

Abstract
Mutations in collagen V are associated with classic Ehlers-Danlos syndrome (EDS). A significant percentage of these mutations result in haploinsufficiency for collagen V. The purpose of this work was to determine if changes in collagen V expression are associated with altered dermal fibroblast behavior contributing to the poor wound healing response. A haploinsufficient Col5a1+/- mouse model of EDS was utilized. In vivo wound healing studies demonstrated that mutant mice healed significantly slower than Col5a1+/+ mice. The basis for this difference was examined in vitro using dermal fibroblast strains isolated from Col5a1+/- and Col5a1+/+ mice. Fibroblast proliferation was determined for each strain by counting cells at different time points after seeding as well as using the proliferation marker Ki-67. Fibroblastattachment to collagens I, III, and fibronectin also was analyzed. In addition, in vitro scratch wounds were used to analyze fibroblast wound closure. Significantly decreased fibroblast proliferation was observed in Col5a1+/- compared to Col5a1+/+ fibroblasts. Our data indicate that the decreased fibroblast number was not due to apoptosis. Wild type Col5a1+/+ fibroblasts attached significantly better to components of the wound matrix (collagens I, III and fibronectin) than Col5a1+/- fibroblasts. A significant difference in in vitro scratch wound closure rates also was observed. Col5a1+/+ fibroblasts closed wounds in 22hr while Col5a1+/- fibroblasts demonstrated ∼80% closure. There were significant differences in closure at all time points analyzed. Our data suggest that decreased fibroblast proliferation, extracellular matrix attachment, and migration contribute to the decreased wound healing response in classic EDS.
Keywords: Ehlers-Danlos syndrome, collagen V, dermal fibroblasts, mouse model, skin
Introduction
Ehlers-Danlos syndrome (EDS) classic type is an autosomal dominant inherited connective tissue disorder. Clinical manifestations include skin hyperelasticity, joint hypermobility, tissue fragility, and poor wound healing (1-4). Patients with classic EDS typically have skin involvement; skin may be thin, fragile, and easily torn. In addition, wound healing is delayed.
During wound healing in skin, new connective tissue is formed to replace the lost tissue. The wound healing process entails four sequential yet overlapping stages: hemostasis and coagulation; inflammation; proliferation and repair; and maturation and remodeling. Disruption in any of these processes may result in a poor wound healing response (5). During the proliferation and repair phase, fibroblasts proliferate and migrate into the wound site and subsequently produce collagen and other extracellular matrix components. Extracellular matrix and granulation tissue are formed, and collagen fibrils are deposited in the extracellular matrix. At the same time, the epidermis re-epithelializes to cover the new tissue. In a later overlapping stage, fibroblasts differentiate into myofibroblasts, which function in wound closure through contraction. Finally during maturation and remodeling, collagen III is replaced by collagen I, and collagen fibers are reorganized and aligned along tension lines (5).
Classic EDS is caused by mutations in collagen V (1,6). Mutations in collagen V have very prominent clinical presentations. There is broad connective tissue involvement with dermis, muscles and tendons among the most affected tissues in classic EDS patients. Collagen V is a fibril-forming collagen that forms heterotypic fibrils with collagen I, the most abundant fibrillar collagen. Although it constitutes less than 5% of collagens in tissues, collagen V plays an important role in the regulation of fibrillogenesis (7,8), and is widely distributed throughout skin (9-12). The collagen V deficient dermis has a disruption in collagen fibrillogenesis with fewer fibrils, abnormal fibril structure, i.e., ‘cauliflower’ shape fibrils with an abnormal diameter distribution, and abnormal packing (13).
Collagen V has been implicated in the wound healing process due to the up-regulated expression during tissue healing (14-16), although the mechanism is not clear. Collagen V is encoded by three genes; COL5A1, COL5A2 and COL5A3, and has multiple isoforms (17). The α1(V)2α2(V) form is ubiquitously expressed in all connective tissues examined. The most common causes of classic EDS are mutations in COL5A1 (18-23). Most of the mutations result in a non-functional COL5A1 allele and thus haploinsufficiency of collagen V (6,18-20,23). A Col5a1+/- mouse model that is Col5a1 haploinsufficient has been created (24). This mouse model has been previously used and demonstrated down-regulated collagen V protein expression (24,25). The mice display the features seen in classic EDS patients (8,24). The mouse skin is thin and fragile, and mice develop spontaneous and non-healing skin wounds. This is an excellent model to study dermal wound healing in classic EDS. Here, this model is used to determine if changes in collagen V expression are associated with altered dermal fibroblast behavior contributing to the poor wound healing response observed in classic EDS.
Methods
Animals
Col5a1+/- mice were created by targeted deletion and have been previously described in detail (24). All animal studies were performed in compliance with Institutional Animal Care and Use Committee (IACUC) approved animal protocols.
In Vivo Wound-Healing
Full thickness wounds were created in the subscapular skin of 60 day old mice using a 4 mm diameter dermal biopsy punch (Acuderm Inc., USA). The wounds were created on day 0, and wound healing was examined during a 10-day post-wounding period. Wounds were photographed using a digital camera with an in-picture ruler for scale. Wound areas were measured using Metamorph Premier for Olympus, Meta Series 7.65 (Olympus America Inc. and MDS Analytical Tech. Inc., USA). Wound healing was expressed as the percentage of wound area relative to the original (day 0) wound size. The original wound sizes were not significantly different. Three independent experiments were analyzed, n=4 for each genotype.
Cells and Cell Culture
Independent dermal fibroblast strains were isolated from 20 day old mice of Col5a1+/+ (wild-type) and Col5a1+/- (classic EDS) and were utilized at passages 2-6. All experiments were repeated using at least 3 different Col5a1+/+ and Col5a1+/- strains. All strains from a given genotype produced comparable results. Differences between strain or passage number were comparable to the differences seen between replicate experiments. Briefly, subscapular dermis (∼2cm × 2cm) was harvested from mice that were cleaned and shaved. Dermal tissue strips were incubated in trypsin at 37°C for 20 min with constant agitation and the epidermis was peeled off. Tissue strips were minced and incubated in 2.5mg/ml collagenase B (Roche 11088807001) in DMEM with HEPES at 150rpm in 37°C shaker for 1.5-2 hours until tissue pieces were mostly digested. The digested tissues were filtered through a cell strainer, and fibroblasts were collected and washed two times with DMEM (Invitrogen, 11995) supplemented with 10% fetal bovine serum (FBS). Fibroblasts were cultured at 37°C in DMEM with or without 10% FBS and 1mM 2-phospho-L-ascorbic acid (Sigma, 49752) according to experiments.
Cell Proliferation
Fibroblasts (3×104) were seeded in 6-well plates and allowed to grow in DMEM with 10% FBS and ascorbic acid at 37°C for 24, 48, 72 and 96 hours. Cells were harvested using trypsin and cell numbers were counted in Trypan Blue with a hemocytometer. The proliferation rate was expressed as a ratio of cell count at different time points versus number of cells plated. Experiments were performed in triplicate for each fibroblast strain. Three fibroblast strains of each genotype were analyzed.
Immunofluorescence Localization of Ki-67
Proliferation also was evaluated using immuno-localization of the proliferation marker Ki-67 (Dako, M7249). Fibroblasts (2×104) were seeded on chamber slides and cultured at 37°C for 24 hours. Cells were washed with PBS (phosphate buffered saline), fixed for 15 min in 4% paraformaldehyde in PBS, permeabilized with 0.2% Triton X-100 in PBS at room temperature for 10 min, blocked in blocking buffer, followed by primary antibody rat anti-mouse Ki-67 at 1:100 overnight at 4°C, and fluorochrome-conjugated secondary antibody goat anti rat Alexa Fluor 568 (Invitrogen, A11077) at 1:250 at room temperature for 1 hour. Nuclei were counterstained using Vectashield mounting solution with DAPI (Vector Laboratories, Inc., H1200). Images were captured using a fluorescence microscope (Leica CTR 5500, Wetzlar, Germany). The percentage of proliferating cells was determined as the total number of Ki67 positive cells compared with all the cells detected by DAPI. Images were randomly chosen in a masked manner for counting for each fibroblast strain (n=10). Three fibroblast strains of each genotype were analyzed.
Apoptosis
Apoptosis was examined using a TACS® 2 TdT DAB In Situ Apoptosis Detection Kit (Trevigen). Briefly, Fibroblasts of 2×104 were seeded on chamber slides and cultured at 37°C for 24 hours. Cells were washed with PBS and fixed for 10 min at room temperature in 3.7% buffered formaldehyde. Cells were washed with PBS and incubated in Proteinase K for 20 min, followed by incubation in labeling mix with TdT enzyme at 37°C for 1hr, and then strep-fluorescein for 20 min. Nuclei were counterstained with Vectashield mounting solution with DAPI and images were captured using a Leica CTR 5500 fluorescence microscope.
Cell Attachment
Fibroblast attachment to extracellular components was analyzed using 96-well bacteriologic plates that were coated overnight with 10 μg/well of collagen I (Invitrogen, A1048301), collagen III (Southern Biotech, 1230-01S), or fibronectin (Invitrogen, 33016-015). Before seeding cells, fibroblasts were harvested using trypsin, centrifuged in trypsin inhibitor (Invitrogen, 17075-029), and resuspended in DMEM with 5 μg/ml of cycloheximide (Sigma 01810). The cells were plated onto the wells at seeding densities of 4×104 cells/well on collagen I, and 2×104 cells/well on collagen III and fibronectin substrates. Cells were incubated at 37°C for 1, 2, or 4 hours, the wells were washed two times with PBS to remove non-adherent cells. Attached cells were fixed with methanol and stained with 0.5% crystal violet solution in 25% methanol for 10 min. Cells were washed several times, dried, and images were acquired with light microscope (4×). To quantify the cells attached, the dye was extracted with 1% triton X-100 and the absorbance at 560nm (A560) was measured using a microplate reader (Synergy HT, Bio-Tek, USA). For each substrate, at least three replicates were analyzed for each fibroblast strain. At least three fibroblast strains of each genotype were analyzed.
In Vitro Scratch Assay
The in vitro scratch assay was modified from a previously described protocol (26). Briefly, at least 3 independently isolated dermal fibroblast strains for each genotype were plated on 12-well plates (1.1 × 105 fibroblasts for Col5a1+/+ and 1.2 × 105 fibroblasts for Col5a1+/-), and allowed to attach and grow to a confluent monolayer. Since proliferation was decreased in Col5a1+/- compared to Col5a1+/+ fibroblasts, more Col5a1+/- fibroblasts were plated to yield confluent monolayers at the same time. To objectively define regions for analysis, markings were made on each plate as reference points before conducting the experiments. Images were acquired on the same fields for each plate (center of the plates) and the same area of each image was measured. A straight line was scraped in the center of the monolayer to create a ‘scratch wound’ with a plastic pipette tip and cells were washed twice with medium to remove debris. Fibroblasts were incubated at 37°C in DMEM with 10% FBS and ascorbic acid. Wound gap closure was monitored and images were captured at 0, 8, 16, 22 and 30 hours using phase contrast microscopy (10×). The wound gap was measured using Metamorph Premier for Olympus, Meta Series 7.65 (Olympus America Inc. and MDS Analytical Tech. Inc., USA). Three areas for each wound gap were measured; wound gap closure was expressed as the percentage of gap distance relative to the respective scrape width and the mean value was utilized for statistical analysis. At least three replicates were analyzed for each fibroblast strain. Three fibroblast strains of each genotype were analyzed.
Statistical Analysis
Numeral data in the results were presented as means ± sd. Student's T test (two tails) were used to evaluate differences. A p<0.05 was considered statistically significant.
Results
In vivo wound healing
To investigate the effect of collagen V deficiency on dermal wound repair, full thickness skin wounds were created in the dorsum of 60 day old mice using 4-mm dermal punches (n=4 for each genotype). There was a significant increase in wound closure in Col5a1+/+ compared with Col5a1+/- mice beginning 2 days post wounding (Figure 1A). At day 5, Col5a1+/+ wound size was 34.5 ± 1.3% of its size at day 0, while Col5a1+/- wounds were 62.5 ± 4.1%. The Col5a1+/+ wound size was about 55% of the Col5a1+/- wounds (Figure 1B). At day 10 post-wounding, Col5a1+/+ mice almost closed their wounds, with wound size 6.5 ± 3.1% of day 0, while Col5a1+/- wounds were 22.8% ± 6.1%. Col5a1+/+ wound size was about 29% of the Col5a1+/- wounds.
Figure 1. In vivo Col5a1+/- mice healed slower than Col5a1+/+ mice.
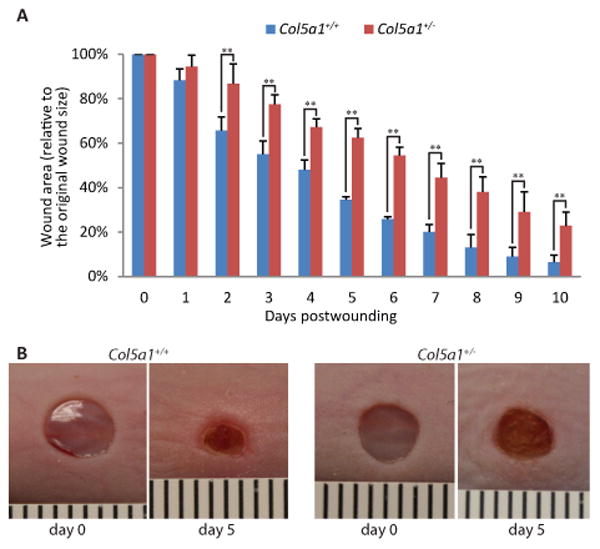
In vivo wound healing studies showed that Col5a1+/- mice healed slower than Col5a1+/+ mice. Full thickness wounds (4 mm diameter) were made in the subscapular skin of 60 days old mice (n=4 for each genotype). Wound closure was photographed every 24hr and wound area was measured. (A) Wound areas were measured daily over a 10-day post wounding period. Wound healing was expressed as the percentage of wound area relative to the day 0 area (punched wound size) (**p<0.01). (B) Representative wound closure of Col5a1+/- and Col5a1+/+ mice at day 0 and day 5 after wounding was shown. A mm scale is pictured.
Fibroblast proliferation rate
A decrease in Col5a1+/- dermal fibroblast proliferation compared to wild type controls was observed at all time points analyzed from 24-96 hours (Figure 2). At time 0, an equal number of fibroblasts were plated, followed byharvest at 24, 48, 72 and 96 hours at which time the relative cell count was calculated. At 24 hours there was no significant difference in the number of Col5a1+/+ fibroblasts compared to Col5a1+/- cells, but Col5a1+/- cells were consistently less. The difference became significant (p<0.05) at 48 hours with relative cell count of 1.45 ± 0.15 for Col5a1+/- fibroblasts and 2.17 ± 0.25 for Col5a1+/+. The relative cell count for Col5a1+/- fibroblasts was 55% (1.70 ± 0.25 vs 3.07 ± 0.23, p<0.01) of that for Col5a1+/+ fibroblasts at 72hr, and 52% (2.00 ± 0.29 vs 3.83 ± 0.37, p<0.01) at 96hr.
Figure 2. EDS dermal fibroblasts proliferated slower than wild type controls.

Representative data demonstrating decreased proliferation of EDS dermal fibroblasts. Proliferation rate was calculated as ratio of cell count at 24, 48, 72 and 96hrs versus number of cells plated (0 hour cell number). There was no significant difference in proliferation at 24 hr. A significant decrease in fibroblast proliferation was observed in Col5a1+/- fibroblasts at 48hrs (*p<0.05), 72hrs and 96hrs (**p<0.01) relative to the Col5a1+/+ fibroblasts. Differences in the proliferation rate from 24 to 96 hrs were seen as a decrease in the slope for the Col5a1+/- fibroblasts. The experiments were repeated 3 times with 3 strains derived from Col5a1+/+ and Col5a1+/- dermis.
The rate of proliferation for classic EDS cells was significantly decreased from 24 to 96 hours and over each 24hr period as well. The decreased proliferation rate is seen as a decrease in the slope of Col5a1+/- cells compared with Col5a1+/+ cells. The growth slope in the first 24 hours was 0.65 for Col5a1+/+ cells compared with 0.22 for Col5a1+/-; Col5a1+/- maintained 0.22 compared to Col5a1+/+ 0.52 from 24hr to 48hr. Col5a1+/+ cells grew significantly faster between 48hr and 72hr with growth slope of 0.90, compared to a growth slope of 0.25 for Col5a1+/- fibroblasts. Between 72hr and 96hr, the Col5a1+/+ growth slope was 0.76 compared to 0.30 for Col5a1+/- cells.
Apoptosis
No significant difference was observed in apoptosis in Col5a1+/- fibroblasts when compared to Col5a1+/+ fibroblasts with less than 1% of either genotype undergoing apoptosis (data not shown). Negative controls in which no TdT enzyme was added in the labeling mix showed no apoptotic cells. One sample treated with nuclease to generate DNA breaks was used as positive control, and virtually all cells exhibited apoptotic fluorescence signal.
Fibroblast proliferation
More proliferating cells were observed in Col5a1+/+ compared to Col5a1+/- fibroblasts using the proliferation marker Ki-67 (27). This result is consistent with the decreased proliferation rate of Col5a1+/- fibroblasts observed (Figure 2). The fraction of Ki-67 positive cells within the whole cell population was analyzed. Cells were immunofluorescently labeled with anti-Ki67 antibody and Ki-67 positive signal was detected within the nucleus for proliferating cells. In three independent experiments, a higher percentage of proliferating cells were observed in Col5a1+/+ cultures compared to Col5a1+/- cultures, with 43.3% ± 5.2% for Col5a1+/+ and 14.5% ± 2.3% for Col5a1+/- (means ± sd, p<0.01) (Figure 3).
Figure 3. Decreased proliferation marker Ki-67 in EDS compared with wild type dermal fibroblasts.
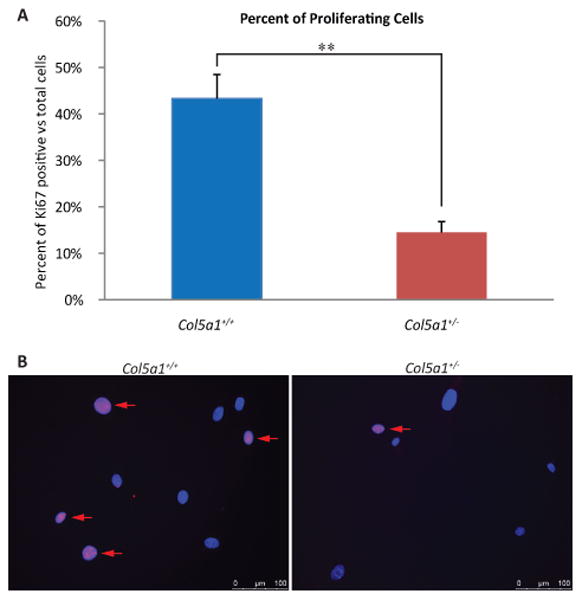
Representative data demonstrating that proliferating Col5a1+/- fibroblasts were significantly decreased compared to Col5a1+/+ fibroblasts. Fibroblasts were incubated at 37°C for 24hr, fixed and labeled with anti-Ki67 antibody. Proliferating cells were detected by anti-Ki67, and nuclei were counterstained with DAPI. Number of Ki67 positive cells and total cells were counted. (A) Percentage of proliferating Col5a1+/- cells (14.5%) was ∼ 30% of that of Col5a1+/+ cells (43.3%) (**p<0.01). Ratio was calculated by number of Ki67 positive cells versus total number of cells. (B) Representative immunofluorescence micrographs showing decreased proliferation in Col5a1+/- compared with Col5a1+/+ fibroblasts. Proliferating fibroblasts were identified using anti-Ki67 (arrows). Anti-Ki-67 (red), nuclear localization with DAPI (blue). The experiments were repeated 3 times with 3 strains derived from Col5a1+/+ and Col5a1+/- dermis.
Fibroblast attachment
When compared to Col5a1+/+ wild type fibroblasts, attachment of Col5a1+/- classic EDS dermal fibroblasts was significantly decreased on three wound substrates collagen I, collagen III and fibronectin (Figure 4). Attachment was visualized microscopically, and the attached cells were quantified spectrophotometrically. The correlation between absorbance and cell numbers was determined using serially diluted wild type fibroblasts on collagen I and fibronectin substrates. In both cases the serially diluted fibroblast A560 values, i.e., cell numbers, formed a straight line (the linear relationship R2 values were between 0.98 and 0.995) (data not shown).
Figure 4. Decreased attachment to wound matrix components in EDS compared with wild type fibroblasts.
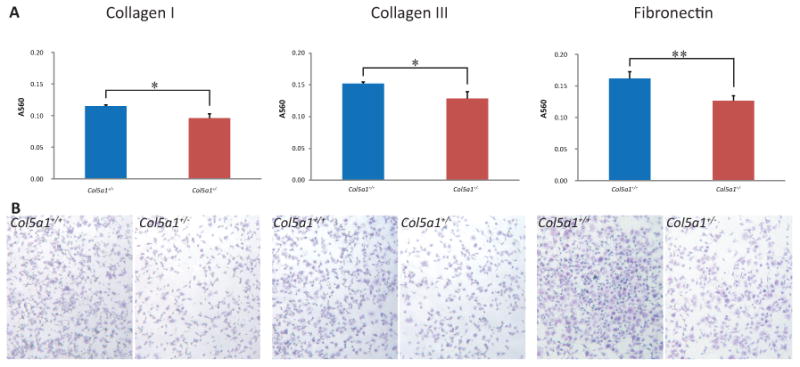
Representative analysis of fibroblast attachment. Fibroblasts were seeded on plates coated with collagen I, collagen III or fibronectin and incubated for 2hrs. Plates were washed and attached cells were stained using Crystal Violet. (A) The dye was extracted and the absorbance was measured at 560nm (A560). A560 values showed that more Col5a1+/+ fibroblasts attached compared to Col5a1+/- fibroblasts at a level of significance of p<0.05(*) on collagen I and collagen III, and p<0.01 (**) on fibronectin at 2hr. (B) Attachment of Col5a1+/- and Col5a1+/+ fibroblasts on collagen I, collagen III, and fibronectin were shown under light microscopy (4×). Attached fibroblasts were visualized in blue color by staining with Crystal Violet. The experiments were repeated 3 times with 3 strains derived from Col5a1+/+ and Col5a1+/- dermis.
More Col5a1+/+ fibroblasts attached when compared to Col5a1+/- fibroblasts for all three proteins (Figure 4). The A560 values for Col5a1+/+ and Col5a1+/- cells were 0.115 ± 0.002 and 0.096 ± 0.007 on collagen I (means ± sd, p<0.05), 0.152 ± 0.003 and 0.129 ± 0.011 on collagen III (p<0.05), and 0.162 ± 0.011 and 0.127 ± 0.008 on fibronectin (p<0.01), respectively. If viewed as a ratio, the A560 ratios of (Col5a1+/+: Col5a1+/-) were 1.20:1 on collagen I, 1.18:1 on collagen III, and 1.29:1 on fibronectin. Three independent experiments and at least three replicates for each experiment were analyzed. Other incubation time points, 1hr and 4hr, were also tested (data not shown). At all the time points studied, there was enhanced attachment of Col5a1+/+ compared to Col5a1+/- fibroblasts on all three proteins. For all three proteins, more fibroblasts attached when incubation time increased within the range of four hours. The change in slopes was relatively sharp before 3hrs, especially before 2hrs, and they became stable between 3hrs and 4hrs, although the change in slopes for three proteins was slightly different (data not shown). Therefore, 2hr incubation time was used as the representative time points (Figure 4).
Attachment of Col5a1+/- and Col5a1+/+ mouse dermal fibroblasts on collagen V, also was examined. Neither Col5a1+/- nor Col5a1+/+ fibroblasts demonstrated substantial attachment to the collagen V substrate over the time period studied (data not shown). This was true even when more substrate (50 μg/well) was used for coating the plates. Collagen V protein from two difference sources was tested, giving similar results. HeLa cells were used for the control. Hela cells attached to collagen V, while Col5a1+/- and Col5a1+/+ mouse dermal fibroblasts did not attach to collagen V over the period studied (data not shown).
In vitro wound healing
In vitro wound healing was assessed using Col5a1+/- and Col5a1+/+ mouse dermal fibroblast strains. A wound was created in the center of the confluent cell monolayer. Wound closure was monitored, and images were captured after culture for 8, 16, 22 and 30 hours. Wound gap width was measured, and is shown as percent of wound gap width relative to the original scratch width. The original wound widths were not significantly different (Figure 5). Fibroblast migration and closure of the scratch wounds was analyzed for Col5a1+/- and Col5a1+/+ mouse dermal fibroblasts. At post-wounding time points analyzed, Col5a1+/- fibroblasts closed wounds significantly slower than control Col5a1+/+ fibroblasts (Figure 5). After incubation for 8 hours, the Col5a1+/+ wound gap was 32.2 ± 3.4% of the original wound width, while the Col5a1+/- was 54.7 ± 6.2%. After 16-hours of incubation, the Col5a1+/+ wound was 11.3 ± 8.5% and Col5a1+/- was 32.9 ± 5.4% of the original wound width. At 22 hours, most of Col5a1+/- wounds were closed, while Col5a1+/- showed ∼80% closure. The mean wound width for Col5a1+/+ was 3.3% ± 5.7%, while Col5a1+/- was 20.6% ± 7.8%. Almost all wounds closed by 30hr. The difference in closure was significant at all time points analyzed: 8hr and 16hr (p<0.01) and 22hr (p<0.05) (Figure 5).
Figure 5. Fibroblast migration and in vitro wound closure is reduced in EDS compared to wild type dermal fibroblasts.

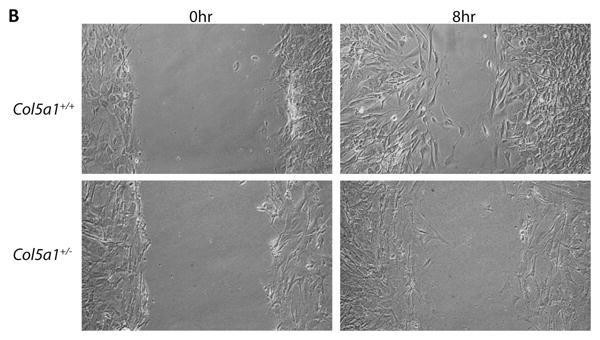
A representative analysis of in vitro scratch wound closure showed a significant difference between Col5a1+/- fibroblasts and Col5a1+/+ fibroblasts. A scratch wound was created on the monolayer of Col5a1+/- and Col5a1+/+ fibroblasts. Images were acquired after culture at 37°C for 0, 8, 16, 22 and 30hrs after scratching and the wound gap width was measured. (A) Wound closure was expressed as percentage of wound gap width relative to the scratch width (shown as 0 hour width). There was a significant difference in closure at all time points analyzed: 8hr and 16hr (**p<0.01) and 22hr (*p<0.05). (B) Representative wound closure of Col5a1+/- and Col5a1+/+ fibroblasts at 0hr and 8hr are shown. The experiments were repeated 3 times with 3 strains derived from Col5a1+/+ and Col5a1+/- dermis.
Discussion
Mutations in collagen V genes lead to classic Ehlers-Danlos syndrome. The most common is haploinsufficiency in COL5A1 which results in an ∼50% reduction in collagen V (24). Collagen V is a key regulator of fibrillogenesis, and thus the maintenance of tissue structure and integrity (24). Through its role in regulating fibrillogenesis, collagen V also plays a critical role in wound healing. The Col5a1+/- mouse model has structural defects in collagen fibrils similar to those seen in patients with classic EDS. Characteristic of the clinical phenotype seen in classic EDS, the mouse model has hyperextensible skin and hypermobile joints. In addition, the mice have fragile skin, and wound easily (8,24). We previously demonstrated, that comparable to classic EDS patients, the tensile strength of both normal and wounded skin was reduced in Col5a1+/- mice (24).
Our data indicate that most, if not all, of the Col5a1+/- mice older than 6 months developed spontaneous and non-healing skin wounds. Dermal wound repair was examined using 60-days old mice and the Col5a1+/- mice closed wounds significantly slower than wild-type mice in our in vivo wound studies. These data are consistent with the delayed wound healing in classic EDS patients. These results implicate collagen V in the dermal wound healing response. Previous studies have demonstrated that during wound healing, collagen V expression increases in connective tissues (14-16) which is consistent with this role.
During cutaneous wound repair, fibroblasts progress through four phases: proliferation, migration, synthesis of extracellular matrix molecules, and then turning into myofibroblasts which are responsible for contraction (5). Previous work suggested that collagen V increases connective tissue contraction through modulating fibroblast behavior, and thus contributes to wound healing (28). This would be consistent with our results demonstrating that reduction in collagen V is associated with delayed wound closure. However, our results suggest that wound contraction is not the sole factor contributing to an abnormal wound healing response in patients with classic EDS and our mouse model of classic EDS. The current work addresses this with an in vitro model using dermal fibroblasts isolated from wild-type and Col5a1+/- mice. We found that haploinsufficiency in Col5a1 which leads to reduced collagen V expression (24) was associated with decreased fibroblast proliferation, reduced attachment to components of the wound matrix, and reduced fibroblast migration.
Fibroblast proliferation was decreased in Col5a1+/- fibroblasts compared to wild type controls. Col5a1+/- fibroblasts demonstrated a significant decrease in proliferation as well as a decrease in the number of cells expressing the proliferation marker Ki-67, a protein not expressed during the G(0) phase (27). Our data suggest that the decreased proliferation was not due to apoptosis. As fibroblast proliferation at the site of injury is essential for the wound healing process, dysfunction of fibroblast proliferation in classic EDS would be consistent with a disruption and/or delay in wound healing. The mechanism whereby collagen V effects fibroblast proliferation is not known. However, altered extracellular matrix structure or substrate attachment may be involved.
Our results also demonstrated a decreased attachment of Col5a1+/- fibroblasts to components of the wound matrix compared to wild type controls. Attachment to collagens I, III and fibronectin was significantly reduced in EDS fibroblasts. Collagen I fibrils are heteropolymers containing collagen V (7,29) and present throughout the injury response. Collagen III is a major extracellular matrix protein during the proliferation phase of wound healing, but it is replaced by collagen I, during the maturation phase (5). Fibronectin plays a very important role and has various functions in wound healing including: in the initial step in extracellular matrix formation, mediating fibroblast attachment to cells and other matrix components, and in fibroblast migration (30). Altered interactions with the wound extracellular matrix would be expected to have effects on wound closure. In addition, a role in migration is anticipated which would be consistent with delayed wound closure as well as in deposition and assembly of a deficient extracellular matrix consistent with decreased wound strength. In addition, the disruption of cell substrate interactions may in turn alter the biosynthetic profiles of the EDS fibroblasts.
Our data indicate decreased fibroblast migration of Col5a1+/- fibroblasts compared to the wild type controls. This altered fibroblast migration of Col5a1+/- fibroblasts is consistent with the delayed wound closure. It is possible that the decreased fibroblast proliferation, and attachment to wound matrix affects migration. In summary, collagen V deficiency was shown to decrease dermal fibroblast proliferation, attachment of fibroblasts to wound matrix components, and fibroblast migration. All would be expected to directly impair wound healing. The data are consistent with the conclusion that collagen V expression affects fibroblast behavior, and alterations in expression are contributing factors that decrease the wound healing response in classic EDS.
Acknowledgments
This study was supported by NIH/NIAMS AR044745 (DEB) and a Summer Research Fellowship from the American Heart Association (JD).
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. The authors certify that their institution has approved the animal protocol for this investigation and all investigations were conducted in conformity with ethical principles of research.
Contributor Information
John DeNigris, Email: jdenigri@health.usf.edu.
Qingmei Yao, Email: qyao@health.usf.edu.
Erika K. Birk, Email: ebirk@colgate.edu.
David E. Birk, Email: dbirk@health.usf.edu.
References
- 1.Malfait F, Wenstrup RJ, De Paepe A. Clinical and genetic aspects of Ehlers-Danlos syndrome, classic type. Genet Med. 2010;12:597–605. doi: 10.1097/GIM.0b013e3181eed412. [DOI] [PubMed] [Google Scholar]
- 2.Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology, Villefranche, 1997. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK) Am J Med Genet. 1998;77:31–37. doi: 10.1002/(sici)1096-8628(19980428)77:1<31::aid-ajmg8>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 3.De Paepe A, Malfait F. The Ehlers-Danlos syndrome, a disorder with many faces. Clin Genet. 2012;82:1–11. doi: 10.1111/j.1399-0004.2012.01858.x. [DOI] [PubMed] [Google Scholar]
- 4.Malfait F, De Paepe A. The Ehlers-Danlos syndrome. Adv Exp Med Biol. 2014;802:129–143. doi: 10.1007/978-94-007-7893-1_9. [DOI] [PubMed] [Google Scholar]
- 5.Reinke JM, Sorg H. Wound repair and regeneration. Eur Surg Res. 2012;49:35–43. doi: 10.1159/000339613. [DOI] [PubMed] [Google Scholar]
- 6.Symoens S, Syx D, Malfait F, Callewaert B, De Backer J, Vanakker O, Coucke P, De Paepe A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum Mutat. 2012;33:1485–1493. doi: 10.1002/humu.22137. [DOI] [PubMed] [Google Scholar]
- 7.Birk DE, Fitch JM, Babiarz JP, Linsenmayer TF. Collagen type I and type V are present in the same fibril in the avian corneal stroma. J Cell Biol. 1988;106:999–1008. doi: 10.1083/jcb.106.3.999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun M, Connizzo BK, Adams SM, Freedman BR, Wenstrup RJ, Soslowsky LJ, Birk DE. Targeted deletion of collagen V in tendons and ligaments results in a classic Ehlers-Danlos syndrome joint phenotype. Am J Pathol. 2015;185:1436–1447. doi: 10.1016/j.ajpath.2015.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Garrone R, Lethias C, Le Guellec D. Distribution of minor collagens during skin development. Microsc Res Tech. 1997;38:407–412. doi: 10.1002/(SICI)1097-0029(19970815)38:4<407::AID-JEMT8>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 10.Chanoki M, Ishii M, Fukai K, Kobayashi H, Hamada T, Muragaki Y, Ooshima A. Immunohistochemical localization of type V collagen in normal human skin. Arch Dermatol Res. 1988;280:145–151. doi: 10.1007/BF00456844. [DOI] [PubMed] [Google Scholar]
- 11.Konomi H, Hayashi T, Nakayasu K, Arima M. Localization of type V collagen and type IV collagen in human cornea, lung, and skin. Immunohistochemical evidence by anti-collagen antibodies characterized by immunoelectroblotting. Am J Pathol. 1984;116:417–426. [PMC free article] [PubMed] [Google Scholar]
- 12.Weber L, Kirsch E, Muller P, Krieg T. Collagen type distribution and macromolecular organization of connective tissue in different layers of human skin. J Invest Dermatol. 1984;82:156–160. doi: 10.1111/1523-1747.ep12259720. [DOI] [PubMed] [Google Scholar]
- 13.Wenstrup RJ, Florer JB, Brunskill EW, Bell SM, Chervoneva I, Birk DE. Type V collagen controls the initiation of collagen fibril assembly. J Biol Chem. 2004;279:53331–53337. doi: 10.1074/jbc.M409622200. [DOI] [PubMed] [Google Scholar]
- 14.Shimomura T, Jia F, Niyibizi C, Woo SL. Antisense oligonucleotides reduce synthesis of procollagen alpha1 (V) chain in human patellar tendon fibroblasts: potential application in healing ligaments and tendons. Connect Tissue Res. 2003;44:167–172. doi: 10.1080/03008200390215872. [DOI] [PubMed] [Google Scholar]
- 15.Niyibizi C, Visconti CS, Kavalkovich K, Woo SL. Collagens in an adult bovine medial collateral ligament: immunofluorescence localization by confocal microscopy reveals that type XIV collagen predominates at the ligament-bone junction. Matrix Biol. 1995;14:743–751. doi: 10.1016/s0945-053x(05)80017-4. [DOI] [PubMed] [Google Scholar]
- 16.Niyibizi C, Kavalkovich K, Yamaji T, Woo SL. Type V collagen is increased during rabbit medial collateral ligament healing. Knee Surg Sports Traumatol Arthrosc. 2000;8:281–285. doi: 10.1007/s001670000134. [DOI] [PubMed] [Google Scholar]
- 17.Smith SM, Birk DE. Focus on molecules: collagens V and XI. Exp Eye Res. 2012;98:105–106. doi: 10.1016/j.exer.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malfait F, De Paepe A. Molecular genetics in classic Ehlers-Danlos syndrome. Am J Med Genet C Semin Med Genet. 2005;139C:17–23. doi: 10.1002/ajmg.c.30070. [DOI] [PubMed] [Google Scholar]
- 19.Symoens S, Malfait F, Renard M, Andre J, Hausser I, Loeys B, Coucke P, De Paepe A. COL5A1 signal peptide mutations interfere with protein secretion and cause classic Ehlers-Danlos syndrome. Hum Mutat. 2009;30:E395–403. doi: 10.1002/humu.20887. [DOI] [PubMed] [Google Scholar]
- 20.Wenstrup RJ, Florer JB, Willing MC, Giunta C, Steinmann B, Young F, Susic M, Cole WG. COL5A1 haploinsufficiency is a common molecular mechanism underlying the classical form of EDS. Am J Hum Genet. 2000;66:1766–1776. doi: 10.1086/302930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Paepe A, Nuytinck L, Hausser I, Anton-Lamprecht I, Naeyaert JM. Mutations in the COL5A1 gene are causal in the Ehlers-Danlos syndromes I and II. Am J Hum Genet. 1997;60:547–554. [PMC free article] [PubMed] [Google Scholar]
- 22.Burrows NP, Nicholls AC, Richards AJ, Luccarini C, Harrison JB, Yates JR, Pope FM. A point mutation in an intronic branch site results in aberrant splicing of COL5A1 and in Ehlers-Danlos syndrome type II in two British families. Am J Hum Genet. 1998;63:390–398. doi: 10.1086/301948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwarze U, Atkinson M, Hoffman GG, Greenspan DS, Byers PH. Null alleles of the COL5A1 gene of type V collagen are a cause of the classical forms of Ehlers-Danlos syndrome (types I and II) Am J Hum Genet. 2000;66:1757–1765. doi: 10.1086/302933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenstrup RJ, Florer JB, Davidson JM, Phillips CL, Pfeiffer BJ, Menezes DW, Chervoneva I, Birk DE. Murine model of the Ehlers-Danlos syndrome. col5a1 haploinsufficiency disrupts collagen fibril assembly at multiple stages. J Biol Chem. 2006;281:12888–12895. doi: 10.1074/jbc.M511528200. [DOI] [PubMed] [Google Scholar]
- 25.Wenstrup RJ, Smith SM, Florer JB, Zhang G, Beason DP, Seegmiller RE, Soslowsky LJ, Birk DE. Regulation of collagen fibril nucleation and initial fibril assembly involves coordinate interactions with collagens V and XI in developing tendon. J Biol Chem. 2011;286:20455–20465. doi: 10.1074/jbc.M111.223693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 27.Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 28.Berendsen AD, Bronckers AL, Smit TH, Walboomers XF, Everts V. Collagen type V enhances matrix contraction by human periodontal ligament fibroblasts seeded in three-dimensional collagen gels. Matrix Biol. 2006;25:515–522. doi: 10.1016/j.matbio.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 29.Birk DE. Type V collagen: heterotypic type I/V collagen interactions in the regulation of fibril assembly. Micron. 2001;32:223–237. doi: 10.1016/s0968-4328(00)00043-3. [DOI] [PubMed] [Google Scholar]
- 30.McDonald JA. Extracellular matrix assembly. Annu Rev Cell Biol. 1988;4:183–207. doi: 10.1146/annurev.cb.04.110188.001151. [DOI] [PubMed] [Google Scholar]