

Abstract
Objective
Oxidative posttranslational modifications of intracellular proteins can potentially regulate signaling pathways relevant to cartilage destruction in arthritis. In this study, oxidation of cysteine residues to form sulfenic acid (S-sulfenylation) was examined in osteo-arthritic (OA) chondrocytes and investigated in normal chondrocytes as a mechanism by which fragments of fibronectin (FN-f) stimulate chondrocyte catabolic signaling.
Methods
Chondrocytes isolated from OA and normal human articular cartilage were analyzed using analogs of dimedone that specifically and irreversibly react with protein S-sulfenylated cysteines. Global S-sulfenylation was measured in cell lysates with and without FN-f stimulation by immunoblotting and in fixed cells by confocal microscopy. S-sulfenylation in specific proteins was identified by mass spectroscopy and confirmed by immunoblotting. Src activity was measured in live cells using a fluorescence resonance energy transfer biosensor.
Results
Proteins in chondrocytes isolated from OA cartilage were found to have elevated basal levels of S-sulfenylation relative to those of chondrocytes from normal cartilage. Treatment of normal chondrocytes with FN-f induced increased levels of S-sulfenylation in multiple proteins, including the tyrosine kinase Src. FN-f treatment also increased the levels of Src activity. Pretreatment with dimedone to alter S-sulfenylation function or with Src kinase inhibitors inhibited FN-f–induced production of matrix metalloproteinase 13.
Conclusion
These results demonstrate for the first time the presence of oxidative posttranslational modification of proteins in human articular chondrocytes by S-sulfenylation. Due to the ability to regulate the activity of a number of cell signaling pathways, including catabolic mediators induced by fibronectin fragments, S-sulfenylation may contribute to cartilage destruction in OA and warrants further investigation.
More than 27 million people in the US have symptomatic osteoarthritis (OA) (1), representing a US economic burden in excess of $60 billion per year (2). There is a critical need for new treatments for OA that target the disease process so that disease progression can be slowed or stopped. In order to develop novel therapeutic interventions, a better understanding of the various mechanisms that promote joint tissue destruction is needed. Destruction of the articular cartilage is a key feature of OA and stems from an imbalance in chondrocyte anabolic and catabolic activities (3,4). This results in excessive production of proteolytic enzymes that break down the extracellular matrix, generating matrix fragments that can further propagate catabolic activity through a positive feedback loop. The goal of the present study was to investigate the mechanisms by which fragments of fibronectin (FN-f) stimulate chondrocyte catabolic signaling pathways to result in increased production of matrix metalloproteinase 13 (MMP-13), a matrix-degrading enzyme.
FN-f have been found in OA cartilage and synovial fluid and have been shown to stimulate cartilage matrix destruction (5–8). The FN-f composed of domains 7–10 of fibronectin is known to bind to the α5β1 integrin receptor and to activate MAP kinase signaling, resulting in the up-regulation of a number of cytokine, chemokine, and MMP genes (9–12). Previous studies have shown that activation of chondrocyte catabolic signaling by FN-f requires reactive oxygen species (ROS) as second messengers; however, the mechanism by which ROS regulate chondrocyte signaling is not completely understood (13).
Redox signaling is an exciting new area of cell signaling research that is beginning to receive increased attention. ROS, such as hydrogen peroxide (H2O2), hydroxyl radicals (HO•), superoxide ( ), and singlet oxygen (1O2), are now known to be important second messengers in a number of cell signaling pathways via oxidative post-translational modifications (Ox-PTMs) of specific proteins that regulate signal transduction (14). The primary mechanism by which these ROS regulate cell signaling is by oxidation of the thiol (R-SH) group of reactive cysteine residues, leading initially to cysteine sulfenic acid (R-SOH) formation (S-sulfenylation). S-sulfenylation can be a stable product of oxidation, or it can be further converted to intra- or intermolecular protein disulfides, S-glutathione, sulfenylamides, and other Ox-PTMs (15,16).
Importantly, S-sulfenylation functions in a reversible manner akin to phosphorylation, thus having a regulatory function for the activity of kinases and phosphatases as has been demonstrated previously (16–19). ROS have been shown to influence cell adhesion and migration via integrin signaling, spatially regulating S-sulfenylation (20,21). Although S-sulfenylation has been recently studied in multiple cell types, e.g., fibroblasts, leukocytes, and tumor cells (22,23), no such protein modification has previously been reported in chondrocytes.
The small molecule 5,5-dimethylcyclohexanedione (dimedone) has been shown to react selectively with sites of S-sulfenylation, forming a stable adduct that can be detected using a variety of techniques (24,25). Our objective in the present study was to use dimedone-based labeling techniques to study S-sulfenylation of proteins in chondrocytes and to determine whether FN-f stimulates S-sulfenylation to regulate downstream production of MMP-13. We found that cells from OA cartilage had increased levels of S-sulfenylation relative to those from healthy cartilage and that FN-f stimulation of S-sulfenylation promoted the increase in MMP-13 production. The protooncogene tyrosine protein kinase Src was identified as one of the chondrocyte proteins undergoing S-sulfenylation. Inhibition of Src blocked FN-f–stimulated MMP-13 production, and pretreatment with the general antioxidant N-acetyl-L-cysteine (NAC) delayed the activation of Src in response to FN-f, suggesting that redox regulation of Src is an important component of FN-f–induced signaling in chondrocytes.
MATERIALS AND METHODS
Tissue acquisition and chondrocyte isolation
Normal human articular cartilage from the ankle joint was obtained through the Department of Biochemistry at Rush University Medical Center (Chicago, IL) from the Gift of Hope Organ and Tissue Donor Network (Elmhurst, IL), or from the National Disease Research Interchange (Philadelphia, PA). Normal cartilage donors (n = 27) had no known history of arthritis. Their age range was 39–90 years (mean ± SD 61.8 ± 13.5 years), and the ratio of men to women was 2:1. Human OA articular cartilage from the knee joint was obtained through the Department of Orthopedic Surgery at Wake Forest Baptist Hospital (Winston-Salem, NC) from patients undergoing total joint replacement because of OA. Samples were obtained from OA patients (n = 4 [3 men and 1 woman]) ranging in age from 69 to 74 years (mean ± SD 72.0 ± 2.1 years).
Chondrocytes were isolated by sequential digestion with Pronase and collagenase, as described previously (26), then plated in high-density monolayers, and used without passaging to ensure maintenance of chondrocyte phenotype. Chondrocytes were incubated for 5–7 days with Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and antibiotics, followed by a switch to serum-free medium overnight, prior to the experiments.
Stimulation with fibronectin fragments
Purified endotoxin-free recombinant human FN-f of 42 kd, consisting of domains 7–10 of native fibronectin, which includes the cell-binding RGD domain (27), were produced using a plasmid expression construct obtained from Dr. Harold Erickson (Duke University, Durham, NC). Endotoxin was removed using high-capacity endotoxin-removal columns (Pierce). Cells were treated with 1 μM FN-f either overnight (for MMP secretion experiments) or for 30 minutes (for signaling studies), except where indicated otherwise. For Src sulfenylation and Src activity experiments, control cells were treated with recombinant human epidermal growth factor (EGF; R&D Systems), a growth factor that is known to both increase Src activity (28,29) and induce S-sulfenylation in other cell types (30).
To detect the effects of dimedone preincubation on signaling and MMP secretion, cells were pretreated with the indicated doses of dimedone (Sigma-Aldrich) for 30 minutes prior to FN-f stimulation. In order to observe H2O2-specific S-sulfenylation, control cells were pretreated with 100 units/ml of polyethylene glycol (PEG)–catalase (Sigma-Aldrich) overnight prior to FN-f stimulation. For Src activity experiments, control cells were pretreated for 30 minutes with the Src inhibitor SU6656 at 3 μM (Cayman Chemical). In MMP-13 secretion experiments, cells were pretreated overnight with 1 of 3 Src inhibitors, 3 μM SU6656, 6 μM Src inhibitor 1 (Sigma-Aldrich), or 10 μM PP2 (Calbiochem). Dosages were chosen based on the relative Ki of each inhibitor (31).
Cells were lysed and processed for immunoblotting of intracellular signaling proteins as previously described (26,32). Src was identified with a rabbit polyclonal antibody from Cell Signaling Technology. For extracellular MMP secretion analysis, conditioned medium was collected after overnight stimulation and immunoblotted as previously described (12,33,34).
S-sulfenylation labeling for immunoblots
S-sulfenylated cysteines were identified by direct labeling either with dimedone (USB) or with the biotin-conjugated dimedone derivative DCP-Bio1 (Kerafast) for affinity capture of proteins with streptavidin. For imaging, samples were labeled with DCP-Rho1 (Kerafast), another dimedone-based reagent conjugated to rhodamine. These reagents and detailed methods for their use have been previously described (35,36). Lysates for evaluation of both global and protein-specific cysteine sulfenylation levels were prepared as previously described (26), in the presence of 200 units/ml of catalase (from bovine liver; Sigma) with either 2 mM dimedone or 5 mM DCP-Bio1. Samples were rotated end-over-end at 4°C for 30 minutes, then 20 mM iodoacetamide (Sigma-Aldrich) was added to block free sulfhydryls. Samples were rotated for an additional 30 minutes at 4°C and then centrifuged at 15,000g for 10 minutes at 4°C to remove the insoluble fraction. Dimedone-labeled proteins were identified with a specific antibody against protein sulfenic acid derivatized with dimedone (EMD Millipore) as described elsewhere (37).
Immunoprecipitation of proteins from dimedone-labeled samples was performed using a GlycoLink immunoprecipitation kit (Thermo Scientific) according to the manufacturer’s specifications. Immunoprecipitated samples were then subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) under nonreducing conditions.
DCP-Bio1–labeled samples were passed through Bio-Gel P-6 Gel columns (Bio-Rad) to remove unbound DCP-Bio1, then loaded onto high-capacity streptavidin agarose resin (Thermo Scientific) to capture the DCP-Bio1–labeled proteins. The beads were washed with 10 column volumes of each of the following (5 minutes per wash at 23°C): Tris buffered saline, pH 7.0, with 0.1% Tween 20 (3 washes), 2M urea (1 wash), 1M NaCl (1 wash), 0.1% SDS supplemented with 10 mM dithiothreitol (DTT, once), and phosphate buffered saline (PBS; 2 washes). DCP-Bio1–labeled proteins were eluted from the beads by boiling for 15 minutes in Laemmli sample buffer with 5% 2-mercaptoethanol (both from Bio-Rad).
S-sulfenylation labeling for imaging
Cells were treated for the indicated times with FN-f. Two minutes prior to the end of the treatment period, DCP-Rho1 was added to a concentration of 5 μM. At the end of that treatment period, iodoacetamide was added to a concentration of 20 mM for an additional 2 minutes to block free sulfhydryls. Cells were then washed 3 times with 20 mM iodoacetamide in PBS, followed by 2 washes with 20 mM iodoacetamide prepared in serum-free medium. Cells were subsequently incubated for 5 minutes at 37°C in a 5% CO2 environment before the addition of 16% paraformaldehyde (diluted to 4% upon addition).
After fixation, cells were permeabilized with 0.1% Tween 20 (Fisher Scientific), and autofluorescence was quenched with 10 mM glycine and 50 mM ammonium chloride. Cells then received a series of washes with bovine serum albumin and normal serum to block nonspecific binding and, finally, 3 washes with PBS. Cells were mounted in Vectashield with DAPI (Vector).
DCP-Rho1 image acquisition and analysis
Briefly, 3 image sets per sample were acquired at 200× magnification with a Zeiss LSM 710 microscope, using a voxel size of 420 nm × 420 nm × 2 μm with spacing of 1 μm between slices, a pixel dwell time of 1.58 μs, and 2× line averaging. Eleven images were acquired per z-stack, resulting in capture of signal through the complete thickness of the samples. Images were analyzed with ImageJ v1.48t software (38). Segmentation was performed by applying a Huang threshold (39) to a maximum-intensity projection of each channel. Segmented regions of interest (defined as foreground) were empirically selected as contiguous thresholded areas ≥1 μm2 for DCP-Rho1 and ≥10 μm2 for DAPI. Intensity values were calculated using the following equation:
where I is the intensity value, IntDenRho1F is the integrated optical density (mean × area) of the foreground of the DCP-Rho1 summed intensity projection, MeanRho1B is the mean of the background of the DCP-Rho1 summed intensity projection, AreaRho1 is the area of the foreground of the DCP-Rho1 summed intensity projection, and AreaDAPI is the area of the foreground of the DAPI maximum intensity projection. This formula corrects for the background present in the DCP-Rho1 images and then corrects for the number of cells per image by normalizing against the nuclear cross-sectional area.
Mass spectrometry
Cells (human chondrocyte cell line C-28/I2) were grown in SILAC medium containing light and heavy isotope–labeled Arg and Lys amino acids: 12C6,14N4-Arg and 12C6,14N2-Lys (light); 13C6,15N4-Arg and 13C6,15N2-Lys (heavy). After 8 passages, 95% of the cells had incorporated the heavy and light isotopes. Cells were stimulated with FN-f for 30 minutes, then lysed in 1 ml of buffer containing 20 mM Tris, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, and 1 μg/ml of leupeptin, and supplemented with bovine liver catalase (200 units/ml) and DCP-Bio1 (2 mM). Labeling with DCP-Bio1 proceeded for 1 hour on ice. Lysates were clarified by centrifugation, and the protein concentration was determined using the bicinchoninic acid assay. Samples were normalized, combined 1:1, and the proteins were precipitated in 200-μl aliquots using a standard methanol/chloroform protocol.
Pellets were resuspended in 1% SDS in PBS, diluted 10-fold with PBS, and enriched in biotinylated proteins using Dynabeads MyOne Streptavidin T1 beads (Invitrogen) by rotating end-over-end overnight at 4°C. Beads were prewashed 3 times with 1 ml of 0.1% (weight/volume) SDS in PBS. After overnight incubation, the supernatant was removed, and the beads were washed 3 times (10 minutes each) with 1 ml of 1M NaCl, 2M urea, and PBS for a total of 9 washes. Biotinylated proteins were released by boiling for 10 minutes in 1× reducing Laemmli sample buffer.
The supernatant was collected, and the proteins were immediately separated by SDS-PAGE and stained with Coomassie brilliant blue (Bio-Rad). Bands were excised, destained, and processed according to standard in-gel digestion protocols. Prior to digestion, proteins were treated with DTT (10 mM; for 1 hour at 56°C) and then with iodoacetamide (17 mM; for 45 minutes at room temperature). Digestions were performed overnight at 37°C using Trypsin Gold (Promega) in 50 mM NH4HCO3. The resulting peptides were extracted from the gel, dried, and analyzed on a Dionex UltiMate 3000 splitless Nano LC system coupled to a Thermo Orbitrap Velos Pro high-resolution mass spectrometer. Peptides were separated using a gradient of buffer A (0.1% formic acid/2% acetonitrile/98% water) and buffer B (0.1% formic acid/20% water/80% acetonitrile) over 160 minutes (2–85% buffer B) at a flow rate of 300 nl/minute with the column held at 35°C.
Eluent was introduced to the mass spectrometer via positive nanospray electrospray ionization with the following settings: capillary temperature 200°C, spray voltage 1.8 kV, spray current 100 mA. The mass spectrometer was operated in data-dependent acquisition mode using Xcalibur v. 2.1 (Thermo). After a full scan (150–2,000 mass/charge range) at high resolution (60,000), the top 10 most intense precursor ions were isolated and fragmented using collision-induced dissociation. Dynamic exclusion was enabled with a repeat duration of 30 seconds and an exclusion duration of 9.5 seconds. The normalized collision energy was set at 35%, activation Q at 0.25, and activation time at 10 msec. Acquired raw data were processed and quantified using Proteome Discoverer v 1.4 (Thermo) with the Mascot search engine and the UniProtKB human database. Cysteine carbamidomethylation and methionine mono-oxidation were selected as dynamic modifications. The results were filtered using a false discovery rate of 1%.
Chondrocyte transfection and fluorescence resonance energy transfer (FRET) analysis
Normal human chondrocytes were transfected using a human chondrocyte nucleofection kit (Lonza) as described previously (11). Briefly, for each sample, 2,000,000 cells were resuspended in transfection reagent and nucleofected with 5 μg of the appropriate plasmid: either SrcC1 (enhanced cyan fluorescent protein [ECFP]/enhanced yellow fluorescent protein [EYFP] Src activity sensor) or SrcRV (ECFP/EYFP nonresponsive mutant negative control) (both obtained from Dr. Yingxiao [Peter] Wang, University of California, San Diego) (40). Cells were then plated in monolayer onto black-walled, coverglass-bottomed petri dishes (SPL Lifesciences), allowing for visualization with a confocal microscope. Coverglass surfaces were functionalized in an air-fed plasma cleaner (Harrick Plasma) for 10 minutes, immediately coated with fibronectin for 1 hour at 37°C, and washed with PBS prior to cell seeding. After a 48-hour recovery period, cells were switched to serum-free medium overnight before stimulation and imaging. Pretreatment with 10 mM N-acetyl-L-cysteine was performed 30 minutes prior to stimulation with FN-f.
Fluorophores were excited at 440 nm using an argon laser. The ECFP signal was collected over an emission range of 463–500 nm, and the EYFP (i.e., FRET) signal was collected over an emission range of 516–553 nm. FRET samples were imaged every 60 seconds. Eight single-cell image sets were acquired per sample at 2,520× magnification using a GaAsP detector on a Zeiss LSM 880 with a voxel size of 800 nm × 800 nm × 8.4 μm, a pixel dwell time of 10 μs, and no averaging. Images were collected for 5 minutes prior to stimulation to establish the baseline Src activity level. FN-f was then added, and samples were imaged for a subsequent 35 minutes.
Images were analyzed in ImageJ using the sensitized FRET method (41). Briefly, images were segmented and subsequently masked by adding the ECFP and EYFP images at each time point and applying a Li threshold (42). Src activity was determined on a pixel-by-pixel basis using the following equation at each time point:
where FRET−1 is the Src activity and CFP and YFP are the intensities from the corresponding ECFP and EYFP image channels. Ten cells per sample were chosen for analysis based on basal FRET−1 levels lying within 1 SD of the mean for that sample, and a lack of blebbing, necrosis, cell detachment, etc. throughout the course of the experiment. Following analysis, a 1-pixel median filter was applied to the FRET-indicated Src activity images for display purposes.
Statistical analysis
Data are presented as the mean ± SEM of 3 independent samples except where indicated otherwise. Analyses of statistical significance were performed using Graph-Pad Prism 6.04. One-way analysis of variance tests were performed, followed by Tukey’s honest significant difference or Dunnett’s multiple comparison post hoc tests.
RESULTS
Increased amounts of S-sulfenylated proteins in OA chondrocytes
To determine the potential relevance of S-sulfenylation signaling to OA, we labeled normal and OA chondrocytes (4 donors each) with dimedone and detected S-sulfenylated proteins by immunoblotting with a specific antibody against dimedone-labeled proteins. While both normal and OA chondrocytes contained S-sulfenylated proteins, OA chondrocytes showed significantly increased levels of S-sulfenylation labeling relative to normal chondrocytes (Figure 1). Samples not treated with dimedone had no immunolabeling, indicating specificity of the antidimedone antibody.
Figure 1.

Increase in basal cysteine S-sulfenylation in osteoarthritic (OA) chondrocytes. Confluent cultures of articular chondrocytes were switched to serum-free medium, and the next day, cells were lysed with lysis buffer containing or not containing dimedone (see Materials and Methods). Samples of cell lysates with equal amounts of total protein were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with an antibody against dimedone-conjugated S-sulfenylated cysteines. A, Immunoblot of a representative normal and OA cartilage sample. B, Quantification of basal S-sulfenylation in normal and OA cartilage. Values are the mean ± SEM of 4 samples per group. ** = P < 0.01 by Student’s unpaired 2-tailed t-test with Welch’s correction.
Increased S-sulfenylation in chondrocytes following FN-f stimulation
We used FN-f as a catabolic stimulus relevant to OA to study chondrocyte S-sulfenylation. Cells that were plated on poly-L-lysine–coated coverslips and labeled with DCP-Rho1 for imaging showed dynamic changes in S-sulfenylation at various time points following FN-f treatment (Figure 2A). The amount of DCP-Rho1 labeling of cells was greatest at 30 and 60 minutes after stimulation (Figure 2B). Pretreatment with PEG-catalase, which converts intracellular H2O2 into H2O and O2, resulted in levels of S-sulfenylation labeling similar to those in cells that were not treated with FN-f. This indicates that H2O2 is the dominant ROS responsible for the FN-f–induced S-sulfenylation.
Figure 2.

Time course of fibronectin fragment (FN-f)–induced S-sulfenylation in normal chondrocytes. Articular chondrocytes were cultured in monolayer and treated with 1 μM FN-f for the indicated times. A, Changes in normal chondrocyte S-sulfenylation after FN-f treatment. S-sulfenylated cysteines were labeled in live cells with DCP-Rho1 for the final 2 minutes of treatment with FN-f (see Materials and Methods). Nuclei were labeled using mounting medium supplemented with DAPI. Images are from a representative donor, with DCP-Rho1 displayed in heatmap format. B, Quantification of the DCP-Rho1 imaging data. Values are the mean ± SEM of 3 independent experiments. ** = P < 0.01; *** = P < 0.001 versus phosphate buffered saline (PBS)–treated samples and versus polyethylene glycol–catalase (PC)–pretreated samples, by one-way analysis of variance followed by Tukey’s honest significant difference post hoc test. C, Dimedone labeling of cell lysates and insoluble cell pellets. Cells were lysed in the presence or absence (control) of dimedone. Samples of the cell lysates with equal amounts of total protein as well as the cell pellets from each sample were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with an antibody against dimedone-conjugated S-sulfenylated cysteines.
Chondrocytes were treated with FN-f for various durations, and dimedone labeling was examined in cell lysates as well as in the insoluble cell pellets. Increased dimedone labeling was noted in several protein bands after FN-f stimulation, with the majority of the labeled proteins being in the soluble lysate fractions (Figure 2C). Some protein bands exhibited decreased dimedone labeling over time. Since dimedone is specific for sites of S-sulfenylation, decreased labeling would occur with either reduction of S-sulfenylated cysteines back to cysteine thiols, reaction with a neighboring cysteine thiol to form a disulfide, glutathiolation of the S-sulfenylated cysteine, or further oxidation resulting in S-sulfinylation (R-SO2H) or S-sulfonylation (R-SO3H).
Regulation of FN-f signaling by S-sulfenylation
To investigate whether FN-f–stimulated MAP kinase activation and MMP production are regulated by S-sulfenylation, we pretreated normal human articular chondrocytes with dime-done prior to FN-f stimulation. This allows the compound to quickly and irreversibly react with proteins as they undergo S-sulfenylation, thereby interfering with or locking in the functional changes due to S-sulfenylation. Dimedone inhibited the FN-f–induced phosphorylation of both JNK-1 and JNK-2, modestly reduced ERK and p65 phosphorylation, and did not affect the phosphorylation of p38 (Figures 3A and B). JNK phosphorylation was inhibited by dimedone in a dose-dependent manner (Figures 3C and D). This dose-dependent response was also reflected downstream in the inhibition of MMP-13 (collagenase 3) production (Figures 3C and D). These findings suggest that JNK and/or another protein upstream of JNK within the signaling pathway activated by fibronectin fragments is regulated by S-sulfenylation.
Figure 3.

Contribution of S-sulfenylation to fibronectin fragment (FN-f) induction of cell signaling and matrix metalloproteinase 13 (MMP-13) production. A, Effects of dimedone (DM) on FN-f–induced phosphorylation of p38, ERK, JNK, and p65. Primary normal human articular chondrocytes were pretreated for 30 minutes with 10 mM dimedone and then stimulated with 500 nM FN-f for 30 minutes. Cell lysates with equal amounts of total protein were immunoblotted with antibodies to the phosphorylated forms of the indicated proteins or to the total proteins (control). B, Quantification of the densitometry data, showing levels of phosphorylated p38, ERK, JNK, and p65 as normalized to the respective total protein levels (p/T). Values are the mean ± SEM of 3 independent experiments. C, Effects of dimedone on FN-f–induced phosphorylation of JNK in cell lysates and on MMP-13 and MMP-2 in conditioned media. Chondrocytes were pretreated with increasing concentrations of dimedone and then stimulated with FN-f for 30 minutes (for JNK phosphorylation in cell lysates) or overnight (for MMP-13 production in conditioned medium). D, Quantification of the densitometry data, showing levels of phosphorylated JNK and MMP-13. Results were normalized to total JNK and MMP-2, respectively. Values in B and D are the mean ± SEM of 3 independent experiments. * = P < 0.05; ** = P < 0.01; *** = P < 0.005 versus corresponding FN-f–treated positive control, by one-way analysis of variance followed by Dunnett’s multiple comparison post hoc test.
Proteomic analysis of FN-f–induced S-sulfenylation in chondrocytes
A proteomics approach was used to discover specific S-sulfenylated proteins in control and FN-f–stimulated cells. For this experiment, the SILAC-labeling technique was used, with immortalized chondrocytes in order to have a large number of cells where the control and FN-f–treated samples could be directly and quantitatively compared by mass spectrometry. More than 1,000 proteins were detected in the control or FN-f–treated cell lysates after streptavidin capture of DCP-Bio1–labeled cells (Supplementary Table 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39326/abstract). Three proteins were enriched >1.9-fold in samples from FN-f–treated cells relative to controls. These were ubiquitin-conjugating enzyme E2 S (2.4× control levels), Src (2.1× control levels), and calpastatin (1.9× control levels). Five proteins were much more abundant in the controls relative to FN-f–treated cells (ratios of 0.32–0.56) (Table 1).
Table 1.
Selected chondrocyte proteins in fibronectin fragment–treated and control cells identified by DCP-Bio1 and mass spectrometry*
| Protein | Ratio of FN-f–treated cells to control cells |
|---|---|
| Ubiquitin-conjugating enzyme E2 S | 2.43 |
| Protooncogene tyrosine protein kinase Src | 2.09 |
| Calpastatin | 1.91 |
| E3 ubiquitin-protein ligase ARIH2 | 0.555 |
| Glucose-6-phosphate 1-dehydrogenase | 0.476 |
| Double-strand break–repair protein MRE11A | 0.397 |
| U3 small nucleolar RNA–interacting protein | 0.388 |
| Cell division cycle 5–like protein | 0.315 |
The fibronectin fragment (FN-f)–to-control ratios were generated by calculating the average ratio of heavy isotope–labeled (FN-f–treated) to light isotope–labeled (control) peptides identified in each protein.
FN-f–induced S-sulfenylation of Src in chondrocytes
Previous studies, including studies relevant to integrin signaling, have shown that Src contains reactive cysteines that can be S-sulfenylated to regulate Src activity (5,23,43). Therefore, we chose Src from the list of candidate proteins discovered in the proteomics study of immortalized chondrocytes for verification studies in primary human chondrocytes. Two separate and complementary techniques were used to identify S-sulfenylation of Src in response to FN-f. S-sulfenylated Src was detected after 30 minutes of FN-f treatment in DCP-Bio1–labeled samples by affinity capture of all labeled proteins using streptavidin, as described above, and then by immunoblotting for Src (Figure 4A). Separately, Src was immunoprecipitated from dimedone-labeled samples treated for various times with FN-f, followed by immunodetection with the antidimedone antibody (Figure 4B). Using these techniques, Src S-sulfenylation was repeatedly observed as early as 1 minute following FN-f treatment and peaked at various time points between 5 and 30 minutes. Donor-to-donor variability was observed at the peak time points for Src S-sulfenylation. We speculate that this is due to variation in, and influence by, the oxidative state of the donor cells at the time of the experiment, which is known to vary (44).
Figure 4.
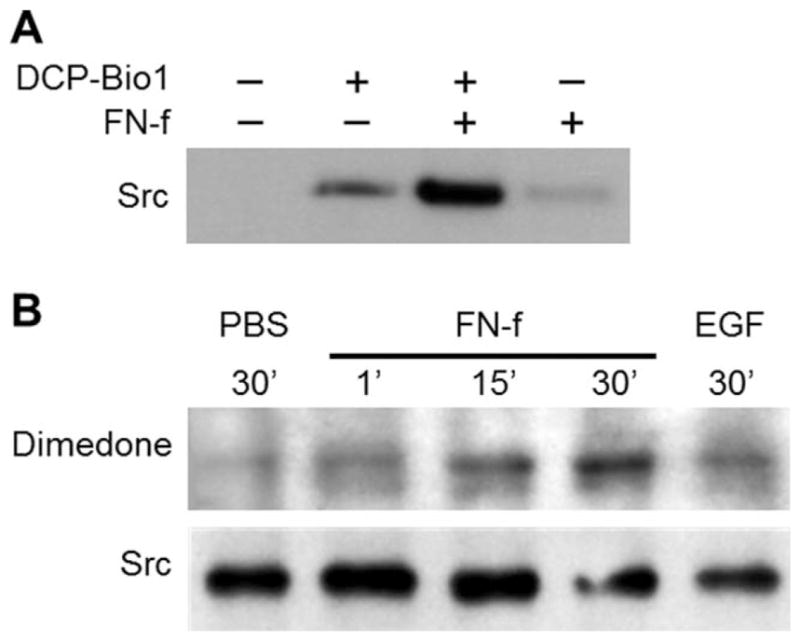
Effects of treatment with fibronectin fragments (FN-f) on S-sulfenylation of Src. A, Normal articular chondrocytes were treated with 1 μM FN-f for 30 minutes and lysed in the presence of DCP-Bio1 to label S-sulfenylated cysteines. Samples of cell lysates with equal amounts of total protein were captured using streptavidin-conjugated beads (see Materials and Methods). The captured proteins were then separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with an antibody to Src. B, Normal articular chondrocytes were treated with phosphate buffered saline (PBS), with 1 μM FN-f for the indicated times, or with human epidermal growth factor (EGF) and lysed in the presence of dimedone to label S-sulfenylated cysteines. Samples of cell lysates with equal amounts of total protein were immunoprecipitated using an antibody to Src and immunoblotted with an antibody to dimedone-conjugated S-sulfenylated cysteines.
FN-f stimulation of Src activity and blocking of FN-f–induced MMP-13 by Src inhibition
To determine the effect of FN-f on Src activity in chondrocytes, we used a genetically encoded FRET biosensor of Src activity (40). With a single-cell analysis technique, Src activity was found to be elevated at 30 minutes poststimulation with FN-f, and stimulation was delayed by pretreatment with the general antioxidant N-acetyl-L-cysteine (Figures 5A and B and Supplementary Video 1, available on the Arthritis & Rheumatology web site at http://onlinelibrary.wiley.com/doi/10.1002/art.39326/abstract). Src activity levels did not change with PBS treatment (control) or with FN-f stimulation in cells that had been pretreated with the Src inhibitor SU6656 or in cells expressing the negative control RV FRET sensor mutant (inhibitor and mutant sensor data not shown).
Figure 5.

Stimulation of Src activity with fibronectin fragments (FN-f) and requirement of Src activity for FN-f induction of matrix metalloproteinase 13 (MMP-13). A, Effect of FN-f on Src activity in normal articular chondrocytes. Chondrocytes were transfected with a fluorescence resonance energy transfer (FRET)–based Src activity biosensor, and Src activity was measured as described in Materials and Methods. Briefly, fluorescence photomicrographs of live cells left unpretreated or pretreated for 30 minutes with N-acetyl-L-cysteine (NAC) were acquired every 60 seconds for 5 minutes prior to treatment, and then for an additional 35 minutes following treatment with equal volumes of phosphate buffered saline (PBS) or FN-f (final concentration 1 μM). Images are from a representative donor, with FRET−1 displayed in heatmap format. B, Quantification of the FRET data from 3 donors. Values are the mean response of 10 individual cells from 3 independent experiments run on each sample. C, Comparison of early (0–15 minutes) and late (15–30 minutes) Src activation rates, as determined by the slope of FRET−1 data presented in B. D, Effect of Src inhibition on FN-f–induced production of MMP-13 and MMP-2. Chondrocytes were pretreated for 30 minutes with the indicated Src inhibitors (SU6656, Src kinase inhibitor 1 [SKI-1], or PP2). Following overnight treatment with PBS, 1 μM FN-f, or 25 μg/ml of interleukin-1β (IL-1β), samples of equal volumes of conditioned medium were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotted with antibody to MMP-13. The blot was then stripped and reprobed with antibody to MMP-2. E, Quantification of the immunoblot data from 3 donors. Values in C and E are the mean ± SEM * = P < 0.05; ** = P < 0.01; **** = P < 0.0001 versus the respective FN-f–treated positive control, by one-way analysis of variance followed by Dunnett’s multiple comparison post hoc test.
After chondrocyte Src S-sulfenylation and activity were found to be regulated by FN-f, 3 different Src inhibitors were used to test whether Src activation was required for FN-f–induced MMP-13 (Figure 5C). Multiple signaling protein inhibitors were used to help control for potential off-target effects of kinase inhibition, which vary from inhibitor to inhibitor. Chondrocytes pretreated with SU6656, Src inhibitor 1, or PP2 were found to produce less MMP-13 than controls, indicating that Src activity is required for FN-f–induced MMP-13 production.
DISCUSSION
This study is the first to demonstrate the presence of the S-sulfenylation oxidative posttranslational modification of proteins in immortalized and primary human articular chondrocytes. S-sulfenylation of multiple proteins was increased in chondrocytes from both OA and normal cartilage treated with the catabolic stimulus FN-f. S-sulfenylation was found to be temporally regulated by FN-f, with both dimedone and DCP-Rho1 labeling methods showing dynamic changes in S-sulfenylation levels in chondrocytes at various time points following FN-f treatment. Moreover, FN-f stimulation was associated with S-sulfenylation in the protein kinase Src, the activation of which was delayed by an antioxidant and was required for the increase in MMP-13 production in response to FN-f.
These findings build upon those of previous studies showing that ROS serve as second messengers in FN-f signaling. Early studies in synovial fibroblasts demonstrated that ROS were required for signaling generated by activation of the α5β1 integrin that resulted in collagenase expression (45), and antioxidants were shown to block the cartilage catabolic effects of FN-f (46). Treatment of chondrocytes with FN-f has been shown to trigger a burst of ROS within the cells, and FN-f–induced MMP production has been shown to be inhibited by pretreatment with anti-oxidants or by overexpression of antioxidant enzymes, including catalase and glutathione peroxidase, which primarily target H2O2 (13).
H2O2, a key mediator of redox signaling, is known to function through S-sulfenylation of cysteine residues in specific protein kinases and phosphatases (16,24,47). We found that pretreatment of chondrocytes with dimedone, which specifically reacts with S-sulfenylated cysteines and thereby alters subsequent S-sulfenylated cysteine functions, blocked the FN-f stimulation of JNK phosphorylation and reduced the phosphorylation of the p65 subunit of NF-κB, 2 signaling components required for the regulation of gene expression in response to FN-f (11,33). Importantly, the disruption in signaling was associated with reduced MMP-13 production. We further demonstrated a role of S-sulfenylation in the production of MMP-13 via the protein kinase Src. Previous studies have also shown regulation of Src kinase activity via S-sulfenylation in response to integrin signaling (23), but this mechanism had not previously been studied in chondrocytes.
The delay of Src activation with antioxidant treatment that we noted in chondrocytes indicates that early activation of Src is more ROS-dependent than later activation is. Src active-site phosphorylation has previously been shown to be enhanced by the 29-kd amino-terminal and 50-kd gelatin-binding domains of FN-f in chondrocytes, and consistent with our results, a Src inhibitor had been found to block MMP-13 production (5). However, we did not observe inhibition of MAP kinase or p65 phosphorylation in the presence of the Src inhibitor, which indicates that Src may regulate other pathways that lead to MMP-13 production (data not shown).
Our results suggest that the formation and subsequent reactivity of S-sulfenylated cysteines may regulate signaling pathways that play a role in OA. We noted that, relative to cells from normal tissue, chondrocytes isolated from OA joints had an increased number of proteins containing S-sulfenylated cysteines in the basal state. OA is closely associated with aging, and previous studies have indicated an age-associated increase in the overall intracellular oxidation state of chondrocytes (as measured by the ratio of intracellular concentrations of oxidized-to-reduced glutathione [GSSG:GSH]) that may contribute to increased S-sulfenylation (44). Cytokines that may contribute to the development of OA, including interleukin-1β (IL-1β) and tumor necrosis factor, stimulate endogenous ROS production and use ROS as second messengers in their signaling pathways via S-sulfenylation (48).
There are some potential limitations to this study. Cartilage from ankle joints was used as a source of normal chondrocytes due to the difficulty of obtaining sufficient numbers of normal cartilage samples from older adult knee joints. In previous studies, however, we showed that the response to FN-f of donor-matched pairs of ankle and knee chondrocytes was similar (12,49) and that inducing oxidative stress in normal ankle chondrocytes resulted in signaling changes that mimicked those seen in chondrocytes from the OA knee (26), suggesting that ankle chondrocytes are appropriate for these experiments. Also, serum-free medium was used for all experiments, and this may not represent physiologic conditions. However, since in vivo chondrocytes would not be exposed to serum, but rather, would be exposed to the endogenous growth factors produced locally in the tissue, it could be argued that serum-free conditions, where chondrocytes are allowed to produce their own growth factors, are more physiologic than serum-containing conditions. In addition, N-acetyl-L-cysteine can have a number of effects in addition to ROS scavenging (e.g., altering cellular concentrations of IL-1α, IL-1β, and IL-2 in a concentration-dependent manner) (50).
Further, the results obtained using chemical inhibitors of Src could be complicated by off-target effects that can sometimes occur with kinase inhibitors. We attempted knockdown of Src using small interfering RNA as an alternative approach, but this led to an increase in basal MMP-13 levels in unstimulated control samples, which made it impossible to interpret Src knockdown in FN-f–stimulated cells (Wood ST, et al: unpublished observations). This suggests a kinase-independent function of Src in the regulation of chondrocyte MMP-13 production that warrants investigation in future studies. Kinase-independent functions of Src have been noted in previous studies (51–54). Since 3 different chemical inhibitors all yielded the same result, making off-target effects seen with a specific inhibitor less likely, we believe that the data presented here sufficiently implicate a role of Src in FN-f–induced production of MMP-13.
In addition to a role of S-sulfenylation of Src as discussed above, it is likely that redox regulation via S-sulfenylation of many other proteins contributes to the altered signaling seen in OA chondrocytes. We therefore used a proteomics approach with an immortalized cell line to generate a list of candidate proteins. Because S-sulfenylation is not unique to catabolic signaling but is also involved in the regulation of certain growth factor signaling pathways, some of the proteins undergoing S-sulfenylation in chondrocytes may promote cartilage homeostasis. Further studies will be needed to identify which of the proteins that undergo S-sulfenylation contribute to OA and to establish the functional consequences of the oxidation of specific proteins. These studies may elucidate new targets for modulation of the signaling events that lead to the development and progression of OA.
Supplementary Material
Acknowledgments
Supported by the NIH (National Cancer Institute grant CA-177461 to Drs. Poole and Furdui, National Institute of Arthritis and Musculoskeletal and Skin Diseases grant AR-049003 to Dr. Loeser, and National Institute on Aging grant AG-044034 to Dr. Loeser). Purchase of the Zeiss LSM 710 confocal microscope was made possible by the NSF (Major Research Instrumentation awards 0722926 and 1039755).
We thank the Gift of Hope Tissue and Organ Donor Network, Dr. Susan Chubinskaya, the National Disease Research Interchange, and the donor families for providing the normal donor tissues, and we thank the Department of Orthopedic Surgery and Surgical Pathology Laboratory at Wake Forest Baptist Hospital (Winston-Salem, NC) for providing the OA tissues. We would also like to acknowledge the Wake Forest University Microscopic Imaging Facility and Confocal Microscopy Center and the University of North Carolina School of Medicine Hooker Imaging Core for use of the confocal microscopy equipment, and Dr. Glen Marrs and Robert Currin for technical assistance in the use thereof. We also acknowledge Dr. Yingxiao (Peter) Wang at the University of California, San Diego for providing the FRET biosensors of Src activity.
Footnotes
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Dr. Loeser had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design. Wood, Long, Reisz, Yammani, Burke, Poole, Furdui, Loeser.
Acquisition of data. Wood, Long, Reisz, Yammani, Burke, Klomsiri, Poole, Furdui, Loeser.
Analysis and interpretation of data. Wood, Reisz, Yammani, Burke, Klomsiri, Poole, Furdui, Loeser.
References
- 1.Lawrence RC, Felson DT, Helmick CG, Arnold LM, Choi H, Deyo RA, et al. for the National Arthritis Data Workgroup. Estimates of the prevalence of arthritis and other rheumatic conditions in the United States: part II. Arthritis Rheum. 2008;58:26–35. doi: 10.1002/art.23176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunn JD, Pill MW. A claims-based view of health care charges and utilization for commercially insured patients with osteoarthritis. Manag Care. 2009;18:44–50. [PubMed] [Google Scholar]
- 3.Loeser RF, Goldring SR, Scanzello CR, Goldring MB. Osteoarthritis: a disease of the joint as an organ [review] Arthritis Rheum. 2012;64:1697–707. doi: 10.1002/art.34453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241–8. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ding L, Guo D, Homandberg GA. Fibronectin fragments mediate matrix metalloproteinase upregulation and cartilage damage through proline rich tyrosine kinase 2, c-Src, NF-κB and protein kinase Cδ. Osteoarthritis Cartilage. 2009;17:1385–92. doi: 10.1016/j.joca.2009.03.024. [DOI] [PubMed] [Google Scholar]
- 6.Homandberg GA, Meyers R, Williams JM. Intraarticular injection of fibronectin fragments causes severe depletion of cartilage proteoglycans in vivo. J Rheumatol. 1993;20:1378–82. [PubMed] [Google Scholar]
- 7.Xie DL, Meyers R, Homandberg GA. Fibronectin fragments in osteoarthritic synovial fluid. J Rheumatol. 1992;19:1448–52. [PubMed] [Google Scholar]
- 8.Homandberg GA, Meyers R, Xie DL. Fibronectin fragments cause chondrolysis of bovine articular cartilage slices in culture. J Biol Chem. 1992;267:3597–604. [PubMed] [Google Scholar]
- 9.Ding L, Guo D, Homandberg GA. The cartilage chondrolytic mechanism of fibronectin fragments involves MAP kinases: comparison of three fragments and native fibronectin. Osteoarthritis Cartilage. 2008;16:1253–62. doi: 10.1016/j.joca.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 10.Homandberg GA, Costa V, Wen C. Fibronectin fragments active in chondrocytic chondrolysis can be chemically cross-linked to the α5 integrin receptor subunit. Osteoarthritis Cartilage. 2002;10:938–49. doi: 10.1053/joca.2002.0854. [DOI] [PubMed] [Google Scholar]
- 11.Pulai JI, Chen H, Im HJ, Kumar S, Hanning C, Hegde PS, et al. NF-κB mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J Immunol. 2005;174:5781–8. doi: 10.4049/jimmunol.174.9.5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Forsyth CB, Pulai J, Loeser RF. Fibronectin fragments and blocking antibodies to α2β1 and α5β1 integrins stimulate mitogen-activated protein kinase signaling and increase collagenase 3 (matrix metalloproteinase 13) production by human articular chondrocytes. Arthritis Rheum. 2002;46:2368–76. doi: 10.1002/art.10502. [DOI] [PubMed] [Google Scholar]
- 13.Del Carlo M, Schwartz D, Erickson EA, Loeser RF. Endogenous production of reactive oxygen species is required for stimulation of human articular chondrocyte matrix metalloproteinase production by fibronectin fragments. Free Radic Biol Med. 2007;42:1350–8. doi: 10.1016/j.freeradbiomed.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schroder K. NADPH oxidases in redox regulation of cell adhesion and migration. Antioxid Redox Signal. 2014;20:2043–58. doi: 10.1089/ars.2013.5633. [DOI] [PubMed] [Google Scholar]
- 15.Chung HS, Wang SB, Venkatraman V, Murray CI, van Eyk JE. Cysteine oxidative posttranslational modifications: emerging regulation in the cardiovascular system. Circ Res. 2013;112:382–92. doi: 10.1161/CIRCRESAHA.112.268680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Klomsiri C, Karplus PA, Poole LB. Cysteine-based redox switches in enzymes. Antioxid Redox Signal. 2011;14:1065–77. doi: 10.1089/ars.2010.3376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–47. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 18.Weerapana E, Wang C, Simon GM, Richter F, Khare S, Dillon MB, et al. Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature. 2010;468:790–5. doi: 10.1038/nature09472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roos G, Messens J. Protein sulfenic acid formation: from cellular damage to redox regulation. Free Radic Biol Med. 2011;51:314–26. doi: 10.1016/j.freeradbiomed.2011.04.031. [DOI] [PubMed] [Google Scholar]
- 20.Frijhoff J, Dagnell M, Godfrey R, Ostman A. Regulation of protein tyrosine phosphatase oxidation in cell adhesion and migration. Antioxid Redox Signal. 2014;20:1994–2010. doi: 10.1089/ars.2013.5643. [DOI] [PubMed] [Google Scholar]
- 21.Chiarugi P, Pani G, Giannoni E, Taddei L, Colavitti R, Raugei G, et al. Reactive oxygen species as essential mediators of cell adhesion: the oxidative inhibition of a FAK tyrosine phosphatase is required for cell adhesion. J Cell Biol. 2003;161:933–44. doi: 10.1083/jcb.200211118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eble JA, de Rezende FF. Redox-relevant aspects of the extracellular matrix and its cellular contacts via integrins. Antioxid Redox Signal. 2014;20:1977–93. doi: 10.1089/ars.2013.5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Giannoni E, Chiarugi P. Redox circuitries driving SRC regulation. Antioxid Redox Signal. 2014;20:2011–25. doi: 10.1089/ars.2013.5525. [DOI] [PubMed] [Google Scholar]
- 24.Paulsen CE, Carroll KS. Cysteine-mediated redox signaling: chemistry, biology, and tools for discovery. Chem Rev. 2013;113:4633–79. doi: 10.1021/cr300163e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Furdui CM, Poole LB. Chemical approaches to detect and analyze protein sulfenic acids. Mass Spectrom Rev. 2014;33:126–46. doi: 10.1002/mas.21384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin W, Park JI, Loeser RF. Oxidative stress inhibits insulin-like growth factor-I induction of chondrocyte proteoglycan synthesis through differential regulation of phosphatidylinositol 3-kinase-Akt and MEK-ERK MAPK signaling pathways. J Biol Chem. 2009;284:31972–81. doi: 10.1074/jbc.M109.056838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leahy DJ, Aukhil I, Erickson HP. 2.0 A crystal structure of a four-domain segment of human fibronectin encompassing the RGD loop and synergy region. Cell. 1996;84:155–64. doi: 10.1016/s0092-8674(00)81002-8. [DOI] [PubMed] [Google Scholar]
- 28.Garcia-Hernandez V, Flores-Maldonado C, Rincon-Heredia R, Verdejo-Torres O, Bonilla-Delgado J, Meneses-Morales I, et al. EGF regulates claudin-2 and -4 expression through STAT3 and Src in MDCK cells. J Cell Physiol. 2015;230:105–15. doi: 10.1002/jcp.24687. [DOI] [PubMed] [Google Scholar]
- 29.Lien GS, Wu MS, Bien MY, Chen CH, Lin CH, Chen BC. Epidermal growth factor stimulates nuclear factor-κB activation and heme oxygenase-1 expression via c-Src, NADPH oxidase, PI3K, and Akt in human colon cancer cells. PLoS One. 2014;9:e104891. doi: 10.1371/journal.pone.0104891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang J, Gupta V, Carroll KS, Liebler DC. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bain J, Plater L, Elliott M, Shpiro N, Hastie CJ, McLauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Long D, Blake S, Song XY, Lark M, Loeser RF. Human articular chondrocytes produce IL-7 and respond to IL-7 with increased production of matrix metalloproteinase-13. Arthritis Res Ther. 2008;10:R23. doi: 10.1186/ar2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Loeser RF, Forsyth CB, Samarel AM, Im HJ. Fibronectin fragment activation of proline-rich tyrosine kinase PYK2 mediates integrin signals regulating collagenase-3 expression by human chondrocytes through a protein kinase C-dependent pathway. J Biol Chem. 2003;278:24577–85. doi: 10.1074/jbc.M304530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reisz JA, Bechtold E, King SB, Poole LB, Furdui CM. Thiol-blocking electrophiles interfere with labeling and detection of protein sulfenic acids. FEBS J. 2013;280:6150–61. doi: 10.1111/febs.12535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klomsiri C, Nelson KJ, Bechtold E, Soito L, Johnson LC, Lowther WT, et al. Use of dimedone-based chemical probes for sulfenic acid detection evaluation of conditions affecting probe incorporation into redox-sensitive proteins. Methods Enzymol. 2010;473:77–94. doi: 10.1016/S0076-6879(10)73003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nelson KJ, Klomsiri C, Codreanu SG, Soito L, Liebler DC, Rogers LC, et al. Use of dimedone-based chemical probes for sulfenic acid detection methods to visualize and identify labeled proteins. Methods Enzymol. 2010;473:95–115. doi: 10.1016/S0076-6879(10)73004-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maller C, Schroder E, Eaton P. Glyceraldehyde 3-phosphate dehydrogenase is unlikely to mediate hydrogen peroxide signaling: studies with a novel anti-dimedone sulfenic acid antibody. Antioxid Redox Signal. 2011;14:49–60. doi: 10.1089/ars.2010.3149. [DOI] [PubMed] [Google Scholar]
- 38.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–5. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang LK, Wang MJ. Image thresholding by minimizing the measures of fuzziness. Pattern Recognit. 1995;28:41–51. [Google Scholar]
- 40.Wang Y, Botvinick EL, Zhao Y, Berns MW, Usami S, Tsien RY, et al. Visualizing the mechanical activation of Src. Nature. 2005;434:1040–5. doi: 10.1038/nature03469. [DOI] [PubMed] [Google Scholar]
- 41.Vilela M, Halidi N, Besson S, Elliott H, Hahn K, Tytell J, et al. Fluctuation analysis of activity biosensor images for the study of information flow in signaling pathways. In: Tetin SY, editor. Fluorescence fluctuation spectroscopy. San Diego: Elsevier Academic Press; 2013. pp. 253–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li CH, Tam PK. An iterative algorithm for minimum cross entropy thresholding. Pattern Recognit Lett. 1998;19:771–6. [Google Scholar]
- 43.Chiarugi P. Src redox regulation: there is more than meets the eye. Mol Cells. 2008;26:329–37. [PubMed] [Google Scholar]
- 44.Del Carlo M, Jr, Loeser RF. Increased oxidative stress with aging reduces chondrocyte survival: correlation with intracellular gluta-thione levels. Arthritis Rheum. 2003;48:3419–30. doi: 10.1002/art.11338. [DOI] [PubMed] [Google Scholar]
- 45.Kheradmand F, Werner E, Tremble P, Symons M, Werb Z. Role of Rac1 and oxygen radicals in collagenase-1 expression induced by cell shape change. Science. 1998;280:898–902. doi: 10.1126/science.280.5365.898. [DOI] [PubMed] [Google Scholar]
- 46.Homandberg GA, Hui F, Wen C. Fibronectin fragment mediated cartilage chondrolysis. I. Suppression by anti-oxidants. Biochim Biophys Acta. 1996;1317:134–42. doi: 10.1016/s0925-4439(96)00046-4. [DOI] [PubMed] [Google Scholar]
- 47.Wani R, Qian J, Yin L, Bechtold E, King SB, Poole LB, et al. Isoform-specific regulation of Akt by PDGF-induced reactive oxygen species. Proc Natl Acad Sci U S A. 2011;108:10550–5. doi: 10.1073/pnas.1011665108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lo YY, Cruz TF. Involvement of reactive oxygen species in cytokine and growth factor induction of c-fos expression in chondrocytes. J Biol Chem. 1995;270:11727–30. doi: 10.1074/jbc.270.20.11727. [DOI] [PubMed] [Google Scholar]
- 49.Forsyth CB, Cole A, Murphy G, Bienias JL, Im HJ, Loeser RF., Jr Increased matrix metalloproteinase-13 production with aging by human articular chondrocytes in response to catabolic stimuli. J Gerontol A Biol Sci Med Sci. 2005;60:1118–24. doi: 10.1093/gerona/60.9.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kelly GS. Clinical applications of N-acetylcysteine. Altern Med Rev. 1998;3:114–27. [PubMed] [Google Scholar]
- 51.Kaplan KB, Swedlow JR, Morgan DO, Varmus HE. c-Src enhances the spreading of src−/− fibroblasts on fibronectin by a kinase-independent mechanism. Genes Dev. 1995;9:1505–17. doi: 10.1101/gad.9.12.1505. [DOI] [PubMed] [Google Scholar]
- 52.Schwartzberg PL, Xing LP, Hoffmann O, Lowell CA, Garrett L, Boyce BF, et al. Rescue of osteoclast function by transgenic expression of kinase-deficient Src in src−/− mutant mice. Genes Dev. 1997;11:2835–44. doi: 10.1101/gad.11.21.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia-Martinez JM, Calcabrini A, Gonzalez L, Martin-Forero E, Agullo-Ortuno MT, Simon V, et al. A non-catalytic function of the Src family tyrosine kinases controls prolactin-induced Jak2 signaling. Cell Signal. 2010;22:415–26. doi: 10.1016/j.cellsig.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 54.Kaplan KB, Bibbins KB, Swedlow JR, Arnaud M, Morgan DO, Varmus HE. Association of the amino-terminal half of c-Src with focal adhesions alters their properties and is regulated by phosphorylation of tyrosine 527. EMBO J. 1994;13:4745–56. doi: 10.1002/j.1460-2075.1994.tb06800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.