

Stacks of nonmuscle myosin IIA filaments form by the expansion of single filaments and concatenation of multiple filaments. Expansion is the dominant mechanism and is characterized by distinct structural steps. It is dependent on both motor activity and actin filament concentration. Expansion and catenation occur in both crawling and dividing cells.
Abstract
Cell movement and cytokinesis are facilitated by contractile forces generated by the molecular motor, nonmuscle myosin II (NMII). NMII molecules form a filament (NMII-F) through interactions of their C-terminal rod domains, positioning groups of N-terminal motor domains on opposite sides. The NMII motors then bind and pull actin filaments toward the NMII-F, thus driving contraction. Inside of crawling cells, NMIIA-Fs form large macromolecular ensembles (i.e., NMIIA-F stacks), but how this occurs is unknown. Here we show NMIIA-F stacks are formed through two non–mutually exclusive mechanisms: expansion and concatenation. During expansion, NMIIA molecules within the NMIIA-F spread out concurrent with addition of new NMIIA molecules. Concatenation occurs when multiple NMIIA-Fs/NMIIA-F stacks move together and align. We found that NMIIA-F stack formation was regulated by both motor activity and the availability of surrounding actin filaments. Furthermore, our data showed expansion and concatenation also formed the contractile ring in dividing cells. Thus interphase and mitotic cells share similar mechanisms for creating large contractile units, and these are likely to underlie how other myosin II–based contractile systems are assembled.
INTRODUCTION
Forces generated by the molecular motor, nonmuscle myosin II (NMII), are essential for cell migration and cytokinesis (Straight et al., 2003; Vicente-Manzanares et al., 2009; Gardel et al., 2010; Ma et al., 2012). Mutations in NMII isoforms lead to an array of human diseases, and changes in NMII activity during processes relying on precise regulation of cell migration and cytokinesis lead to developmental defects and disease progression (Tullio et al., 1997; Kelley et al., 2000; Seri et al., 2000; Hu et al., 2002; Vicente-Manzanares et al., 2009; Beach et al., 2011; Tuzovic et al., 2013; Hamdan et al., 2014; Ma and Adelstein, 2014; Vasquez et al., 2014). Furthermore, increased cellular contractility—which is primarily driven by NMII—is increasingly being recognized as an emergent property of tumor cells (Samuel et al., 2011). Highlighting this, overexpression of the NMIIA isoform is correlated with increased metastasis and poor patient prognosis in a variety of cancers (Ma et al., 2003; Xia et al., 2012). Despite the biological importance of, and wealth of genetic data surrounding NMIIA-associated diseases, how NMIIA-based contractile units form inside of cells is a surprisingly poorly understood process. This lack of understanding precludes a thorough understanding of NMIIA-related disease states and prevents potential therapeutic strategies.
A single NMII molecule is a heterohexamer composed of two heavy chains, two essential light chains, and two regulatory light chains (Vicente-Manzanares et al., 2009). The N-terminal motor domains of the heavy chains contain both the actin-binding and ATPase activity (Adelstein et al., 1971; Vicente-Manzanares et al., 2009). In vitro biochemical and structural studies have elegantly shown that C-terminal electrostatic interactions of the heavy chains create bipolar filaments, positioning the N-terminal motor domains on opposite sides of the filament (Figure 1A, steps 1–4; Pollard, 1975; Ricketson et al., 2010). In this orientation, the motor domains of NMII bipolar filaments can grab onto and contract actin filaments to generate force. Of importance, a single NMII bipolar filament contains multiple NMII molecules, each of which can bind to and contract actin filaments (Billington et al., 2013). The number of NMII molecules added to a NMII filament (NMII-F) is limited by steric hindrance, however, and thus actin-binding and force-generating capabilities of a single NMII-F are limited (Pollard, 1975; Billington et al., 2013). Thus, in order to increase the scale of force generation, cells organize NMII-Fs into larger arrays referred to as “stacks” (Figure 1A, step 5; Verkhovsky and Borisy, 1993; Verkhovsky et al., 1995; Svitkina et al., 1997). The mechanism of NMII stack formation has eluded previous efforts. This is in part due to the relatively small size (∼300 nm in width) of NMII-Fs, making to difficult to obtain structurally dynamic information using conventional imaging modalities (i.e., diffraction-limited microscopy), and because they do not readily form in vitro. Thus current models on NMII stack formation are based on fixed snapshots using electron microscopy, which lack dynamic information (Verkhovsky et al., 1999).
FIGURE 1:

Organization of NMIIA-Fs at the leading edge. (A) Classic model of the formation of a 2-motor-group NMIIA-F. An autoinhibited NMIIA molecule (1) becomes polymerization competent (2) after phosphorylation of its regulatory light chain. Two polymerization-competent NMIIA molecules then form a mini-NMIIA-F (3) through electrostatic interactions of their rod domains. Addition of more NMIIA molecules to the minifilament drives the growth of a 2-motor-group NMIIA-F (4). Steric hindrance limits the number of molecules that can be added to a 2-motor-group NMIIA-F (Billington et al., 2013). Note that a smaller number of myosins than the ∼15 on each side of the filament are drawn for simplicity. Of importance, how larger contractile arrays (i.e., stacks) form containing more myosins capable of generating more contractile forces is not understood (5). (B) Time-lapse wide-field epifluorescence recording of the motor domains, NMIIA-(N-terminal)-mEGFP, at the leading edge of a U2-OS cell. (C) Representative montage of NMIIA-F in the box expanding into a stack. (D) SIM of U2-OS cell expressing NMIIA heavy chain fused to mEmerald-N-terminal (magenta) and mApple-C-terminal (green). (E) High-magnification views of 2-, 3-, and 2-motor-group NMIIA-Fs (1–3, respectively). (F) Representative examples of 2-, 3-, 4-, and >4-motor-group NMIIA-Fs. (G) Representative images of the ∼30% of filaments that were too close (i.e., overlapping) for analysis. (H) Percentage of endogenous NMIIA-Fs (RLC and NMIIA antibodies) in a 2-, 3-, 4-, or >4-motor-group organization at the edge of the cell. Total of 246 NMIIA-Fs from 37 cells over three experiments. (I) X/Y and Z/X views of 2-motor-group NMIIA-Fs imaged with 3D PALM. (J) X/Y and Z/X views of 3-motor-group NMIIA-Fs imaged with 3D PALM. Molecular probability refers to the cumulative probability per unit volume (nm3) of all single molecules (mEOS2-NMIIA) detected within any given motor group (cluster of single-molecule localizations). The certainty for the location of each probe in a given image frame depends on the number of photons detected for each mEOS2 molecule and the background parameters of the specimen and camera (Betzig et al., 2006). Scale bars, 5 μm (B), 2 μm (D), 200 nm (E–G), 100 nm (I–J). Error bars in H represent SEM.
To address these issues, we took advantage of recently developed live-cell superresolution imaging techniques to investigate the dynamic mechanism of NMII stack formation. Of interest, we found that at the leading edge of crawling cells, NMII stacks predominantly form via expansion and concatenation of smaller NMII filaments, which is in contrast to current models of NMII stack formation (Verkhovsky et al., 1999). This expansion depends on NMII motor activity and actin filament availability. Intriguingly, NMII expansion and concatenation also underlie the formation of the contractile ring in dividing cells. Thus we present a new model of NMIIA stack formation that underlies the formation of contractile units in distinct cellular processes and cell types.
RESULTS
We used the flat, leading edge of motile U2-OS cells (human osteosarcoma) as a model system to define the structural dynamics underlying NMII-F stack formation. NMII-Fs comprising the NMII isoform NMIIA are formed at the leading edge of cells and then move away from the edge in a process called retrograde flow, making room for new NMIIA-Fs to form (Verkhovsky et al., 1995; Hotulainen and Lappalainen, 2006; Vallenius, 2013). This continuous treadmill allowed us to monitor NMIIA-F formation using high-resolution, wide-field fluorescence time-lapse microscopy (Figure 1B and Supplemental Video S1). Examination of the data revealed that newly formed NMIIA-Fs increased in intensity over time (Figure 1C). This is consistent with NMIIA molecules being added to the NMIIA-F. The increase in intensity was followed by a growth in size of the NMIIA-F (Figure 1C). Conventional fluorescence imaging was not able, however, to resolve the underlying structural changes during the expansion of NMIIA-Fs. Thus we turned to structured illumination microscopy (SIM; Gustafsson et al., 2008), which has a resolution (∼110 nm) that can separate the motor and rod domains in the NMIIA-F (Burnette et al., 2014).
To study the structure of NMIIA-Fs during expansion, we visualized motor and rod domains with SIM using two different localization methods. First, we exogenously expressed NMIIA heavy chain fused to mEmerald on the N-terminus and mApple on the C-terminus (Burnette et al., 2014). This construct allowed us to image the motor and rod domains, respectively (Figure 1D). We also localized endogenous NMII-Fs with antibodies specific to the NMIIA rod domain and myosin regulatory light chain (RLC; Supplemental Figure S1). RLC is positioned in the neck region of NMIIA and was not distinguishable from a fluorescent protein tag on the N-terminus of the heavy chain using SIM (Supplemental Figure S1). Of importance, our data revealed similar NMIIA-F organization as previously described by electron microscopy studies (Verkhovsky and Borisy, 1993; Shutova et al., 2012). Thus we could use the relative localization of motor and rod domains to group together structurally similar NMIIA-Fs.
We defined the structure of an individual NMIIA-F by measuring the number of discrete “motor-groups” that could be resolved by SIM (Figure 1, E and F, and Supplemental Figure S1-1). We use the term “motor-groups” because each contained multiple NMIIA motor domains, which were required for detection, as our SIM microscope did not have single-molecule imaging sensitivity. In fixed cells, we found that the majority (71.1 ± 11.0%) of individual NMIIA-Fs at the leading edge fell into one of four structural categories: 2-, 3-, 4-, and >4-motor-group NMIIA-Fs. Morphologies of the remaining ∼30% of NMIIA-Fs were difficult to define mainly because they overlapped with other NMIIA-Fs (Figure 1G). Of the individually resolvable NMIIA-Fs, 2-motor-group NMIIA-Fs were the most common (35.0 ± 1.7%) and had one motor-group on each side of the rod domains (Figure 1, E, 1 and 3, F, and H). Of interest, the second-most-common organization was an asymmetric 3-motor-group NMIIA-F (25.2 ± 1.4%), which had two motor groups on one side of the rod domains and one on the other side (Figure 1, E, 2, F, and H). In contrast, 4-motor-group NMIIA-Fs (19.8 ± 1.6%) were relatively symmetrical, with two motor groups on each side of the rod domain (Figure 1, F and H).
FIGURE 3:

NMIIA-F expansion requires motor activity. (A) Immunolocalized RLC showing motor-group distribution and NMIIA showing rod-domain localization in control and cells treated with 500 nM, 5 μM, and 50 μM blebbistatin for 1 h. (B–E) Distribution of 2-motor-groups (B), 3-motor-groups (C), 4-motor-groups (D), and >4-motor-groups (E) in cells treated with increasing amounts of blebbistatin. Control: 225 NMIIA-Fs, 37 cells, three experiments; 500 nM blebbistatin: 421 NMIIA-Fs, 27 cells, three experiments; 5 μM blebbistatin: 435 NMIIA-Fs, 32 cells, three experiments; 50 μM blebbistatin: 237 NMIIA-Fs, 20 cells, three experiments. See Materials and Methods and Supplemental Figure S3-1 for a detailed description of the analysis. (F) Length of NMIIA-F stacks as measured from the NMIIA rod-domain localization in cells treated with increasing amounts of blebbistatin and 10 μM Y-27632 (ROCK inhibitor). Control: 9145 NMIIA-Fs, 48 cells, three experiments; 500 nM blebbistatin: 5807 NMIIA-Fs, 38 cells, three experiments; 5 μM blebbistatin: 11049 NMIIA-Fs, 48 cells, three experiments; 50 μM blebbistatin: 1873 NMIIA-Fs, 37 cells, three experiments; 10 μM Y-27632: 1357 NMIIA-Fs, 28 cells, three experiments. See Materials and Methods and Supplemental Figure S3-2 for a detailed description of the analysis. (G) RLC/NMIIA rod domains in a cell treated with 10 μM Y-27632 for 1 h. (H) Density of NMIIA-Fs. Numbers are the same as in F. (I) Western blotting showing the absence of NMIIA from Hap1-knockout cells compared with control (Supplemental Figure S3-3). (J) NMIIA rod domains localized in Hap1-knockout cells expressing wild-type or N93K NMIIA. (K) Length of NMIIA-F stacks in Hap1-knockout cells transfected with wild-type or N93K NMIIA. (L) Density of NMIIA-Fs in Hap1-knockout cells transfected with wild-type or N93K NMII. *p < 0.001 and #p < 0.05 compared with control. Scale bars, 2 μm. Error bars in B–F, H, K, and L indicate SEM.
FIGURE 2:

NMIIA-Fs expand into stacks. (A) Cartoon of the network contraction model showing a 3-motor-group NMII-F transitioning into a 2-motor-group NMII-F by walking toward the barbed ends of actin filaments (adapted from Figure 7 of Verkhovsky et al., 1999). (B) Time montage from a live-cell SIM acquisition of NMIIA-(N-terminal)-mEmerald showing a 2-motor-group NMIIA-F expanding into a 3-motor-group and collapsing into a 2-motor-group NMIIA-F. (C) Percentage of collapsing (i.e., network contracting) NMIIIA-Fs. (D) Time montage of NMIIA-(N-terminal)-mEmerald showing a 2-motor-group NMIIA-F expanding into a 3-motor-group and then into a 4-motor-group NMIIA-F. (E) Percentage of 2-motor-group NMIIA-Fs that expand. (F) Time montage of NMIIA-(N-terminal)-mEmerald from a higher–temporal resolution acquisition, showing intermediate structures during NMIIA-F expansion. Yellow arrowhead denotes transition between 2-motor-group (open) and 3-motor-group NMIIA-Fs (closed). Open blue arrowhead denotes transition between 3-motor-group and 4-motor-group NMIIA-Fs (closed blue arrowhead). (G) Time montage from a dual-color acquisition of NMIIA-(N-terminal)-mEmerald/(C-terminal)-mApple showing that rod domains expand concurrently with the initial asymmetric expansion of the motor domains (asterisks). (H) Photoconverted (red) and unconverted (green) NMIIA-mEOS2 at 0 and 2 min after conversion of the leading edge (white dashed line). Time montage from the yellow box shows the unconverted (green), converted (red), and overlay. Arrowheads denote expanding NMIIA-F. Note that the converted NMIIA-mEOS2 (red) spread out during expansion. (I) Time montage showing unconverted NMIIA-mEOS2 (green) and a single converted NMIIA-mEOS2 molecule (red) localized with sptPALM. Yellow dashed line shows the boundary of NMIIA-Fs, and cross-hairs denote the centroid of the NMIIA-F. The molecule moved 353 ± 25 nm within the NMIIA-F. The average velocity of converted NMIIA-mEOS2 molecules was 0.75 ± 0.25 μm/min (∼12.5 nm/s). (J) Quantification of the number of molecules that moved in NMIIA-Fs. (K) Montage showing a concatenation of expanding NMIIA-Fs into a larger stack. (L) Quantification of the percentage of concatenation events 2 μm from the edge. For C, E, and L, there were 211 NMIIA-Fs from 32 cells over five independent experiments. For I–J, there were 18 NMIIA molecules from 11 cells over three independent experiments. *p < 0.001. Scale bars, 200 nm (B, D, F, G, I, K), 5 μm (H, low magnification), 1 μm (H, high magnification). Live-cell 3D SIM data were acquired by taking four images with 125-nm Z-steps and creating a maximum projection of each time point. All images are oriented with leading edge toward the top. Error bars in C, E, J, and L indicate SEM.
Several of these NMIIA-F organizations were reported by electron microscopy (EM) studies of fixed cells (Verkhovsky and Borisy, 1993; Verkhovsky et al., 1995; Shutova et al., 2012). Of note, EM showed the existence of the 3-motor-group NMIIA-Fs in both crawling cells and in vitro in a variety of salt and ATP concentrations (Verkhovsky et al., 1995; Billington et al., 2013). Of importance, these EM data show that in the 3-motor-group orientation, NMII molecules of the same 3-motor-group filament are “linked” via interacting motor domain groups, which is consistent with our fluorescence data. However, there was still the possibility that the 3-motor-group NMIIA-F in our SIM data was not a single unit but was two distinct filaments. Previous EM studies yielded inherently two-dimensional (2D) views of NMII-Fs, and the lateral and axial resolution of our three-dimensional (3D) SIM data (∼110 and ∼250 nm, respectively) might not be able to resolve two NMIIA-Fs close together, which also might result in an overestimation of the percentage of 3-motor-group filaments in our cells. Therefore we turned to 3D PALM (Betzig et al., 2006; Brown et al., 2011) of U2-OS cells expressing NMIIA fused to mEOS2 on its N-terminus (i.e., motor). Our image strategy yielded average single-molecule localization precisions of μx = 11 ± 5 nm and μz = 20 ± 11 nm (Supplemental Figures S1-2A), which afforded us higher spatial resolution than SIM to test whether the 3-motor-group filaments indeed had three groups of motors as opposed to four if they were composed of two distinct filaments. We observed that the groups of motors in the 2-motor-group NMIIA-Fs were similar in dimension to those previously shown by 2D PALM (Burnette et al., 2014; Figure 1I and Supplemental Figure S1-2B). Of importance, 3D PALM revealed isolated 3-motor-group localization clusters spaced by similar dimensions as 2-motor-group clusters. Each motor cluster visualized by 3D PALM displayed a unimodal X, Y, and Z distribution, consistent with previous EM evidence and the interpretation of our SIM data (Figure 1H and Supplemental Figure S1-2C).
The 3-motor-group filaments were the basis of previously proposed models of NMII stack formation (Verkhovsky et al., 1999). Previous studies proposed a spatial and temporal relationship between “open” NMII-Fs like the 3-motor-group and “closed” NMII-Fs like the 2-motor-group NMII-Fs to explain how actin filament networks contract (i.e., actin filaments being moved closer together; Verkhovsky et al., 1999). The “network contraction” model predicts that a 3-motor-group NMIIA-F would transition into a 2-motor-group NMIIA-F similar to the way in which a pair of scissors closes (Figure 2A; Verkhovsky et al., 1999). In theory, the two motor groups on the larger side of the 3-motor-group would pull different actin filaments together. Whether this type of transition occurs inside of cells or contributes to NMIIA-F expansion was unknown. Therefore we turned to live-cell SIM to monitor the structural and dynamic changes during NMIIA-F stack formation (Supplemental Video S2).
We imaged NMIIA motor domains and found that 2-motor-group NMIIA-Fs unfolded to become 3-motor-group NMIIA-Fs (Figure 2, B and D). Surprisingly, only a small percentage (5.3 ± 2.5%) of 3-motor-group NMIIA-Fs went back or “collapsed” to being a 2-motor-group NMIIA-F as the “network contraction” model predicted (Figure 2, B and C). The vast majority (85.8 ± 4.1%) of newly formed NMIIA-Fs expanded (Figure 2, D and E). These data indicated that the 2-, 3-, and 4-motor-groups represented distinct structural steps underlying the formation of NMIIA-F stacks. We next wanted to explore the physical mechanisms underlying stack formation.
By increasing our sampling rate to 15-s intervals, we were able to detect transitional structures between 2- to 3-motor group and 3- to 4-motor-group NMIIA-Fs (Figure 2, F and G). New motor-groups did not simply appear next to preexisting motor-groups. Instead, a preexisting motor-group extended (Figure 2, F and G, open yellow arrowheads) before the appearance of a new motor-group (Figure 2, F and G, closed yellow arrowheads). These data led us to hypothesize that a subset of NMIIA molecules were moving away from one side of the 2-motor-group NMIIA-F. We also detected this type of extension during the transition from a 3- to a 4-motor-group NMIIA-F (Figure 2G, blue arrowheads). In addition, rod domains extended, and this occurred with the extension of the first motor-group of a 2-motor-group NMIIA-F (Figure 2G, asterisk, and Supplemental Video S3).
We next used photoconversion of NMII-A-mEOS2 (Burnette et al., 2014), labeled on the N-terminal motor domains, to test whether NMIIA molecules within filaments were spreading out in space as the extension of the motor-groups revealed by SIM (Figure 2, F and G) suggested. EOS is a green fluorescent protein, which can be photoconverted into red by exposure to ultraviolet (UV) light (Wiedenmann et al., 2004). Using this method, we converted NMIIA-mEOS2 at the edge of cells and thus specifically marked NMIIA molecules that were incorporated into NMIIA-Fs (Figure 2H, red channel). A subset of NMIIA-mEOS2 molecules were not converted and allowed us to monitor the expansion of stacks in the green channel (Figure 2H, arrowheads). Note that the converted pool of NMIIA-mEOS2 also spread out during expansion (Figure 2H, arrowheads), which is consistent with NMIIA molecules within the NMIIA-F spreading out in space. An alternative interpretation of this result is that converted NMIIA-mEOS2 molecules are being removed from NMIIA-Fs or photoconverted in the cytoplasmic pool and then being added to expanding stacks. Although this seems unlikely, as we did not detect incorporation of red-labeled NMIIA-mEOS2 in NMIIA-F outside of the region of conversion during the short time frame of the experiment, we still wanted to confirm with another method that NMIIA molecules could change position within individual NMIIA-Fs. In addition, it is important to point out that, because this was done on a wide-field (i.e., diffraction-limited) microscope, we can only confidently detect expansion of the larger NMIIA filaments, such as those shown in Figure 2H.
To test further whether NMIIA molecules could move within the NMIIA-F, we monitored the precise movements of single molecules of NMIIA-mEOS2 (N-terminal motor domain) within NMIIA-Fs, using single-particle tracking photoactivated localization microscopy (sptPALM; Betzig et al., 2006; Manley et al., 2008; Figure 2, I and J). sptPALM allows for the precise localization and tracking of a single molecule which photoconverted from green into red. Using sptPALM, we converted single NMIIA-mEOS2 molecules within NMIIA-Fs at the leading edge of cells to test whether individual NMIIA molecules could spread out in space within an NMIIA-F. To measure this, we drew a mask around the perimeter of a diffraction-limited image of the unconverted mEOS2 molecules (i.e., green channel) within an NMIIA-F for every image in the acquisition. We then tracked the movement of a single converted NMIIA-mEOS2 molecule within this NMIIA-F. Any molecule that moved a diffraction-limited distance (i.e., 250 nm) within the NMIIA-F mask was counted as moving. Indeed, sptPALM data revealed that 32.8 ± 4.3% of single NMIIA-mEOS2 molecules changed their position within their respective NMIIA-F (Figure 2, I and J). Of interest, of the moving single NMIIA-mEOS2 molecules, our analysis revealed that all but one moved relatively closer to the leading edge by moving more slowly than the respective expanding NMIIA-F (Figure 2I). Taken together, our SIM, photoconversion, and PALM results show that the creation of stacks from NMIIA-Fs involved NMIIA molecules spreading out from preexisting 2-motor-group NMIIA-Fs. We next asked whether NMIIA-F stacks are formed by other mechanisms.
In vitro experiments have shown that rabbit skeletal myosin II can align into stack-like orientations in the presence of actin filaments. Mathematical modeling suggested that this could arise as a result of distinct myosin II filaments coming closer together by contracting actin filaments (i.e., concatenation; Stachowiak et al., 2012). To test whether concatenation occurs inside of a living cell, we quantified how many NMIIA-Fs moved together (Figure 2, K and L). We found that ∼10% of NMIIA-Fs concatenated with other NMIIA-Fs, and this was independent of whether the NMIIA-Fs were expanding (Figure 2L). Concatenation was defined as two separate NMIIA-Fs aligning their rod domains within 4 pixels (160 nm) of each other (Figure 2K). Thus, although concatenation did occur at a low rate at the leading edge, most NMIIA-F stacks arose from expansion. However, our data do not preclude network contraction and/or concatenation as dominant mechanisms of NMIIA-F dynamics in parts of the cell other than the leading edge where NMIIA-F densities were too high for us to resolve individual NMIIA-Fs.
We next wanted to test the molecular mechanisms driving NMIIA-F stack formation. Our data show that both expansion and concatenation involve the movement of NMIIA-Fs, and movement of NMII-Fs in crawling cells is dependent on NMII motor activity (Wilson et al., 2010). Therefore we tested whether motor activity is required for stack formation, by inhibiting the ATPase activity of the NMII motor domain using increasing concentrations of blebbistatin (Straight et al., 2003; Figure 3A). Cells treated with 5 and 50 μM blebbistatin showed an increased percentage of 2-motor-group NMIIA-Fs compared with control cells (Figure 3B), whereas the percentage of 3-, 4-, and >4-motor-group NMIIA-Fs decreased (Figure 3, C, D, and E). Using a low concentration of blebbistatin, 500 nM, we observed that the percentage of larger NMIIA-F groups (4- and >4-motor-groups) declined (Figure 3, D and E), the 3-motor-group NMIIA-F percentage remained similar (Figure 3C), and the 2-motor-group NMIIA-F percentage increased with respect to control cells (Figure 3B). Thus NMIIA-F expansion depended on NMII motor activity (i.e., either binding to or generating force on actin filaments). Furthermore, even in low concentrations of blebbistatin, NMIIA-Fs could expand, but not to the same extent as in cells that maintained full contractile potential.
The decrease in 4- and >4-motor-groups suggested that NMIIA-F stack formation was inhibited by blebbistatin. Therefore we directly quantified stack lengths by measuring the length of the rod domains as localized with an NMIIA-specific antibody (Figure 3F). Consistent with the measured decrease in 4- and >4-motor-group filaments, we found that stack lengths decreased with increasing amounts of blebbistatin. We also inhibited Rho-associated kinase (ROCK) with 10 μM Y-27632 (Uehata et al., 1997), which indirectly reduces NMII contractility, and found a similar decrease in stack lengths as with 50 μM blebbistatin (Figure 3, F and G). Of interest, the ability to form NMIIA-Fs (e.g., 2-motor-groups) was less sensitive to NMII ATPase inhibition than NMIIA stack expansion. For example, we found that 5 μM blebbistatin did not significantly reduce the density of NMIIA-Fs even though it did reduce overall NMIIA-F stack lengths (Figure 3, F and H). Of importance, we found the widths of NMIIA-Fs (i.e., motor-group to motor-group across the rod domain) to be similar in all experimental conditions. This is not surprising, as the widths of NMIIA filaments in vitro are also extremely similar (Billington et al., 2013).
To further test whether motor activity is required for NMIIA-F stack formation, we expressed either wild-type or a N93K mutant NMIIA–monomeric enhanced green fluorescent protein (mEGFP) in a human fibroblast–like cell line with its endogenous NMIIA knocked out with clustered regularly-interspaced short palindromic repeats (CRISPR)/Cas9 (Figure 3, I–L). The N93K mutant has the ability to bind actin filaments but has reduced motor activity compared with wild type (Hu et al., 2002; Vicente-Manzanares et al., 2007). Consistent with motor activity driving stack formation, we found that the N93K mutant NMIIA created shorter stacks, with a slight reduction in filament density (Figure 3, K and L). Given that NMIIA motors pull on actin filaments, we next tested whether the availability of actin filaments could also regulate stack formation.
To test whether the availability of actin filaments regulated NMIIA stack formation, we sought a way to reduce actin filament density. We treated cells with cytochalasin B (CB), a molecule that binds to the plus end of actin filaments and thus reduces the rate of actin filament polymerization (Smith et al., 1967; MacLean-Fletcher and Pollard, 1980). High concentrations of CB (1–5 μM) can inhibit the majority of actin filament polymerization and result in the removal of most of the actin filament cytoskeleton (Forscher and Smith, 1988). However, subsaturating concentrations of CB have been shown to remove specific populations of actin filaments, such as the actin bundles that make the core of filopodia in neuronal growth cones (Burnette et al., 2007). Therefore we tested several concentrations of CB and found that 100 nM specifically and significantly reduced the actin filaments in the region where NMIIA-Fs form (i.e., right behind the protrusive lamellipodium; Figure 4, A and B, arrow), with no effect on the amount of actin filament in the protrusive lamellipodium (Figure 4A, asterisks). Furthermore, cells treated with 100 nM CB also had a significant reduction in NMIIA-F stack length (Figure 4, C and D). These results were consistent with the concept that actin filament densities regulate the ability of NMIIA-Fs to expand into stacks. Taken together, our data support a model in which NMIIA molecules split from an NMIIA-F through the interactions of motor domains and actin filaments. We next tested whether the formation of contractile systems in other cellular contexts could also involve NMIIA-F expansion.
FIGURE 4:
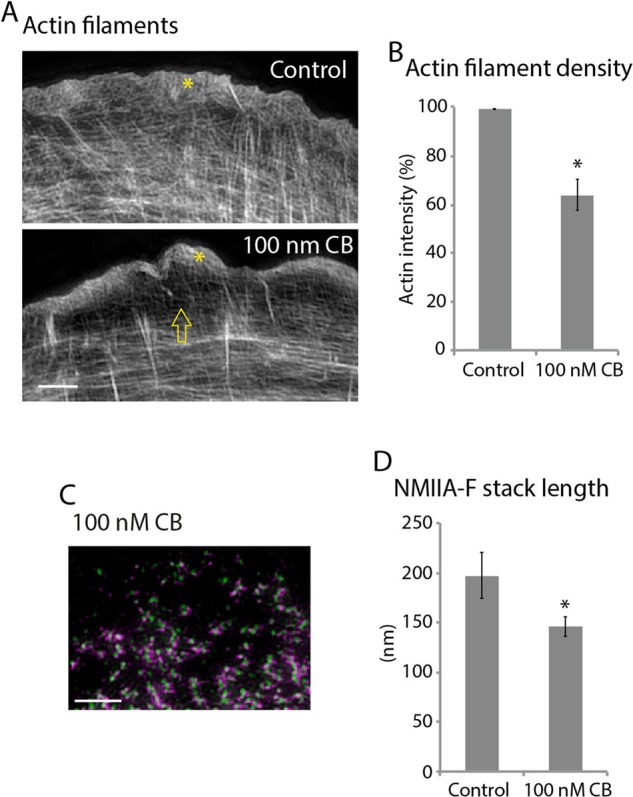
Reducing the actin filament density reduced the NMIIA-F stacks. (A) Actin filaments in a control U2-OS cell and a cell treated with 100 nM CB for 1 h. Arrow denotes the drop in intensity behind the lamellipodium. (B) Quantification of the drop in intensity of actin filaments treated with 100 nM CB compared with control. Control: three measurements each from 42 cells over three experiments; 100 nm CB: three measurements each from 46 cells over three experiments. (C) RLC/NMIIA rod domains in a cell treated with 100 nM CB for 1 h. (D) Lengths of NMIIA-Fs in control cells and cell treated with 100 nM CB. Control: 9145 NMIIA-Fs, 48 cells, three experiments; 100 nm CB: 925 NMIIA-Fs, 20 cells, three experiments. *p < 0.001 compared with control. Scale bars, 2 μm. Error bars in B and D indicate SEM.
NMII is largely responsible for the forces driving ingression of the cleavage furrow during cytokinesis of vertebrate cells (Straight et al., 2003; Ma et al., 2012). Furthermore, there is electron microscopy evidence from both classic and recent studies that strongly suggest the presence of NMII stacks in the cleavage furrow (Sanger and Sanger, 1980; Ong et al., 2014). NMIIA is recruited to the membrane of dividing cells during the transition between anaphase and telophase, and this recruitment is followed by cleavage furrow ingression (Matsumura et al., 1998). We started by confirming this, using spinning-disk confocal microscopy of dividing HeLa cells expressing NMIIA-(N-terminal)-mEGFP and histone 2B-mCherry to visualize chromosomes (Figure 5A). As expected, NMIIA-(N-terminal)-mEGFP localized to the membrane at the cleavage furrow during ingression (Figure 5A, arrowheads). We next tested whether there were similar NMIIA-F dynamics (e.g., expansion) during contractile ring formation in HeLa cells to what we found at the leading edge of crawling U2-OS cells.
FIGURE 5:

NMIIA-F stacks expand in the cleavage furrow. (A) Time-lapse montage of DIC and confocal section showing H2B-mCherry (magenta) and NMIIA-(N-terminal)-mEGFP in HeLa cell between anaphase and G1. NMIIA accumulation occurs at the membrane from anaphase to telophase (yellow arrowheads). Arrows show NMIIA at sites of blebbing. (B) Time-lapse wide-field epifluorescence imaging of NMIIA-(N-terminal)-mEGFP at the bottom of the cleavage furrow. Time montage from the yellow box shows NMIIA-F expansion (yellow arrow). Note that expanding NMIIA-Fs appear to align into larger stacks. (C) SIM of NMIIA-(N-terminal)-mEmerald and immunolocalized NMIIA rod domains. Whole-cell and zoomed views of the cleavage furrow. (D) Stack lengths at the leading edge of U2-OS cells (9145 NMIIA-Fs, 48 cells, three experiments) and cleavage furrow of HeLa cells (362 NMIIA-Fs, 11 cells, eight experiments). (E) SIM of immunolocalized NMIIA rod domains in a control and 20 μM blebbistatin–treated cleavage furrow. Note that the loss of linear structures in the 20 μM–treated furrow. (F, G) Length of NMIIA-F stacks (F) and density of NMIIA-Fs (G) in the cleavage furrow of control cells (362 NMIIA-Fs, 11 cells, eight experiments) and cells treated with 20 μM blebbistatin (94 NMIIA-Fs, five cells, three experiments). *p < 0.001 compared with control. Scale bars, 10 μm (A, B), 2 μm (C, E). Error bars in D, F, and G indicate SEM.
To test whether there was NMIIA-F expansion in the cleavage furrow, we acquired high-resolution time-lapse images of NMIIA-(N-terminal)-mEGFP of the very bottom of the forming contractile ring (Figure 5B and Supplemental Video S4). We observed NMIIA-Fs both expanded and also lined up creating NMIIA-F stack-like arrays (Figure 5B, yellow arrows). To confirm the presence of NMIIA-F stacks, we imaged the motor domains with NMIIA-(N-terminal)-mEmerald and rod domains with an antibody in the cleavage furrow using SIM. Indeed, HeLa cells had prominent NMIIA-F stacks spanning the cleavage furrow (Figure 5C and Supplemental Figure S5-1). NMIIA-Fs within stacks were organized parallel to the division plane, and the lengths of stacks were perpendicular to the division plane, consistent with a model in which the furrow closes through a purse-string mechanism. Of interest, the NMIIA-F stack lengths in the cleavage furrow of HeLa cells were the same as at the leading edge of U2-OS crawling cells (Figure 5D). Taken together, these data show that NMIIA-F expansion occurs in the cleavage furrow and results in a similar organization as found at the leading edge of a crawling cell during interphase.
We next wanted to determine whether NMIIA-F stacks formed in the cleavage furrow by similar mechanisms as at the leading edge. Because NMIIA-F stack length in interphase cells can be modulated by varying the concentration of blebbistatin (Figure 3F), we analyzed the effect of treating cytokinetic cells with a concentration of blebbistatin that would not halt furrow ingression. As expected, a high concentration of blebbistatin (50 μM) stops furrow ingression (Straight et al., 2003; Figure S5-2). Cells treated with 20 μM blebbistatin, however, had significantly slower ingression rates but still divided (Figure S5-2). Therefore we used SIM to image and then quantify the NMIIA-F stack lengths in the cleavage furrows of control cells and those treated with 20 μM blebbistatin. Cells treated with 20 μM blebbistatin showed a decrease in overall NMIIA-F stack length (Figure 5, E and F), even though the number of filaments as defined by the number of rod localizations was not significantly different (Figure 5G). Thus the same number of NMIIA-Fs formed, but they failed to elongate into stacks. Thus NMIIA-F stack formation was sensitive to NMII ATPase inhibition in both interphase and mitosis.
DISCUSSION
We present evidence for a new model of NMIIA stack formation that occurs at both the leading edge of crawling cells and in the cleavage furrow of dividing cells. Through a combination of high-resolution and superresolution light microscopy techniques, we show that NMIIA stacks form from two non–mutually exclusive mechanisms—expansion and concatenation (Figure 2). Pharmacological and genetic perturbations showed that NMIIA motor activity, cellular contractility, and actin filament availability regulate NMIIA stack formation (Figures 3 and 4). Furthermore, we show that a slowing of cleavage furrow ingression is associated with a loss of large organized NMIIA stacks. As such, NMIIA filament expansion and concatenation could represent a universal mechanism of contractile unit assembly.
The initial steps of NMIIA-F formation have been well characterized (Vicente-Manzanares et al., 2009). Autoinhibited NMIIA molecules become polymerization competent through phosphorylation (Figure 6A, steps 1 and 2) and form a 2-motor-group filament through electrostatic interactions of their rod domains (Figure 6A, steps 2–4; Pollard, 1975; Craig et al., 1983; Ricketson et al., 2010). Steric hindrance limits the number of NMII molecules that can be added to a growing 2-motor-group NMIIA-F (Pollard, 1975; Billington et al., 2013). Thus a likely role for the formation of NMIIA-F stacks would be to circumvent this inherent limitation in order to create stronger contractile units. Our data show that NMIIA-F stacks form by both expansion of a single 2-motor-group NMIIA-F and concatenation of multiple NMIIA-F/NMIIA-F stacks (Figure 6, B and C). During expansion, NMIIA molecules within an NMIIA-F spread out in space (cartoon in Figure 6B, steps 1–3, and data in Figure 2, D–I). We envision that “pioneer” myosins could move relative to the filament in a number of mechanisms. Pioneers are seemingly left behind by moving more slowly than other NMII molecules in an NMIIA-F (Figure 2, I and J). This could be a result of their binding more strongly to actin filaments, walking along actin filaments (i.e., as NMIIB is capable of; Norstrom et al., 2010; Nagy et al., 2013), or being pulled out by another molecular motor. The spreading out of the original NMIIA molecules is accompanied by addition of new NMIIA molecules to the growing stack (Figure 6B, steps 4 and 5), as revealed in our live-cell data by an increase in fluorescence signal during expansion (Figures 1B and 2, F and G).
FIGURE 6:

Model of NMIIA-F stack formation. (A) Classic model of the formation of a 2-motor-group NMIIA-F. An autoinhibited NMIIA molecule (1) becomes polymerization competent (2) after phosphorylation of its regulatory light chain. Two polymerization-competent NMIIA molecules then form a mini-NMIIA-F (3) through electrostatic interactions of their rod domains. Addition of more NMIIA molecules to the minifilament drives the growth of a 2-motor-group NMIIA-F (4). Steric hindrance limits the number of molecules that can be added to a 2-motor-group NMIIA-F (Billington et al., 2013). Note that a smaller number of myosins than the ∼15 on each side of the filament are drawn for simplicity. Of importance, how larger contractile arrays (i.e., stacks) containing more myosins capable of generating more contractile forces form is not understood (5). (B) Model of NMIIA-F expansion. After the formation of a 2-motor-group NMIIA-F (1), a subset of “pioneer” myosins separate themselves from one side of the filament (2). “Pioneer” myosins are then followed by myosins from the other side of the filament (3) to create a template for a stack (4). Growth of these templates is driven by addition of new NMIIA molecules (5). We have drawn this addition at the end for simplicity, but it could occur as soon as the “pioneer” myosins have separated themselves enough from the 2-motor-group NMIIA-F to fit in new molecules as in (2). (C) Stacks can also form through concatenation of multiple NMIIA-Fs. Expansion and concatenation are not mutually exclusive, as expanding NMIIA-Fs can concatenate with other NMIIA-Fs as in Figure 2K.
Exactly why expansion of NMIIA filaments occurs, as opposed to only addition of NMIIA molecules from a soluble cytoplasmic pool adjacent to a 2-motor-group filament, is not understood, but our data point to a few plausible explanations. Previous data showed that a majority of NMIIA molecules are in a soluble, unpolymerized pool (i.e., not in a bipolar filament; Breckenridge et al., 2009; Shutova et al., 2012). In addition, a finite number of NMIIA molecules make up an individual NMIIA bipolar filament before steric hindrance restricts addition of more NMIIA molecules (Niederman and Pollard, 1975; Pollard, 1975; Billington et al., 2013). Thus expansion and separation of a NMIIA 2-motor-group filament would reduce steric hindrance in the filament and allow for addition of more NMIIA molecules, increasing the force-generating capabilities of the ensemble. Continued expansion and addition would result in larger NMIIA ensembles (i.e., a NMIIA stack). This mechanism would conceivably be more efficient than polymerizing a 2-motor-group filament de novo adjacent to a previously formed 2-motor-group filament.
Surprisingly, we found that only a small percentage of NMIIA filaments exhibit a network contraction–like behavior (Figure 2A; Verkhovsky et al., 1999). Despite this small percentage, we do not rule out a role for this population of NMIIA filaments in either crawling or dividing cells or in other cellular contexts. For example, this small percentage of NMIIA filaments could locally remodel actin filaments at the leading edge of crawling cells, just as the network contraction model predicts. The larger percentage of expanding NMIIA filaments could be involved in NMII-required processes, such as adhesion maturation and actin arc translocation, which are required for efficient cell migration (Medeiros et al., 2006; Vicente-Manzanares et al., 2007).
NMIIA-F stacks have inherently more NMIIA molecules within them than nascent 2-motor-group filaments. Thus stacks are likely to be able to generate more force. This may be vital to the function of the sarcomere-like stress fibers driving the movement of crawling and the separation of dividing cells (Supplemental Figure S4; Sanger and Sanger, 1980; Verkhovsky et al., 1995; Burnette et al., 2014). Another possible role for NMIIA-F stack formation could be to template the formation of other myosin isoforms with different biophysical properties, such as NMIIB, which has been shown to incorporate into NMIIA-Fs over time (Beach et al., 2014; Shutova et al., 2014). Indeed, we found that reducing NMIIA in U2-OS cells with RNA interference does tend to abolish NMIIB filament stacks (Supplemental Figure S6).
Our SIM analysis of NMIIA organization in cleaving cells also provides insight into the force-generating machinery that drives cytokinesis. Constriction of an annular actomyosin array is arguably the dominant model, but others are equally plausible (Wang, 2005; Eggert et al., 2006). Debate has continued in part because we lack information on how actomyosin is arranged within the cleavage furrow. In fission yeast, this problem was addressed through electron microscopic analysis of time-dependent changes in the 3D arrangement of F-actin throughout cytokinesis (Kamasaki et al., 2007). Consistent with a purse-string mechanism, actin filaments circumscribe the cortex, forming a circular structure composed of filaments oriented parallel to the plane of division (Supplemental Figure S5-1). Our data clearly show that furrow NMIIAs form stacks that orient perpendicular to the plane of division (Figure 5C), suggesting that they slide an array of actin filaments that are also arranged in parallel relative to the direction of ingression. Of interest, the stack length and density of NMIIA correlate with the speed of ingression (Figures 5, E–G). Thus, although our analysis does not formally prove that furrow-localized NMIIA generates the forces that drive cell cleavage, it is consistent with the notion that cytokinesis in human cells is also driven by a purse-string mechanism.
MATERIALS AND METHODS
Cell culture and chemicals
U2-OS (HTB-96; American Type Culture Collection, Manassas, VA) and HeLa (CCL-2; ATTC) cells were cultured in growth medium comprising DMEM (10-013-CV; Mediatech, Manassas, VA) containing 4.5 g/l l-glutamine, l-glucose, and sodium pyruvate and supplemented with 10% fetal bovine serum (F2442; Sigma-Aldrich, St. Louis, MO). Wild-type HAP1 cells and HAP1 cells with MYH9 (myosin IIA) knocked out using CRISPR were purchased from Haplogen Genomics (Vienna, Austria). The company confirmed knockout with PCR, and we confirmed it using Western blotting (Figure 3I and Supplemental Figure S3-3). HAP1 cells were cultured in IMDM (12440-053; Life Technologies, Grand Island, NY). Cells were kept at 37°C and 5% CO2. Growth substrates were prepared by coating #1.5 glass coverslips (D35C4-20-1.5N or D35-20-1.5N; In Vitro Scientific, Mountain View, CA) with 25 μg/ml laminin (L2020; Sigma-Aldrich) in phosphate-buffered saline (PBS; 46-013-CM; Mediatech) at 37°C for 1 h. Cells were plated on a growth substrate, and experiments were performed the next day. For protein expression experiments, cells were transiently transfected with FuGENE 6 (E2691; Promega, Madison, WI) according to the manufacturer’s instructions overnight in a 25-cm2 cell culture flask (25-207; Genessee Scientific Corporation, San Diego, CA) before plating on a growth substrate. Blebbistatin (B0560), cytochalasin B (C6762), Y-27632 (Y0503), paclitaxel (T7402), phalloidin (P2141), RO-3306 (SML0569), and mouse anti-RLC (M4401) were purchased from Sigma-Aldrich. Alexa Fluor 488–phalloidin (A12379), Alexa Fluor 488–goat anti-mouse (A11029), Alexa Fluor 568–goat anti-rabbit (A11011), and Alexa Fluor 647–goat anti-rabbit (A21244) antibodies were purchased from Life Technologies (Grand Island, NY). Rabbit anti–myosin IIA (909801) was purchased from BioLegends (San Diego, CA). Paraformaldehyde (15710) was purchased from Electron Microscopy Sciences (Hatfield, PA). Triton X-100 (BP151100) was purchased from Fischer Scientific (Suwanee, GA). Knockdown of nonmuscle myosin IIA was performed as previously, with the Accell SMARTpool siRNA to human MYH9 or Accell scrambled control purchased from Thermo Fisher Scientific (Waltham, MA) and used according to the manufacturer’s instructions.
Plasmids
The plasmids encoding NMIIA-(N-terminal)-mEGFP (11347; Addgene, Cambridge, MA) with mEGFP on the N-terminus of NMIIA heavy chain were used as described previously (Chua et al., 2009). NMIIA was in an enhanced GFP-N3 vector (Takara Bio, Otsu, Japan) with a cytomegalovirus promoter to drive expression. The plasmids encoding mEmerald-myosinIIA-C-18 (i.e., NMIIA-(N-terminal)-mEmerald; 54190; Addgene) or mEos2-MyosinIIA-C-18 (i.e., NMIIA-(N-terminal)-mEOS2; 57403; Addgene) with mEmerald or mEOS2 on the N-terminus of NMIIA heavy chain were used as described previously (Burnette et al., 2014). Briefly, cDNA (Blue Heron) was ligated into a mEmerald-C1 or mEOS2 cloning vector (Clontech style). The plasmid encoding mEmerald-C-16-mApple-N-14-MyosinIIA (i.e., NMIIA-(N-terminal)-mEmerald/(C-terminal)-mApple) with mEmerald and mApple on the N- and C-termini of NMIIA heavy chain, respectively, was used as described previously (Burnette et al., 2014). Briefly, mEmerald was PCR amplified and ligated into a mApple-MyosinIIA-N-14. The plasmid encoding NMIIA-N93K-mEGFP (Vicente-Manzanares et al., 2007) was a generous gift from Alan Horwitz (Department of Cell Biology, University of Virginia).
Pharmacological treatments
Growth medium was equilibrated at 37°C and 5% CO2 for 1 h before adding drugs. For blebbistatin, cytochalasin B, and Y-27632, drugs were diluted in medium, and cells were treated for 1 h at 37°C and 5% CO2, followed by immediate fixation (see Fixation and immunohistochemistry). For cell division studies, plates were treated with RO-3306 to catch more cells in telophase (Vassilev et al., 2006). RO-3306 was used as described (Vassilev et al., 2006). Briefly, after plating on growth substrate and attachment, cells were incubated with 9 μM RO-3306 for 12 h and washed out twice with preequilibrated medium. Cells were fixed 45 min after washout.
Fixation and immunohistochemistry
U2-OS cells were fixed with 4% paraformaldehyde (PFA) in PBS at room temperature for 20 min and then extracted for 5 min with 1% Triton X-100 and 4% PFA in PBS as previously described (Burnette et al., 2014). Cells were washed three times in 1× PBS. HeLa cells were live-cell extracted before fixation as described previously (Burnette et al., 2011) to remove unincorporated NMIIA, as this caused a significant background, especially in early ingression stages. Note that the “hazy” signal in Figure 4A diminishes as the contractile ring forms. Briefly, a cytoskeleton-stabilizing live-cell extraction buffer was made fresh containing 2 ml of stock solution (500 mM 1,4-piperazinediethanesulfonic acid, 25 mM ethylene glycol tetraacetic acid, 25 mM MgCl2), 4 ml of 10% polyoxyethylene glycol (PEG; 35,000 molecular weight), 4 ml H2O, and 100 μl of Triton X-100, 10 μM paclitaxel, and 10 μM phalloidin. Cells were treated with this extraction buffer for 1 min, followed by a 1-min wash with wash buffer (extraction buffer without PEG or Triton X-100). Cells were then fixed with 4% PFA for 20 min.
After fixation, the following labeling procedures were used: for actin visualization, phalloidin-488 in 1× PBS (15 μl of stock phalloidin per 200 μl of PBS) was used for 3 h at room temperature. For immunofluorescence experiments, cells were blocked in 10% bovine serum albumin (BSA) in PBS. Primary antibodies were diluted in 10% BSA. Myosin IIA antibody was used at 1:1000. RLC antibody was used at 1:200. Secondary antibodies were diluted in 10% BSA at 1:100 and centrifuged at 13,000 rpm for 10 min before use. The DNA in HeLa cells was also stained with Hoechst stain for 25 min.
Fluorescence, photoconversion, and phase microscopy
High-resolution wide-field fluorescence images were acquired on a Nikon Eclipse Ti equipped with a Nikon 100× Plan Apo 1.45 numerical aperture (NA) oil objective and a Nikon DS-Qi2 CMOS camera. Images were acquired every 10 s using 100- to 300-ms integration using a fluorescein isothiocyanate (FITC) filter cube. Photoconversion of NMIIA-mEOS2 was accomplished by closing the Eclipse Ti’s field diaphragm and using a 4’,6-diamidino-2-phenylindole filter cube to shine UV light on the cell’s leading edge for 10 s as previously described (Baker et al., 2010). This was immediately followed by opening the field diaphragm and imaging with FITC and Tex Red filter cubes every 1 min. Phase imaging was performed on a Nikon Eclipse Ti equipped with a Nikon 20× Plan 0.40 NA air objective and Nikon DS-Qi2 cMOS camera. Images were acquired every 1-min using 50-ms integration. A custom-built incubator was constructed to keep cells cultured in L-15 medium (BW12700Q; Fisher Scientific) at 37°C. An orange glass filter was placed on top of the condenser to prevent the inactivation of blebbistatin (Sakamoto et al., 2005).
Spinning-disk confocal microscopy
For Figure 4, confocal and differential interference contrast (DIC) images were taken on a Leica DMi8 inverted microscope equipped with a Leica 100× Plan Apo 1.44 NA oil objective and an Andor DSD2 “laser-free” spinning- disk confocal with LED excitation source and an Andor Zyla 5.5 scientific complementary metal-oxide-semiconductor (sCMOS) camera. Fluorescence images were acquired with 300-ms integration. Cells were incubated in L-15 medium within a heated chamber at 37°C.
Structured illumination microscopy
SIM imaging and processing was performed on a GE Healthcare DeltaVision OMX equipped with a 60×/1.42 NA oil objective and sCMOS camera. For live-cell studies, cells were incubated in L-15 medium within a heated chamber at 37°C.
Live-cell photoactivated localization microscopy
Live-cell PALM imaging was performed as previously described (Manley et al., 2008). Total internal reflection microscopy was performed with a Nikon N-STORM microscope using a Nikon 100× Plan Apo 1.49 NA objective and an iXon Ultra electron-multiplying charge-coupled device (EMCCD) camera (Andor, Belfast, United Kingdom). An image of the unconverted green channel (excited with 488 nm) and converted red channel (excited with 561 nm) was acquired every 5 s. Integration times for the green channel varied between 100 and 150 ms, and the red channel integration time was 150 ms. Camera gain was set to 400. We did not use the 405-nm laser because imaging the green channel with the 488-nm laser alone was sufficient to convert enough molecules for analysis, as previously described (Sengupta et al., 2011). The precise localization of single converted NMIIA-mEOS2 molecules was determined with the ThunderSTORM ImageJ plug-in as previously described (Ovesny et al., 2014). The average localization precision for the NMIIA-mEOS2 molecules analyzed was 24.6 ± 4.0 nm.
Three-dimensional photoactivated localization microscopy
The 3D-PALM measurements were performed as previously described (Betzig et al., 2006; Brown et al., 2011; Xu et al., 2012). Briefly, U2OS cells expressing NMIIA-mEOS2 were fixed with 4% PFA and 0.25% glutaraldehyde and imaged with a custom-built dual objective (4Pi; Nikon 1.49 NA total internal reflection fluorescence objectives; Melville, NY) epifluorescence microscope. Astigmatism was introduced on both objective images using deformable mirrors (Brown et al., 2011) and was collected on separate EMCCD cameras (Andor iXon Ultra, DU-897). The z-coordinate parameters were extracted using the ellipticity of image localizations for multiple gold fiducials embedded in the coverslip (Hestzig, Leesburg, VA). Images from each objective were independently drift corrected in all dimensions and registered with one another using gold fiducials (typically, <10–20 nm of residual error persisted after these corrections). Localization pairs were spatially correlated in each image frame, and those with <0.3-pixel (133.3-nm pixels) deviations were used to recalculate the number of photons collected for each localization. Those localizations that did not display a correlating image from the other objective were discarded (typically, 10–20% of the total localizations). All images were rendered using custom Matlab software (Natick, MA) using the positional uncertainty as a probability distribution for each localization dimension (Betzig et al., 2006). With our imaging approach, discrete clusters of NMIIA-mEOS2 localizations yielded 30- to 40- and 60- to 90-nm full-width at half-maximum in the X/Y- and Z-dimensions, respectively.
Selection of cells for this study
Polarized migrating U2-OS cells were chosen for this study by finding cells that had a curved leading edge and characteristic protrusive elements (i.e., lamellipodium), as described previously (Burnette et al., 2014). HeLa cells in telophase were chosen based on chromosome position/morphology and their having an ingressed furrow but not a midbody.
Data quantification
ImageJ (National Institutes of Health, Bethesda, MD), SlideBook 6 (Intelligent Imaging Innovations, Denver, CO), and Nikon Elements software were used for data quantification. For 2-, 3-, and 4-motor-group and stack length quantification, regions of interest were used and placed at the leading edge of a migrating cell. For 2-, 3-, and 4-motor-group and filament density quantification, a region of interest extending 1.5 μm from the leading edge was defined (Supplemental Figure S3-1), and every discrete myosin II ensemble that could be quantified as a 2-, 3-, 4-, or >4-motor-group in this region was counted. Increasing the distance from the edge of the region of interest further increased the number of overlapping NMIIA-Fs, whose structures could not be defined. For stack length quantification in U2OS cells, a 15 × 7–μm box (determined empirically as the best size for all cells measured) was placed, starting with the first NMIIA-F in the protrusive area of a migrating cell, and all NMIIA-Fs were measured (Supplemental Figure S3-2). For stack length quantification in the cleavage furrow of HeLa cells, an 8 × 6–μm box (determined empirically as the best size for all cells measured) was placed in the middle of the cleavage furrow, and every NMIIA-F was measured. For actin filament intensities, all cells were imaged with the same laser intensity and exposure, and maximum projections of the cells were analyzed. Three 1 × 1–μm boxes were used per cell and averaged to measure intensity. Boxes were placed 2 μm behind the cell edge between the dorsal stress fibers, which do not contain myosin II (Hotulainen and Lappalainen, 2006) and were unchanged by cytochalasin B treatment (Figure 4). The p values from Student’s t tests and SEM were calculated using Excel (Microsoft, Redmond, WA).
Supplementary Material
Acknowledgments
We thank the M. Tyska and R. Ohi labs at Vanderbilt for invaluable discussions and reagents and M. Tyska and V. Farmer for comments on the manuscript. We give a special thanks to Sean Schaffer of the Cell Imaging Shared Resources at Vanderbilt University for help with and maintenance of the GE Healthcare OMX and Anthony Tharp for the CDB CORE Equipment. This work was supported by Vanderbilt University School of Medicine Molecular Biophysics Training Grant T32 GM08320 to A.M.F. and Career Development Award from the National Cancer Institute SPORE in GI Cancer P50 CA095103 to D.T.B.
Abbreviations used:
- NMII
nonmuscle myosin II
- NMIIA
nonmuscle myosin IIA
- NMIIA-F
nonmuscle myosin IIA filament
- NMII-F
nonmuscle myosin II filament
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E15-10-0725) on March 9, 2016.
REFERENCES
- Adelstein RS, Pollard TD, Kuehl WM. Isolation and characterization of myosin and two myosin fragments from human blood platelets. Proc Natl Acad Sci USA. 1971;68:2703–2707. doi: 10.1073/pnas.68.11.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker SM, Buckheit RW, 3rd, Falk MM. Green-to-red photoconvertible fluorescent proteins: tracking cell and protein dynamics on standard wide-field mercury arc-based microscopes. BMC Cell Biol. 2010;11:15. doi: 10.1186/1471-2121-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach JR, Hussey GS, Miller TE, Chaudhury A, Patel P, Monslow J, Zheng Q, Keri RA, Reizes O, Bresnick AR, et al. Myosin II isoform switching mediates invasiveness after TGF-beta-induced epithelial-mesenchymal transition. Proc Natl Acad Sci USA. 2011;108:17991–17996. doi: 10.1073/pnas.1106499108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beach JR, Shao L, Remmert K, Li D, Betzig E, Hammer JA., 3rd Nonmuscle myosin II isoforms coassemble in living cells. Curr Biol. 2014;24:1160–1166. doi: 10.1016/j.cub.2014.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- Billington N, Wang A, Mao J, Adelstein RS, Sellers JR. Characterization of three full-length human nonmuscle myosin II paralogs. J Biol Chem. 2013;288:33398–33410. doi: 10.1074/jbc.M113.499848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge MT, Dulyaninova NG, Egelhoff TT. Multiple regulatory steps control mammalian nonmuscle myosin II assembly in live cells. Mol Biol Cell. 2009;20:338–347. doi: 10.1091/mbc.E08-04-0372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF, Clayton DA. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol Cell Biol. 2011;31:4994–5010. doi: 10.1128/MCB.05694-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette DT, Manley S, Sengupta P, Sougrat R, Davidson MW, Kachar B, Lippincott-Schwartz J. A role for actin arcs in the leading-edge advance of migrating cells. Nat Cell Biol. 2011;13:371–381. doi: 10.1038/ncb2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnette DT, Schaefer AW, Ji L, Danuser G, Forscher P. Filopodial actin bundles are not necessary for microtubule advance into the peripheral domain of Aplysia neuronal growth cones. Nat Cell Biol. 2007;9:1360–1369. doi: 10.1038/ncb1655. [DOI] [PubMed] [Google Scholar]
- Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, et al. A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. J Cell Biol. 2014;205:83–96. doi: 10.1083/jcb.201311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua J, Rikhy R, Lippincott-Schwartz J. Dynamin 2 orchestrates the global actomyosin cytoskeleton for epithelial maintenance and apical constriction. Proc Natl Acad Sci USA. 2009;106:20770–20775. doi: 10.1073/pnas.0909812106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Smith R, Kendrick-Jones J. Light-chain phosphorylation controls the conformation of vertebrate non-muscle and smooth muscle myosin molecules. Nature. 1983;302:436–439. doi: 10.1038/302436a0. [DOI] [PubMed] [Google Scholar]
- Eggert US, Mitchison TJ, Field CM. Animal cytokinesis: from parts list to mechanisms. Annu Rev Biochem. 2006;75:543–566. doi: 10.1146/annurev.biochem.74.082803.133425. [DOI] [PubMed] [Google Scholar]
- Forscher P, Smith SJ. Actions of cytochalasins on the organization of actin filaments and microtubules in a neuronal growth cone. J Cell Biol. 1988;107:1505–1516. doi: 10.1083/jcb.107.4.1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Mechanical integration of actin and adhesion dynamics in cell migration. Annu Rev Cell Dev Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson MG, Shao L, Carlton PM, Wang CJ, Golubovskaya IN, Cande WZ, Agard DA, Sedat JW. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys J. 2008;94:4957–4970. doi: 10.1529/biophysj.107.120345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Srour M, Capo-Chichi JM, Daoud H, Nassif C, Patry L, Massicotte C, Ambalavanan A, Spiegelman D, Diallo O, et al. De novo mutations in moderate or severe intellectual disability. PLoS Genet. 2014;10:e1004772. doi: 10.1371/journal.pgen.1004772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. J Cell Biol. 2006;173:383–394. doi: 10.1083/jcb.200511093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu A, Wang F, Sellers JR. Mutations in human nonmuscle myosin IIA found in patients with May-Hegglin anomaly and Fechtner syndrome result in impaired enzymatic function. J Biol Chem. 2002;277:46512–46517. doi: 10.1074/jbc.M208506200. [DOI] [PubMed] [Google Scholar]
- Kamasaki T, Osumi M, Mabuchi I. Three-dimensional arrangement of F-actin in the contractile ring of fission yeast. J Cell Biol. 2007;178:765–771. doi: 10.1083/jcb.200612018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley MJ, Jawien W, Ortel TL, Korczak JF. Mutation of MYH9, encoding non-muscle myosin heavy chain A, in May-Hegglin anomaly. Nat Genet. 2000;26:106–108. doi: 10.1038/79069. [DOI] [PubMed] [Google Scholar]
- Ma X, Adelstein RS. A point mutation in Myh10 causes major defects in heart development and body wall closure. Circ Cardiovasc Genet. 2014;7:257–265. doi: 10.1161/CIRCGENETICS.113.000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Kovacs M, Conti MA, Wang A, Zhang Y, Sellers JR, Adelstein RS. Nonmuscle myosin II exerts tension but does not translocate actin in vertebrate cytokinesis. Proc Natl Acad Sci USA. 2012;109:4509–4514. doi: 10.1073/pnas.1116268109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean-Fletcher S, Pollard TD. Mechanism of action of cytochalasin B on actin. Cell. 1980;20:329–341. doi: 10.1016/0092-8674(80)90619-4. [DOI] [PubMed] [Google Scholar]
- Manley S, Gillette JM, Patterson GH, Shroff H, Hess HF, Betzig E, Lippincott-Schwartz J. High-density mapping of single-molecule trajectories with photoactivated localization microscopy. Nat Methods. 2008;5:155–157. doi: 10.1038/nmeth.1176. [DOI] [PubMed] [Google Scholar]
- Matsumura F, Ono S, Yamakita Y, Totsukawa G, Yamashiro S. Specific localization of serine 19 phosphorylated myosin II during cell locomotion and mitosis of cultured cells. J Cell Biol. 1998;140:119–129. doi: 10.1083/jcb.140.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medeiros NA, Burnette DT, Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat Cell Biol. 2006;8:215–226. doi: 10.1038/ncb1367. [DOI] [PubMed] [Google Scholar]
- Nagy A, Takagi Y, Billington N, Sun SA, Hong DK, Homsher E, Wang A, Sellers JR. Kinetic characterization of nonmuscle myosin IIb at the single molecule level. J Biol Chem. 2013;288:709–722. doi: 10.1074/jbc.M112.424671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederman R, Pollard TD. Human platelet myosin. II. In vitro assembly and structure of myosin filaments. J Cell Biol. 1975;67:72–92. doi: 10.1083/jcb.67.1.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norstrom MF, Smithback PA, Rock RS. Unconventional processive mechanics of non-muscle myosin IIB. J Biol Chem. 2010;285:26326–26334. doi: 10.1074/jbc.M110.123851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong K, Wloka C, Okada S, Svitkina T, Bi E. Architecture and dynamic remodelling of the septin cytoskeleton during the cell cycle. Nat Commun. 2014;5:5698. doi: 10.1038/ncomms6698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ovesny M, Krizek P, Borkovec J, Svindrych Z, Hagen GM. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics. 2014;30:2389–2390. doi: 10.1093/bioinformatics/btu202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD. Electron microscopy of synthetic myosin filaments. Evidence for cross-bridge. Flexibility and copolymer formation. J Cell Biol. 1975;67:93–104. doi: 10.1083/jcb.67.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricketson D, Johnston CA, Prehoda KE. Multiple tail domain interactions stabilize nonmuscle myosin II bipolar filaments. Proc Natl Acad Sci USA. 2010;107:20964–20969. doi: 10.1073/pnas.1007025107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamoto T, Limouze J, Combs CA, Straight AF, Sellers JR. Blebbistatin, a myosin II inhibitor, is photoinactivated by blue light. Biochemistry. 2005;44:584–588. doi: 10.1021/bi0483357. [DOI] [PubMed] [Google Scholar]
- Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, Munro J, Schroder E, Zhou J, Brunton VG, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanger JM, Sanger JW. Banding and polarity of actin filaments in interphase and cleaving cells. J Cell Biol. 1980;86:568–575. doi: 10.1083/jcb.86.2.568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta P, Jovanovic-Talisman T, Skoko D, Renz M, Veatch SL, Lippincott-Schwartz J. Probing protein heterogeneity in the plasma membrane using PALM and pair correlation analysis. Nat Methods. 2011;8:969–975. doi: 10.1038/nmeth.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seri M, Cusano R, Gangarossa S, Caridi G, Bordo D, Lo Nigro C, Ghiggeri GM, Ravazzolo R, Savino M, Del Vecchio M, et al. Mutations in MYH9 result in the May-Hegglin anomaly, and Fechtner and Sebastian syndromes. The May-Heggllin/Fechtner Syndrome Consortium. Nat Genet. 2000;26:103–105. doi: 10.1038/79063. [DOI] [PubMed] [Google Scholar]
- Shutova M, Yang C, Vasiliev JM, Svitkina T. Functions of nonmuscle myosin II in assembly of the cellular contractile system. PLoS One. 2012;7:e40814. doi: 10.1371/journal.pone.0040814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutova MS, Spessott WA, Giraudo CG, Svitkina T. Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Curr Biol. 2014;24:1958–1968. doi: 10.1016/j.cub.2014.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GF, Ridler MA, Faunch JA. Action of cytochalasin B on cultured human lymphocytes. Nature. 1967;216:1134–1135. doi: 10.1038/2161134a0. [DOI] [PubMed] [Google Scholar]
- Stachowiak MR, McCall PM, Thoresen T, Balcioglu HE, Kasiewicz L, Gardel ML, O’Shaughnessy B. Self-organization of myosin II in reconstituted actomyosin bundles. Biophys J. 2012;103:1265–1274. doi: 10.1016/j.bpj.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003;299:1743–1747. doi: 10.1126/science.1081412. [DOI] [PubMed] [Google Scholar]
- Svitkina TM, Verkhovsky AB, McQuade KM, Borisy GG. Analysis of the actin-myosin II system in fish epidermal keratocytes: mechanism of cell body translocation. J Cell Biol. 1997;139:397–415. doi: 10.1083/jcb.139.2.397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, Westphal H, Preston YA, Adelstein RS. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc Natl Acad Sci USA. 1997;94:12407–12412. doi: 10.1073/pnas.94.23.12407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuzovic L, Yu L, Zeng W, Li X, Lu H, Lu HM, Gonzalez KD, Chung WK. A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice. Rare Dis. 2013;1:e26144. doi: 10.4161/rdis.26144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Vallenius T. Actin stress fibre subtypes in mesenchymal-migrating cells. Open Biol. 2013;3:130001. doi: 10.1098/rsob.130001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez CG, Tworoger M, Martin AC. Dynamic myosin phosphorylation regulates contractile pulses and tissue integrity during epithelial morphogenesis. J Cell Biol. 2014;206:435–450. doi: 10.1083/jcb.201402004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev LT, Tovar C, Chen S, Knezevic D, Zhao X, Sun H, Heimbrook DC, Chen L. Selective small-molecule inhibitor reveals critical mitotic functions of human CDK1. Proc Natl Acad Sci USA. 2006;103:10660–10665. doi: 10.1073/pnas.0600447103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Borisy GG. Non-sarcomeric mode of myosin II organization in the fibroblast lamellum. J Cell Biol. 1993;123:637–652. doi: 10.1083/jcb.123.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Svitkina TM, Borisy GG. Myosin II filament assemblies in the active lamella of fibroblasts: their morphogenesis and role in the formation of actin filament bundles. J Cell Biol. 1995;131:989–1002. doi: 10.1083/jcb.131.4.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkhovsky AB, Svitkina TM, Borisy GG. Network contraction model for cell translocation and retrograde flow. Biochem Soc Symp. 1999;65:207–222. [PubMed] [Google Scholar]
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol. 2009;10:778–790. doi: 10.1038/nrm2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Manzanares M, Zareno J, Whitmore L, Choi CK, Horwitz AF. Regulation of protrusion, adhesion dynamics, and polarity by myosins IIA and IIB in migrating cells. J Cell Biol. 2007;176:573–580. doi: 10.1083/jcb.200612043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YL ( The mechanism of cortical ingression during early cytokinesis: thinking beyond the contractile ring hypothesis. Trends Cell Biol. 2005;15:581–588. doi: 10.1016/j.tcb.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Rocker C, Salih A, Spindler KD, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. Proc Natl Acad Sci USA. 2004;101:15905–15910. doi: 10.1073/pnas.0403668101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CA, Tsuchida MA, Allen GM, Barnhart EL, Applegate KT, Yam PT, Ji L, Keren K, Danuser G, Theriot JA. Myosin II contributes to cell-scale actin network treadmilling through network disassembly. Nature. 2010;465:373–377. doi: 10.1038/nature08994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia ZK, Yuan YC, Yin N, Yin BL, Tan ZP, Hu YR. Nonmuscle myosin IIA is associated with poor prognosis of esophageal squamous cancer. Dis Esophagus. 2012;25:427–436. doi: 10.1111/j.1442-2050.2011.01261.x. [DOI] [PubMed] [Google Scholar]
- Xu K, Babcock HP, Zhuang X. Dual-objective STORM reveals three-dimensional filament organization in the actin cytoskeleton. Nat Methods. 2012;9:185–188. doi: 10.1038/nmeth.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.