

Abstract
Purpose
The purpose of this study was to investigate the mechanisms by which miR-183 may contribute to the phenotypic alterations associated with stress-induced senescence of human trabecular meshwork (HTM) cells.
Methods
Changes in gene expression induced by miR-183 in HTM cells were evaluated by gene array analysis, confirmed by quantitative-PCR (Q-PCR), and analyzed by MetaCore pathway analysis. Effects of miR-183 on cell proliferation were assessed by incorporation of bromodeoxyuridine incorporation, and DNA damage by CometAssay after ultraviolet (UV) irradiation in primary HTM cells, and confirmed in human diploid fibroblasts (HDF) and HeLa cells. A plasmid expressing KIAA0101 without its 3′-untranslated region (3′-UTR) was cotransfected with miR-183 to evaluate the role of KIAA0101 on the effects induced by miR-183.
Results
miR-183 affected the expression of multiple genes involved in cell cycle regulation and DNA damage response in HTM cells. Forced expression of miR-183 in HTM and HDF resulted in a significant decrease in proliferation in primary HTM and HDF cells but not in HeLa cells. In all cell types tested, overexpression of miR-183 resulted in increased DNA damage under UV irradiation. Expression of KIAA0101 lacking the 3′-UTR region partially prevented the effects of miR-183 on cell proliferation and completely reversed the effects on UV-induced DNA damage.
Conclusions
Our results suggest that the observed up-regulation of miR-183 after stress-induced senescence in HTM cells may contribute to reinforce cellular senescence by inhibiting cell cycle progression through multiple gene targets and limiting the DNA repair mechanisms through inhibition of KIAA0101.
Keywords: cell proliferation, cellular senescence, DNA damage repair, miR-183, trabecular meshwork
Senescent cells display phenotypic alterations that could contribute to disruption of tissue homeostasis in aging.1,2 Increased accumulation of senescent cells in the human trabecular meshwork (HTM) as a result of aging, and chronic oxidative damage has been hypothesized to contribute to the abnormal increase in resistance to aqueous humor outflow associated with primary open-angle glaucoma.3–8 In the past, we have reported the up-regulation of miR-183 after induction of cellular senescence by exposure to H2O2 in both HTM and human diploid fibroblasts (HDF),9 suggesting that miR-183 could also contribute to the phenotypic changes associated with stress-induced senescence.10 miR-183 (HGNC:MIR183; HUGO Gene Nomenclature Committee [www.genenames.org]) is a multifunctional microRNA (miRNA) that contributes to the regulation of a variety of biological processes including development, physiology, and pathology of the inner ear11–15 and retina.16–19 miR-183 is also believed to regulate cancer progression and metastasis.20,21 However, the role of this miRNA in the regulation of the cell cycle and its potential contribution to reinforce the senescent phenotype is controversial because it has been reported to exert both pro- and antiproliferative effects. Although miR-183 has been reported to promote cell and tumor growth,22–24 there is also evidence for a role of miR-183 as an inhibitor of cell proliferation and cancer growth.25–33 Effects of miR-183 on apoptosis and cell survival are similarly conflicting. Although down-regulation of miR-183 inhibits apoptosis and enhances cell survival of endometrial stromal cells in endometriosis,34 up-regulation of miR-183 has been reported to inhibit TGF-beta 1–induced apoptosis by down-regulation of PDCD4 expression in human hepatocellular carcinoma cells.35
In order to gain a better understanding of the biological roles of miR-183 and its contribution to the senescent phenotype of HTM cells, we analyzed by gene array the effects of miR-183 over-expression on the gene expression profile of HTM cells, validated experimentally four novel targets and conducted functional analysis of the effects on cell proliferation and ultra violet (UV) induced DNA damage repair mediated by targeting of the recently identified target KIAA010132 by miR-183 in HTMs. The functional effects of miR-183 were further confirmed in primary HDFs and HeLa cells.
Materials and Methods
Cell Culture and Transfection
Human tissue for generation of trabecular meshwork cell cultures was obtained from the Eye Bank for Sight Restoration, Inc. (New York, NY, USA) according to the tenets of the Declaration of Helsinki. The study was approved by the Duke University Health System Institutional Review Board. HTM, HDF, and HeLa cells were cultured and maintained following methods previously described.9 Transfections of miRNAs (183 mimic [183M] and negative miRNA control mimic [ConM]) or plasmids (KIAA0101 vector [pKIAA0101] or pcDNA3.1 vector [pCon]; Invitrogen, Carlsbad, CA, USA) to HTM or HDF cells were performed using the Nucleofector system (Amaxa, Inc., Gaithersburg, MD, USA) as previously described.9,10 HeLa cells were transfected using Effectene transfection reagent (Qiagen, Valencia, CA, USA) following the manufacturer instruction.
RNA Isolation
Total RNA was isolated using an RNeasy kit (Qiagen) following the manufacturer's protocol. RNA yields were measured using RiboGreen fluorescent dye (Molecular Probes, Eugene, OR, USA). Briefly, HTM cells were transfected with miR-183 mimic (183M) or scramble control mimic (ConM), using the Nucleofector system (Amaxa, Inc.). Three days after transfection, HTM cells were harvested by adding lysis buffer from the kit and homogenization using a QIAshredder spin column (Qiagen). DNA was removed using DNase I (Qiagen). RNA was selectively isolated from the column flow through use of an RNeasy mini-spin column (Qiagen). Twenty microliters of RNase-free water was added to the spin column to recover RNA for each sample. The RNA quantity was analyzed using the RNA Nanodrop with the Bioanalyzer 2100 system (Agilent, Santa Clara, CA, USA).
Affymetrix GeneChip Microarray and Data Analysis
High-quality RNA, 200 ng, was used to generate the probe for hybridization with the Ambion message amplification kit (Thermo Fisher Scientific, Grand Island, NY, USA). Fragmented cRNA, 10 μg, was hybridized to the Affymetrix Human Genome U133 2.0 Array at Duke University Microarray facility (Durham, NC, USA). This array covers the Human Genome U133 Set plus 6500 additional genes for analysis of more than 47,000 transcripts. Raw data were log2 transformed and normalized using the default Robust Mult-chip Average method in GeneSpring 10 software (Silicon Genetics, Wilmington, DE, USA). Genes were filtered to their intensities in the control channel. A paired Student's t-test was performed (P values ≤0.05 were considered significant), and changes less than 2-fold were excluded. The list of genes that were significantly down-regulated was compared to those in three databases that predict targets for miRNAs: Microcosm (http://www.ebi.ac.uk/enright-srv/microcosm/htdocs/targets/v5/), TargetScan (http://www.targetscan.org), and PicTar-Vert (http://pictar.mdc-berlin.de/). To analyze the biological significance and regulatory pathways involved in the changes observed in the arrays, we performed GeneGo pathway map analysis of genes more than 2-fold differentially expressed (P value ≤0.05, Metacore pathway analysis [GeneGo, St. Joseph, MI, USA]).
Quantitative Polymerase Chain Reaction
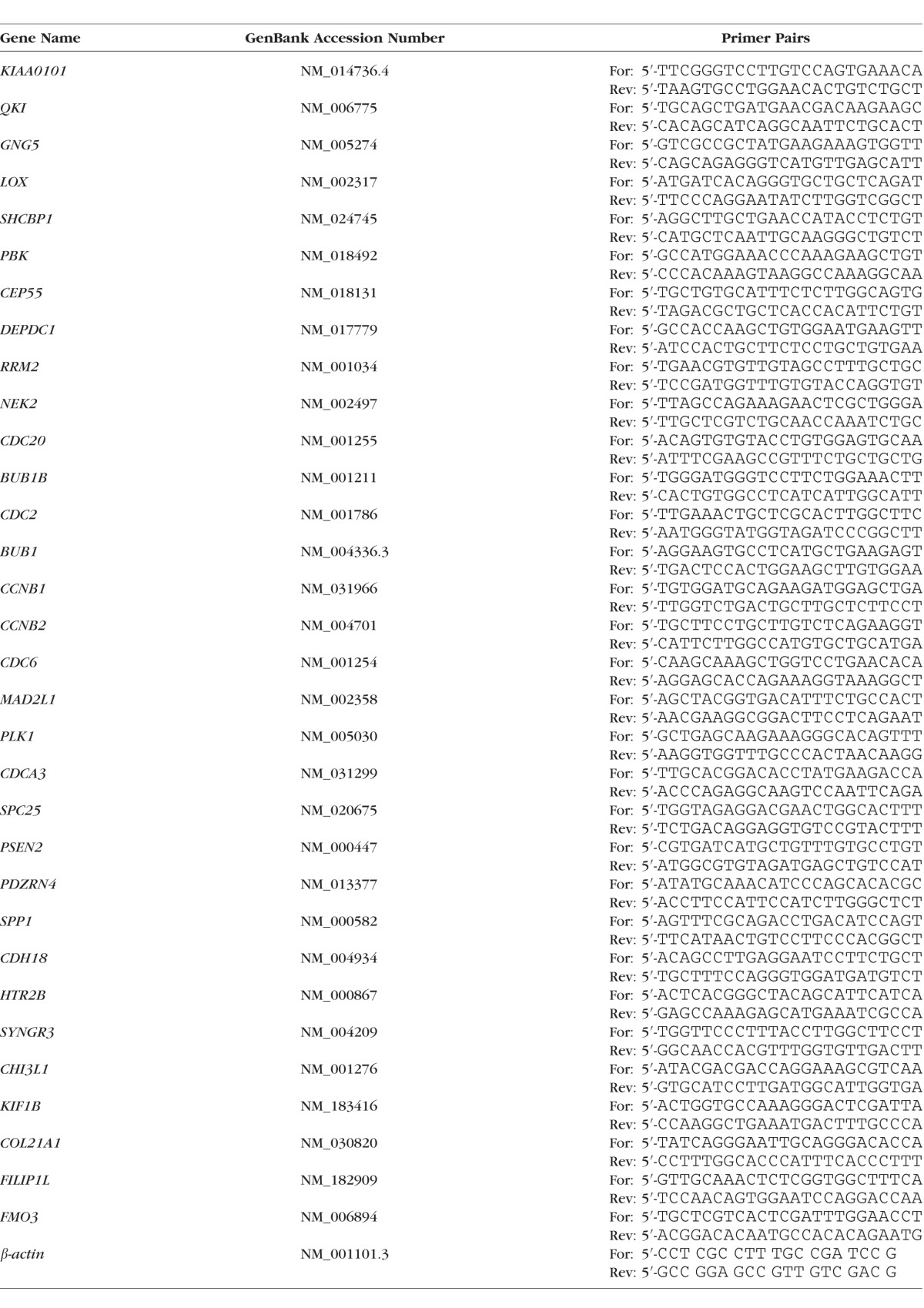
After total RNA isolation, first-strand cDNA was synthesized from total RNA (1 μg) by reverse transcription by using oligo(dT) and Superscript II reverse transcriptase (Invitrogen). Quantitative polymerase chain reaction (Q-PCR) analyses were performed in a 20-μL mixture that contained 1 μL of the cDNA preparation and 1× iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA), using the following PCR parameters: 95°C for 3 minutes followed by 40 cycles of 95°C for 10 seconds, 60°C for 30 seconds, plus melting curve 65°C to 95°C increment 0.5°C for 5 seconds. The fluorescence threshold value (Ct) was calculated using the iCycle system software (Bio-Rad). The absence of nonspecific products was confirmed by both the analysis of the melt curves and by electrophoresis in 3% Super AcrylAgarose gels. β-actin was used as an internal standard of RNA expression to normalize gene expressions. The specific primer pairs used were listed in Table 1.
Table 1.
Primer Pairs Used To Quantify Gene Expressions in HDF Cells
Protein Extraction and Western Blotting
Cultured cells were washed twice in cold phosphate-buffered saline (PBS). Total protein was extracted using radioimmunoprecipitation assay buffer (150 mM NaCl, 10 mM Tris, pH 7.2, 0.1% SDS, 1.0% Triton X-100, 5 mM EDTA, pH 8.0) containing a 1× protease inhibitor cocktail (Roche, Basel, Switzerland). Protein concentration was determined by using Micro BCA protein assay kit (Pierce, Rockford, IL, USA). Total protein extracts were separated by 12% SDS-PAGE gels and transferred to polyvinylidene fluoride membrane (Bio-Rad). Membranes were blocked with 5% nonfat dry milk and incubated overnight with an anti-KIAA0101 primary antibody (Abcam, Cambridge, MA, USA) and then with a secondary antibody conjugated to horseradish peroxidase. Immunoreactive proteins were visualized using enhanced chemiluminescence substrate (ECL Plus; GE Healthcare, Pittsburgh, PA, USA). For detection of endogenous control, the membrane was stripped with stripping buffer (25 mM glycine, pH 3.0, plus 1% SDS) and then incubated with anti-β-tubulin (product SC-9935; Santa Cruz Biotechnology, Dallas, TX, USA).
Generation of a KIAA0101 Expression Plasmid
The expression vector of pKIAA0101 lacking the 3′-UTR was generated using forward primer (5′-GCA GTCGAC GAAC ATG GTG CGG ACT AAA GCA GAC AGT G) containing SalI restriction site and reverse primer (5′-TAT GCGGCCGC ACC AGG GTA AAC AAG GAG ACG TT) containing NotI restriction site. The fragment amplified by this primer pair was cloned into pCR2.1 vector for sequence confirmation. A clone with correct sequence was then cut with SalI and BamH1 to release the fragment containing the complete KIAA0101 open reading frame. The fragment was then ligated to pcDNA3.1(+) vector (Invitrogen) as a pKIAA0101 expression vector (pKIAA0101). An empty pcDNA3.1(+) was used as a control vector (pCon). To confirm the ability of this vector to overexpress KIAA0101 even in the presence of miR-183, HeLa cells were cotransfected with ConM or 183M (60 pmol) and pCon or pKIAA0101 (0.1 μg), and the expression of KIAA0101 was analyzed by Western blotting 24 hours after transfection.
Cell Proliferation Assay
Cell growth was quantified using a bromodeoxyuridine (BrdU) cell proliferation assay kit (Calbiochem, San Diego, CA, USA) according to the manufacturer's instructions. Briefly, 100 μL of 183M- or conM-transfected cells at 8 × 104 cells/mL were seeded into a 96-well culture dish and incubated for 48 hours (for HTM and HDF cells). HeLa cells at 1 × 104 cells/well were plated the day before 183M or ConM transfection and incubated for 24 hours. Culture medium was then replaced with 100 μL fresh DMEM containing 10% FBS and BrdU, 1:10,000 dilution. After overnight (HTM and HDF cells) or 2 hours (HeLa cells) incubation, the cells were fixed and BrdU incorporation was measured using anti-BrdU antibody and reconstituted Peroxidase Goat Anti-Mouse IgG HRP Conjugate. The color was then developed by adding Substrate Solution to each well. After 15 minutes of incubation in the dark at room temperature, Stop Solution was added to each well, and absorbance was measured using a spectrophotometric plate reader at dual wavelengths of 450 to 540 nm.
CometAssay
DNA damage following UV irradiation was detected using CometAssay kit for single cell gel electrophoresis assay (Trevigen, Inc., Gaithersburg, MD, USA) following the manufacture's instruction. Briefly, HTM, HDF and HeLa cells were co-transfected with ConM and pCon, 183M and pCon, or 183M and pKIAA0101. Forty eight hours (HTM and HDF cells) or 24 hours (HeLa cells) after transfection, cells were irradiated with UV-C (10 J/m2) at 50% to 70% confluency in the presence of 5 mL medium/100 mm petri dish without the lid. Two hours after UV irradiation, cells were collected and resuspended at 1 × 105 cells/mL in cold PBS. The cells were then loaded to molten LMAgarose (at 37°C) at a ratio of 1:10 (v/v), and 50 μL of the mixture was pipette into CometSlide. Slides were incubated at 4°C in the dark for 10 minutes and then immersed in pre-chilled Lysis Solution for 30 minutes at 4°C, Alkaline Solution for 20 minutes at RT in the dark. The slides were then subjected to TBE electrophoresis for 10 minutes. After drying, the samples were stained with SYBR Green I. The image was visualized and captured using fluorescence microscopy.
Statistical Analysis
Data are mean ± SD. Statistical significance between groups was assessed by the Mann-Whitney U test. A P value <0.05 was considered statistically significant.
Results
Effects of miR-183 on Gene Expression in HTM Cells
HTM1073-07-26 cells at passage 6 were transfected with miR-183 mimic (183M) or control mimic (ConM). Three days after transfection, RNAs were extracted, and Affymetrix gene array was conducted. Results were analyzed using GeneSpring version 10 software (Agilent), which indicated that 121 genes were significantly 2-fold up- or down-regulated more than by miR-183 (Supplementary Table S1). Pathway analysis indicated strong involvement of miR-183 in the regulation of cell cycle progression and DNA damage response (Fig. 1). To validate the gene array results, we conducted the experiments in three additional individual primary HTM cell lines (HTM616-09-61, HTM681-09-27, and HTM330-08-50) by Q-PCR of 32 genes. As shown in Table 2, 25 of these genes showed consistent and significant up- or down-regulation in all 3 HTM cell lines. The inhibitory effects of miR-183 on expression of the KIAA0101 protein, the most highly down-regulated gene and recently validated target,32 was confirmed by Western blot analysis in HTM, HDF, and HeLa cells transfected with 183M (Fig. 2).
Figure 1.
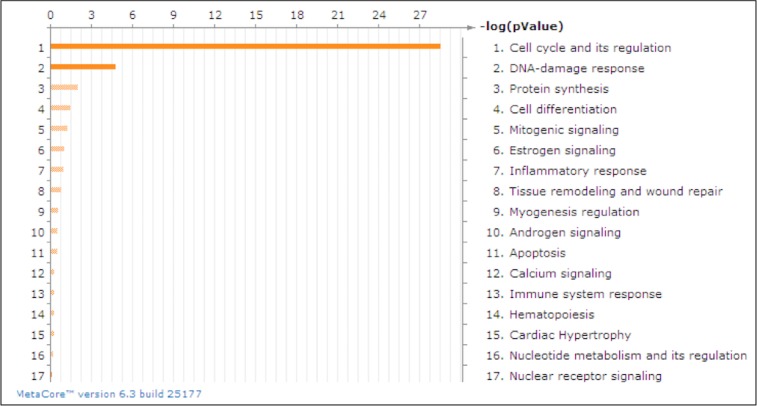
MetaCore pathway analysis of the Affymetrix gene array. GeneGo pathways were identified by MetaCore analysis as the most significantly affected by miR-183. Cell cycle and its regulation were the highest ranking in terms of P value and Gene Ontology interpretation.
Table 2.
Q-PCR Results in Three HTM Cell Lines Transfected With miR-183 and Normalized by β-Actin
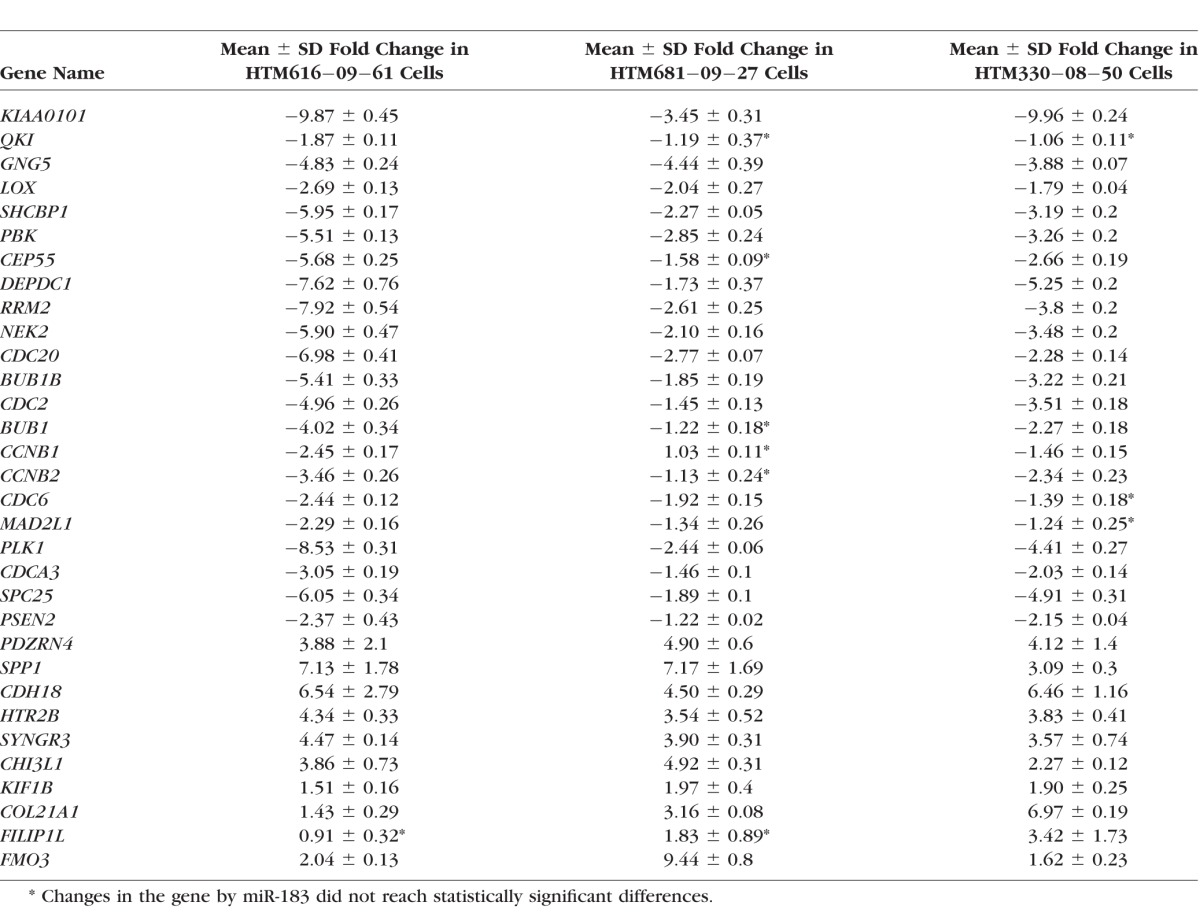
Figure 2.
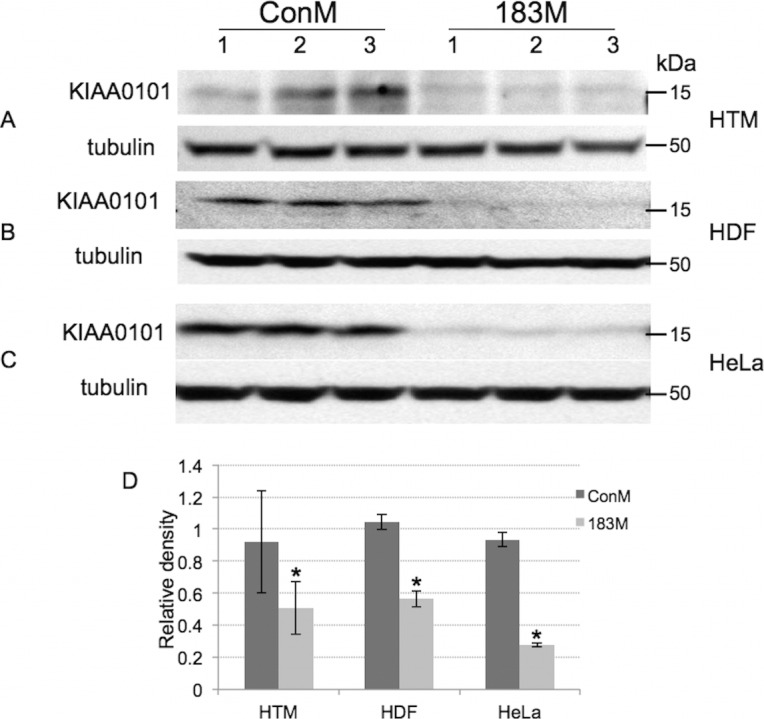
Inhibition of KIAA0101 protein expression by miR-183. HDF, HTM, and HeLa cells were transfected with either 183M or conM, and the production of KIAA0101 protein was evaluated 3 days (HTM and HDF cells) or 1 day (HeLa cells) post transfection by Western blot analysis. A total of 60 μg of proteins was loaded to be able to detect KIAA0101 production in HTM and HDF cells, and 40 μg in HeLa cells. (A) HTM cells; (B) HDF cells; and (C) HeLa cells; (D) Densitometry of Figure 3A, 3B, and 3C.
Targeting of KIAA0101 Contributes to Inhibition of Cell Proliferation Induced by miR-183
To evaluate the role of KIAA0101 on the effects mediated by miR-183, we generated a plasmid expressing KIAA0101 without its 3′-UTR (Fig. 3). Forced expression of miR-183 resulted in significant inhibition of cell proliferation in both primary HTM cells and HDFs but had no effect on the proliferation of HeLa cells (Fig. 4). Since KIAA0101 is known to regulate cell proliferation19,25 we investigated whether these effects were mediated by targeting KIAA0101. For this purpose we generated an expression plasmid (pKIAA0101), containing the entire coding region of KIAA0101 and lacking the 3′-UTR. Expression of KIAA0101 lacking its 3′-UTR was capable of reverting completely the inhibitory effects of miR-183 on cell growth in HTM cells and significantly decreased the effects of miR-183 in HDFs.
Figure 3.
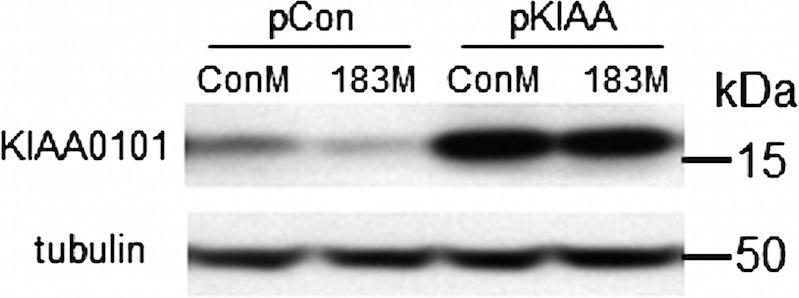
Confirmation of KIAA0101 expression by the pKIAA0101 plasmid. To demonstrate overexpression of KIAA0101 driven by the pKIAA0101 plasmid even in the presence of miR-183, HeLa cells were cotransfected with ConM plus 183M (60 pmol) and pCon plus pKIAA0101 (0.1 μg). Expression of KIAA0101 was evaluated 24 hours after transfection by Western blot analysis.
Figure 4.
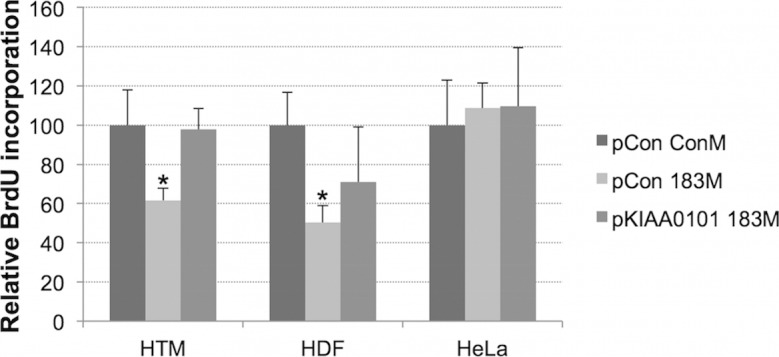
KIAA0101 partially mediated the effects on cell proliferation induced by miR-183. To evaluate the effects of miR-183 on cell proliferation and determine whether these effects were dependent on KIAA0101 inhibition, HTM, HDF, and HeLa cells were co-transfected with either ConM plus pCon, 183M plus pCon, ConM plus pKIAA0101, or 183M plus pKIAA0101. At 48 hours (HTM, HDF) or 24 hours (HeLa) after transfection, BrdU was loaded into the cells and cultures were incubated overnight (HTM, HDF) or for 2 hours (HeLa cells). Cells were then fixed, and proliferation was determined by using BrdU cell proliferation assay kit. Data are percentages of BrdU incorporation compared to that in control (ConM + pCon). n = 4 to 6; *P < 0.05, Mann-Whitney U test.
Targeting of KIAA0101 by miR-183 Leads To Increased Susceptibility to UV-Induced DNA Damage
Because of the documented role of KIAA0101 in DNA damage repair,36,37 we first investigated whether miR-183 had effects on the levels of DNA damage induced by UV radiation. As shown in Figure 5, forced expression of miR-183 resulted in a significant increase in UV-induced DNA damage in HTM, HDF, and HeLa cells. Expression of KIAA0101 lacking its 3′-UTR was enough to restore levels of DNA damage to control levels with and without over-expression of miR-183 in all three cell types (Fig. 5).
Figure 5.

Overexpression of miR-183 led to increased UV-induced DNA damage through inhibition of KIAA0101. To evaluate the effects of miR-183 on UV-induced DNA damage and to determine whether these effects were dependent on KIAA0101 inhibition, HTM (A), HDF (B), and HeLa cells (C) were co-transfected with either ConM plus pCon, 183M plus pCon, ConM plus pKIAA0101, or 183M plus pKIAA0101. At 48 hours (HTM, HDF) or 24 hours (HeLa) after transfection, cells were exposed to low dose of UV irradiation. Two hours after UV irradiation, cells were collected, and DNA damage was determined using CometAssay kit for single-cell gel electrophoresis assay. Quantitative data were generated by image analysis of the length of the comet tail from 75 to 115 random individual cells for each group. Data are percentages of DNA damage compared to those in the control (mean ± SD ConM plus pCon). *P < 0.001, Mann-Whitney U test).
Discussion
Our results demonstrated extensive changes in gene expression induced by miR-183 in HTM cells including down-regulation of multiple genes involved in the regulation of cell cycle and DNA damage responses, such as the recently identified target KIAA0101,32 as well as PLK, CCNB1, PSEN2, and SPC25, which were confirmed direct targets of miR-183.
KIAA0101 was the gene most notably down-regulated by miR-183 in HTM cells, and its down-regulation in HTM, HDF, and HeLa cells was confirmed by Q-PCR and Western blot analysis. These results suggested that targeting of KIAA0101 could potentially play an important role on the biological effects mediated by miR-183. KIAA0101 is a proliferating cell nuclear antigen (PCNA)-associated factor that binds with PCNA and regulates cell proliferation.36,38 Over-expression of KIAA0101 has been demonstrated to promote cell growth, whereas attenuation of expression by small interfering RNA (siRNA) leads to decreased cell proliferation.38 Expression of KIAA0101 is regulated at the transcriptional level by the Rb/E2F complex, and loss of Rb/E2F-mediated repression during the G1/S transition phase leads to the up-regulation of KIAA0101, which facilitates DNA synthesis and S phase progression.38 Consistent with its growth-promoting role, KIAA0101 is over-expressed in multiple types of human cancer.36,38 Its over-expression is associated with poor patient outcome39 and has been recognized as an independent risk factor associated with high-grade tumors.36
Our results showed that miR-183 significantly inhibited proliferation in HTM cells and that expression of KIAA0101 lacking its 3′-UTR was able to recover proliferation to control levels, suggesting that targeting of KIAA0101 may be a primary mechanism of miR-183 for inhibition of cell growth in HTM cells. Although miR-183 also inhibited cell proliferation in HDFs at levels similar to those in HTM cells, the rescue of this effect by KIAA0101 was only partial, suggesting that other targets are likely to contribute to the inhibition of cell growth in these cells. In contrast to the study by Roche et al.,32 miR-183 failed to exert any significant effect on proliferation of HeLa cells in our experiments. However, we evaluated cell proliferation by BrdU incorporation only 24 hours after transfection with miR-183, whereas Roche et al.32 evaluated the effects of miR-183 on cell proliferation by daily counting of viable GFP expressing cells and were evident only 2 days after transfection. Therefore, differences in methodology may account for this discrepancy. In any case, our results indicate that inhibitory effects of miR-183 on proliferation may be cell type dependent, and are only partially dependent on the targeting of KIAA0101.
In addition to its role in cell cycle regulation, KIAA0101 was reported to control DNA repair. KIAA0101 has been shown to be up-regulated after UV irradiation.36,37 KIAA0101 localizes in the same multiprotein complex as the tumor suppressor p33ING1B, and the associations of KIAA0101 and p33ING1b with PCNA are enhanced after UV irradiation.36 Furthermore, KIAA0101 is a direct transcriptional target of ATF3, and expression of ATF3 and KIAA0101 are sufficient to trigger the DNA repair machinery against UV damage. Consistently, inhibition of KIAA0101 expression leads to modifications in the DNA repair process, rendering cells more sensitive to UV-induced cell death.37
Our results demonstrated that expression of miR-183 leads to increased susceptibility to UV-induced DNA damage and that this effect could be completely abolished by restoring expression of KIAA0101. These results were similar in primary HTM cells, HDFs, and HeLa cells in contrast to the effects in cell proliferation, suggesting that targeting of KIAA0101 by miR-183 and the associated increase in susceptibility to DNA damage may be a common feature in multiple cell types.
It is important to acknowledge that, in addition to KIAA0101, our results also showed that miR-183 had the ability to target the 3′-UTRs and inhibit transcript expression of PLK1, CCNB1, SPC25, and PSEN2 (Supplementary Table S2, Supplementary Fig. S1). These genes can also potentially contribute to functional effects of miR-183 on cell cycle and DNA repair. PLK1 has multiple important roles in cell cycle progression and in cellular response to DNA damage40–43; it strongly promotes progression of the cell cycle and is responsible for aggressive proliferation of tumor cells44; and its over-expression enables cells to override checkpoints, leading to genomic instability and promoting cell transformation.45 Similarly, CCNB1 is one of the principal mitotic cyclins in animals.46 Although knockdown of CCNB1 by siRNA only delays temporarily chromatin condensation, nuclear CCNB1 cooperates with cyclin A2 to the early mitotic events and appears to be particularly critical for the maintenance of the mitotic state.47 CCNB1 expression is frequently elevated in human cancers and is associated with tumor aggressiveness and poor clinical outcome.48 SPC25 is a component of the NDC80 kinetochore complex required, along with SPC24, to establish and maintain kinetochore-microtubule attachment.49,50 Finally, PSEN2 is one of the enzymatic components of the γ-secretase complex that cleaves amyloid precursor protein, as well as other proteins.51 Inhibition of PSEN2 appears to have different effects on cancer cells. While siRNA-mediated inhibition of presenilin 2 inhibits glioma cell growth and invasion,52 similar inhibition by siRNA has been demonstrated to increased lung cancer cell growth.53 Therefore, the inhibition of PSEN2 by miR-183 could also contribute to the differences in the effects of this microRNA on the growth of different types of cancer cells.
A limitation of this study is that we only focused on the role of miR-183 on HTM cell proliferation and DNA damage by targeting KIAA0101. However, the role this miRNA in the regulation of cell cycle and its potential contribution to reinforce the senescent phenotype may be cell type specific and also the consequence of the combined effects of multiple target genes. As discussed above, PLK1, CCNB1, SPC25, and PSEN2, as well as additional as yet unidentified targets of miR-183 are likely to play important roles in DNA repair and cell cycle regulation. Further studies aimed at a more complete identification and functional analysis of miR-183 targets will be necessary to better understand the physiologic role of miR-183.
In summary, miR-183 affects the expression of multiple genes associated with cell cycle and DNA repair in HTM cells. Inhibition of cell proliferation by miR-183 appears to be only partially mediated by targeting of KIAA0101. Our results also showed that miR-183 exerts significant inhibitory effects on UV-induced DNA damage repair mechanisms and these effects are mediated primarily by targeting of KIAA0101. Overall, these results are consistent with a role for miR-183 in reinforcing cellular senescence by inhibiting cell cycle progression, and at the same time limiting the DNA repair mechanisms, which could lead to increase rate of mutations.
Supplementary Material
Acknowledgments
Supported by Research to Prevent Blindness Grants NEI EY 23287 and EY 05722.
Disclosure: G. Li, None; C. Luna, None; P. Gonzalez, None
References
- 1. Campisi J. Aging, cellular senescence, and cancer. Ann Rev Physiol. 2013; 75: 685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ohtani N,, Hara E. Roles and mechanisms of cellular senescence in regulation of tissue homeostasis. Cancer Sci. 2013; 104: 525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liton PB,, Luna C,, Challa P,, Epstein DL,, Gonzalez P. Genome-wide expression profile of human trabecular meshwork cultured cells nonglaucomatous and primary open angle glaucoma tissue. Mol Vis. 2006; 12: 774–790. [PMC free article] [PubMed] [Google Scholar]
- 4. Sacca SC,, Izzotti A. Oxidative stress and glaucoma: injury in the anterior segment of the eye. Prog Brain Res. 2008; 173: 385–407. [DOI] [PubMed] [Google Scholar]
- 5. Morgan JT,, Raghunathan VK,, Chang YR,, Murphy CJ,, Russell P. The intrinsic stiffness of human trabecular meshwork cells increases with senescence. Oncotarget. 2015; 6: 15362–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pulliero A,, Seydel A,, Camoirano A,, Sacca SC,, Sandri M,, Izzotti A. Oxidative damage and autophagy in the human trabecular meshwork as related with ageing. PLoS One. 2014; 9: e98106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Babizhayev MA,, Yegorov YE. Senescent phenotype of trabecular meshwork cells displays biomarkers in primary open-angle glaucoma. Curr Mol Med. 2011; 11: 528–552. [DOI] [PubMed] [Google Scholar]
- 8. Yu AL,, Birke K,, Moriniere J,, Welge-Lussen U. TGF-{beta}2 induces senescence-associated changes in human trabecular meshwork cells. Invest Ophthalmol Vis Sci. 2010; 51: 5718–5723. [DOI] [PubMed] [Google Scholar]
- 9. Li G,, Luna C,, Qiu J,, Epstein DL,, Gonzalez P. Alterations in microRNA expression in stress-induced cellular senescence. Mech Ageing Dev. 2009; 130: 731–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Li G,, Luna C,, Qiu J,, Epstein DL,, Gonzalez P. Targeting of integrin beta1 and kinesin 2alpha by microRNA 183. J Biol Chem. 2010; 285: 5461–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li H,, Kloosterman W,, Fekete DM. MicroRNA-183 family members regulate sensorineural fates in the inner ear. J Neurosci. 2010; 30: 3254–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li H,, Fekete DM. MicroRNAs in hair cell development and deafness. Curr Opin Otolaryngol Head Neck Surg. 2010; 18: 459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gu C,, Li X,, Tan Q,, Wang Z,, Chen L,, Liu Y. MiR-183 family regulates chloride intracellular channel 5 expression in inner ear hair cells. Toxicol In Vitro. 2013; 27: 486–491. [DOI] [PubMed] [Google Scholar]
- 14. Patel M,, Cai Q,, Ding D,, Salvi R,, Hu Z,, Hu BH. The miR-183/Taok1 target pair is implicated in cochlear responses to acoustic trauma. PLoS One. 2013; 8: e58471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang Q,, Liu H,, McGee J,, Walsh EJ,, Soukup GA,, He DZ. Identifying microRNAs involved in degeneration of the organ of corti during age-related hearing loss. PLoS One. 2013; 8: e62786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pierce ML,, Weston MD,, Fritzsch B,, Gabel HW,, Ruvkun G,, Soukup GA. MicroRNA-183 family conservation and ciliated neurosensory organ expression. Evol Dev. 2008; 10: 106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lumayag S,, Haldin CE,, Corbett NJ,, et al. Inactivation of the microRNA-183/96/182 cluster results in syndromic retinal degeneration. Proc Natl Acad Sci U S A. 2013; 110: E507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu Q,, Sun W,, Okano K,, et al. Sponge transgenic mouse model reveals important roles for the microRNA-183 (miR-183)/96/182 cluster in postmitotic photoreceptors of the retina. J Biol Chem. 2011; 286: 31749–31760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Loscher CJ,, Hokamp K,, Kenna PF,, et al. Altered retinal microRNA expression profile in a mouse model of retinitis pigmentosa. Genome Biol. 2007; 8: R248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Larne O,, Ostling P,, Haflidadottir BS,, et al. miR-183 in prostate cancer cells positively regulates synthesis and serum levels of prostate-specific antigen. Eur Urol. 2015; 68: 581–588. [DOI] [PubMed] [Google Scholar]
- 21. Zhang QH,, Sun HM,, Zheng RZ,, et al. Meta-analysis of microRNA-183 family expression in human cancer studies comparing cancer tissues with noncancerous tissues. Gene. 2013; 527: 26–32. [DOI] [PubMed] [Google Scholar]
- 22. Ueno K,, Hirata H,, Shahryari V,, et al. microRNA-183 is an oncogene targeting Dkk-3 and SMAD4 in prostate cancer. Brit J Cancer. 2013; 108: 1659–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tanaka H,, Sasayama T,, Tanaka K,, et al. MicroRNA-183 upregulates HIF-1alpha by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J Neurooncol. 2013; 111: 273–283. [DOI] [PubMed] [Google Scholar]
- 24. Sarver AL,, Li L,, Subramanian S. MicroRNA miR-183 functions as an oncogene by targeting the transcription factor EGR1 and promoting tumor cell migration. Cancer Res. 2010; 70: 9570–9580. [DOI] [PubMed] [Google Scholar]
- 25. Lodrini M,, Oehme I,, Schroeder C,, et al. MYCN and HDAC2 cooperate to repress miR-183 signaling in neuroblastoma. Nucleic Acids Res. 2013; 41: 6018–6033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang J,, Wang X,, Li Z,, Liu H,, Teng Y. MicroRNA-183 suppresses retinoblastoma cell growth, invasion and migration by targeting LRP6. FEBS J. 2014; 281: 1355–1365. [DOI] [PubMed] [Google Scholar]
- 27. Zhu J,, Feng Y,, Ke Z,, et al. Down-regulation of miR-183 promotes migration and invasion of osteosarcoma by targeting Ezrin. Am J Pathol. 2012; 180: 2440–2451. [DOI] [PubMed] [Google Scholar]
- 28. Mu Y,, Zhang H,, Che L,, Li K. Clinical significance of microRNA-183/Ezrin axis in judging the prognosis of patients with osteosarcoma. Med Oncol. 2014; 31: 821. [DOI] [PubMed] [Google Scholar]
- 29. Zhao H,, Guo M,, Zhao G,, et al. miR-183 inhibits the metastasis of osteosarcoma via downregulation of the expression of Ezrin in F5M2 cells. Int J Mol Med. 2012; 30: 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Li J,, Liang S,, Jin H,, Xu C,, Ma D,, Lu X. Tiam1 negatively regulated by miR-22, miR-183 and miR-31, is involved in migration, invasion and viability of ovarian cancer cells. Oncol Rep. 2012; 27: 1835–1842. [DOI] [PubMed] [Google Scholar]
- 31. Lowery AJ,, Miller N,, Dwyer RM,, Kerin MJ. Dysregulated miR-183 inhibits migration in breast cancer cells. BMC Cancer. 2010; 10: 502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Roche M,, Wierinckx A,, Croze S,, et al. Deregulation of miR-183 and KIAA0101 in aggressive and malignant pituitary tumors. Front Med (Lausanne). 2015; 2: 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang G,, Mao W,, Zheng S. MicroRNA-183 regulates Ezrin expression in lung cancer cells. FEBS Lett. 2008; 582: 3663–3668. [DOI] [PubMed] [Google Scholar]
- 34. Shi XY,, Gu L,, Chen J,, Guo XR,, Shi YL. Downregulation of miR-183 inhibits apoptosis and enhances the invasive potential of endometrial stromal cells in endometriosis. Int J Mol Med. 2014; 33: 59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li J,, Fu H,, Xu C,, et al. miR-183 inhibits TGF-beta1-induced apoptosis by downregulation of PDCD4 expression in human hepatocellular carcinoma cells. BMC Cancer. 2010; 10: 354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Simpson F,, Lammerts van Bueren K,, Butterfield N,, et al. The PCNA-associated factor KIAA0101/p15(PAF) binds the potential tumor suppressor product p33ING1b. Exp Cell Res. 2006; 312: 73–85. [DOI] [PubMed] [Google Scholar]
- 37. Turchi L,, Fareh M,, Aberdam E,, et al. ATF3 and p15PAF are novel gatekeepers of genomic integrity upon UV stress. Cell Death Differ. 2009; 16: 728–737. [DOI] [PubMed] [Google Scholar]
- 38. Chang CN,, Feng MJ,, Chen YL,, Yuan RH,, Jeng YM. p15(PAF) is an Rb/E2F-regulated S-phase protein essential for DNA synthesis and cell cycle progression. PLoS One. 2013; 8: e61196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kato T,, Daigo Y,, Aragaki M,, Ishikawa K,, Sato M,, Kaji M. Overexpression of KIAA0101 predicts poor prognosis in primary lung cancer patients. Lung Cancer. 2012; 75: 110–118. [DOI] [PubMed] [Google Scholar]
- 40. Bahassi el M. Polo-like kinases and DNA damage checkpoint: beyond the traditional mitotic functions. Exp Biol Med. 2011; 236: 648–657. [DOI] [PubMed] [Google Scholar]
- 41. Strebhardt K,, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer. 2006; 6: 321–330. [DOI] [PubMed] [Google Scholar]
- 42. Archambault V,, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol. 2009; 10: 265–275. [DOI] [PubMed] [Google Scholar]
- 43. Petronczki M,, Lenart P,, Peters JM. Polo on the rise-from mitotic entry to cytokinesis with Plk1. Dev Cell. 2008; 14: 646–659. [DOI] [PubMed] [Google Scholar]
- 44. Yuan J,, Horlin A,, Hock B,, Stutte HJ,, Rubsamen-Waigmann H,, Strebhardt K. Polo-like kinase, a novel marker for cellular proliferation. Am J Pathol. 1997; 150: 1165–1172. [PMC free article] [PubMed] [Google Scholar]
- 45. Eckerdt F,, Yuan J,, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene. 2005; 24: 267–276. [DOI] [PubMed] [Google Scholar]
- 46. Murphy M,, Stinnakre MG,, Senamaud-Beaufort C,, et al. Delayed early embryonic lethality following disruption of the murine cyclin A2 gene. Nat Genet. 1997; 15: 83–86. [DOI] [PubMed] [Google Scholar]
- 47. Gong D,, Ferrell JE,, Jr. The roles of cyclin A2 B1, and B2 in early and late mitotic events. Mol Biol Cell. 2010; 21: 3149–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nam HJ,, van Deursen JM. Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol. 2014; 16: 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bharadwaj R,, Qi W,, Yu H. Identification of two novel components of the human NDC80 kinetochore complex. J Biol Chem. 2004; 279: 13076–13085. [DOI] [PubMed] [Google Scholar]
- 50. McCleland ML,, Kallio MJ,, Barrett-Wilt GA,, et al. The vertebrate Ndc80 complex contains Spc24 and Spc25 homologs, which are required to establish and maintain kinetochore-microtubule attachment. Curr Biol. 2004; 14: 131–137. [DOI] [PubMed] [Google Scholar]
- 51. Shen J. Function and dysfunction of presenilin. Neurodegener Dis. 2014; 13: 61–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liu B,, Wang L,, Shen LL,, et al. RNAi-mediated inhibition of presenilin 2 inhibits glioma cell growth and invasion and is involved in the regulation of Nrg1/ErbB signaling. Neurooncol. 2012; 14: 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yun HM,, Park MH,, Kim DH,, et al. Loss of presenilin 2 is associated with increased iPLA2 activity and lung tumor development. Oncogene. 2014; 33: 5193–5200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.