

Sorting nexin 9 is a multifunctional scaffold protein that coordinates endocytic trafficking, activation of RhoGTPases, and actin nucleation via N-WASP to modulate cancer cell invasion and metastasis.
Abstract
Despite current advances in cancer research, metastasis remains the leading factor in cancer-related deaths. Here we identify sorting nexin 9 (SNX9) as a new regulator of breast cancer metastasis. We detect an increase in SNX9 expression in human breast cancer metastases compared with primary tumors and demonstrate that SNX9 expression in MDA-MB-231 breast cancer cells is necessary to maintain their ability to metastasize in a chick embryo model. Conversely, SNX9 knockdown impairs this process. In vitro studies using several cancer cell lines derived from a variety of human tumors reveal a role for SNX9 in cell invasion and identify mechanisms responsible for this novel function. We show that SNX9 controls the activation of RhoA and Cdc42 GTPases and also regulates cell motility via the modulation of well-known molecules involved in metastasis, namely RhoA-ROCK and N-WASP. In addition, we find that SNX9 is required for RhoGTPase-dependent, clathrin-independent endocytosis, and in this capacity can functionally substitute to the bona fide Rho GAP, GTPase regulator associated with focal adhesion kinase (GRAF1). Taken together, our data establish novel roles for SNX9 as a multifunctional protein scaffold that regulates, and potentially coordinates, several cellular processes that together can enhance cancer cell metastasis.
INTRODUCTION
Breast cancer, the most common cancer in women, accounts for 25% of all cancer cases and is responsible of 15% of cancer-related deaths worldwide: 90% of these are due to metastases (Gupta and Massague, 2006; Torre et al., 2015). Hence the ability of cancer cells to escape the primary tumor, or metastasis, determines, in part, tumor aggressiveness and disease prognosis. Metastatic spread of malignant cells is a multistep program that requires the coordination of a several cellular processes that contribute to loss of adhesiveness, increased invasiveness in surrounding tissues, and entry and exit from blood/lymph vessels. Only cells that develop survival abilities in all of these different environments are able to colonize distant organs and establish tumor metastases (Bacac and Stamenkovic, 2008; Hanahan and Weinberg, 2011).
RhoGTPases are key regulators that translate and coordinate external cues into signals driving cell motility (Vega and Ridley, 2008). The main members of the RhoGTPase family—Rac1, Rho (A/C), and Cdc42—have been extensively studied in the context of cell invasion through their downstream regulation of actin cytoskeleton remodeling. RhoGTPases themselves are regulated by ∼85 GTPase exchange factors (GEFs) and ∼80 GTPase-activating proteins (GAPs) that are responsible for their activation and inactivation, respectively. Active GTPases transmit signals upon direct binding to their various effectors (Van Aelst and D’Souza-Schorey, 1997; Schmidt and Hall, 2002). Perturbed activation of these GTPases, often attributed to alterations in the expression of specific GEFs and/or GAPs, highly influences cell motility in vitro and metastasis in vivo (Sahai and Marshall, 2002; Vega and Ridley, 2008).
In addition to their role in regulating actin dynamics, cell morphology, and motility, RhoGTPases also regulate a plethora of other cellular processes, including both clathrin-dependent and clathrin-independent endocytic pathways (Lamaze et al., 1996). For example, GTPase regulator associated with focal adhesion kinase (GRAF1) is a conventional GAP for both RhoA and Cdc42 (Hildebrand et al., 1996) and a major regulator of clathrin-independent endocytosis (CIE; Lundmark et al., 2008). GRAF1 also encodes an N-terminal Bin–amphiphysin–Rvs (BAR) domain that can sense and/or generate curvature (Lundmark et al., 2008). Several other BAR domain–containing proteins—for example, TOCA1 and TUBA—have been implicated in clathrin-dependent and/or -independent endocytosis and reported to directly regulate RhoGTPases or serve as binding platforms for activated GTPases at the plasma membrane (PM) (de Kreuk and Hordijk, 2012).
Sorting nexin 9 (SNX9) is a ubiquitously expressed BAR-domain protein and functions as a scaffold that, through its SH3 and low-complexity (LC) domains, binds proteins involved in many cellular processes. First identified as a major binding partner of the GTPase dynamin through its SH3 domain, SNX9 also binds AP2 and clathrin through its LC domain and hence functions as a scaffold in clathrin-mediated endocytosis (CME; Lundmark and Carlsson, 2004, 2005, 2009; Soulet et al., 2005). Other studies implicated SNX9 in actin polymerization via neural Wiskott–Aldrich syndrome protein (N-WASP) binding and activation and in platelet-derived growth factor–induced formation of actin-rich structures (Yarar et al., 2007, 2008), which can result in clathrin-independent bulk endocytosis. SNX9 has also been identified as a direct partner for the adaptor protein DOCK1, the Drosophila homologue of the adaptor protein NCK1 (Worby et al., 2002). Finally, SNX9 binds to and potentially regulates matrix proteases and signaling molecules, namely a disintegrin and metalloprotease (ADAM) 9 and 15 at the Golgi (Howard et al., 1999). Given that changes in SNX9 expression have been detected in many cancers, including breast cancer (www.nextbio.com, www.oncomine.org), we were motivated to explore the role of SNX9 in cancer cell behavior using well-established in vitro and in vivo assays for cell invasion and metastasis.
RESULTS
SNX9 regulates CIE of the stemness marker CD44
The GAP-BAR domain protein GRAF1 functions both as a curvature generator/sensor and as a regulator of RhoGTPases. GRAF1 is required for CIE (Lundmark et al., 2008; Doherty et al., 2011), which can be measured by the uptake of CIE-dependent cargo, such as Glycophosphatidylinositol (GPI)-anchored proteins (Doherty and McMahon, 2009) or, more selectively, CD44, a transmembrane receptor for the extracellular matrix component hyaluronic acid (Howes et al., 2010; Chaudhary et al., 2014). CD44 surface expression contributes not only to adhesive and motile properties of cancer cells, but also to transduction of a signaling cascade downstream of substrate binding (Bourguignon, 2008). mCherry-SNX9 dynamically localizes with GPI–green fluorescent protein (GFP) at the PM (Yarar et al., 2007), suggesting that it also might be involved in CIE. To test this directly, we measured the internalization rates of CD44 in the breast cancer cell line MDA-MB-231 treated with small interfering RNA (siRNA) against SNX9 (here referred to as 231-siSNX9) compared with cells treated with siRNA against GRAF1 (231-siGRAF1) used as a positive control. Cells treated with nonspecific siRNA (231-siCTR) were used as negative controls. In parallel, we measured rates of CIE in MDA-MB-231 stably expressing low amounts of GFP-tagged SNX9, generated through transfection followed by antibiotic selection (here referred to as 231-oxSNX9 cells), compared with untreated control cells (231-CTR; see Materials and Methods; Supplemental Figure S1A).
SNX9 depletion inhibited CD44 internalization to the same extent as the siRNA-mediated knockdown of GRAF1 in MDA-MB-231 cells (Figure 1A). As expected, inhibition of CIE correlated with an increase in surface expression of CD44 (Supplemental Figure S1B). Moreover, in cells overexpressing SNX9, knockdown of GRAF1 had little effect (Figure 1B), indicating that SNX9 overexpression can compensate for GRAF1 function in CIE. These data confirm that SNX9 is indeed involved in CIE (Yarar et al., 2007) and further define the endocytic pathway involved. Of interest, SNX9 knockdown under these conditions only mildly inhibits CME relative to the dynamin-2–knockdown positive control (Figure 1C), whereas SNX9 overexpression has no effect (Figure 1D). From this, we propose that SNX9 has overlapping functions with GRAF1 in CIE.
FIGURE 1:
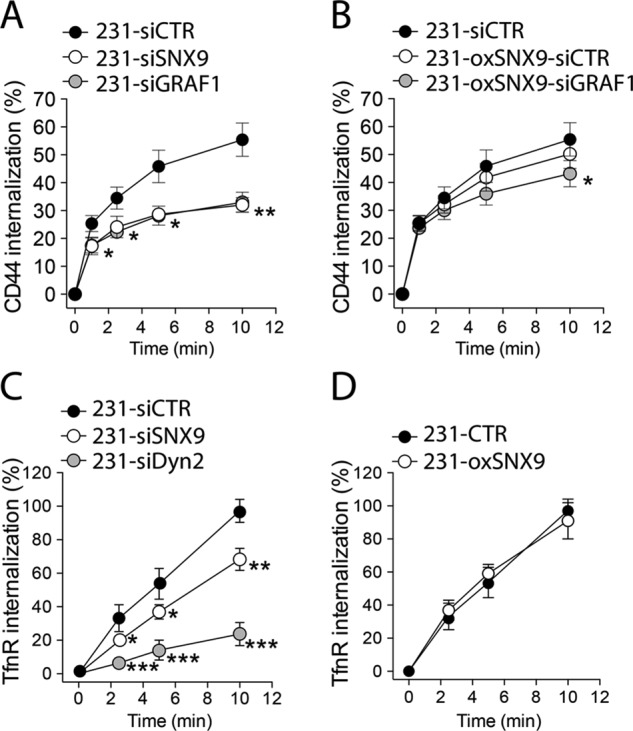
SNX9 regulates clathrin independent endocytosis of CD44. (A, B) Kinetics of clathrin-independent endocytosis of CD44 measured by internalization of an anti-CD44 antibody (see Materials and Methods) in siRNA-treated parental MDA-MB-231 (A) or siRNA-treated 231-oxSNX9 (B). GRAF1 depletion was used as a positive control for CIE perturbation. n = 3, *p < 0.05, ***p < 0.001. (C, D) Kinetics of clathrin-mediated endocytosis of the TfnR measured by internalization of an anti-TfnR antibody (see Materials and Methods) in 231-siSNX9 (C) or 231-oxSNX9 (D) vs. 231-si dynamin 2 (siDyn2) or controls. Dynamin 2 depletion was used as a positive control for CME perturbation. n = 6 and 3, respectively. *p < 0.05, **p < 0.001, ***p < 0.0001.
SNX9 expression regulates Cdc42 and RhoA activation
It is well established that CIE requires actin network remodeling mediated by RhoGTPases. Indeed, GRAF1 is a conventional GAP for both RhoA and Cdc42 in vitro (Hildebrand et al., 1996), although it has a greater affinity for RhoA than for Cdc42 in cells (Taylor et al., 1999; Doherty et al., 2011). We thus hypothesized that modulation of SNX9 expression could alter the activation of RhoA, Cdc42, and possibly other Rho family members.
Only GTP-bound, that is, active RhoA/B/C, Cdc42, and Rac1 can bind and activate their effectors, Rhotekin, WASP, and PAK1, respectively. Using beads coupled to the G protein–binding domains of Rhotekin, WASP, or PAK, we measured active forms of RhoA/B/C, Cdc42, and Rac1, respectively, in MDA-MB-231 cells either depleted of or overexpressing SNX9 (Figure 2, A and B, and Supplemental Figure S2, A and B). Active RhoA was significantly increased upon SNX9 knockdown. Conversely, RhoA-GTP was decreased in 231-oxSNX9. These effects of SNX9 expression were specific to RhoA, as the activation level of neither Rac1 nor RhoC, which shares 85% protein sequence identity with RhoA (Wheeler and Ridley, 2004), was affected. We were unable to detect RhoB due to its low expression in MDA-MB-231 cells. Consistent with RhoA activation, we detected increased phosphorylation of both myosin light chain 2 (MLC2) and cofilin (Figure 2C and Supplemental Figure S2C), known to be phosphorylated downstream of Rho-associated protein kinase (ROCK), a direct effector for RhoA.
FIGURE 2:

Differential expression of SNX9 affects the activation of RhoGTPases. (A, B) Active forms of the indicated Rho-family GTPases were pulled down using beads coupled to respective effector domains that can only bind the GTP-bound (i.e., active) form of the GTPases (see Supplemental Figure S2, A and B, and Materials and Methods). Bar chart shows quantification of active forms of RhoA, RhoC, Cdc42, and Rac1 in 231-siSNX9 (A) or 231-oxSNX9 (B) after normalization to respective control cells. n = 3–6; *p = 0.02. (C) Myosin light chain (MLC2) and cofilin are phosphorylated downstream of RhoA-ROCK activation. The bar chart compares the phosphorylation of MLC2 and cofilin in control and SNX9-depleted cells. n = 3; *p = 0.05. (D) Representative Western blot of His-SNX9 interaction with GST-Cdc42 or GST-RhoA in vitro. Before transferring to nitrocellulose membranes, protein loading was measured on Stain-Free gels (see Materials and Methods) to ensure that comparable amounts of GST- vs. GST-Cdc42 or GST-RhoA beads were used in each condition. Blot is representative of three independent experiments. (E, F) Pi production after GTP hydrolysis by RhoA (E) or Cdc42 (F) either alone or incubated with SNX9 and/or p50GAP. p50GAP alone was used as a positive control for Pi production by the GTPases. n = 4; ****p < 0.0001.
We also noted small but reciprocal changes in Cdc42 activation with SNX9 underexpression and overexpression (Figure 2, A and B, and Supplemental Figure S2, A and B). On the basis of these results, we hypothesized that SNX9 might directly interact with RhoA and at least a subpopulation of Cdc42. To test for interactions between SNX9 and these Rho-family GTPases, we used glutathione S-transferase (GST)–RhoA, GST-Cdc42, or GST immobilized on agarose beads as bait to pull down histidine (His)-SNX9 and confirmed the direct interaction between SNX9 and both RhoA and Cdc42 (Figure 2D). Of note, Rac1 did not bind SNX9 under similar conditions (Supplemental Figure S2D). We also confirmed interactions between endogenous SNX9 and GFP-tagged Cdc42 and RhoA in cell lysates (Supplemental Figure S2, E and F) but failed to detect any interaction between GFP-SNX9 and RhoC or Rac1 (Supplemental Figure S2G). Together these data suggest that SNX9 specifically interacts with and regulates Cdc42 and RhoA both in vivo and in vitro.
RhoGTPases have basal rates of GTP hydrolysis and GDP/GTP exchange, which are enhanced by the presence of GAPs and GEFs, respectively. Some GAPs and GEFs, such as GRAF1 and TUBA, also possess a BAR domain (Salazar et al., 2003; Lundmark et al., 2008). BAR-domain proteins that do not have GAP or GEF domains have been reported as indirect RhoGTPase regulators, including CIP4, PACSIN2, and TOCA-1 (for review see de Kreuk and Hordijk, 2012). On the basis of this and the direct interaction of SNX9 with RhoA and Cdc42, we hypothesized that SNX9 might regulate their GTPase activities. We first asked whether SNX9 could act as a GEF or modulate the GEF activity of the generic RhoGEF domain of DBL’s big sister toward RhoA or Cdc42; however, we were unable to detect any effect of SNX9 using in vitro GTP exchange assays. We next tested whether SNX9 could act as a GAP or modulate a GAP activity toward RhoA or Cdc42, using a colorimetric assay that measures the release of inorganic phosphate (Pi) after GTP hydrolysis by RhoA or Cdc42. We used p50GAP as a positive control for both GTPases. SNX9 addition to RhoA alone or to RhoA plus p50GAP did not affect Pi release (Figure 2E), showing that SNX9 is not acting as a direct GAP for RhoA and does not regulate p50GAP. However, when we performed the GAP assay on Cdc42 under the same conditions, we detected a significant and specific decrease in p50GAP-stimulated Cdc42 GTPase activity in the presence of either GST-SNX9 (Figure 2F) or His-tagged SNX9 (Supplemental Figure S2H). Consistent with the increase of Cdc42-GTP measured in 231-oxSNX9 cells (Figure 2B), these data demonstrate that SNX9, by inhibiting a GAP activity, can stabilize Cdc42 in its active state.
SNX9 regulates cancer cell invasiveness
Cell motility can be affected by both alterations in RhoGTPase activity (Van Aelst and D’Souza-Schorey, 1997) and CIE (Doherty and McMahon, 2009). Therefore we assessed the effect of SNX9 knockdown and overexpression on the ability of MDA-MB-231 cells to invade through a three-dimensional collagen I matrix, using an inverted invasion assay in which cells invade upward, attracted by a gradient of serum present in the overlying culture medium (Sanz-Moreno et al., 2008; Smith et al., 2008; Arsic et al., 2012). Nuclei were subsequently stained with Hoechst and their distribution measured in Z-sections (Figure 3A). The invasion efficiency of 231-siSNX9 cells was severely impaired (Figure 3B). This result was confirmed using a second siRNA directed against the 3′ untranslated region (UTR) of SNX9 (Supplemental Figure S3, A and B). We also reproduced the SNX9-dependent impairment of cell invasion in other cancer cell lines, including non–small cell lung cancer-H1299, fibrosarcoma-HT1080, and melanoma-MV3 cells (Supplemental Figure S3, C–F), indicating that the role of SNX9 in cell invasion is not cell line specific. Correspondingly, even low levels of overexpression of GFP-SNX9 (Figure 3, C–E) dramatically increased the ability of stably transformed 231-oxSNX9 cells to invade through collagen. Given the pronounced effects of SNX9 overexpression even at these low levels, we were concerned that the large GFP tag might somehow activate the protein; however, we obtained similar results with low levels of overexpression of HA-SNX9 (Supplemental Figure S3, G and H). Finally, to confirm that these effects were indeed due to SNX9 overexpression, we showed that the invasive activity of 231-oxSNX9 could be restored to control levels upon specific depletion of GFP-SNX9 with siRNA against GFP (Figure 3, D and E). On the basis of these results, we propose that SNX9 is a limiting factor for cancer cell invasion.
FIGURE 3:
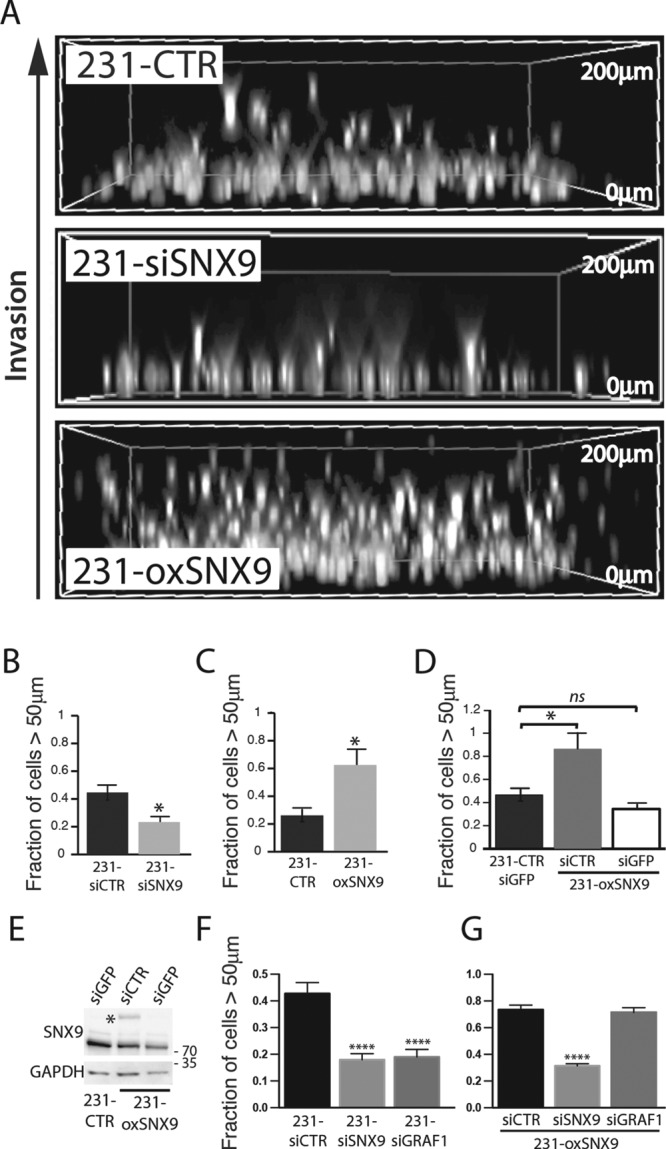
SNX9 regulates the ability of MDA-MB-231 cells to invade through collagen matrix. (A) 231-CTR, -siSNX9, or -oxSNX9 cells were subjected to an inverted three-dimensional cell invasion assay through a bovine collagen I matrix (see Materials and Methods). Representative images of the positions of the nuclei of invading cells detected by Hoechst staining under the indicated conditions. (B, C) Quantification of nuclei distribution in inverted invasion assay of control cells vs. 231-siSNX9 (B) or 231-oxSNX9 (C). n = 10 and 6, respectively; *p = 0.02. (D) Quantification of cell invasion after specific depletion of exogenous SNX9, using an siGFP treatment of 231-oxSNX9. n = 4; *p = 0.04; ns, nonsignificant. (E) Western blot analysis of SNX9 expression in cell lines used in D. GAPDH was used as loading control. Blot is representative of three independent experiments. (F, G) Quantification of cell invasion of siRNA-treated parental MDA-MB-231 (F) or in siSNX9- or siGRAF1-treated 231-oxSNX9 (G). n = 3; ****p < 0.0001.
Several reports have described roles for CIE, GRAF1, and the cargo molecule CD44 in cell motility (Bretscher et al., 1997; Howes et al., 2010). Given the overlapping functions of SNX9 and GRAF1 in CIE, we next tested whether GRAF1 also functions in cell invasion and, if so, whether SNX9 overexpression could compensate for GRAF1 loss. We found both to be true. GRAF1 knockdown decreased cell invasiveness (Figure 3F), and SNX9 overexpression rescued the effect of GRAF1 depletion on cell motility (Figure 3G), demonstrating that GRAF1 and SNX9 have overlapping functions in the regulation of cell motility.
Regulators of RhoGTPases not only stimulate GTP loading or hydrolysis but also participate in the recruitment of the GTPases to specific domains of the PM (Bourguignon, 2008). SNX9 contains BAR and Phox homology (PX) domains, which function as a unit to mediate its binding to phosphatidylinositol lipids on liposomes and at the PM (Yarar et al., 2008; Lundmark and Carlsson, 2009; Figure 4A). The BAR domain is also necessary for SNX9 dimerization (Yarar et al., 2008). Mutations in the PX (PXmut) and BAR domains (BARmut) of SNX9 have been shown to impair binding to liposomes and/or dimerization, respectively (Yarar et al., 2008). Both membrane binding and SNX9 dimerization are required to fully activate N-WASP in vitro (Yarar et al., 2008). To address the importance of membrane binding in SNX9’s new functions, we generated stable cell lines expressing low levels of mCherry-tagged wild-type WT-, BARmut-, or PXmut-SNX9 (Figure 4, A and B). We verified the impaired membrane-binding properties of both SNX9 mutants using subcellular fractionation experiments (Figure 4, C and D) and then tested the function of these SNX9 mutants using the inverted invasion assay. Similarly to WT-SNX9, the PXmut significantly enhanced cell invasion compared with control (Figure 4E). This result indicates that membrane binding is not necessary for SNX9 function in cell invasion, although others have shown that the same PX mutant is unable to support SNX9 function in endocytosis (Posor et al., 2013). In contrast, SNX9 BARmut failed to increase cell invasion, revealing that dimerization, rather than membrane binding, is important for this function (Figure 4E).
FIGURE 4:

Membrane binding is not important for SNX9 function in cell invasion. (A) Schematic representation of SNX9 protein domains, indicating the mutations of BARmut and PXmut (from Yarar et al., 2008). (B) Western blot analysis of SNX9 protein expression in cell lines stably expressing WT- or mutant-mCherry-SNX9. Asterisk indicates mCherry-SNX9 band. GAPDH was used as loading control. Blot is representative of three independent experiments. (C) Fractionation experiment using the cell lines in B, showing WT- or mutant-mCherry-SNX9 in cytoplasm (S, supernatant) vs. membranes (P, pellet). The absence of actin was used as purity readout of pellet fractions. Blot is representative of three independent experiments. (D) Quantification of SNX9 distribution in conditions used in C. (E) Quantification of cell invasion abilities of the stable cell lines in B. n = 3; *p = 0.025, ***p = 0.0001; ns, nonsignificant.
Taken together, these results suggest that the role of SNX9 in cell invasion likely reflects its role as an inhibitor of RhoA, which appears to be independent of its ability to bind membranes and hence its role in CIE.
SNX9 expression levels control cancer cell metastasis
Metastasis is triggered when cancer cells first develop the ability to invade the surrounding tissues. The activation of RhoGTPases, regulated by altered expression of GAPs and GEFs, has been associated with enhanced metastatic potential of cancer cells (Sahai and Marshall, 2002). Given that our in vitro findings establish a role for SNX9 in regulating the activity of RhoGTPases, we monitored the effects of SNX9 expression levels on the metastatic activity of MDA-MB-231 cells. The chick embryo model has been widely used to study the metastatic spread of human cancer cells after they are directly injected intravenously into the chorioallantoic membrane. In addition to higher throughput, the chick embryo model has several advantages over mouse models (Lokman et al., 2012), and, of importance, results obtained using this system have been shown to replicate those derived from mouse models (Azoitei et al., 2014; Pommier et al., 2015; Virtakoivu et al., 2015). To facilitate the detection of metastases, we infected 231-CTR or 231-oxSNX9 cells with retroviruses driving the stable expression of zsGreen. These cells, treated or not with siRNA against SNX9, were injected intravenously into chick embryos. The formation of metastases throughout the embryo was quantified 4 d after the injection (see Materials and Methods). In accordance with our in vitro measurement of cell invasion, SNX9 depletion dramatically decreased, whereas SNX9 overexpression significantly increased, the number of detected metastases (Figure 5, A and B).
FIGURE 5:

SNX9 protein expression enhances metastatic activity and is increased in human breast cancer metastases. (A) Representative image of metastases detected after injection of zsGreen-231-siSNX9 or zsGreen-231-oxSNX9, compared with their respective controls, into the chorioallantoic membrane of chicken embryos. Stromal cells were visualized with Lectin-Dylight-649 and metastases detected throughout the embryo by zsGreen fluorescence. Bar, 20 μm. (B) Quantification of metastatic efficiency of cells and conditions described in A. n = 14–16 for each condition. Results represent three independent experiments. ****p < 0.0001, **p = 0.0012. (C) Representative image of immunohistochemical staining of SNX9 in primary human breast tumors vs. their corresponding metastases. (D) Quantification of SNX9 expression levels corresponding to C. n = 7 for matched samples. Bar, 100 μm. **p = 0.0097.
Having shown that SNX9 is a regulator of metastasis, we investigated whether the expression level of SNX9 changes in primary or secondary sites of breast cancer by performing an immunohistochemical survey of SNX9 expression in a human tissue array containing seven matched breast tumor samples and their corresponding lymph node metastases. A representative staining is shown in Figure 5C for a set of matched samples, and quantification of all data confirms higher expression levels of SNX9 in metastases versus primary tumors (Figure 5D). Taken together, these findings provide strong in vivo support for our in vitro findings that SNX9 expression levels regulate cellular processes (CIE, invasion, RhoGTPase activation) to enhance cancer cell metastasis.
SNX9 controls cell invasion through the RhoA-ROCK pathway and N-WASP
To better understand the mechanism behind SNX9-driven metastasis, we directly manipulated signaling pathways that might be downstream of SNX9. Given that cell invasiveness in vitro has been highly predictive of in vivo metastasis (Figures 3, A–C, and 5A), we assessed the effect of these signaling pathways using the cell invasion assay. The GTPase RhoA triggers downstream activation of the kinase ROCK, leading to rearrangements of the actomyosin network. Inhibition of the RhoA-ROCK pathway enhances cell motility in some cancer cell lines (Vial et al., 2003; Simpson et al., 2004; Arsic et al., 2012). Therefore we hypothesized that the increase in RhoA-GTP after SNX9 knockdown might be responsible for impeding cell invasion. For the invasion assay, we directly added the ROCK inhibitor Y27632 into the collagen matrix. The invasion ability of 231-siCTR cells was not affected under these conditions (Figure 6A), although, a modest increase in cell invasion was observed when cells were pretreated with Y27632 before plating (unpublished data). In contrast, ROCK inhibition increased the invasiveness of 231-siSNX9 cells, suggesting that activation of the RhoA-ROCK pathway in 231-siSNX9 cells is detrimental and partially responsible for their low capacity to invade through collagen (Figure 6A).
FIGURE 6:
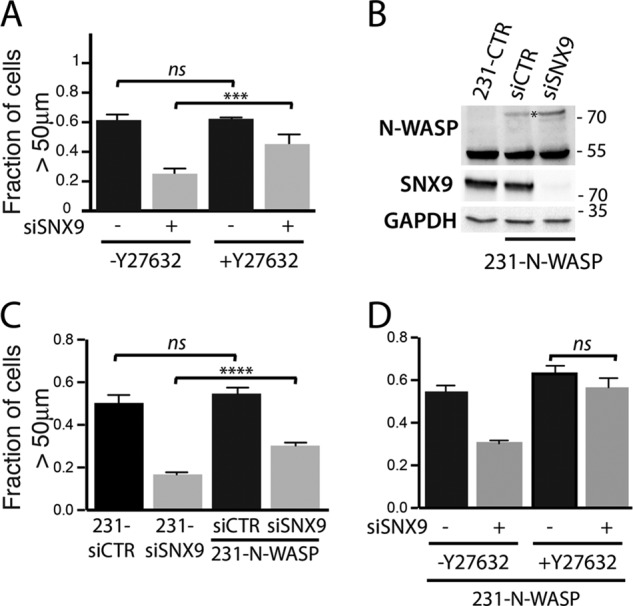
SNX9 controls cell invasion through the RhoA-ROCK pathway and N-WASP. (A) Bar chart representing quantification of cell invasiveness of 231-siCTR and 231-siSNX9 either mock treated or treated with ROCK inhibitor, Y27632. n = 4; ***p = 0.002. (B) Western blot analysis of N-WASP protein expression in cell lines used in C. Asterisks indicate exogenous N-WASP. GAPDH was used as loading control. Blot is representative of three independent experiments. (C) Quantification of cell invasion after SNX9 knockdown in MDA-MB-231 parental cells or N-WASP–overexpressing cells. n = 4, ****p < 0.0001. ns, nonsignificant. (D) Similar experiment as in C, except using cells that were either mock treated or treated with ROCK inhibitor.
N-WASP is an actin nucleator that is activated downstream of Cdc42 (Van Aelst and D’Souza-Schorey, 1997) but is also directly activated by SNX9 (Yarar et al., 2008). N-WASP depletion in cancer cells impairs their ability to invade in vitro and in vivo (Gligorijevic et al., 2012; Yu et al., 2012). Correspondingly, the effects of SNX9 depletion on cell invasion phenocopies N-WASP knockdown in MDA-MB-231 cells (Yu et al., 2012). We reasoned that if the effects of SNX9 depletion were due to decreased N-WASP activation, then N-WASP overexpression should rescue these phenotypes. To test this, we generated MDA-MB-231 cell lines stably expressing N-WASP-mCherry (231-N-WASP; Figure 6B). Consistent with this hypothesis, we found that even low levels of overexpressed N-WASP partially but significantly attenuated the inhibitory effect of SNX9 depletion on the ability of 231-N-WASP cells to invade (Figure 6C). We thus propose that the effects of SNX9 on cell invasion are, at least in part, dependent on its ability to directly and/or indirectly, via Cdc42, activate N-WASP. Finally, we hypothesized that a combination of ROCK inhibition and N-WASP overexpression might fully rescue this phenotype. Indeed, cell invasion of SNX9-depleted 231-N-WASP cells was restored to control levels after ROCK inhibition (Figure 5D). On the basis of these results, we conclude that SNX9 regulates cell invasion in vitro, and eventually metastasis, through its dual and parallel effects on two pathways known to separately affect metastasis, namely the RhoA-ROCK and N-WASP pathways.
DISCUSSION
We report new roles for the multifunctional scaffolding protein SNX9 as a regulator of RhoGTPases and cell invasion. SNX9 expression levels can affect cancer cell invasiveness and their metastatic potential both in vitro and in vivo. Increased expression of SNX9 has a prometastatic effect on breast cancer cells measured using a chick embryo model. Moreover, breast cancer metastases express higher levels of SNX9 than do primary tumors from the same patient, consistent with the proinvasive role of SNX9. At the molecular level, our in vitro data establish that SNX9 can simultaneously integrate endocytic membrane trafficking and RhoGTPase regulation to control cancer cell behavior (Figure 7). Our results reveal novel functions for SNX9 as a regulator of the metastatic potential of cancer cells.
FIGURE 7:

SNX9 is a multifunctional scaffold regulating CIE, cell invasion, and metastasis. Model placing the findings described in this article (solid arrows) in the context of existing knowledge (dashed arrows). (A) SNX9 overexpression decreases active RhoA and increases active Cdc42, consequently enhancing in vitro cancer cell invasion and leading to increased metastasis. (B) SNX9 depletion impairs CD44 internalization and Cdc42 and N-WASP activation and enhances RhoA activation. In this condition, in vitro cell invasion and metastasis are also decreased. The question marks indicate SNX9 functions whose exact mechanisms remain unknown. Our results summarized here are in accordance with the higher expression of SNX9 in human metastases than in matched primary breast tumors.
It is well established that RhoGTPases are involved in tumorigenesis and cancer cell metastasis (Van Aelst and D’Souza-Schorey, 1997; Sahai and Marshall, 2002; Vega and Ridley, 2008). RhoGTPases play a central role in regulating not only actin cytoskeleton dynamics and subsequent cell motility but also in oncogenic transformation and cancer survival (Sahai and Marshall, 2002; Vega and Ridley, 2008). Their activation could be achieved either directly by overexpression or indirectly via changes in the expression levels of their respective GEFs and GAPs (Sahai and Marshall, 2002; Vega and Ridley, 2008). We present both in vitro and in vivo evidence that SNX9 functions as a scaffold that directly interacts with both RhoA and Cdc42 and their regulators (e.g., p50GAP) to alter their activation state in vivo. High expression of RhoA in breast primary tumors has been positively associated with tumor grade, proliferative activity, and size (Bellizzi et al., 2008). Therefore we speculate that the lower expression of SNX9 in primary tumors can contribute to increased RhoA activation and lead to aggressive features of the malignancy. Conversely, it has been reported that depletion of the RhoA GEF obscurin, which leads to decreased RhoA activation in normal breast cells, increases their ability to invade and their survival when in suspension (Perry et al., 2014). Thus the decrease in RhoA activation downstream of SNX9 overexpression might also influence cell survival in a hostile environment.
We previously reported that SNX9 activates the actin nucleator N-WASP (Yarar et al., 2007). Using a combination of RhoA-ROCK inhibition and N-WASP overexpression, we restored the invasion abilities of SNX9-deficient cells to control levels, suggesting that the mechanism of action of SNX9 on the regulation of cell invasion is through RhoA-ROCK inhibition and N-WASP activation directly and/or indirectly through Cdc42. Of interest, it has been shown that N-WASP loss in cancer cells impairs their ability to invade in vitro and in vivo (Gligorijevic et al., 2012; Yu et al., 2012), supporting our finding that SNX9 and N-WASP cooperate in regulating cancer cell invasiveness.
We also report that SNX9 functions in the same clathrin-independent pathway as the N-BAR and RhoGAP domain–containing protein GRAF1. Of interest, at higher levels of expression, SNX9 can substitute for GRAF1 depletion. However, despite this apparent functional overlap, the PX-BAR domain of SNX9 has very different curvature-generating properties than the N-BAR domain of GRAF1 (Qualmann et al., 2011). Therefore we assume that the ability of SNX9 to compensate for loss of GRAF1 reflects its ability to activate RhoA rather than a direct role in deforming the membrane. Similarly, because a PX-domain mutant of SNX9 defective in membrane binding (Yarar et al., 2008) and endocytosis (Posor et al., 2013) retains its ability to enhance cell invasion, this function is also likely related to regulation of RhoA. Consistent with this, we show that the effects of SNX9 knockdown on cell invasion are rescued by N-WASP overexpression and ROCK inhibition, which together point to signaling pathways downstream of RhoA and Cdc42. Thus we believe that the overlapping functions of SNX9 and GRAF1 as regulators of Rho-family GTPases account for the ability of SNX9 overexpression to compensate for the loss of GRAF1 in both CIE and invasion.
A role for SNX9 in cancer cell biology through the regulation of or by cancer-related molecules is suggested by other observations. For example, SNX9 is degraded downstream of EGFR internalization (Childress et al., 2006), and, conversely, contributes to epidermal growth factor receptor (EGFR) degradation after SNX9 is phosphorylated by Cdc42-activated kinase 2 (Lin et al., 2002). SNX9 also binds to the matrix proteases and signaling molecules ADAM9 and 15 and contributes to their trafficking and/or maturation in the Golgi (Howard et al., 1999). Differences in SNX9 expression levels or changes in its subcellular localization likely contribute to alterations of signaling involving EGFR and ADAM proteins. Of interest, RhoGTPases, including Cdc42, are also sensors for and are activated in response to EGF stimulation (for review, see Sahai and Marshall, 2002). These observations, together with our data, suggest a feedback loop between Cdc42 activation and SNX9 function.
Taken together, our data establish that SNX9 is a multifunctional scaffold that integrates key cellular processes (RhoGTPase activity, endocytic trafficking, and actin remodeling downstream of ROCK and N-WASP), all of which have established roles in cancer cell invasion and metastasis. We also identified the altered molecular pathways downstream of SNX9 that leads to differences in cell invasion (Figure 7). Further studies on SNX9 function and stage-specific expression levels in human tumors could potentially reveal additional correlations between differences in SNX9 expression levels and disease progression.
MATERIALS AND METHODS
Cell lines and transfections
MDA-MB-231 and MV3 cells were a gift from R. Brekken and from S. Morrison, respectively (UT Southwestern Medical Center, Dallas, TX). HT1080 and SCC61 cells were a gift from S. Courtneidge (Sanford Burnham, San Diego, CA). H1299 cells were a gift from J. Minna (Hamon Center for Therapeutic Oncology Research, Dallas, TX). MDA-MB-231, HT1080, and MV3 cells were cultivated in DMEM (Invitrogen, Carlsbad, CA)/10% fetal bovine serum (FBS; Sigma-Aldrich, St. Louis, MO). H1299 cells were cultivated in RPMI/5% FBS. Cells were tested negative for mycoplasma contaminations. Lipofectamine RNAiMAX and Lipofectamine 2000 (Life Technologies, Carlsbad, CA) were used for siRNA and plasmid delivery into cells, respectively. Stable cell lines were generated by transfection of appropriate plasmids, followed by antibiotic selection using 1 mg/ml G418 (Life Technologies). We generated and used three independent nonclonal cell lines stably expressing GFP-SNX9. Throughout the article, MDA-MB-231 parental cells are referred as 231-CTR, whereas MDA-MB-231 cells stably expressing GFP-, mCherry-, or HA-tagged WT-SNX9 are referred to as 231-oxSNX9 cells.
siRNA and plasmids
We used AllStars negative control siRNA in all knockdown experiments (Qiagen, Valencia, CA). SNX9 siRNA sense, UAAGCACUUUGACUGGUUAUU (Yarar et al., 2007); and SNX9-3′UTR siRNA sense, GGGACUUUGUAGAGAAUUU. GRAF1 siRNA sense, UUUGAAACUGGUACAUCAUGAGUGG (Lundmark et al., 2008). GFP siRNA sense, GCAAGCTGACCCTGAAGTTC. Dynamin 2 siRNAs (mix of 1:1) sense: Dyn2_1, CCGAAUCAAUCGCAUCUUCUU; and Dyn2_2, GACAUGAUCCUGCAGUUCAUU.
SNX9 and N-WASP constructs were previously described (Yarar et al., 2007). Cdc42-WT, Rhotekin-RBD, and PAK1-PBD bacterial expression vectors were obtained from Addgene (Cambridge, MA; deposited by G. Bokoch, M. Schwartz, and J. Chernoff, respectively) and WASP-GBD plasmid was a gift from M. Rosen (UT Southwestern Medical Center). All RhoA and Cdc42 mammalian expression vectors were a gift from B. Lemmers (Institut de Génétique Moléculaire de Montpellier, Montpellier, France).
Antibodies and reagents
We used the following antibodies: anti-SNX9 polyclonal serum (Yarar et al., 2007, 2008) for Western blotting and anti-SNX9 (HPA031410; Sigma-Aldrich) for immunohistochemistry; anti–N-WASP (ab126626; Abcam, Cambridge, MA), anti-TfnR (hybridoma clone HTR-D65; Schmid and Smythe, 1991), anti-His (27471001; GE Healthcare, Pittsburgh, PA), anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH; G9545) and anti-actin (A1978; both from Sigma-Aldrich), anti-RhoC (D40E4), anti-cofilin (D3F9), anti-MLC2 (D18E2), and anti–p-MLC2 (p-Thr18/Ser19; 3674; all from Cell Signaling, Danvers, MA), anti-RhoA (sc-418), anti-Cdc42 (B-8), and anti-p-Cofilin (hSer3 sc-12912-R; all purchased from Santa Cruz Biotechnology, Dallas, TX); anti-Rac1 (clone-102) and anti-CD44 (G44-26; both from BD Biosciences, San Jose, CA), and horseradish peroxidase (HRP)–conjugated antibodies (Invitrogen). Y27632, paraformaldehyde (PFA), and o-phenylenediamine dihydrochloride (OPD; P1536) were purchased from Selleckchem (Houston, TX), Electron Microscopy Sciences (Hatfield, PA), and Sigma-Aldrich, respectively. Recombinant GST-RhoA-wt was a gift from B. Lemmers.
Cell imaging
Fixed cells were imaged using a microscope with 60× or 100×/1.49 numerical aperture objective (Nikon, Melville, NY) mounted on a Ti-Eclipse inverted microscope. Images were collected using a charge-coupled device camera (CoolSNAP HD2; Roper Scientific, Tucson, AZ) driven by MicroManager software.
Invasion through collagen was imaged using a 20× Plan-Fluor objective on a Nikon Ti microscope with perfect focus, motorized stage for parallel data acquisition, Zyla sCMOS camera, SOLA solid-state white-light excitation system, and 4′,6-diamidino-2-phenylindole excitation and emission filters for invasion assay. The microscope was operated by Nikon Elements Software.
Inverted invasion assay
Experiments were performed according to Arsic et al. (2012). Invasion assays were performed in 96-well dishes (PerkinElmer, Waltham, MA). In brief, cells were suspended in 2.3 mg/ml serum-free liquid bovine collagen I (Advanced Biomatrix, San Diego, CA) at 5 × 104 cells/ml, and 100-μl aliquots were dispensed into the plates. Plates were centrifuged at 1000 rpm and incubated in a 37°C/5% CO2 tissue-culture incubator. After collagen polymerization, 30 μl of medium containing 5% fetal calf serum was added on top of the collagen plug. After 36 h, cells were fixed with 4% formaldehyde (final concentration) and stained with 2 μg/ml Hoechst-33342 (Invitrogen). For ROCK inhibition, Y27632 was directly added to the collagen and to the medium at a final concentration of 10 μM. For quantification, 25 adjacent images were acquired in each well, yielding a total of ∼5 × 103 cells imaged per well. Nuclei labeled with Hoechst from 0 μm (bottom of the plate) to 150 μm into the collagen plug, with a 50-μm step, were detected with the object counts feature of Nikon Elements (Figure 3) or with custom Matlab software (Natick, MA; Figure 3 and Supplemental Figure S3). Invasion ratio was calculated as the sum of cell counts at 50, 100, and 150 μm over cell counts at 0 μm. Results were obtained from at least three independent experiments including five replicates on each day. Bar charts are plotted as mean of all experiments ± SEM.
Pull-down and recombinant protein interactions
Pull-down assays were performed using GFP-trap beads (Chromotek, Planegg-Martinsried, Germany) according to manufacturer’s instructions, as reported by Arsic et al. (2012). After washes, beads were resuspended in Laemmli buffer for Western blot analysis. Input fraction represents 1/20 of total lysate. In vitro binding assays were performed with bacterially produced GST-fused (RhoA, Cdc42, SNX9) or His-fused (SNX9 or Rac1) constructs. RhoA, Cdc42, and SNX9 proteins were purified on glutathione-conjugated agarose beads (Agarose Beads Technology, Torrejon de Ardoz, Spain) according to standard methods (Yarar et al., 2008). RhoA or Cdc42 Beads were incubated in NP40 buffer (10 mM Tris/HCl. pH 7.5, 150 mM NaCl, 0.5 mM EDTA, 0.05% NP-40) with His-SNX9 for 2 h at 4°C, extensively washed with the same buffer, and resuspended in Laemmli buffer. Input fraction represents 1/40 of total recombinant SNX9. Acrylamide gels were loaded on a Stain-Free precast polyacrylamide gel (Bio-Rad, Hercules, CA). Before the transfer on nitrocellulose membrane, total protein was visualized and quantified using ultraviolet exposure on a G-Box (Syngene, Frederick, MD) and used as a loading control. We followed the same experimental procedure using GST-SNX9 beads with His-Rac1.
Pull down of active RhoGTPases
GST-Rhotekin-RBD–, GST-WASP-CBD–, and GST-PAK1-PBD–agarose beads were prepared according to Wittchen and Burridge (2008) and used as in Borm et al. (2005). For active RhoA/B/C experiments, cells were lysed in the buffer 50 mM Tris, pH 7.5, 1% Triton X-100, 0.5% Na deoxycholate, 500 mM NaCl2, and 1 mM dithiothreitol. For Cdc42 and Rac1, cells were homogenized in 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.5, 1% NP-40, 5% glycerol, 100 mM NaCl2, and 10 mM MgCl2. Cell lysates were incubated with 30 μg of appropriate beads for 1 h at 4°C. After SDS–PAGE and Western blotting using anti-RhoA or anti-RhoC antibodies, we used ImageJ software (National Institutes of Health, Bethesda, MD) to measure band intensities. The activation index was calculated as pull down/input/GAPDH intensity ratio.
In vitro GAP assay
Experiments were performed using a commercial kit for RhoGAP (BK105; Cytoskeleton, Denver, CO), following the manufacturer’s instructions. The enzymatic reactions of the conditions described in Results were carried out at 37°C for 60 min using bacterially expressed GST-SNX9 at equimolar ratio with p50GAP. Absorbance at 650 nm, proportional to Pi release, was measured using a Biotek Synergy (Vinooski, VT) H1 Hybrid Reader.
Cell fractionation assay
We used a fractionation buffer (0.1 M 2-(N-morpholino)ethanesulfonic acid, pH 6.5, 0.2 mM ethylene glycol tetraacetic acid, 0.5 mM MgCl2, 0.02% NaN3) to separate membranes from the cytoplasm as in Borner et al. (2012). Cells from different conditions were detached using 5 mM EDTA/phosphate-buffered saline (PBS), followed by cell counting. The same amounts (106) of cells were resuspended in 200 μl of cold fractionation buffer and homogenized with a 27-gauge, 0.5-in. needle. Cell lysates were cleared from cell nuclei by centrifugation at 3000 RPM for 10 min. After transfer to a fresh tube, the cell lysates were further spun at 100,000 × g for 45 min. The supernatants (cytosolic fraction) were then separated from pellets containing the membranes by transfer to fresh tubes. The pellets (membranes) were washed using the fractionation buffer. Laemmli buffer was then added, and 1/10 of each fraction was loaded into acrylamide gel, followed by Western blotting against SNX9. We used actin as a marker for the cytosolic fraction.
TfnR internalization
Quantitative endocytic in-cell enzyme-linked immunosorbent assays were used to measure TfnR uptake (Elkin et al., 2015). Briefly, 96-well plates were coated with gelatin as in Diaz et al. (2013), and 3 × 104 cells/well were allowed to adhere for 3 h at 37°C and chilled at 4°C, followed by incubation with 5 μg/ml TfnR antibody and diluted in PBS4+ (PBS supplemented with 1 mM MgCl2, 1 mM CaCl2, 5 mM glucose, and 0.2% bovine serum albumin) for 1 h at 4°C to label surface-associated receptors. Cells were then moved to 37°C for the indicated times to allow internalization of TfnR–antibody complex. To measure total surface binding, some wells (surface TfnR) were incubated at 4°C and washed (5× PBS4+) to remove unbound antibody. All wells (except surface TfnR) were washed with an acidic solution (5 × 2 min 0.2 M acetic acid, 0.2 M NaCl, pH 2.5) to remove remaining antibodies from the surface. Cells were washed three times with PBS and then fixed in 4% PFA and further permeabilized with 0.1% Triton X-100. Internalized TfnR monoclonal antibody (mAb) was assessed using a secondary HRP-conjugated antibody and further developed with OPD, and the reaction was stopped by using 5 M H2SO4. The absorbance was read at 490 nm using a BioTek Synergy H1 Hybrid Reader. Internalized ligand was expressed as the percentage of the total surface-bound ligand at 4°C (i.e., cells incubated with TfnR mAb in cold without an acid wash step), measured in parallel.
CD44 internalization
The same procedure as just given was used to measure CD44 internalization, with some modifications: cells were incubated with 0.2 μg/ml anti-CD44 mAb at 37°C for the indicated times without prechilling. Cells were then immediately cooled (4°C) to arrest internalization. In parallel, we also labeled cell surface CD44 by incubating prechilled cells in presence of anti-CD44 antibody at 4°C. Subsequent steps were performed as in the preceding subsection, and the results are presented as percentage internalized/total surface bound.
Experimental metastases formation in avian model
We used a well-characterized chick embryo model for human cancer cell metastasis (Deryugina and Quigley, 2008; Lokman et al., 2012; Azoitei et al., 2014; Mudduluru et al., 2015; Pommier et al., 2015), following standard procedures. Briefly, to detect metastases, all cells were stably infected with retrovirus encoding zsGreen. We used MDA-MB-231 control cells (231-CTR), 231-oxSNX9 cells, and 231-siCTR and 231-siSNX9 cells, which had been transfected with either control or siRNA against SNX9, respectively, 24 and 48 h before the experiment. To assay metastasis, cells were trypsinized and washed three times with PBS, counted, and resuspended at 106 cells/ml. A 100-μl amount of cells was intravenously injected into the chorioallantoic membrane of day 14 chicken embryos, using a disposable micropipette syringe as described previously (Leong et al., 2012). Four days postinjection, Dylight-649 lectin and Hoescht were injected into the vein of the chicken embryo to label the vasculature, and zsGreen-positive metastases detected throughout the embryo chorioallantoic membrane were imaged and scored using a Nikon upright confocal microscope. From 12 to 16 chicken embryos were injected for each construct in three independent experiments.
Immunohistochemistry
Paraffin-embedded tissue microarrays of breast cancer primary tumors, metastases, and normal tissues were obtained from SuperBioChips. The tumors were classified as infiltrating duct carcinomas according to the seventh TNM Classification of Malignant Tumors: four were T2N3aM0, and one each was classified as T3N3aM0, T2N1aM0, or T2N2aM0. Among them, five were classified as stage IIIC, one as stage IIIA, and one as stage IIB. For the labeling, anti-SNX9 antibody was used after antigen retrieval in citrate buffer and according to Human Protein Atlas procedures (www.proteinatlas.org/ENSG00000130340-SNX9/antibody). Slides were scanned using virtual microscopy (Nanozoomer; Hamamatsu Photonics, Bridgewater, NJ), and H-score was evaluated in the paired primary tumors versus their corresponding metastases.
Statistical analysis
All experiments were repeated at least three times. Data are reported as arithmetic means ± SEM. Statistical analyses were performed using either nonparametric Mann–Whitney or column statistics (Figure 2, normalized values) tests, using GraphPad (La Jolla, CA) software. Statistical significance was defined as p ≤ 0.05.
Supplementary Material
Acknowledgments
We are grateful to G. Gadea and K. Reed for reading and commenting on the manuscript. We thank Ashley Lakoduk, Aparna Mohanakrishnan, and Kim Reed for help in recombinant protein production, Saipraveen Srinivasan for His-SNX9 proteins, and Wesley Burford for help in retrovirus production and fluorescence-activated cell sorting. We thank Robert Hipskind for sharing reagents. We are grateful for Joëlle Simony-Lafontaine and Florence Boissière for their help in validating the immunohistochemical staining. This work was supported by National Institutes of Health Grants GM42455 and MH61345 to S.L.S. N.B. was partially supported by Cancer Prevention Research Institute of Texas Training Grant RP140110.
Abbreviations used:
- ADAM
a disintegrin and metalloprotease
- BAR
Bin–amphiphysin–Rvs
- CIE
clathrin-independent endocytosis
- CME
clathrin-mediated endocytosis
- GAP
GTPase-activating protein
- GEF
GTPase exchange factor
- GRAF1
GTPase regulator associated with focal adhesion kinase
- MLC2
myosin light chain 2
- Pi
inorganic phosphate
- PM
plasma membrane
- PX
Phox homology
- ROCK
Rho-associated protein kinase
- SNX9
sorting nexin 9.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E16-02-0101) on March 9, 2016.
REFERENCES
- Arsic N, Bendris N, Peter M, Begon-Pescia C, Rebouissou C, Gadea G, Bouquier N, Bibeau F, Lemmers B, Blanchard JM. A novel function for Cyclin A2: control of cell invasion via RhoA signaling. J Cell Biol. 2012;196:147–162. doi: 10.1083/jcb.201102085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoitei N, Diepold K, Brunner C, Rouhi A, Genze F, Becher A, Kestler H, van Lint J, Chiosis G, Koren J, 3rd, et al. HSP90 supports tumor growth and angiogenesis through PRKD2 protein stabilization. Cancer Res. 2014;74:7125–7136. doi: 10.1158/0008-5472.CAN-14-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacac M, Stamenkovic I. Metastatic cancer cell. Annu Rev Pathol. 2008;3:221–247. doi: 10.1146/annurev.pathmechdis.3.121806.151523. [DOI] [PubMed] [Google Scholar]
- Bellizzi A, Mangia A, Chiriatti A, Petroni S, Quaranta M, Schittulli F, Malfettone A, Cardone RA, Paradiso A, Reshkin SJ. RhoA protein expression in primary breast cancers and matched lymphocytes is associated with progression of the disease. Int J Mol Med. 2008;22:25–31. [PubMed] [Google Scholar]
- Borm B, Requardt RP, Herzog V, Kirfel G. Membrane ruffles in cell migration: indicators of inefficient lamellipodia adhesion and compartments of actin filament reorganization. Exp Cell Res. 2005;302:83–95. doi: 10.1016/j.yexcr.2004.08.034. [DOI] [PubMed] [Google Scholar]
- Borner GH, Antrobus R, Hirst J, Bhumbra GS, Kozik P, Jackson LP, Sahlender DA, Robinson MS. Multivariate proteomic profiling identifies novel accessory proteins of coated vesicles. J Cell Biol. 2012;197:141–160. doi: 10.1083/jcb.201111049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourguignon LY. Hyaluronan-mediated CD44 activation of RhoGTPase signaling and cytoskeleton function promotes tumor progression. Semin Cancer Biol. 2008;18:251–259. doi: 10.1016/j.semcancer.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretscher A, Reczek D, Berryman M. Ezrin: a protein requiring conformational activation to link microfilaments to the plasma membrane in the assembly of cell surface structures. J Cell Sci. 1997;110:3011–3018. doi: 10.1242/jcs.110.24.3011. [DOI] [PubMed] [Google Scholar]
- Chaudhary N, Gomez GA, Howes MT, Lo HP, McMahon KA, Rae JA, Schieber NL, Hill MM, Gaus K, Yap AS, et al. Endocytic crosstalk: cavins, caveolins, and caveolae regulate clathrin-independent endocytosis. PLoS Biol. 2014;12:e1001832. doi: 10.1371/journal.pbio.1001832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress C, Lin Q, Yang W. Dimerization is required for SH3PX1 tyrosine phosphorylation in response to epidermal growth factor signalling and interaction with ACK2. Biochem J. 2006;394:693–698. doi: 10.1042/BJ20050576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kreuk BJ, Hordijk PL. Control of Rho GTPase function by BAR-domains. Small GTPases. 2012;3:45–52. doi: 10.4161/sgtp.18960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deryugina EI, Quigley JP. Chick embryo chorioallantoic membrane model systems to study and visualize human tumor cell metastasis. Histochem Cell Biol. 2008;130:1119–1130. doi: 10.1007/s00418-008-0536-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz B, Yuen A, Iizuka S, Higashiyama S, Courtneidge SA. Notch increases the shedding of HB-EGF by ADAM12 to potentiate invadopodia formation in hypoxia. J Cell Biol. 2013;201:279–292. doi: 10.1083/jcb.201209151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty JT, Lenhart KC, Cameron MV, Mack CP, Conlon FL, Taylor JM. Skeletal muscle differentiation and fusion are regulated by the BAR-containing Rho-GTPase-activating protein (Rho-GAP), GRAF1. J Biol Chem. 2011;286:25903–25921. doi: 10.1074/jbc.M111.243030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty GJ, McMahon HT. Mechanisms of endocytosis. Annu Rev Biochem. 2009;78:857–902. doi: 10.1146/annurev.biochem.78.081307.110540. [DOI] [PubMed] [Google Scholar]
- Elkin SR, Bendris N, Reis C, Zhou Y, Xie Y, Huffman KE, Minna JD, Schmid SL. A systematic analysis reveals heterogeneous changes in the endocytic activities of cancer cells. Cancer Res 75, 4640–4650. 2015 doi: 10.1158/0008-5472.CAN-15-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gligorijevic B, Wyckoff J, Yamaguchi H, Wang Y, Roussos ET, Condeelis J. N-WASP-mediated invadopodium formation is involved in intravasation and lung metastasis of mammary tumors. J Cell Sci. 2012;125:724–734. doi: 10.1242/jcs.092726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Taylor JM, Parsons JT. An SH3 domain-containing GTPase-activating protein for Rho and Cdc42 associates with focal adhesion kinase. Mol Cell Biol. 1996;16:3169–3178. doi: 10.1128/mcb.16.6.3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard L, Nelson KK, Maciewicz RA, Blobel CP. Interaction of the metalloprotease disintegrins MDC9 and MDC15 with two SH3 domain-containing proteins, endophilin I and SH3PX1. J Biol Chem. 1999;274:31693–31699. doi: 10.1074/jbc.274.44.31693. [DOI] [PubMed] [Google Scholar]
- Howes MT, Kirkham M, Riches J, Cortese K, Walser PJ, Simpson F, Hill MM, Jones A, Lundmark R, Lindsay MR, et al. Clathrin-independent carriers form a high capacity endocytic sorting system at the leading edge of migrating cells. J Cell Biol. 2010;190:675–691. doi: 10.1083/jcb.201002119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamaze C, Chuang TH, Terlecky LJ, Bokoch GM, Schmid SL. Regulation of receptor-mediated endocytosis by Rho and Rac. Nature. 1996;382:177–179. doi: 10.1038/382177a0. [DOI] [PubMed] [Google Scholar]
- Leong HS, Chambers AF, Lewis JD. Assessing cancer cell migration and metastatic growth in vivo in the chick embryo using fluorescence intravital imaging. Methods Mol Biol. 2012;872:1–14. doi: 10.1007/978-1-61779-797-2_1. [DOI] [PubMed] [Google Scholar]
- Lin Q, Lo CG, Cerione RA, Yang W. The Cdc42 target ACK2 interacts with sorting nexin 9 (SH3PX1) to regulate epidermal growth factor receptor degradation. J Biol Chem. 2002;277:10134–10138. doi: 10.1074/jbc.M110329200. [DOI] [PubMed] [Google Scholar]
- Lokman NA, Elder AS, Ricciardelli C, Oehler MK. Chick chorioallantoic membrane (CAM) assay as an in vivo model to study the effect of newly identified molecules on ovarian cancer invasion and metastasis. Int J Mol Sci. 2012;13:9959–9970. doi: 10.3390/ijms13089959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark R, Carlsson SR. Regulated membrane recruitment of dynamin-2 mediated by sorting nexin 9. J Biol Chem. 2004;279:42694–42702. doi: 10.1074/jbc.M407430200. [DOI] [PubMed] [Google Scholar]
- Lundmark R, Carlsson SR. Expression and properties of sorting nexin 9 in dynamin-mediated endocytosis. Methods Enzymol. 2005;404:545–556. doi: 10.1016/S0076-6879(05)04048-6. [DOI] [PubMed] [Google Scholar]
- Lundmark R, Carlsson SR. SNX9—a prelude to vesicle release. J Cell Sci. 2009;122:5–11. doi: 10.1242/jcs.037135. [DOI] [PubMed] [Google Scholar]
- Lundmark R, Doherty GJ, Howes MT, Cortese K, Vallis Y, Parton RG, McMahon HT. The GTPase-activating protein GRAF1 regulates the CLIC/GEEC endocytic pathway. Curr Biol. 2008;18:1802–1808. doi: 10.1016/j.cub.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mudduluru G, Abba M, Batliner J, Patil N, Scharp M, Lunavat TR, Leupold JH, Oleksiuk O, Juraeva D, Thiele W, et al. A systematic approach to defining the microrna landscape in metastasis. Cancer Res. 2015;75:3010–3019. doi: 10.1158/0008-5472.CAN-15-0997. [DOI] [PubMed] [Google Scholar]
- Perry NA, Vitolo MI, Martin SS, Kontrogianni-Konstantopoulos A. Loss of the obscurin-RhoGEF downregulates RhoA signaling and increases microtentacle formation and attachment of breast epithelial cells. Oncotarget. 2014;5:8558–8568. doi: 10.18632/oncotarget.2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier RM, Gout J, Vincent DF, Alcaraz LB, Chuvin N, Arfi V, Martel S, Kaniewski B, Devailly G, Fourel G, et al. TIF1gamma suppresses tumor progression by regulating mitotic checkpoints and chromosomal stability. Cancer Res. 2015;75:4335–4350. doi: 10.1158/0008-5472.CAN-14-3426. [DOI] [PubMed] [Google Scholar]
- Posor Y, Eichhorn-Gruenig M, Puchkov D, Schoneberg J, Ullrich A, Lampe A, Muller R, Zarbakhsh S, Gulluni F, Hirsch E, et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature. 2013;499:233–237. doi: 10.1038/nature12360. [DOI] [PubMed] [Google Scholar]
- Qualmann B, Koch D, Kessels MM. Let’s go bananas: revisiting the endocytic BAR code. EMBO J. 2011;30:3501–3515. doi: 10.1038/emboj.2011.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer. 2002;2:133–142. doi: 10.1038/nrc725. [DOI] [PubMed] [Google Scholar]
- Salazar MA, Kwiatkowski AV, Pellegrini L, Cestra G, Butler MH, Rossman KL, Serna DM, Sondek J, Gertler FB, De Camilli P. Tuba, a novel protein containing bin/amphiphysin/Rvs and Dbl homology domains, links dynamin to regulation of the actin cytoskeleton. J Biol Chem. 2003;278:49031–49043. doi: 10.1074/jbc.M308104200. [DOI] [PubMed] [Google Scholar]
- Sanz-Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E, Marshall CJ. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. doi: 10.1016/j.cell.2008.09.043. [DOI] [PubMed] [Google Scholar]
- Schmid SL, Smythe E. Stage-specific assays for coated pit formation and coated vesicle budding in vitro. J Cell Biol. 1991;114:869–880. doi: 10.1083/jcb.114.5.869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- Simpson KJ, Dugan AS, Mercurio AM. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 2004;64:8694–8701. doi: 10.1158/0008-5472.CAN-04-2247. [DOI] [PubMed] [Google Scholar]
- Smith HW, Marra P, Marshall CJ. uPAR promotes formation of the p130Cas-Crk complex to activate Rac through DOCK180. J Cell Biol. 2008;182:777–790. doi: 10.1083/jcb.200712050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulet F, Yarar D, Leonard M, Schmid SL. SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:2058–2067. doi: 10.1091/mbc.E04-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor JM, Macklem MM, Parsons JT. Cytoskeletal changes induced by GRAF, the GTPase regulator associated with focal adhesion kinase, are mediated by Rho. J Cell Sci. 1999;112:231–242. doi: 10.1242/jcs.112.2.231. [DOI] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS Lett. 2008;582:2093–2101. doi: 10.1016/j.febslet.2008.04.039. [DOI] [PubMed] [Google Scholar]
- Vial E, Sahai E, Marshall CJ. ERK-MAPK signaling coordinately regulates activity of Rac1 and RhoA for tumor cell motility. Cancer Cell. 2003;4:67–79. doi: 10.1016/s1535-6108(03)00162-4. [DOI] [PubMed] [Google Scholar]
- Virtakoivu R, Mai A, Mattila E, De Franceschi N, Imanishi SY, Corthals G, Kaukonen R, Saari M, Cheng F, Torvaldson E, et al. Vimentin-ERK signaling uncouples Slug gene regulatory function. Cancer Res. 2015;75:2349–2362. doi: 10.1158/0008-5472.CAN-14-2842. [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Ridley AJ. Why three Rho proteins? RhoA, RhoB, RhoC, and cell motility. Exp Cell Res. 2004;301:43–49. doi: 10.1016/j.yexcr.2004.08.012. [DOI] [PubMed] [Google Scholar]
- Wittchen ES, Burridge K. Analysis of low molecular weight GTPase activity in endothelial cell cultures. Methods Enzymol. 2008;443:285–298. doi: 10.1016/S0076-6879(08)02014-4. [DOI] [PubMed] [Google Scholar]
- Worby CA, Simonson-Leff N, Clemens JC, Huddler D, Jr, Muda M, Dixon JE. Drosophila Ack targets its substrate, the sorting nexin DSH3PX1, to a protein complex involved in axonal guidance. J Biol Chem. 2002;277:9422–9428. doi: 10.1074/jbc.M110172200. [DOI] [PubMed] [Google Scholar]
- Yarar D, Surka MC, Leonard MC, Schmid SL. SNX9 activities are regulated by multiple phosphoinositides through both PX and BAR domains. Traffic. 2008;9:133–146. doi: 10.1111/j.1600-0854.2007.00675.x. [DOI] [PubMed] [Google Scholar]
- Yarar D, Waterman-Storer CM, Schmid SL. SNX9 couples actin assembly to phosphoinositide signals and is required for membrane remodeling during endocytosis. Dev Cell. 2007;13:43–56. doi: 10.1016/j.devcel.2007.04.014. [DOI] [PubMed] [Google Scholar]
- Yu X, Zech T, McDonald L, Gonzalez EG, Li A, Macpherson I, Schwarz JP, Spence H, Futo K, Timpson P, et al. N-WASP coordinates the delivery and F-actin-mediated capture of MT1-MMP at invasive pseudopods. J Cell Biol. 2012;199:527–544. doi: 10.1083/jcb.201203025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.