

Forces on JAM-A activate RhoA to increase cell stiffness. Activation of RhoA requires GEF-H1 and p115 RhoGEF activation downstream of FAK/ERK and Src family kinases, respectively.
Abstract
Junctional adhesion molecule A (JAM-A) is a broadly expressed adhesion molecule that regulates cell–cell contacts and facilitates leukocyte transendothelial migration. The latter occurs through interactions with the integrin LFA-1. Although we understand much about JAM-A, little is known regarding the protein’s role in mechanotransduction or as a modulator of RhoA signaling. We found that tension imposed on JAM-A activates RhoA, which leads to increased cell stiffness. Activation of RhoA in this system depends on PI3K-mediated activation of GEF-H1 and p115 RhoGEF. These two GEFs are further regulated by FAK/ERK and Src family kinases, respectively. Finally, we show that phosphorylation of JAM-A at Ser-284 is required for RhoA activation in response to tension. These data demonstrate a direct role of JAM-A in mechanosignaling and control of RhoA and implicate Src family kinases in the regulation of p115 RhoGEF.
INTRODUCTION
Adhesion molecules on the surface of endothelial cells serve as ligands for circulating leukocytes. Interactions between these adhesion molecules and their corresponding receptors facilitate transendothelial migration of the leukocytes to regions of inflammation. As the leukocyte crawls atop the endothelial cell, mechanical forces are imposed on the endothelial cell, resulting in activation of the small GTPase RhoA and an increase in cell stiffness (Liu et al., 2010; Stroka and Aranda-Espinoza, 2011; Heemskerk et al., 2014; Lessey-Morillon et al., 2014; Schaefer and Hordijk, 2015). In a similar manner, homodimerization of adhesion molecules at cell–cell contacts regulates zone-specific contractility through regulation of RhoA (Nelson et al., 2004; Bazellieres et al., 2015; Priya et al., 2015).
RhoA, like other small GTPases, cycles between a GTP-bound, active state and a GDP-bound, inactive state. Addition of GTP is regulated by guanine nucleotide exchange factors (GEFs), whereas hydrolysis of GTP to GDP, which inactivates the protein, is promoted by GTPase-activating proteins (GAPs; Marjoram et al., 2014). When activated, RhoA promotes actomyosin-based contractility, thus regulating cytoskeletal organization (Chrzanowska-Wodnicka and Burridge, 1996). Recent work has demonstrated that mechanical force, in the form of tension, imposed on individual adhesion molecules is sufficient to activate RhoA (Zhao et al., 2007; Guilluy et al., 2011b; Collins et al., 2012; Lessey-Morillon et al., 2014; Schaefer et al., 2014; Barry et al., 2015; Bazellieres et al., 2015). Of interest, the kinetics of RhoA activation and its associated GEFs is unique for individual adhesion molecules, implying pathway specificity and a spatiotemporal response.
Junctional adhesion molecule A (JAM-A) belongs to the immunoglobulin (Ig) superfamily of adhesion molecules. Originally described as a platelet receptor (Naik et al., 1995), the protein is also expressed on endothelial and epithelial cells, as well as in most leukocyte subsets (Martin-Padura et al., 1998). JAM-A participates in a number of cellular functions, including formation and maintenance of cell–cell contacts (Martin-Padura et al., 1998; Aurrand-Lions et al., 2001b), is a reovirus receptor (Barton et al., 2001; Campbell et al., 2005), and is a ligand for the leukocyte-expressed LFA-1integrin dimer (Ostermann et al., 2002). At cell–cell contacts, JAM-A forms cis- and trans-homodimers (Severson et al., 2008; Monteiro et al., 2014), which have been implicated in supporting tension between cells (Bazellieres et al., 2015; Tornavaca et al., 2015). These signaling events require the protein’s short C-terminus, which contains a PDZ-binding domain and at least two phosphorylation sites (Severson and Parkos, 2009; Iden et al., 2012; Naik et al., 2014). Through its interactions with LFA-1, JAM-A is recognized as a critical regulator of leukocyte transendothelial migration, and mice lacking endothelial expression of JAM-A display impaired immune responses (Woodfin et al., 2009; Lakshmi et al., 2012; Schmitt et al., 2014). It is unknown whether tension on JAM-A, such as that found at cell–cell contacts or imposed by a crawling leukocyte, can be mechanically transmitted to support RhoA activation.
The work described here shows that tension on JAM-A activates RhoA, which results in increased cell stiffness. Activation of RhoA is mediated by GEF-H1 and p115 RhoGEF in a phosphoinositide 3-kinase (PI3K)–dependent manner. Tension on JAM-A activates focal adhesion kinase (FAK) and extracellular signal-regulated kinase (ERK) to control GEF-H1, whereas p115 RhoGEF is regulated by Src family kinases (SFKs). Finally, activation of RhoA in response to tension on JAM-A requires phosphorylation of S284 on the protein’s C-terminus. These data demonstrate that JAM-A supports tension-induced outside-in signaling to control RhoA and increase cellular stiffness.
RESULTS
Tension imposed on JAM-A activates RhoA via P13K
Tension imposed on cell surface receptors regulates cellular stiffness through activation of RhoA (Matthews et al., 2006; Guilluy et al., 2011b; Collins et al., 2012; Lessey-Morillon et al., 2014). To determine whether JAM-A could support similar signaling responses, we used models of continuous and pulsatile force as outlined in Figure 1. Paramagnetic beads coated with a monoclonal antibody that recognizes the first Ig-like domain of JAM-A (Mandell et al., 2004) were added to cells, and pulsatile forces were applied using magnetic tweezers. Cell stiffening was determined by measuring bead displacement using single-particle analysis between successive pulling events. Alternatively, continuous force was generated on JAM-A by suspending a magnet in parallel to the cells and followed by biochemical analysis.
FIGURE 1:
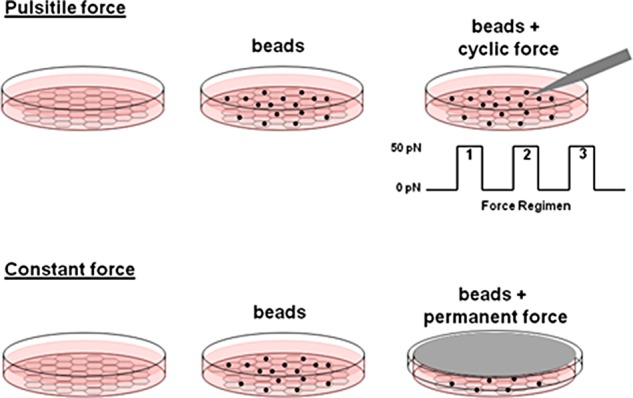
Overview of tension models. Cells were grown on fibronectin-coated substratum, and anti-JAM-A–coated paramagnetic beads were added at approximately a 3:1 bead-to-cell ratio. For application of pulsatile forces, the pole tip of a magnetic tweezers was lowered to 25 μm above the bead and force applied using a defined regimen. Bead displacement was imaged at 30 frames/s, and bead movement was tracked using custom software as described in Materials and Methods. For application of continuous force, a magnet was suspended parallel to the apical surface of the cells for the determined time. Cells were then lysed and processed for biochemical analysis as needed.
We first wanted to determine whether forces through JAM-A activated RhoA. Tension on anti–JAM-A coated beads increased RhoA activity, whereas addition of beads alone had no effect (Figure 2A). As a control, tension on poly-l-lysine (PLL)–coated beads did not activate RhoA (Figure 2B). Quantification of RhoA activation in response to tension on JAM-A or PLL is shown in Figure 2, C and D, respectively. Previous reports also showed that tension on PLL does not activate RhoA (Collins et al., 2012) or increase cell stiffness in response to force (Collins et al., 2012; Barry et al., 2015). Because tension on JAM-A increased RhoA activity, we determined the effect on cell stiffness. Tension imposed on JAM-A increased cell stiffness, as evidenced by decreased bead displacement between pulse 1 and subsequent pulses (Figure 2E). As seen in Figure 2, F and G, inhibition of RhoA or Rho-associated protein kinase (Rho-associated, coiled-coil–containing protein kinase [ROCK]) prevented the decrease in bead displacement. These data indicate that tension on JAM-A activates RhoA to regulate cell stiffness.
FIGURE 2:

Tension on JAM-A activates RhoA to increase cell stiffness. RhoA activity was measured using RBD-pull-down assays on untreated HUVECs, HUVECs treated with anti-JAM-A–coated beads, HUVECs with the same beads plus 3 min of continuous force (A), or the same regimen with PLL-coated beads (B). Data are representative of at least three separate experiments and are quantified as means ± SEM in C and D. *p < 0.01 vs. untreated as determined by t test. To determine cell stiffness, HUVECs were untreated (E) or treated with 1 mg/ml C3 transferase for 60 min (F) or 10 μM Y-27632 for 30 min (G) before force application on anti-JAM-A–coated magnetic beads with magnetic tweezers. *p < 0.01 vs. pulse 1 as determined by t test.
Previous studies showed that tension imposed on some adhesion molecules activates RhoA via phosphoinositide 3 kinase (PI3K; Collins et al., 2012), and recent reports indicate that JAM-A regulates PI3K signaling (Nava et al., 2011; Tuncay et al., 2015). To determine whether PI3K participated in signaling in response to tension on JAM-A, we assessed colocalization between beads and the PI3K sensor green fluorescent protein (GFP)–Akt-PH in the presence or absence of force. Tension imposed on JAM-A recruited GFP-Akt-PH but not GFP alone to the beads, indicating activation of PI3K (Figure 3, A and B). No recruitment of GFP-Akt-PH occurred around PLL beads in the presence or absence of force. To confirm that PI3K was activated in response to force on JAM-A, we examined phosphorylation of Akt. As seen in Figure 3C, tension imposed on JAM-A rapidly increased phosphorylation of Akt. We next wanted to see whether PI3K signaling was required for activation of RhoA. As seen in Figure 3, D and E, the PI3K inhibitor LY294002 prevented RhoA activation downstream of force on JAM-A. Together these data demonstrate that force on JAM-A activates RhoA in a PI3K-dependent manner.
FIGURE 3:

JAM-A activates PI3K upstream of RhoA. HUVECs were transfected with GFP or GFP-Akt-PH and incubated with anti-JAM-A– or PLL–coated magnetic beads in the presence or absence of 1 min of continuous force. Cells were fixed with paraformaldehyde, and enrichment of GFP to the area around the bead was determined. (A) Representative images. (B) Quantification. Data are mean ± SEM of >25 cells/experiment from three independent experiments. *p < 0.05 vs. control by t test. HUVECs were incubated with anti–JAM-A beads, and force was applied for 0–10 min. Akt phosphorylation, used as a marker of PI3K activation, was determined by Western blot (C). RhoA activity in response to force on JAM-A–coated beads was measured in HUVECs with or without incubation with the PI3K inhibitor LY294002 (10 μM, 30 min). (D) Representative blots. (E) Means ± SEM from four experiments. *p < 0.05 vs. control by t test.
Tension imposed on JAM-A activates GEF-H1 and p115 RhoGEF to regulate RhoA activity
Rho family GTPases are regulated by the activity of GEFs and GAPs (Schmidt and Hall, 2002; Lessey et al., 2012). To determine which GEFs were activated in response to tension imposed on JAM-A, we used a nucleotide-free RhoA pull-down assay (Garcia-Mata et al., 2006; Guilluy et al., 2011a). As seen in Figure 4, A and B, tension imposed on JAM-A increased the activity of GEF-H1 (ARHGEF2) and p115 RhoGEF (ARHGEF1) but not that of LARG, p190 RhoGEF, or PDZ RhoGEF. To determine whether GEF-H1 and p115 RhoGEF were responsible for RhoA activation in response to tension on JAM-A, we knocked down the expression of these GEFs individually or together. As seen in Figure 5, A and B, small interfering RNA (siRNA)–mediated knockdown of GEF-H1 and p115 RhoGEF individually did not prevent RhoA activation in response to tension on JAM-A. However, knockdown of both GEFs concomitantly prevented RhoA activation in this system. Of importance, knockdown of GEF-H1 and/or p115 RhoGEF did not alter JAM-A expression levels.
FIGURE 4:
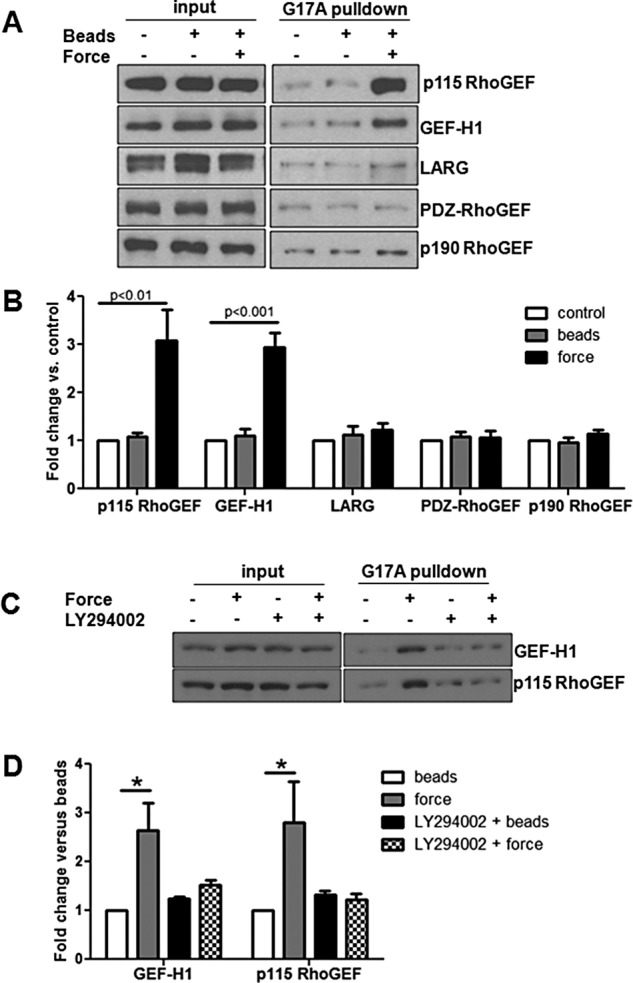
Tension on JAM-A activates GEF-H1 and p115 RhoGEF via PI3K. HUVECs were exposed to anti-JAM-A–coated beads, and tension was imposed with a permanent magnet for 3 min. Activation of RhoA GEFs was determined using the GST-RhoAG17A pull-down assay. (A) Representative Western blots. (B) Means ± SEM from at least three experiments. White bars are untreated control, gray bars are bead only, and black bars are beads plus 3 min of tension. To test for a role of PI3K in activation of GEFs, some cells were treated with LY294002 (10 μM, 30 min) before addition of beads. GEF activity was assessed using GST-RhoAG17A pull-down assay as described as methods (C). (D) White bars are bead only, gray bars are beads plus 3 min of tension, black bars are LY294002 plus beads, and checkered bars are LY29004 plus beads with tension for 3 min. Statistical analysis was conducted by t test.
FIGURE 5:
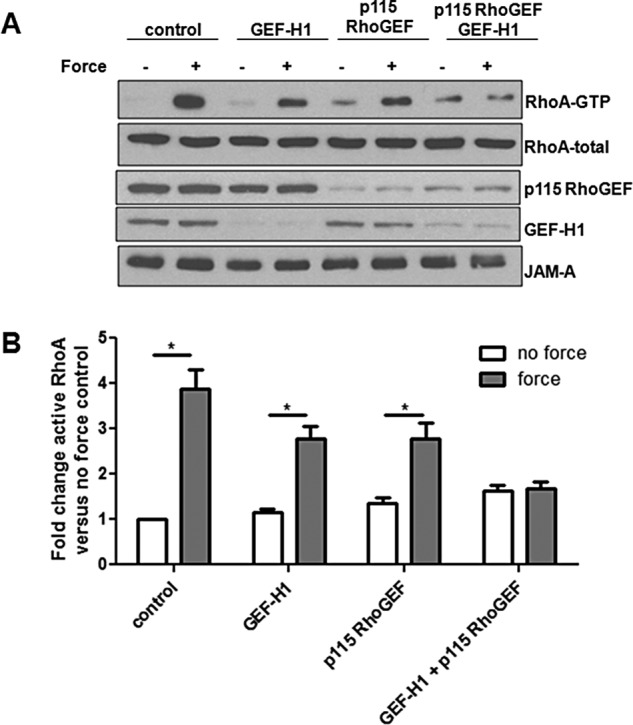
JAM-A–mediated RhoA activation requires both GEF-H1 and p115 RhoGEF. HUVECs were transfected with control siRNA or oligos designed against GEF-H1or p115 Rho GEF. Transfected cells were incubated with anti-JAM-A–coated magnetic beads, and some cells were exposed to force for 3 min. RhoA activity was determined by GST-RBD pull down. Representative Western blots (A) and quantifications (B) from six separate experiments. *p < 0.05 vs. no force sample for each condition as determined by t test.
Because inhibition of PI3K blocked activation of RhoA downstream of tension on JAM-A, we next investigated the protein’s role in the activation of GEF-H1 and p115 RhoGEF. As seen in Figure 5, C and D, inhibition of Akt with the inhibitor LY294002 prevented activation of the two GEFs. These data demonstrate that tension on JAM-A activates GEF-H1 and p115 RhoGEF, both of which are required for RhoA activation, all of which occur downstream of PI3K activation.
Tension imposed on JAM-A activates GEF-H1 through FAK/ERK and p115 RhoGEF through Src family kinases
We next wanted to determine what pathways were leading to GEF-H1 and p115 RhoGEF activation downstream of tension on JAM-A. Previous reports showed that GEF-H1 can be activated downstream of FAK/ERK signaling in response to mechanical forces (Fujishiro et al., 2008; Guilluy et al., 2011b; Collins et al., 2012). To determine whether a similar pathway was operating in our system, we examined phosphorylation of FAK and ERK in response to tension on JAM-A and found increased phosphorylation of both proteins (Figure 6A). As shown in Figure 6B, the upstream inhibitor of ERK, U0126 (mitogen-activated protein kinase kinase [MEK] inhibitor), and FAK inhibitor 14 both inhibited GEF-H1 but had no effect on p115 RhoGEF activation. These results are quantified in Figure 6, C (GEF-H1) and D (p115 RhoGEF).
FIGURE 6:

GEF-H1 is activated downstream of FAK/ERK in response to tension on JAM-A. (A) HUVECs were incubated with anti-JAM-A–coated magnetic beads, and tension was applied for 0–5 min. Cells were lysed and analyzed for ERK and FAK phosphorylation by Western blot analysis. (B) HUVECs were incubated with anti-JAM-A–coated magnetic beads, and some cells were pretreated with the MEK inhibitor U0126 (25 μM) or FAK inhibitor 14 (2 μM) for 30 min before addition of beads, with some samples experiencing 3 min of force. RhoA GEF activity was assessed by GST-RhoAG17A pull-down assay. (C, D) Activation of GEF-H1 and p115 RhoGEF, respectively. Data are mean ± SEM from at least three experiments. *p < 0.05 vs. no-force control for each condition by t test.
It was reported that p115 RhoGEF can be activated downstream of JAK2 and protein kinase Cα (PKCα; Guilluy et al., 2010; Peng et al., 2011), and so we examined the involvement of these kinases in the activation of p115 RhoGEF in response to tension on JAM-A. As seen in Figure 7A, neither inhibition of JAK2 with AG 490 nor inhibition of PKCα with Gö6976 prevented p115 RhoGEF activation in response to tension on JAM-A. To further confirm that PKCα was not required for p115 RhoGEF activation in response to tension on JAM-A, we used siRNA to knock down expression of the protein. As seen in Supplemental Figure S1, knockdown of PKCα did not prevent p115 RhoGEF activation in response to tension on JAM-A. Other candidates for activating p115 RhoGEF are the SFKs, which are known to be activated by mechanical tension (Kostic and Sheetz, 2006; Guilluy et al., 2011b). Inhibition of SFKs with Su6656 caused a significant reduction in p115 RhoGEF activation in response to tension on JAM-A (Figure 7, A and B). These data demonstrate that tension on JAM-A regulates GEF-H1 via FAK/ERK and p115 RhoGEF via SFKs.
FIGURE 7:
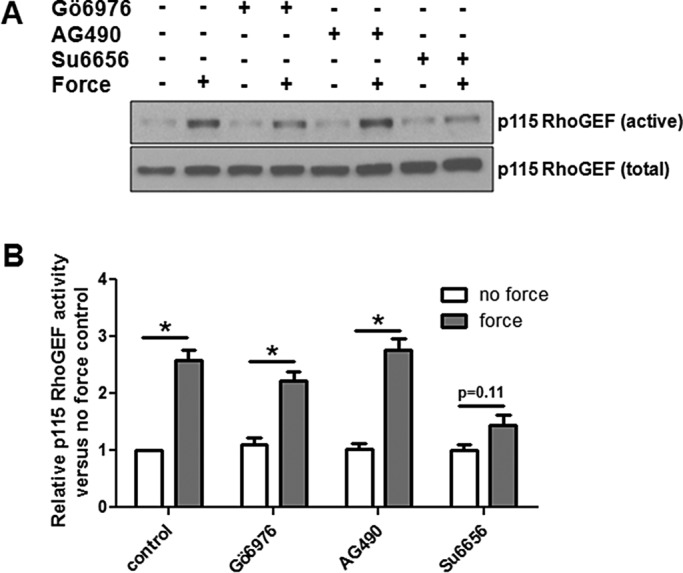
Src family kinases activate p115 RhoGEF in response to tension on JAM-A. (A) HUVECs were incubated with inhibitors against JAK2 (AG 490, 25 μM), PKCα (Gö6976, 10 μM), or Src family kinases (su6656, 5 μM) for 30 min, followed by addition of anti-JAM-A– coated magnetic beads. Some cells also experienced force for 3 min. p115 RhoGEF activity was determined by RhoAG17A pull-down assay. (B) Means ± SEM of at least three separate experiments *p < 0.05 vs. no-force control for each condition by t test.
JAM-A phosphorylation is regulated by direct and global mechanical forces
JAM-A is phosphorylated at S285 in mice (S284 in humans) when the protein is localized to tight junctions (Iden et al., 2012). Because tight junctions are regions of high RhoA activity (Priya et al., 2015), we hypothesized that increased tension on JAM-A homodimers within these zones could regulate this phosphorylation event. To test this hypothesis, we examined JAM-A phosphorylation in response to tension on anti-JAM-A beads. Phosphorylation of JAM-A S284 increases rapidly in response to tension on JAM-A (Figure 8A). We next tested to see whether this response was specific to tension on JAM-A or was a general response to mechanical forces. With fluid shear stress as a model, JAM-A S284 phosphorylation rapidly increased before returning to levels at or below baseline within 30 min of shear stress onset (Figure 8B). These data demonstrate that JAM-A phosphorylation is stimulated by mechanical forces.
FIGURE 8:

JAM-A S284 phosphorylation is regulated by mechanical forces. (A) HUVECs were incubated with anti-JAM-A–coated magnetic beads and exposed to force for 0–10 min. (B) HUVECs were exposed to shear stress for 0–60 min. For both experiments, cells were lysed, and Western blot analysis of total and phosphorylated JAM-A was conducted. Blots are representative of at least three independent experiments
JAM-A phosphorylation controls RhoA activation in response to tension
To determine whether S284 phosphorylation is required for RhoA activation in response to force on JAM-A, we generated a phosphodeficient S284A mutant and expressed it along with empty vector and wild-type JAM-A in CHO cells that lack endogenous JAM-A. Wild-type human JAM-A expressed in CHO-K1 cells is phosphorylated at S284 (Figure 9A), indicating that the necessary molecular components to control this modification are present. Further, expression of wild-type but not S284A JAM-A increased barrier function in CHO cells (Figure 9B), as previously reported (Iden et al., 2012), indicating functional differences between the mutant and wild-type protein. We first tested to see whether RhoA activation occurred in the JAM-A S284A mutant. Tension imposed on wild-type JAM-A but not on JAM-A S284A resulted in increased RhoA activity (Figure 9, C and D). As a control, anti-JAM-A beads were added to empty vector–transfected CHO cells, with no RhoA activation observed. Force imposed on wild-type JAM-A resulted in cell stiffening, whereas force imposed on cells expressing the S284A mutant did not (Figure 9, E and F). Attempts were made to measure bead displacement in empty vector–transfected cells, but this measurement was not possible because the beads did not attach to the cell surface and were drawn to the magnetic tweezers instantly. Similar to what was observed in human umbilical vein endothelial cells (HUVECs), cell stiffening in CHO cells could be inhibited by C3-transferase and Y-27632 (Figure 9, F and G, respectively), indicating that RhoA was responsible. These data demonstrate that JAM-A S284 phosphorylation is required for RhoA activation in response to tension on the protein.
FIGURE 9:

JAM-A S284 phosphorylation is required for RhoA activation and cell stiffening in response to force on JAM-A. (A) CHO cells were transfected with empty vector, JAM-A, or JAM-A S284A, and expression of JAM-A and phosphorylated JAM-A was determined by Western blot. (B) Barrier function of CHO cells expressing empty vector (EV), JAM-A, or JAM-A S284 was determined by FITC-dextran flux. Data are representative of three experiments run in triplicate. (C) CHO cells expressing EV, JAM-A, or JAM-A S284A were incubated with anti-JAM-A–coated magnetic beads, and some cells were exposed to force for 5 min. RhoA activity was determined by the RBD pull-down assay. (D) Means ± SEM of four independent experiments, with *p < 0.05 vs. no-force control by t test. Cell stiffness was determined in cells expressing (E) JAM-A, (F) JAM-A S284A, (G) JAM-A and incubated with 10 μM Y-27632, or (H) expressing JAM-A and incubated with 1 mg/ml C3-transferase as described in Materials and Methods.
JAM-A is phosphorylated at S284 through the actions of PKCζ (Iden et al., 2012). Previous reports showed that PKCζ is regulated by mechanical forces (Disatnik et al., 2002; Suzuma et al., 2002; Heo et al., 2011), is regulated via PI3K (Mas et al., 2003; Sarkar et al., 2006), and controls RhoA activity in some systems (Dovas et al., 2006). Therefore we investigated a role for PKCζ in activation of RhoA downstream of tension on JAM-A. As seen in Figure 10A, tension imposed on JAM-A increased phosphorylation of PKCζ. To test for a role in RhoA activation, we used a short-peptide inhibitor of PKCζ to block downstream signaling. As seen in Figure 10B, inhibition of PKCζ prevented RhoA activation in response to force on JAM-A. Similarly, inhibition of PKCζ prevents cell stiffening in response to force on JAM-A.
FIGURE 10:
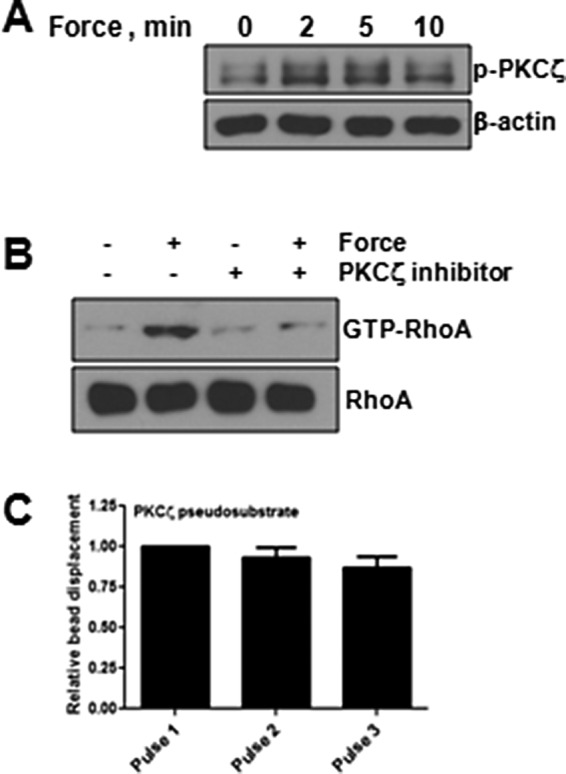
Force on JAM-A regulates PKCζ to activate RhoA. HUVECs were incubated with anti–JAM-A beads in the presence or absence of forces, and PKCζ phosphorylation was determined by Western blot (A). HUVECs were incubated with anti-JAM-A–coated magnetic beads, and some cells were exposed to force for 3 min. Some cells had been pretreated with PKCζ inhibitory peptide. RhoA activity was determined by the RBD pull-down assay (B). Cell stiffness was determined in HUVECs after tension was imposed on anti-JAM-A–coated beads in the presence of PKCζ inhibitor as described in Materials and Methods (C).
DISCUSSION
Before this study, a role for JAM-A in mechanosignaling was unknown. As diagrammed in Figure 11, we have shown that tension on JAM-A activates RhoA to control cell stiffness (Figure 2). Activation of RhoA in this system requires PI3K (Figure 3) and the combined activities of GEF-H1 and p115 RhoGEF (Figures 4 and 5). Activation of GEF-H1 depends on FAK/ERK (Figure 6), whereas activation of p115 RhoGEF depends on SFKs (Figure 7). Tension imposed on JAM-A or exposure to shear stress increases phosphorylation of JAM-A at S284 (Figure 8). Phosphorylation of JAM-A at S284 is required for activation of RhoA and increased cell stiffness in response to tension on the protein (Figure 9). Finally, PKCζ is required for activation of RhoA in response to force on JAM-A (Figure 10). Together these results identify JAM-A as a direct transducer of mechanical force, which activates RhoA to regulate cell stiffness.
FIGURE 11:
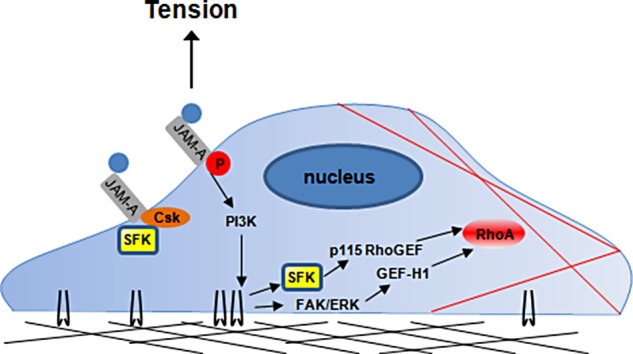
Working model of RhoA activation in response to force on JAM-A. Anti-JAM-A–coated magnetic beads engage JAM-A on the cell surface. In response to force, SFKs dissociate from the protein’s C-terminus. In short order, PI3K likely signals for the activation of PKCζ, as well as of GEF-H1 and p115 RhoGEF. Activation of GEF-H1 and p115 RhoGEF also requires FAK/ERK- and SFK-mediated pathways, respectively. Finally, RhoA is activated by either GEF to regulate actomyosin-based cellular contractility and cell stiffness.
JAM-A exists in cells as monomers and cis- and trans-homodimers. At cell–cell junctions, JAM-A homodimers control barrier dynamics (Aurrand-Lions et al., 2001a; Severson et al., 2008; Monteiro et al., 2014). Regulation of barrier function requires interactions between JAM-A’s extracellular domain as well as binding partner interactions mediated by the protein’s C-terminal PDZ-binding domain (Monteiro and Parkos, 2012). As a monomer, JAM-A has been shown to inhibit integrin signaling (Peddibhotla et al., 2013; Naik et al., 2014), a function that is lost when the protein dimerizes. A possible explanation for the observations described here is that a monomeric form of JAM-A binds to the anti–JAM-A magnetic beads. Once placed under tension, the protein behaves as a homodimer similar to those found at cell–cell junctions. When localized within tight junctions, which are under high levels of tension (Priya et al., 2015), JAM-A is phosphorylated (Iden et al., 2012). Similarly, in our system, JAM-A S284 phosphorylation is rapidly increased by force imposed on the protein.
Monomeric JAM-A inhibits integrin signaling through interaction with CD9. Peddibhotla et al. (2013) reported that induction of JAM-A dimers results in a disruption of a JAM-A/CD9/αvβ3 complex, leading to increased cell migration. Previous work also showed that activation of platelets results in increased JAM-A dimerization and phosphorylation, as well as decreased interaction with CD9 and αIIbβ3 (Sobocka et al., 2004). Recently Naik et al. (2014) found that JAM-A inhibits αIIbβ3 by suppressing SFK signaling. In this model, monomeric JAM-A forms a complex with c Src-kinase (Csk) to inhibit integrin-Src complexes. Csk negatively regulates SFK family members via phosphorylation of a conserved tyrosine residue in the protein’s C-terminus (Chong et al., 2005). In the context of the present work, tension on JAM-A would increase SFK signaling through dissociation of Csk and integrins. From another perspective, decreasing JAM-A expression should therefore increase similar signaling networks due to loss of monomer-associated signaling inhibition. Indeed, Tornavaca et al. (2015) showed that focal adhesions are more abundant in cells in which JAM-A has been knocked down. We observed a decrease in phosphorylation of Src at Y527, the site regulated by Csk, and an increase in FAK phosphorylation (Y397) in HUVECs in which JAM-A had been knocked down (Supplemental Figure S2). Increases in FAK phosphorylation in response to JAM-A knockdown could indicate increased RhoA activity or a disruption in focal adhesion turnover by regulating integrin recycling. With regard to this possibility, knockdown of JAM-A could lead to activation of RhoA through activation of p115 RhoGEF. This raises the possibility that distinct pools of JAM-A exist to control spatiotemporal control of cellular contractility. Thus loss of JAM-A–mediated suppression of SFKs would result in increased RhoA activity or at least redistribution of active RhoA. Knockdown of JAM-A could also lead to increased FAK phosphorylation through decreased integrin recycling. Indeed, previous work showed a deficiency in β1 integrin recycling in JAM-A–null neutrophils, resulting in impaired chemotaxis (Cera et al., 2009).
Previous reports demonstrated that cis- (Severson et al., 2008; Peddibhotla et al., 2013) and trans-dimerization (Monteiro et al., 2014) mutants of JAM-A control the protein’s function. In our model, JAM-A was engaged using an antibody that recognizes the first Ig-like domain, the region involved in dimerization. Alternatively, LFA-1 binds JAM-A in the protein’s second Ig-like domain. Binding of LFA-1 to JAM-A has been shown to destabilize homophilic interactions, possibly due to the fact that LFA-1/JAM-A binding can support more tension than JAM-A dimers (Wojcikiewicz et al., 2009). Future studies using cis- and trans-dimerization mutants engaged with JAM-A/fc–coupled beads, anti–JAM-A–coupled beads, and LFA-1 I-domain beads engaged to the same proteins would provide further insight into the modes of mechanical forces supported by JAM-A.
The data in Figure 6 demonstrate that SFKs control p115 RhoGEF activity. This is not surprising, because SFKs are known to be regulated by mechanical forces. SFK family members Fyn (Kostic and Sheetz, 2006; Chiu et al., 2008; Guilluy et al., 2011b; Fiore et al., 2015), Src (Chaturvedi et al., 2007; Wijetunge and Hughes, 2007), Yes (Niediek et al., 2012), and Lyn (Alessandri-Haber et al., 2008; Hughan et al., 2014) are all activated in response to mechanical forces. Because SFKs are activated by force and Su6656 inhibits multiple SFKs (Blake et al., 2000), it is difficult to determine which kinase(s) are involved without extensive investigation. Fyn has been associated with activation of the RhoGEF LARG in response to force on integrins (Guilluy et al., 2011b). In our system, LARG was not activated in response to tension on JAM-A, but this does not necessarily rule out a role for Fyn in the activation of p115 RhoGEF, as the ligand and cell type used were different between these studies. Previous work showed that p115 RhoGEF is activated in response to integrin engagement to fibronectin (Dubash et al., 2007), a process that likely involves a force component. Although this earlier study demonstrated that p115 RhoGEF was required for cell spreading onto fibronectin, it did not elucidate the mechanism of activation. Determining whether SFKs are responsible for p115 RhoGEF activation after integrin engagement will be pursued in future work.
The present findings may be relevant for several physiological processes. When expressed on endothelial cells, JAM-A participates in leukocyte transendothelial migration by binding to LFA-1. It is known that crawling leukocytes impose forces on endothelial cells, resulting in regional stiffening responses (Schaefer and Hordijk, 2015). The present work demonstrates that forces imposed on JAM-A activate RhoA to elevate cell stiffness. This suggests that LFA-1/JAM-A interactions would elicit RhoA activation and promote leukocyte migration. A similar pathway was demonstrated for ICAM-1, which also increases RhoA activity and cell stiffening in response to tension (Lessey et al., 2012). It is therefore not surprising that endothelial JAM-A is required for normal leukocyte trafficking to regions of inflammation (Woodfin et al., 2009; Lakshmi et al., 2012; Schmitt et al., 2014). Thus JAM-A and its downstream signaling network could represent a novel therapeutic target for controlling inflammatory responses.
JAM-A also plays a critical role when expressed at cell–cell junctions, as absence of the protein affects the mechanical properties of the cell (Hughan et al., 2014; Tornavaca et al., 2015). Breast cancer provides an intriguing model for the present work. There are conflicting reports on a role for JAM-A in breast cancer. Whereas several studies demonstrated that elevated JAM-A expression is a predictive marker for invasive cellular behavior and poor clinical outcomes, other studies reached opposite conclusions (Naik et al., 2008; McSherry et al., 2009; Murakami et al., 2011; Huang et al., 2014). These studies focused exclusively on total JAM-A expression and did not examine posttranslational modifications such as S284 phosphorylation. Ebnet’s group discovered that JAM-A S284 phosphorylation is regulated by PKCζ and PP2A (Iden et al., 2012), which are often dysregulated in cancer (Switzer et al., 2011; Seshacharyulu et al., 2013; Yin et al., 2014). Thus cells expressing normal levels of JAM-A but that cannot regulate phosphorylation of the protein would behave similarly to cells lacking JAM-A expression. Investigating the levels of JAM-A phosphorylation and correlating these with cell behavior and clinical outcomes are further warranted.
This article has demonstrated that tension on JAM-A activates RhoA to regulate cell stiffness. Activation of RhoA requires GEF-H1 and p115 RhoGEF, as well as phosphorylation of JAM-A at S284. SFKs have been identified as novel regulators of p115 RhoGEF activation. It will be interesting to determine whether changes in JAM-A phosphorylation occur during diseases in vivo, such as in cancers of epithelial origin and in vascular disease. Further studies are needed to identify the SFKs required for p115 RhoGEF activation and to determine whether SFKs activate this GEF in other situations in which it is responsible for activating RhoA.
MATERIALS AND METHODS
Cell lines and reagents
HUVECs were cultured in EBM2+ BulletKit (cells and media from Lonza, Rockville, MD). CHO-K1 cells were obtained from the American Type Culture Collection (Manassas, VA) and grown in high-glucose DMEM supplemented with 10% fetal bovine serum and antibiotic–antimycotic solution (all from Life Technologies/ThermoFisher Scientific, Grand Island, NY). Y-27632 (ROCK inhibitor) was purchased from Millipore (Billerica, MA). Cell-permeable C3 (RhoA inhibitor) transferase was purchased from Cytoskeleton (Denver, CO). U0126 (MEK inhibitor), LY294002 (PI3K inhibitor), Su6656 (Src family kinase inhibitor), AG 490 (JAK2 inhibitor), Gö6976 (PKCα inhibitor), FAK inhibitor 14, and PKCζ pseudosubstrate were from Tocris (Minneapolis, MN). All other reagents were from Sigma-Aldrich (St. Louis, MO) unless otherwise noted.
siRNA, DNA constructs, and transfections
All cells were transfected using Lipofectamine 2000 (ThermoFisher Scientific) according to the manufacturer’s protocol. Human JAM-A expressed in pCDNA3.1 was previously described (Naik et al., 2001), and GFP-PH-Akt was a gift from Tamas Balla (Addgene plasmid 51465). Point mutagenesis to generate S284A used the following primers: forward, 5′-AGAAGGTGATTTACAGCCAGCCTGCTGCCCGAAGTGA-3′, and reverse, 5′-TCACTTCGGGCAGCAGGCTGGCTGTAAATCACCTTCT-3′. siRNA target sequences are as follows: GEF-H1 duplex 1 sense, 5′-UUUAAGAGAUCGUAGGCAA-3′; GEF-H1 duplex 2 sense, 5′-AGACAGAGGAUGAGGCUUA-3′; p115 RhoGEF duplex 1 sense, 5′-GGGCUGAGGAUGAGGAUUU-3′; p115 RhoGEF duplex 2 sense, 5′-CCACAGAACGGGAGAAAGU-3′; JAM-A duplex 1 sense, 5′CGAGUAAGAAGGUGAUUUA-3′; JAM-A duplex 2 sense, 5′-AGGCGCAAGUCGAGAGGAA-3′; PKCα duplex, 5′-AAGAAGAAGGUGAGUACUA-3′; and control sense, 5′-UAAGGCUAUGAAGAGAUAC-3′.
Antibodies
The following antibodies were used for Western blot analysis: anti–GEF-H1 (4076), anti-p115RhoGEF (3669), anti-RhoA (2117), anti–phospho-FAK (3283), anti–phospho-Erk1/2 (4370), phospho-Akt (2965), FAK (3285), and ERK (9102) were purchased from Cell Signaling (Danvers, MA), anti-JAM-A (612120) was from BD Transduction Laboratories (Forest Lakes, NJ), and anti–phospho-JAM-A (sc-17430-R) was from Santa Cruz Biotechnology (Dallas, TX). Antibodies against LARG and PDZ-RhoGEF were custom produced by Pocono Rabbit Farms and Laboratories (Canadenesis, PA) and were previously described (Guilluy et al., 2011b). Anti-p190 RhoGEF was a generous gift of David Schlaepfer (University of California at San Diego, La Jolla, CA).
Application of continuous force
Magnetic beads were prepared as previously described (Lessey-Morillon et al., 2014). Briefly, anti–JAM-A clone J10.4 (sc-53623; Santa Cruz Biotechnology) was conjugated to 4.5-μm tosyl-activated Dynabeads (ThermoFisher Scientific) in 0.1 M borate buffer, pH 9.5, according to the manufacturer’s protocol. After overnight incubation at 37°C with rotation, free sites were quenched by incubation with 0.1% fatty acid–free bovine serum albumin (BSA) for 1 h. For biochemical experiments, a continuous force calculated at ∼10 pN was applied to beads using a permanent ceramic magnetic (K&J Magnetics, Jamison, PA) as previously described (Guilluy et al., 2011b).
Detection of cellular stiffening
The same beads used for the application of continuous force were added to cells 10 min before being engaged using the University of North Carolina three-dimensional force microscope. The magnetic tweezers pole tip was positioned 25 μm above the monolayer, and a force regimen of 3 s of 50-pN force followed by 4 s of no force was applied for repeated cycles. Bead movement was captured using a 40× objective (Olympus UplanLN 40×/0.75) on an Olympus IX81-ZDC2 inverted microscope (Olympus, Waltham, MA) equipped with a high-speed Rolera EM-C2 camera (QImaging, Surrey, BC, Canada), using MetaMorph software at 30 frames/s. Bead movements were tracked by Video Spot Tracker (http://cismm.cs.unc.edu). From 11 to 27 beads per condition were tracked from at least two separate experiments. Data are presented as mean ± SEM relative to the first pull for each condition tested.
Shear stress
HUVECs were subjected to shear stress as previously described (Dardik et al., 2005). Briefly, cells were grown in six-well plates to confluence, switched to 3 ml of serum-free medium for 2 h, and then rotated at 210 rpm for the indicated times. Lysates were collected for Western blot analysis.
RhoA activity assay
Cells were lysed for analysis of RhoA (10 mM MgCl2, 500 mM NaCl, 50 mM Tris, pH 7.6, 1% Triton X-100, 0.1% SDS, 0.5% deoxycholate, 1 mM phenylmethylsulfonyl fluoride [PMSF], and 10 μg/ml aprotinin and leupeptin) and cleared at 14,000 × g for 5 min. Lysates were incubated with 50 μg of glutathione-Sepharose–bound glutathione S-transferase (GST)–RBD (Rhotekin-binding domain) for 30 min at 4°C with gentle rocking. Beads were then washed three times in 50 mM Tris, pH 7.6, 10 mM MgCl2, 150 mM NaCl, 1% Triton X-100, 1 mM PMSF, and 10 μg/ml aprotinin and leupeptin. Released proteins and reserved input control were subjected to Western blot analysis as described later.
GEF activity assay
Active RhoA GEFs were assayed using GST-RhoA G17A as described previously (Guilluy et al., 2011a). Cells were lysed in 150 mM NaCl, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.6, 10 mM MgCl2, 1% Triton X-100, 1 mM PMSF, and 10 μg/ml aprotinin and leupeptin and incubated with 50 μg/ml glutathione-Sepharose–bound GST-RhoAG17A for 60 min at 4°C and washed in the lysis buffer. Samples were then analyzed by Western blotting as described later.
Barrier function analysis
Cells were seeded (5 × 104 cells/well) onto fibronectin-coated (10 μg/ml) 0.4-μm polycarbonate Transwell membranes (Corning). Forty-eight hours after plating, fluorescein isothiocyanate (FITC)–dextran (10 kDa; Sigma-Aldrich) at a final concentration of 1 mg/ml was added to the upper chamber. After 2 h of incubation, medium from the bottom chamber was collected. Medium was transferred to a black-walled 96-well microtiter plate (Corning), and fluorescence intensity was analyzed using a plate reader (excitation 485 nm, emission 520 nm; Tecan).
Western blotting
Samples were resolved on 12% (RhoA) or 8% (all other proteins) polyacrylamide gels (Sambrook and Russell, 2001) in the presence of SDS. Resolved gels were transferred onto nitrocellulose membranes, blocked with 5% BSA in Tris-buffered saline (25 mM Tris, pH 7.6, 150 mM NaCl) plus 0.1% Tween-20 (TBST) and incubated with primary antibody overnight at 4°C with gentle rocking. Blots were washed extensively in TBST before being incubated with species-appropriate horseradish peroxidase–conjugated secondary antibody (Jackson Laboratories) for 1 h at room temperature. Blots were again washed in TBST, and fluorescence was detected using enhanced chemiluminescent reagent (ThermoFisher Scientific) and x-ray film.
PI3K activation
Two days after transfection, cells were plated on fibronectin-coated (10 μg/ml) glass coverslips for 6 h before being incubated with beads. Some cells experienced force for 1 min. Cells were fixed in 4% paraformaldehyde, and images were captured on a Zeiss Axiovert 200 M microscope equipped with a Hamamatsu ORCA-ERAG digital camera.
Statistical analysis
For analysis of cell stiffness, Student’s t test between pulse 1 and each subsequent pulse was calculated. For GFP-PH localization, Student’s t test between beads only and beads plus force was calculated for each construct or ligand. In all instances, p < 0.05 was considered significant, and all calculations were conducted in GraphPad Prism 5.1.
Supplementary Material
Acknowledgments
We thank Richard Superfine and Timothy O’Brien, III for access and assistance with magnetic tweezers experiments. This work was supported by American Heart Association Postdoctoral Fellowship 15POST24470070 to D.W.S. and National Institutes of Health Grants GM029860, GM103723, and HL114388 to K.B.
Abbreviations used:
- FAK
focal adhesion kinase
- GEF
guanine nucleotide exchange factor
- HUVEC
human umbilical vein endothelial cell
- JAM-A
junctional adhesion molecule-A
- PI3K
phosphoinositide 3-kinase
- ROCK
Rho-associated protein kinase/Rho-associated, coiled-coil–containing protein kinase
- SFK
Src family kinase.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-12-0833) on March 16, 2016.
REFERENCES
- Alessandri-Haber N, Dina OA, Joseph EK, Reichling DB, Levine JD. Interaction of transient receptor potential vanilloid 4, integrin, and SRC tyrosine kinase in mechanical hyperalgesia. J Neurosci. 2008;28:1046–1057. doi: 10.1523/JNEUROSCI.4497-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aurrand-Lions M, Duncan L, Ballestrem C, Imhof BA. JAM-2, a novel immunoglobulin superfamily molecule, expressed by endothelial and lymphatic cells. J Biol Chem. 2001a;276:2733–2741. doi: 10.1074/jbc.M005458200. [DOI] [PubMed] [Google Scholar]
- Aurrand-Lions M, Johnson-Leger C, Wong C, Du Pasquier L, Imhof BA. Heterogeneity of endothelial junctions is reflected by differential expression and specific subcellular localization of the three JAM family members. Blood. 2001b;98:3699–3707. doi: 10.1182/blood.v98.13.3699. [DOI] [PubMed] [Google Scholar]
- Barry AK, Wang N, Leckband DE. Local VE-cadherin mechanotransduction triggers long-ranged remodeling of endothelial monolayers. J Cell Sci. 2015;128:1341–1351. doi: 10.1242/jcs.159954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001;104:441–451. doi: 10.1016/s0092-8674(01)00231-8. [DOI] [PubMed] [Google Scholar]
- Bazellieres E, Conte V, Elosegui-Artola A, Serra-Picamal X, Bintanel-Morcillo M, Roca-Cusachs P, Munoz JJ, Sales-Pardo M, Guimera R, Trepat X. Control of cell-cell forces and collective cell dynamics by the intercellular adhesome. Nat Cell Biol. 2015;17:409–420. doi: 10.1038/ncb3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake RA, Broome MA, Liu X, Wu J, Gishizky M, Sun L, Courtneidge SA. SU6656, a selective src family kinase inhibitor, used to probe growth factor signaling. Mol Cell Biol. 2000;20:9018–9027. doi: 10.1128/mcb.20.23.9018-9027.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JA, Schelling P, Wetzel JD, Johnson EM, Forrest JC, Wilson GA, Aurrand-Lions M, Imhof BA, Stehle T, Dermody TS. Junctional adhesion molecule a serves as a receptor for prototype and field-isolate strains of mammalian reovirus. J Virol. 2005;79:7967–7978. doi: 10.1128/JVI.79.13.7967-7978.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, Reichel CA, Krombach F, Pardi R, Dejana E. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci. 2009;122:268–277. doi: 10.1242/jcs.037127. [DOI] [PubMed] [Google Scholar]
- Chaturvedi LS, Marsh HM, Basson MD. Src and focal adhesion kinase mediate mechanical strain-induced proliferation and ERK1/2 phosphorylation in human H441 pulmonary epithelial cells. Am J Physiol Cell Physiol. 2007;292:C1701–C1713. doi: 10.1152/ajpcell.00529.2006. [DOI] [PubMed] [Google Scholar]
- Chiu YJ, McBeath E, Fujiwara K. Mechanotransduction in an extracted cell model: Fyn drives stretch- and flow-elicited PECAM-1 phosphorylation. J Cell Biol. 2008;182:753–763. doi: 10.1083/jcb.200801062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong YP, Mulhern TD, Cheng HC. C-terminal Src kinase (CSK) and CSK-homologous kinase (CHK)–endogenous negative regulators of Src-family protein kinases. Growth Factors. 2005;23:233–244. doi: 10.1080/08977190500178877. [DOI] [PubMed] [Google Scholar]
- Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–1415. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C, Guilluy C, Welch C, O’Brien ET, Hahn K, Superfine R, Burridge K, Tzima E. Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr Biol. 2012;22:2087–2094. doi: 10.1016/j.cub.2012.08.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardik A, Chen L, Frattini J, Asada H, Aziz F, Kudo FA, Sumpio BE. Differential effects of orbital and laminar shear stress on endothelial cells. Eur J Vasc Surg. 2005;41:869–880. doi: 10.1016/j.jvs.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Disatnik MH, Boutet SC, Lee CH, Mochly-Rosen D, Rando TA. Sequential activation of individual PKC isozymes in integrin-mediated muscle cell spreading: a role for MARCKS in an integrin signaling pathway. J Cell Sci. 2002;115:2151–2163. doi: 10.1242/jcs.115.10.2151. [DOI] [PubMed] [Google Scholar]
- Dovas A, Yoneda A, Couchman JR. PKCbeta-dependent activation of RhoA by syndecan-4 during focal adhesion formation. J Cell Sci. 2006;119:2837–2846. doi: 10.1242/jcs.03020. [DOI] [PubMed] [Google Scholar]
- Dubash AD, Wennerberg K, Garcia-Mata R, Menold MM, Arthur WT, Burridge K. A novel role for Lsc/p115 RhoGEF and LARG in regulating RhoA activity downstream of adhesion to fibronectin. J Cell Sci. 2007;120:3989–3998. doi: 10.1242/jcs.003806. [DOI] [PubMed] [Google Scholar]
- Fiore VF, Strane PW, Bryksin AV, White ES, Hagood JS, Barker TH. Conformational coupling of integrin and Thy-1 regulates Fyn priming and fibroblast mechanotransduction. J Cell Biol. 2015;211:173–190. doi: 10.1083/jcb.201505007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujishiro SH, Tanimura S, Mure S, Kashimoto Y, Watanabe K, Kohno M. ERK1/2 phosphorylate GEF-H1 to enhance its guanine nucleotide exchange activity toward RhoA. Bioche Biophys Res Commun. 2008;368:162–167. doi: 10.1016/j.bbrc.2008.01.066. [DOI] [PubMed] [Google Scholar]
- Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol. 2006;406:425–437. doi: 10.1016/S0076-6879(06)06031-9. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, Henrion D, Scalbert E, Bril A, Torres RM, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16:183–190. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Dubash AD, Garcia-Mata R. Analysis of RhoA and Rho GEF activity in whole cells and the cell nucleus. Nat Protoc. 2011a;6:2050–2060. doi: 10.1038/nprot.2011.411. [DOI] [PubMed] [Google Scholar]
- Guilluy C, Swaminathan V, Garcia-Mata R, O’Brien ET, Superfine R, Burridge K. The Rho GEFs LARG and GEF-H1 regulate the mechanical response to force on integrins. Nat Cell Biol. 2011b;13:722–727. doi: 10.1038/ncb2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heemskerk N, van Rijssel J, van Buul JD. Rho-GTPase signaling in leukocyte extravasation: an endothelial point of view. Cell Adh Migr. 2014;8:67–75. doi: 10.4161/cam.28244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo KS, Lee H, Nigro P, Thomas T, Le NT, Chang E, McClain C, Reinhart-King CA, King MR, Berk BC, et al. PKCzeta mediates disturbed flow-induced endothelial apoptosis via p53 SUMOylation. J Cell Biol. 2011;193:867–884. doi: 10.1083/jcb.201010051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JY, Xu YY, Sun Z, Wang ZN, Zhu Z, Song YX, Luo Y, Zhang X, Xu HM. Low junctional adhesion molecule A expression correlates with poor prognosis in gastric cancer. J Surg Res. 2014;192:494–502. doi: 10.1016/j.jss.2014.06.025. [DOI] [PubMed] [Google Scholar]
- Hughan SC, Spring CM, Schoenwaelder SM, Sturgeon S, Alwis I, Yuan Y, McFadyen JD, Westein E, Goddard D, Ono A, et al. Dok-2 adaptor protein regulates the shear-dependent adhesive function of platelet integrin alphaIIbbeta3 in mice. J Biol Chem. 2014;289:5051–5060. doi: 10.1074/jbc.M113.520148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, Robenek H, Suzuki A, Ebnet K. aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol. 2012;196:623–639. doi: 10.1083/jcb.201104143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic A, Sheetz MP. Fibronectin rigidity response through Fyn and p130Cas recruitment to the leading edge. Mol Biol Cell. 2006;17:2684–2695. doi: 10.1091/mbc.E05-12-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lakshmi SP, Reddy AT, Naik MU, Naik UP, Reddy RC. Effects of JAM-A deficiency or blocking antibodies on neutrophil migration and lung injury in a murine model of ALI. Am J Physiol Lung Cell Mol Physiol. 2012;303:L758–L766. doi: 10.1152/ajplung.00107.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessey EC, Guilluy C, Burridge K. From mechanical force to RhoA activation. Biochemistry. 2012;51:7420–7432. doi: 10.1021/bi300758e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessey-Morillon EC, Osborne LD, Monaghan-Benson E, Guilluy C, O’Brien ET, Superfine R, Burridge K. The RhoA guanine nucleotide exchange factor, LARG, mediates ICAM-1-dependent mechanotransduction in endothelial cells to stimulate transendothelial migration. J Immunol. 2014;192:3390–3398. doi: 10.4049/jimmunol.1302525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Sniadecki NJ, Chen CS. Mechanical forces in endothelial cells during firm adhesion and early transmigration of human monocytes. Cell Mol Bioeng. 2010;3:50–59. doi: 10.1007/s12195-010-0105-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell KJ, McCall IC, Parkos CA. Involvement of the junctional adhesion molecule-1 (JAM1) homodimer interface in regulation of epithelial barrier function. J Biol Chem. 2004;279:16254–16262. doi: 10.1074/jbc.M309483200. [DOI] [PubMed] [Google Scholar]
- Marjoram RJ, Lessey EC, Burridge K. Regulation of RhoA activity by adhesion molecules and mechanotransduction. Curr Mol Med. 2014;14:199–208. doi: 10.2174/1566524014666140128104541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, et al. Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol. 1998;142:117–127. doi: 10.1083/jcb.142.1.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mas VM, Hernandez H, Plo I, Bezombes C, Maestre N, Quillet-Mary A, Filomenko R, Demur C, Jaffrezou JP, Laurent G. Protein kinase Czeta mediated Raf-1/extracellular-regulated kinase activation by daunorubicin. Blood. 2003;101:1543–1550. doi: 10.1182/blood-2002-05-1585. [DOI] [PubMed] [Google Scholar]
- Matthews BD, Overby DR, Mannix R, Ingber DE. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J Cell Sci. 2006;119:508–518. doi: 10.1242/jcs.02760. [DOI] [PubMed] [Google Scholar]
- McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, Dervan PA, Hopkins AM, Gallagher WM. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer. 2009;125:1343–1351. doi: 10.1002/ijc.24498. [DOI] [PubMed] [Google Scholar]
- Monteiro AC, Luissint AC, Sumagin R, Lai C, Vielmuth F, Wolf MF, Laur O, Reiss K, Spindler V, Stehle T, et al. Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol Biol Cell. 2014;25:1574–1585. doi: 10.1091/mbc.E14-01-0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AC, Parkos CA. Intracellular mediators of JAM-A-dependent epithelial barrier function. Ann NY Acad Sci. 2012;1257:115–124. doi: 10.1111/j.1749-6632.2012.06521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, Cocito A, d’Ario G, Mazzarol G, Confalonieri S, et al. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PloS One. 2011;6:e21242. doi: 10.1371/journal.pone.0021242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Caplan JL, Naik UP. Junctional adhesion molecule-A suppresses platelet integrin alphaIIbbeta3 signaling by recruiting Csk to the integrin-c-Src complex. Blood. 2014;123:1393–1402. doi: 10.1182/blood-2013-04-496232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik UP, Ehrlich YH, Kornecki E. Mechanisms of platelet activation by a stimulatory antibody: cross-linking of a novel platelet receptor for monoclonal antibody F11 with the Fc gamma RII receptor. Biochem J. 1995;310:155–162. doi: 10.1042/bj3100155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik UP, Naik MU, Eckfeld K, Martin-DeLeon P, Spychala J. Characterization and chromosomal localization of JAM-1, a platelet receptor for a stimulatory monoclonal antibody. J Cell Sci. 2001;114:539–547. doi: 10.1242/jcs.114.3.539. [DOI] [PubMed] [Google Scholar]
- Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008;68:2194–2203. doi: 10.1158/0008-5472.CAN-07-3057. [DOI] [PubMed] [Google Scholar]
- Nava P, Capaldo CT, Koch S, Kolegraff K, Rankin CR, Farkas AE, Feasel ME, Li L, Addis C, Parkos CA, et al. JAM-A regulates epithelial proliferation through Akt/beta-catenin signalling. EMBO Rep. 2011;12:314–320. doi: 10.1038/embor.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CM, Pirone DM, Tan JL, Chen CS. Vascular endothelial-cadherin regulates cytoskeletal tension, cell spreading, and focal adhesions by stimulating RhoA. Mol Biol Cell. 2004;15:2943–2953. doi: 10.1091/mbc.E03-10-0745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niediek V, Born S, Hampe N, Kirchgessner N, Merkel R, Hoffmann B. Cyclic stretch induces reorientation of cells in a Src family kinase- and p130Cas-dependent manner. Eur J Cell Biol. 2012;91:118–128. doi: 10.1016/j.ejcb.2011.10.003. [DOI] [PubMed] [Google Scholar]
- Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C. JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol. 2002;3:151–158. doi: 10.1038/ni755. [DOI] [PubMed] [Google Scholar]
- Peddibhotla SS, Brinkmann BF, Kummer D, Tuncay H, Nakayama M, Adams RH, Gerke V, Ebnet K. Tetraspanin CD9 links junctional adhesion molecule-A to alphavbeta3 integrin to mediate basic fibroblast growth factor-specific angiogenic signaling. Mol Biol Cell. 2013;24:933–944. doi: 10.1091/mbc.E12-06-0481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng J, He F, Zhang C, Deng X, Yin F. Protein kinase C-alpha signals P115RhoGEF phosphorylation and RhoA activation in TNF-alpha-induced mouse brain microvascular endothelial cell barrier dysfunction. J Neuroinflammation. 2011;8:28. doi: 10.1186/1742-2094-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R, Gomez GA, Budnar S, Verma S, Cox HL, Hamilton NA, Yap AS. Feedback regulation through myosin II confers robustness on RhoA signalling at E-cadherin junctions. Nat Cell Biol. 2015;17:1282–1293. doi: 10.1038/ncb3239. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2001. [Google Scholar]
- Sarkar S, Bananis E, Nath S, Anwer MS, Wolkoff AW, Murray JW. PKCzeta is required for microtubule-based motility of vesicles containing the ntcp transporter. Traffic. 2006;7:1078–1091. doi: 10.1111/j.1600-0854.2006.00447.x. [DOI] [PubMed] [Google Scholar]
- Schaefer A, Hordijk PL. Cell-stiffness-induced mechanosignaling—a key driver of leukocyte transendothelial migration. J Cell Sci. 2015;128:2221–2230. doi: 10.1242/jcs.163055. [DOI] [PubMed] [Google Scholar]
- Schaefer A, Te Riet J, Ritz K, Hoogenboezem M, Anthony EC, Mul FP, de Vries CJ, Daemen MJ, Figdor CG, van Buul JD, et al. Actin-binding proteins differentially regulate endothelial cell stiffness, ICAM-1 function and neutrophil transmigration. J Cell Sci. 2014;127:4470–4482. doi: 10.1242/jcs.154708. [DOI] [PubMed] [Google Scholar]
- Schmidt A, Hall A. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes Dev. 2002;16:1587–1609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, Rademakers T, van Zandvoort MA, Hackeng TM, Koenen RR, Weber C. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation. 2014;129:66–76. doi: 10.1161/CIRCULATIONAHA.113.004149. [DOI] [PubMed] [Google Scholar]
- Seshacharyulu P, Pandey P, Datta K, Batra SK. Phosphatase: PP2A structural importance, regulation and its aberrant expression in cancer. Cancer Lett. 2013;335:9–18. doi: 10.1016/j.canlet.2013.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Jiang L, Ivanov AI, Mandell KJ, Nusrat A, Parkos CA. Cis-dimerization mediates function of junctional adhesion molecule A. Mol Biol Cell. 2008;19:1862–1872. doi: 10.1091/mbc.E07-09-0869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severson EA, Parkos CA. Mechanisms of outside-in signaling at the tight junction by junctional adhesion molecule A. Ann NY Acad Sci. 2009;1165:10–18. doi: 10.1111/j.1749-6632.2009.04034.x. [DOI] [PubMed] [Google Scholar]
- Sobocka MB, Sobocki T, Babinska A, Hartwig JH, Li M, Ehrlich YH, Kornecki E. Signaling pathways of the F11 receptor (F11R; a.k.a. JAM-1, JAM-A) in human platelets: F11R dimerization, phosphorylation and complex formation with the integrin GPIIIa. J Recept Signal Transduct Res. 2004;24:85–105. doi: 10.1081/rrs-120034252. [DOI] [PubMed] [Google Scholar]
- Stroka KM, Aranda-Espinoza H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase-dependent cell contraction. Blood. 2011;118:1632–1640. doi: 10.1182/blood-2010-11-321125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuma I, Suzuma K, Ueki K, Hata Y, Feener EP, King GL, Aiello LP. Stretch-induced retinal vascular endothelial growth factor expression is mediated by phosphatidylinositol 3-kinase and protein kinase C (PKC)-zeta but not by stretch-induced ERK1/2, Akt, Ras, or classical/novel PKC pathways. J Biol Chem. 2002;277:1047–1057. doi: 10.1074/jbc.M105336200. [DOI] [PubMed] [Google Scholar]
- Switzer CH, Glynn SA, Ridnour LA, Cheng RY, Vitek MP, Ambs S, Wink DA. Nitric oxide and protein phosphatase 2A provide novel therapeutic opportunities in ER-negative breast cancer. Trends Pharmacol Sci. 2011;32:644–651. doi: 10.1016/j.tips.2011.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornavaca O, Chia M, Dufton N, Almagro LO, Conway DE, Randi AM, Schwartz MA, Matter K, Balda MS. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208:821–838. doi: 10.1083/jcb.201404140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuncay H, Brinkmann BF, Steinbacher T, Schurmann A, Gerke V, Iden S, Ebnet K. JAM-A regulates cortical dynein localization through Cdc42 to control planar spindle orientation during mitosis. Nat Commun. 2015;6:8128. doi: 10.1038/ncomms9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijetunge S, Hughes AD. Src family tyrosine kinases mediate contraction of rat isolated tail arteries in response to a hyposmotic stimulus. J Hypertens. 2007;25:1871–1878. doi: 10.1097/HJH.0b013e328255e8f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcikiewicz EP, Koenen RR, Fraemohs L, Minkiewicz J, Azad H, Weber C, Moy VT. LFA-1 binding destabilizes the JAM-A homophilic interaction during leukocyte transmigration. Trends Pharmacol Sci. 2009;96:285–293. doi: 10.1529/biophysj.108.135491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodfin A, Voisin MB, Imhof BA, Dejana E, Engelhardt B, Nourshargh S. Endothelial cell activation leads to neutrophil transmigration as supported by the sequential roles of ICAM-2, JAM-A, and PECAM-1. Blood. 2009;113:6246–6257. doi: 10.1182/blood-2008-11-188375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Liu Z, Li H, Sun J, Chang X, Liu J, He S, Li B. Association of PKCzeta expression with clinicopathological characteristics of breast cancer. PloS One. 2014;9:e90811. doi: 10.1371/journal.pone.0090811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XH, Laschinger C, Arora P, Szaszi K, Kapus A, McCulloch CA. Force activates smooth muscle alpha-actin promoter activity through the Rho signaling pathway. J Cell Sci. 2007;120:1801–1809. doi: 10.1242/jcs.001586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.