, and in the later stages of dry AMD progression, geographic atrophy. Although the role that drusen play in the pathogenesis of AMD is not currently known, manifestation of these yellow deposits between the RPE layer and Bruch's membrane is one of the hallmark clinical signs of AMD development.4 Drusen can be characterized as hard or soft.5 Hard small drusen are typically less than 65 μm in diameter and have discrete borders. Intermediate drusen range in size from 64 to 125 μm. Soft drusen are commonly greater than 125 μm in diameter and typically have the property of fuzzy indistinct borders.
Many environmental factors have been associated with increased risk of AMD, such as age, race, smoking, obesity, and hypertension.3,6–14 In addition to the factors listed above, genetic studies have successfully identified common genetic variation in genes such as CFH, HTRA1/ARMS2, C2/CFB, and 15 other loci in sample populations of European ancestry.15–30
Of recent interest is the use of these environmental and genetic risk factors to create prognostic models of AMD risk.31–41 Such models could eventually allow the practice of precision medicine, modulating AMD treatment in response to particular risk profiles. One practical application of identifying the underlying risk factors for AMD is building models of disease progression. These models have focused on grouping samples broadly into progressors or nonprogressors, using categories typically defined by pathological changes in drusen progression, GA, or neovascularization. This approach does not take into account the importance of the rate of disease progression when progression is defined as a dichotomous endpoint rather than a quantitative measure of disease. By understanding how the environmental and genetic components of AMD contribute to the rate of progression, we have the potential to understand how to better tailor and administer treatment regimens and recommend more appropriate eye evaluation intervals.
Although significant contributions have been made in the treatment of neovascular AMD with the advent of anti-VEGF injections, there are currently no clinical treatments available for dry AMD outside of risk factor management.31,42 Therefore, it is important to develop models of AMD progression that emphasize characteristics common to dry AMD such as changes in drusen load over time. Here we examine the impact of a cumulative genetic risk score using 19 common AMD risk variants on drusen progression using data made available through the age-related eye disease study (AREDS), and a combined dataset from Case Western Reserve University (CWRU) and the University of Miami John P. Hussmann Institute for Human Genomics (HIHG).30
Materials and Methods
AREDS Dataset
The age-related eye disease study was a clinical trial that examined the impact of zinc and antioxidants on incidence and progression of AMD.31,42 Using AREDS phenotype data tables made available through the database of genotypes and phenotypes (dbGAP) on a subset of participants in the AREDS, we examined subjects with intermediate AMD without the presence of central geographic atrophy or neovascular AMD. Evaluation of the phenotype data was restricted to the 595 subjects that had longitudinal color fundus photographs deposited in dbGAP. Imaging data were available on subjects at 2-year intervals with a maximum of 12 years of follow up. We selected subject eyes that received consecutive diagnoses of intermediate AMD (AREDS category 3) over a course of more than 2 years based on the AREDS dbGAP phenotype tables. A category of 3 for AMD is represented by the presence of one of the following: one or more large drusen, greater than 20 average-sized drusen in the presence of soft drusen, 65 average-sized drusen in the absence of soft indistinct drusen, or noncentral geographic atrophy. Imaging data were included up to the progression of intermediate AMD to a severe grade of central geographic atrophy or neovascular AMD per eye. Extensive details about the AREDS grade categorization, study design, and subject information can be found in AREDS Report No. 1.42 In dbGAP, 30° color fundus photos are available on three separate fields of the retina. Field 1M is centered on the temporal margin of the optic disc. Field 2M is centered on the macula and field 3M is centered temporal to the macula. For this study, Joint Photographic Expert Group (JPEG) images were downloaded from dbGAP related to field 2M. Images were manually and computationally inspected for quality and poor images were removed from the dataset. The major features used in determining quality of images included the observation of images with poor focus, images that had the early treatment diabetic retinopathy study (ETDRS) grid still attached to the photo, no optic nerve present within the photo, incorrect alignment of the camera leading to images containing differential color hues around the periphery of the fundus image, and uneven illumination across the image.
CWRU Dataset and HIHG Dataset
All cases were ascertained through the retinal clinics at the Vanderbilt Eye Institute (VEI) or the Bascom Palmer Eye Institute (BPEI) as part of a longitudinal study examining progression and response to treatment of severe AMD. We took 50° color fundus photos at these visits using either a Zeiss 450 (Carl Zeiss, Jena, Germany) camera at the VEI, or a Topcon TRC 50IX camera (Topcon Corp., Tokyo, Japan) at BPEI. Participants were graded by a retinal specialist on a 1 to 5 scale modified from AREDS at each visit with visit intervals ranging from 1, 4, 6, 8, 10, and 12 months.43,44 A grade of 1 or 2 represented controls, grade 3 represented early/intermediate AMD, and grades 4 and 5 represented late AMD (geographic atrophy and choroidal neovascularization, respectively). Subjects were retrospectively examined for visits with color fundus photos graded as a 3 on the modified AREDS grading scale. Subjects that presented with continuous intermediate AMD for 1 year or more in the absence of geographic atrophy or neovascular AMD had their imaging data examined. A minimum interval of 6 months between images was used in the CWRU/HIHG dataset.42–44 In the situation that image intervals were less than 6 months apart, the higher quality image was selected from the range while still maintaining a 6-month separation between visits.
All procedures followed the tenets of the Declaration of Helsinki and were approved by the institutional review boards of the University of Miami Miller School of Medicine, CWRU, and Vanderbilt University. Informed consent was obtained from all research subjects involved in this study.
Drusen Quantification
Drusen quantification was completed using a previously developed automated drusen detection and quantification algorithm.45 Details about the detection algorithm are defined elsewhere but briefly described here.45 Images were resized to a radius of 650 pixels for the nonblack region of the image to obtain a standard resolution across all images. Anatomical structures were detected including the optic disc, fovea, and vessels; and image quality assessment for calculation of features for drusen detection was carried out. Drusen candidate pixels were extracted using a pixel classification algorithm. Next, drusen candidate regions were segmented using dynamic programming and the information obtained from the pixel classification and drusen candidates were classified as being true druse using a large set of features including contrast, color, shape, and intensity changes within the image. Quantification was completed based on a threshold drusen probability map, which is used to generate a binary drusen map to calculate drusen area. An Early Treatment Diabetic Retinopathy Study (ETDRS) grid normalized to a 3000 μm radius from the fovea to the edge of the optic disc for each image was automatically placed. Although this is a fully automated process, each image was manually reviewed for proper placement of the ETDRS grid. Drusen surface area was quantified in each image's grid as mm2 and used for downstream analyses.
Genotyping
Genotyping data for both the CWRU/HIHG and AREDS datasets were made available through the International AMD Genomics Consortium (IAMDGC) and through permission of Emily Chew at the National Eye Institute (NEI).46 Details about the quality control procedures carried out by the IAMDGC can be found elsewhere.46 Nineteen common variants previously associated with AMD were selected either through direct genotyping on the IAMDGC exome chip array or through surrogate SNPs found to be in high linkage disequilibrium (LD; r2 > 0.8) with these variants if not directly genotyped.
Drusen Progression Rate Estimation
Since our data are represented by multiple visits per subject and drusen measurements in either one or two eyes, we employed a linear mixed effects model (LMEM) to estimate changes in drusen area over time using a similar modeling scheme to what has been presented for estimating GA progression rates.32 Modeling of drusen progression rates was carried out using the R software package (R, version 3.02) and the R-library “linear mixed-effects models using Eigen and S4” (lme4, version 1.1-7). In brief, LMEM allows for an estimation of a population mean regression line known as a fixed-effect and deviations from that mean slope and intercept through estimation of random effects. This multilevel modeling also allows for incorporation of information on both eyes for a subject if available, giving us a single measure of drusen growth. Values of P were generated with the R package “tests in linear effects models” (lmerTest, version 2.0-20) using Satterthwaite's approximations.
Analysis of Drusen Progression Against Demographic, Endpoint Severity, and Treatment Category
To examine the role of sex, smoking (more than 1 year in lifetime), AREDS treatment category, and age, the following statistical analysis plan was carried out. We first examined the impact of smoking, age, and treatment category independently within the mixed model of drusen progression to see the necessity of including these factors as covariates for the genetic analysis. Age was examined as a measure of correlation with drusen progression by way of a Pearson's correlation coefficient. Sex and smoking were examined within independent models. To understand the potential role of drusen growth on the progression of AMD from intermediate to neovascular AMD or geographic atrophy, study subject's endpoint severity was categorized into either “drusen only,” “geographic atrophy,” or “neovascular AMD.” These severity categories were determined by using AREDS severity score information made available through the AREDS dbGAP data tables. Subjects were categorized based upon first instance of severe progression. In the case that subjects did not progress to geographic or neovascular AMD by the end of the full AREDS clinical trial and natural history study, subjects were categorized as having drusen only. Endpoint severity categories were coded as dummy variables and the drusen only category was set as referent within the model. This was completed similarly in the CWRU/HIHG dataset.
Cumulative Genetic Risk Score Analysis and Independent SNP Tests
For our primary analysis, the 19 variants or their respective surrogate SNPs were included in a genetic risk score (GRS) representing common variations significantly contributing to AMD genetic risk.46 Each variant was weighted by the log of the odds ratio as reported by the AMD gene consortium and multiplied by the number of risk alleles present at that locus. More details about the weighting scheme and application of this risk score can be found elsewhere.30,46,47 To examine the effect of the genetic risk score on drusen progression, a Pearson's correlation coefficient was calculated using each subject's progression slope versus their genetic risk score. Examination of each of the common variants' influence on drusen progression was carried out independently in unadjusted single variant analyses.
Pathway Analysis
To examine the role of drusen progression in a pathway-based analysis, an unadjusted linear regression was carried out in Plink using drusen progression rates as an outcome variable for the AREDS subjects against the genome-wide SNP data present on the exome-chip array.48 A minor allele frequency cutoff of 0.05 was used. Pathway analysis was performed on the results of the single variant tests using the pathway analysis by randomization incorporating structure (PARIS) algorithm, and restricted to the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, which contains 199 defined pathways.49 The algorithm PARIS assigns variants from genome-wide association study (GWAS) results into features that are grouped by pathways as defined by the KEGG database. Significance of a pathway is obtained through permutation of the genome instead of affection status; thus accounting for LD, SNP coverage of pathways, and gene size.
Results
Demographics
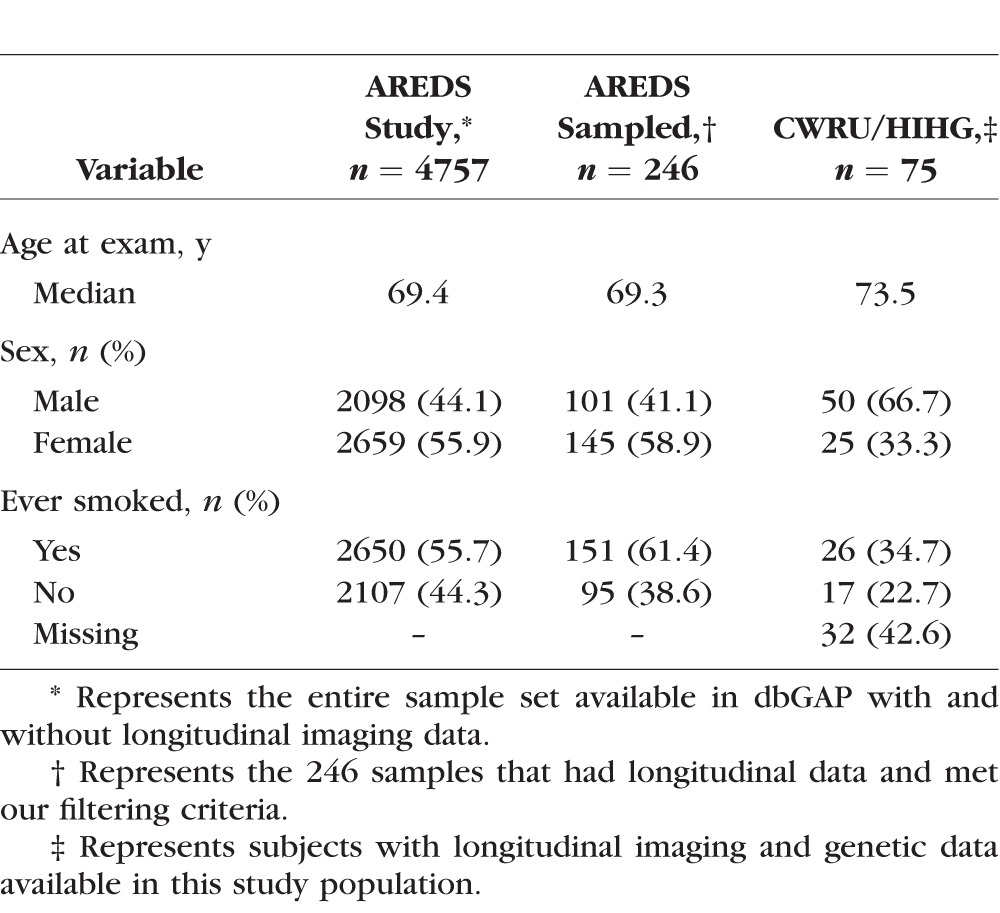
In the AREDS dataset, 246 of the 595 subjects available through dbGAP had either a baseline grade of intermediate drusen or progressed to intermediate drusen and had follow-up visits with grades of intermediate AMD for at least 2 years (Table 1). In the dataset CWRU/HIHG, 75 of 500 subjects examined met the selection criteria for further analysis (Table 1). Analysis in the AREDS and CWRU/HIHG datasets was restricted to the first 6.5 years of follow up and represents the median duration of the AREDS clinical trial. Although imaging data were available for the natural history portion of AREDS, a large proportion of the samples we selected for the study had progressed to severe AMD after the 6.5-year time point and thus did not have usable longitudinal data after that stage. Due to the limited number of observations in the natural history phase of the study, our analysis focused on observations within the clinical trial portion of the study. The median number of visits with quantifiable images including baseline was 3.4 with a total of 973 images being used in the AREDS analysis and a total of 272 images and a median number of three visits for the CWRU/HIHG dataset. Of the 246 AREDS subjects selected, 88 subjects had imaging data that fit our criteria (intermediate AMD) in both eyes, and 158 subjects had imaging data available for one eye. In the CWRU/HIHG dataset, we observed 24 subjects with bilateral intermediate AMD and 51 subjects with imaging data on one eye.
Table 1.
Study Population Demographics
Clinical Presentation and Progression
We observed high correlation in drusen area between eyes of subjects with bilateral intermediate AMD at baseline (ρ = 0.857, P < 2.20 × 106). Correlation of bilateral drusen area progression was also significant (ρ = 0.300, P = 0.004) within these 88 subjects. We observed significantly higher drusen area in subjects with both eyes present in the study versus subjects that had just one eye in our study (P = 0.024). We did not observe a significant association of drusen area progression with sex, age at first exam, smoking status or treatment category.
Genetic Data
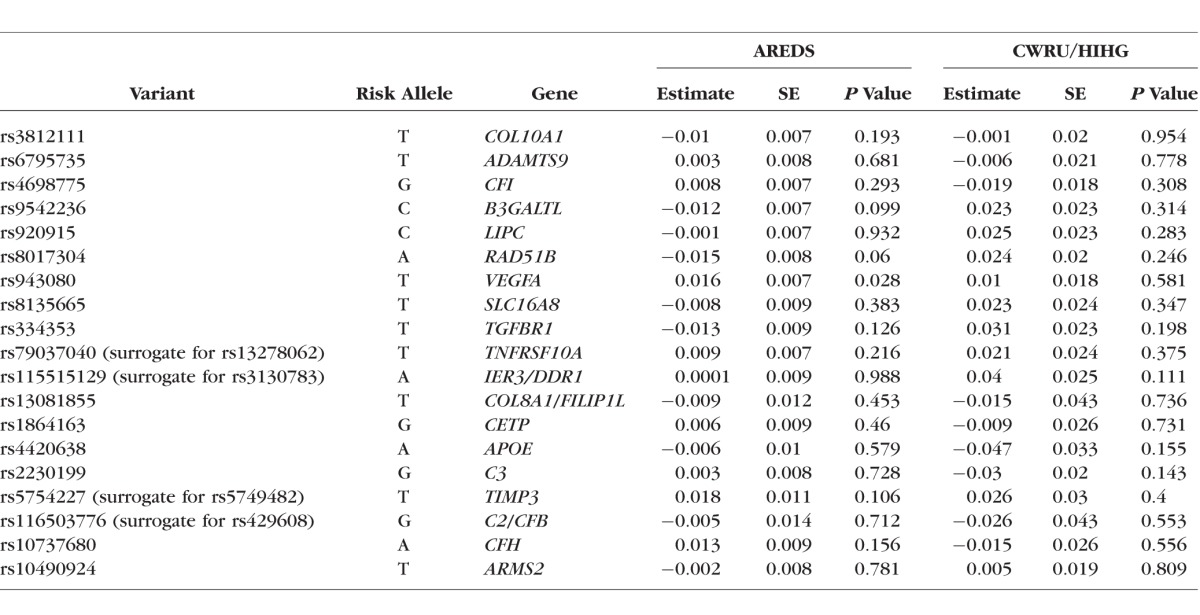
We observed a correlation with baseline drusen area and the GRS in the AREDS dataset (ρ = 0.175, P = 0.006). We did not find a significant correlation between the GRS and drusen progression in the AREDS dataset (ρ = 0.039; P = 0.543). In single marker tests of the SNPs that make up the risk score, we found a nominally significant association with rs943080 in VEGFA within the AREDS dataset (P = 0.028; Table 2); this does not replicate in the CWRU/HIHG dataset. Pathway analysis was performed using the results of the quantitative GWAS based on the IAMDGC exome-chip array and the AREDS dataset. In total, values of P were generated for 252,376 directly genotyped variants that met the minor allele frequency cutoff of 0.05 and a genotyping efficiency of 95% (Supplementary Figs. S1, S2; Supplementary Tables S1, S2). Of the 199 pathways that were interrogated as part of the KEGG database, the most highly associated pathway that passed Bonferroni correction was the cell adhesion molecules pathway (corrected P = 0.02; Supplementary Table S3).
Table 2.
Results of Single-Variant Association Analysis With Progression
Discussion
Age-related macular degeneration is a complex disease characterized by phenotypic heterogeneity with respect to the combinatorial presence of drusen, geographic atrophy, and neovascular AMD. In this analysis, we examined the potential influence of 19 genetic risk loci on drusen growth during the intermediate stages of the disease in the absence of neovascular AMD and geographic atrophy. Since the bulk of studies examining drusen have treated this phenotype as a binary response, we attempted to refine the examination of this phenotype by treating it as a quantitative variable through a previously established drusen quantification algorithm that takes advantage of color fundus photos.45
In a recent study performed by the AREDS research group, it was found that higher drusen severity at baseline was a significant predictor for progressing to geographic atrophy and neovascular AMD.42 Study eyes that maintained intermediate AMD throughout the course of the AREDS had significantly lower average total drusen area compared with subjects that had baseline intermediate and progressed to geographic atrophy and neovascular AMD within a 6.5-year period. This observation is present in the CWRU/HIHG dataset as well.
Although history of ever smoking and age are major risk factors for AMD, they do not appear to have a major influence on drusen progression or baseline area within these datasets. It is important to note that we do not differentiate subjects based on whether they were current smokers during the time of the AREDS or by pack years of smoking exposure. Age was limited to those aged 55 years or older at baseline and subjects that already have presence of intermediate AMD. We observed a highly significant correlation of drusen area within subjects that present with bilateral intermediate AMD (ρ = 0.847, P < 0.0001). This observation may be inflated, as we are not including subject eyes that have either severe AMD in the fellow eye, or not enough medium to large drusen to be classified as intermediate AMD. These correlation findings have also been observed by other groups looking at bilateral drusen using optical coherence tomography and thus our results are consistent with these previous findings.50 When examining correlation in bilateral drusen progression within these same subjects, we found significant although reduced correlation (ρ = 0.300, P = 0.004).
Genotype data were chosen based on previous work carried out by the IAMDGC consortium that identified 19 common AMD risk variants that may explain up to 65% of the variation seen in AMD.30 For our primary genetic analysis, these 19 variants were aggregated into a genetic risk score to examine its impact on drusen at baseline and progression. The risk score analysis revealed significant correlation with drusen area at baseline (ρ = 0.149, P = 0.020) and no correlation with drusen progression (ρ = 0.018; P = 0.794). It is important to note that SNP weights assigned to the variants used in the risk score are based on the AMD gene consortium analysis that performed a cross-sectional analysis of risk, and as such, these weights may not be representative of their role in progression. In light of this, we examined the impact of an unweighted GRS using the 19 variants on drusen progression and did not observe a significant effect (data not shown). In a secondary analysis, we attempted to dissect the role of these variants to drusen progression but did not find any single variant that substantially contributes to drusen progression (Table 2).
As we are presenting a case only analysis in two smaller datasets, power is a significant concern. Post hoc power calculations show that we had 80% power to detect an effect size of increase in drusen area of 0.0196 mm2/y when correcting for independent tests of each of the four major loci. We had 56% power to detect a significant association in the cumulative genetic risk score progression analysis based on the estimated effect size and sample size in the AREDS dataset. Variant rs943080, which is near VEGFA, was the only variant nominally associated with drusen progression out of 19 loci in the AREDS dataset that make up the cumulative genetic risk score. This variant did not replicate in the CWRU/HIHG dataset.
A second exploratory approach is to aggregate potential effects not using a genetic risk score, but on functional relatedness. We performed a pathway-based analysis to see whether any functional pathways within the KEGG database were enriched for drusen progression. The top pathway that was enriched was the cell adhesion molecule pathway (KEGG database id: hsa04514; Supplementary Table S3). We identified three genes driving the signal. These genes were neurofascin (NFASC) on chromosome 1 (P = 0.0004); CD226 molecule (CD226) on chromosome 18 (P < 0.0002); and neurexin 1 (NRXN1) on chromosome 2 (P = 0.0006). The statistical significance of these genes was confirmed using the gene enrichment program: versatile gene-based association study. Molecules in this pathway play a role in a wide array of functions including inflammation and immune response. Previous work has shown that inflammation between the RPE layer and the Bruch's membrane may play a role in AMD associated drusen formation mainly through cellular debris trapped between these layers.51,52 In another study examining the impact of cell adhesion molecules on AMD, it was shown that soluble vascular cell adhesion molecule 1 associates with increased incidence of early AMD.53 These findings highlight that although the 19 risk loci may not contribute significantly to drusen progression within our study, there may be other variants of functional importance directly or indirectly impacting drusen growth during the intermediate stage of disease.
One of the limitations that we found during the course of this study was the quality of images available from the AREDS dbGAP dataset. Images of the AREDS made available in dbGAP are not original images, but digitized copies of slide transparency film. Photographs taken at the VEI and BPEI were native digital images. All images from both the native digital images obtained in the CWRU/HIHG dataset and the digitized photos from the AREDS were assessed for quality using an automated algorithm that assigns a quantitative quality score to the image based on a number of metrics including blurriness, contrast in the image, and ability to delineate blood vessels surrounding the macula.44 A large proportion of the AREDS images were considered poor by this algorithm, which did not necessarily reflect the qualitative assessment of the images by a retinal specialist. Assessing the quality of the directly digitized images from the CWRU/HIHG data we observed most images falling into the high quality range both quantitatively and qualitatively. We must also be aware that the overall low quality of the AREDS images when examined in a longitudinal format may be introducing too much variation across time points to accurately measure drusen progression. This may be somewhat reflected between differences in correlation between intrasubject drusen at baseline and bilateral drusen progression, where we see a highly correlated baseline measurement, but reduced bilateral progression correlation. Thus, if the effect sizes of the major common risk loci are more modest for drusen growth, we may not be able to overcome the signal to noise ratio introduced by image quality. Although the CWRU/HIHG images were of superior quality, the size of the dataset limited our power to detect a true association with drusen at baseline or drusen progression. While we were able to retrospectively collect previous retinal visits on subjects within the progression and treatment study, the major ascertainment criteria for these participants was severe AMD, thus longitudinal data was sparse prior to the presence of reduction in visual acuity.
In conclusion, AMD continues to be a high priority complex disease for genetic studies due to its high heritability and its socioeconomic impact. Although a large proportion of the genetic variation explained by AMD has been identified, its contribution is largely put in the context of disease risk. Elucidation of potential rare and common genetic contributors to AMD rate of progression may lead to better understanding of the mechanisms involved in AMD pathogenesis and strides in AMD clinical management.
Supplementary Material
Acknowledgments
We thank the community members and family participants for agreeing to participate in this ongoing study. Some of the samples used in this study were collected while authors were faculty members at Duke University (WKS, MAP-V) and Vanderbilt University (JLH).
Supported by Grants A6019085, EY012118, AG019726, and AG044089.
Disclosure: J.D. Hoffman, None; M.J.J.P. van Grinsven, None; C. Li, None; M. Brantley, Jr, None; J. McGrath, None; A. Agarwal, None; W.K. Scott, None; S.G. Schwartz, None; J. Kovach, None; M. Pericak-Vance, None; C.I. Sanchez, None; J.L. Haines, None
References
- 1. Friedman DS,, O'Colmain BJ,, Muñoz B,, et al. Prevalence of age-related macular degeneration in the United States. Arch Ophthalmol. 2004; 122: 564–572. [DOI] [PubMed] [Google Scholar]
- 2. Haung D,, Kaiser PK,, Lowder CY,, Traboulsi E. Retinal Imaging. 1st ed. Maryland Heights, MO: Mosby; 2006. [Google Scholar]
- 3. Klein R,, Klein BE,, Knudstson MD,, Meuer SM,, Swift M,, Gangnon RE. Fifteen-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2007; 114: 253–262. [DOI] [PubMed] [Google Scholar]
- 4. Abdelsalam A,, Del Priore PL,, Zarbin MA. Drusen in age-related macular degeneration: pathogenesis natural course, and laser photocoagulation-induced regression. Surv Ophthalmol. 1999; 44: 1–29. [DOI] [PubMed] [Google Scholar]
- 5. Klein R,, Davis MD,, Magli YL,, Segal P,, Klein BE,, Hubbard L. The Wisconsin age-related maculopathy grading system. Ophthalmology. 1991; 98: 1128–1134. [DOI] [PubMed] [Google Scholar]
- 6. Evans JR,, Fletcher AE,, Wormald RP. 28000 cases of age related macular degeneration causing visual loss in people aged 75 years and above in the United Kingdom may be attributable to smoking. Br J Ophthalmol. 2005; 89: 550–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Khan JC,, Thurlby DA,, Shahid H,, et al. Smoking and age related macular degeneration: the number of pack years of cigarette smoking is a major determinant of risk for both geographic atrophy and choroidal neovascularisation. Br J Ophthalmol. 2006; 90: 75–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Klein R,, Klein BE,, Tomany SC,, Meuer SM,, Huang GH. Ten-year incidence and progression of age-related maculopathy: the Beaver Dam Eye Study. Ophthalmology. 2002; 109: 1767–1779. [DOI] [PubMed] [Google Scholar]
- 9. Klein R,, Klein BE,, Knudtson MD,, et al. Prevalence of age-related macular degeneration in 4 racial/ethnic groups in the multi-ethnic study of atherosclerosis. Ophthalmology. 2006; 113: 373–380. [DOI] [PubMed] [Google Scholar]
- 10. Klein R,, Knudtson MD,, Lee KE,, Gangnon RE,, Klein BE. Age-period-cohort effect on the incidence of age-related macular degeneration: the Beaver Dam Eye Study. Ophthalmology. 2008; 115: 1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schaumberg DA,, Christen WG,, Hankinson SE,, Glynn RJ. Body mass index and the incidence of visually significant age-related maculopathy in men. Arch Ophthalmol. 2001; 119: 1259–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Smith W,, Assink J,, Klein R,, et al. Risk factors for age-related macular degeneration: pooled findings from three continents. Ophthalmology. 2001; 108: 697–704. [DOI] [PubMed] [Google Scholar]
- 13. Thornton J,, Edwards R,, Mitchell P,, Harrison RA,, Buchan I,, Kelly SP. Smoking and age-related macular degeneration: a review of association. Eye (Lond). 2005; 19: 935–944. [DOI] [PubMed] [Google Scholar]
- 14. Thylefors B,, Négrel AD,, Pararajasegaram R,, Dadzie KY. Global data on blindness. Bull World Health Organ. 1995; 73: 115–121. [PMC free article] [PubMed] [Google Scholar]
- 15. Edwards AO,, Ritter R,, 3rd,, Abel KJ,, Manning A,, Panhuysen C,, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science. 2005; 308: 421–424. [DOI] [PubMed] [Google Scholar]
- 16. Gold B,, Merriam JE,, Zernant J,, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nat Genet. 2006; 38: 458–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hageman GS,, Anderson DH,, Johnson LV,, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A. 2005; 102: 7227–7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haines JL,, Hauser MA,, Schmidt S,, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science. 2005; 308: 419–421. [DOI] [PubMed] [Google Scholar]
- 19. Jakobsdottir J,, Conley YP,, Weeks DE,, Mah TS,, Ferrell RE,, Gorin MB. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet. 2005; 77: 389–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Klein RJ,, Zeiss C,, Chew EY,, et al. Complement factor H polymorphism in age-related macular degeneration. Science. 2005; 308: 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Maller J,, George S,, Purcell S,, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age-related macular degeneration. Nat Genet. 2006; 38: 1055–1059. [DOI] [PubMed] [Google Scholar]
- 22. Maller JB,, Fagerness JA,, Reynolds RC,, Neale BM,, Daly MJ,, Seddon JM. Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet. 2007; 39: 1200–1201. [DOI] [PubMed] [Google Scholar]
- 23. Rivera A,, Fisher SA,, Fritsche LG,, et al. Hypothetical LOC387715 is a second major susceptibility gene for age-related macular degeneration, contributing independently of complement factor H to disease risk. Hum Mol Genet. 2005; 14: 3227–3236. [DOI] [PubMed] [Google Scholar]
- 24. Schmidt S,, Hauser MA,, Scott WK,, et al. Cigarette smoking strongly modifies the association of LOC387715 and age-related macular degeneration. Am J Hum Genet. 2006; 78: 852–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Souied EH,, Leveziel N,, Richard F,, et al. Y402H complement factor H polymorphism associated with exudative age-related macular degeneration in the French population. Mol Vis. 2005; 11: 1135–1140. [PubMed] [Google Scholar]
- 26. Spencer KL,, Hauser MA,, Olson LM,, et al. Protective effect of complement factor B and complement component 2 variants in age-related macular degeneration. Hum Mol Genet. 2007; 16: 1986–1992. 17576744 [Google Scholar]
- 27. Spencer KL,, Olson LM,, Anderson BM,, et al. C3 R102G polymorphism increases risk of age-related macular degeneration. Hum Mol Genet. 2008; 17: 1821–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Yang Z,, Camp NJ,, Sun H,, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science. 2006; 314: 992–993. [DOI] [PubMed] [Google Scholar]
- 29. Yates JR,, Sepp T,, Matharu BK,, et al. Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med. 2007; 357: 553–561. [DOI] [PubMed] [Google Scholar]
- 30. Fritsche LG,, Chen W,, Schu M,, et al. Seven new loci associated with age-related macular degeneration. Nat Genet. 2013; 45: 433–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Clemons TE,, Milton RC,, Klein R,, Seddon JM,, Ferris FL, 3rd; Age-Related Eye Disease Study Research Group. Risk factors for the incidence of advanced age-related macular degeneration in the age-related eye disease study (AREDS) AREDS report no. 19. Ophthalmology. 2005; 112: 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Dreyhaupt J,, Mansmann U,, Pritsch M,, Dolar-Szczasny J,, Bindewald A,, Holz FG. Modelling the natural history of geographic atrophy in patients with age-related macular degeneration. Ophthalmic Epidemiol. 2005; 12: 353–362. [DOI] [PubMed] [Google Scholar]
- 33. Farwick A,, Wellmann J,, Stoll M,, Pauleikhoff D,, Hense HW. Susceptibility genes and progression in age-related maculopathy: a study of single eyes. Invest Ophthalmol Vis Sci. 2010; 51: 731–736. [DOI] [PubMed] [Google Scholar]
- 34. Fleckenstein M,, Adrion C,, Schmitz-Valckenberg S,, et al. Concordance of disease progression in bilateral geographic atrophy due to AMD. Invest Ophthalmol Vis Sci. 2010; 51: 637–642. [DOI] [PubMed] [Google Scholar]
- 35. Fleckenstein M,, Schmitz-Valckenberg S,, Adrion C,, et al. Tracking progression with spectral-domain optical coherence tomography in geographic atrophy caused by age-related macular degeneration. Invest Ophthalmol Vis Sci. 2010; 51: 3846–3852. [DOI] [PubMed] [Google Scholar]
- 36. Fleckenstein M,, Schmitz-Valckenberg S,, Adrion C,, et al. Progression of age-related geographic atrophy: role of the fellow eye. Invest Ophthalmol Vis Sci. 2011; 52: 6552–6557. [DOI] [PubMed] [Google Scholar]
- 37. Francis PJ,, Hamon SC,, Ott J,, Weleber RG,, Klein ML. Polymorphisms in C2, CFB and C3 are associated with progression to advanced age related macular degeneration associated with visual loss. J Med Genet. 2009; 46: 300–307. [DOI] [PubMed] [Google Scholar]
- 38. Oliver-Fernandez A,, Bakal J,, Segal S,, Shah GK,, Dugar A,, Sharma S. Progression of visual loss and time between initial assessment and treatment of wet age-related macular degeneration. Can J Ophthalmol. 2005; 40: 313–319. [DOI] [PubMed] [Google Scholar]
- 39. Seddon JM,, Cote J,, Davis N,, Rosner B. Progression of age-related macular degeneration: association with body mass index, waist circumference, and waist-hip ratio. Arch Ophthalmol. 2003; 121: 785–792. [DOI] [PubMed] [Google Scholar]
- 40. Wang JJ,, Rochtchina E,, Smith W,, et al. Combined effects of complement factor H genotypes, fish consumption, and inflammatory markers on long-term risk for age-related macular degeneration in a cohort. Am J Epidemiol. 2009; 169: 633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Seddon JM,, Reynolds R,, Yu Y,, Daly MJ,, Rosner B. Risk models for progression to advanced age-related macular degeneration using demographic, environmental, genetic, and ocular factors. Ophthalmology. 2011; 118: 2203–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. The age-related eye disease study (AREDS): design implications. AREDS report no. 1. Control Clin Trials. 1999; 20: 573–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schmidt S,, Saunders AM,, de la Paz MA,, et al. Association of the apolipoprotein E gene with age-related macular degeneration: possible effect modification by family history, age, and gender. Mol Vis. 2000; 6: 287–293. [PubMed] [Google Scholar]
- 44. Seddon JM,, Sharma S,, Adelman RA. Evaluation of the clinical age-related maculopathy staging system. Ophthalmology. 2006; 113: 260–266. [DOI] [PubMed] [Google Scholar]
- 45. van Grinsven MJ,, Lechanteur YT,, van de Ven JP,, et al. Automatic drusen quantification and risk assessment of age-related macular degeneration on color fundus images. Invest Ophthalmol Vis Sci. 2013; 54: 3019–3027. [DOI] [PubMed] [Google Scholar]
- 46. Fritsche LG,, Igl W,, Bailey JN,, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants 3. Nat Genet. 2016; 48: 134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hoffman JD,, JN, Cooke Bailey,, D'Aoust L,, et al. Rare complement factor H variant associated with age-related macular degeneration in the Amish. Invest Ophthalmol Vis Sci. 2014; 55: 4455–4460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Purcell S,, Neale B,, Todd-Brown K,, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007; 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yaspan BL,, Bush WS,, Torstenson ES,, et al. Genetic analysis of biological pathway data through genomic randomization. Hum Genet. 2011; 129: 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Diniz B,, Rodger DC,, Chavali VR,, et al. Drusen and RPE atrophy automated quantification by optical coherence tomography in an elderly population. Eye (Lond). 2015; 29: 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Hageman GS,, Luthert PJ, Victor Chong NH, Johnson LV, Anderson DH, Mullins RF. An integrated hypothesis that considers drusen as biomarkers of immune-mediated processes at the RPE-Bruch's membrane interface in aging and age-related macular degeneration. Prog Retin Eye Res. 2001; 20: 705–732. [DOI] [PubMed] [Google Scholar]
- 52. Anderson DH,, Mullins RF,, Hageman GS,, Johnson LV. A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol. 2002; 134: 411–431. [DOI] [PubMed] [Google Scholar]
- 53. Klein R,, Myers CE,, Cruickshanks KJ,, et al. Markers of inflammation, oxidative stress, and endothelial dysfunction and the 20-year cumulative incidence of early age-related macular degeneration: the Beaver Dam Eye Study. JAMA Ophthalmol. 2014; 132: 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.