

Abstract
Pain is a complex disease which can progress into a debilitating condition. The effective treatment of pain remains a challenge as current therapies often lack the desired level of efficacy or tolerability. One therapeutic avenue, the modulation of ion channel signaling by small molecules, has shown the ability to treat pain. However, of the 215 ion channels that exist in the human genome, with 85 ion channels having a strong literature link to pain, only a small number of these channels have been successfully drugged for pain. The focus of future research will be to fully explore the possibilities surrounding these unexplored ion channels. Toward this end, a greater understanding of ion channel modulation will be the greatest tool we have in developing the next generation of drugs for the treatment of pain.
Keywords: clinical compounds, drug discovery, ion channels, pain, sodium channels
Introduction
The effective and safe treatment of pain remains an immense clinical challenge. Current therapies often lack the desired level of efficacy or tolerability to offer optimal pain management. This unmet medical need has driven huge research efforts toward the improved understanding of the physiology and pathophysiology of pain mechanisms, with the aim of providing more effective treatment options.
Modulation of ion channel signaling has the potential to effectively treat pain. Ion channel modulators such as Lidocaine (1)1 and Carbamazepine (2)2 (Fig. 1) act on voltage-gated sodium channels and are known to reduce pain in both the clinical and pre-clinical setting. However, modulators such as these identified early in the era of modern drug discovery, generally lack ion channel selectivity which makes firm conclusions about the roles of individual ion channels difficult to draw. More recently, evidence for the role of ion channels in pain has come from the study of monogenic pain related diseases caused by mutations that affect the function of specific ion channels. These include studies into both loss and gain of function mutations in the voltage gated sodium channel Nav1.7 mutations, which cause profound pain insensitivity3 and sensitivity4,5 respectively, in individuals with such mutations.
Figure 1.
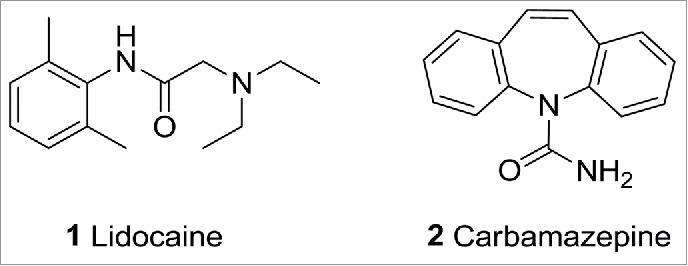
Lidocaine (1) and Carbamazepine (2) are known ion channel modulators.
Ion channels are integral membrane proteins with a gated, water-filled pore that regulates voltage potential across cell membranes via control of ion flow between the intracellular and extracellular environments. Ion flow results in the production of an electrical signal, causing adjacent voltage-sensitive channels to open in a chain-reaction fashion, thereby creating a self-propagating electrical signal that can traverse the entire length of a human nerve cell, a distance of as much as one meter, within milliseconds. Ion channels are broadly classified into voltage or ligand gated families, depending on the primary factors that lead to channel opening and closing. Within family types, ion channels are further categorized into sub-types, based on various factors that include the location and function of the specific channel. This review focuses on the current status of ion channel modulators and their application toward pain relief. It also discusses some of the drawbacks of current therapies and potential directions for improved treatment of the human pain condition.
Current Ion Channel Modulators for Pain Therapy
Of the 215 ion channels that exist in the human genome, 85 ion channels have strong literature links with pain, many of which are linked to multiple pain types.6 Some common ion channel-targeting drugs for pain are highlighted in Table 1. The number of discrete channels that have been successfully drugged for pain is very small compared to the number of ion channels that could have therapeutic potential.
Table 1.
Common Ion Channel Drugs for Pain Indications
| Drug | Target Channel | Disease Target | Year of First Clinical Usage |
|---|---|---|---|
| Gabapentin | Cav (α2δ) | Pain | 1994 |
| Pregabalin | Cav (α2δ) | Pain | 2004 |
| Ziconotide | Cav 2.2 | Severe Pain | 2004 |
| Lidocaine | Nav | Local Anesthetic | 1949 |
| Bupivacaine | Nav | Local Anesthetic | 1987 |
| Lamotrigine | Nav | Off label neuropathic pain | 1994 |
| Lacosamide | Nav | Seizures and Pain | 2008 |
| Carbamazepine | Nav | Epilepsy and off label for pain | 1963 |
| Eslicarbazepine | Nav | Epilepsy | 2009 |
Most of the currently available ion channel pain therapeutics were discovered more than a decade ago and some are over 50 y old. For example, Carbamazepine (2) is a first generation anticonvulsant developed in the 1950s for the treatment of trigeminal neuralgia, epilepsy, and mania.2 Drugs of this era were often discovered using “phenotypic” methods such as in vivo efficacy models or isolated tissue preparations designed to mimic a component of the clinical condition. In this way, a definitive characterization of which protein target(s) the ligand engaged with often came much later on. Carbamazepine is now known to inhibit sustained repetitive firing by blocking sodium channels in a use-dependent fashion with pain relief resulting from synaptic transmission blockade in the trigeminal nucleus. Carbamazepine also blocks calcium channels and GABA receptors at high micromolar levels of potency. This pan-ion channel inhibition profile likely drives Carbamazepines' broad list of indications (including antiarrhythmic, antidepressant, neuromuscular blocking, and sedative effects). Additional older drugs in this class are local anesthetics exemplified by lidocaine (1), which have been used in surgical procedures carried out on peripheral tissues, to reverse acute pain, or to treat chronic pain.1 These anesthetics are administered at relatively high doses to primarily block voltage gated sodium channels, but also block potassium and calcium channels.7 As with many compounds possessing polypharmacology, safety side effects of non-selective agents limit their chronic usage.8
A commercially successful compound, Gabapentin (3)9 (Fig. 2), was discovered using this “phenotypic” method. Gabapentin was originally developed to treat epilepsy and is currently also used in the treatment of neuropathic pain. As a lipophilic analog of GABA, Gabapentin was originally thought to increase GABA levels by activating glutamate decarboxylase and was found to be efficacious as an anti-convulsant. It was not until much later that Gabapentin's true mechanism of action was discovered, namely an interaction with the α2δ subunit of voltage gated calcium channels.10 A follow up drug discovery effort from Pfizer8 has since delivered Pregabalin (4), a compound with improved pharmacokinetics over Gabapentin that has become the gold standard for the treatment of chronic pain associated with diabetic neuropathy.
Figure 3.

Nav1.7 compounds.
Figure 4.

Nav1.8 compounds.
Figure 2.
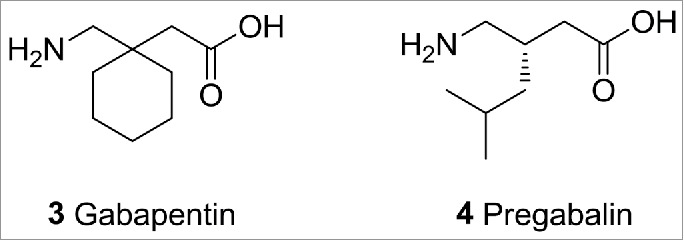
Gabapentin and Pregabalin.
The relative lack of success in bringing new ion channel pain therapies to market in recent years is notable. The reasons for this include failure to deliver clear efficacy and/or safety differentiation over the current standard of care therapies. This lack of return upon investment has driven new approaches in pain research. The strategy to select and validate pain targets is moving away from those supported by preclinical pain models (which are largely unsuccessful in predicting clinical efficacy for novel pain medicines) to an approach in which human data, including genetic data and human in vitro pharmacology data from patient-derived cells, define confidence in mechanism. These human data are coupled with an increased focus on the delivery of translatable biomarkers to enable effective clinical dose-setting. In addition to assessing the efficacy of new mechanisms, it is hoped that such human cell platforms will enable a more phenotypic approach to the identification of new pain targets. The increased investment in human biology, human-relevant in vitro screening platforms, and translational biomarkers is hoped to lead to greater success in Phase 2 and 3 clinical trials.
Ion Channel Modulators for Pain Therapy in Clinical Development
As described above, non-selective sodium channel blockers have been used for many years in the treatment of acute pain, but their utility is limited due to the functional consequences of inhibiting sodium channels other than those expressed in nociceptors, e.g. in the heart and CNS. A more recent understanding of which sodium channels are expressed in nociceptors and the generation of experimental and/or genetic data linking specific Nav receptors to a pain phenotype, has spurred the pain research community to deliver selective sodium inhibitors for clinical characterization. Advances in automated in vitro electrophysiology screening platforms8 have greatly facilitated these research efforts, enabling high quality Nav isoform data to be generated in a high through-put fashion.
In 2006, a seminal publication by Cox et al.3 demonstrated that congenital insensitivity to pain (CIP) could be conferred by missense mutations in Nav1.7. In addition to this, gain-of-function Nav1.7 mutations have been described in the rare, extreme pain conditions of inherited primary erythromelalgia (IEM)5 and paroxysmal extreme pain disorder (PEPD).4 These findings initiated a huge pharmaceutical investment that has delivered a range of reportedly selective Nav 1.7 blockers. A number of these compounds have now entered clinical trials for pain (Fig. 3), including the proline derivative 5 from GSK/Convergence,11 a spiro-oxindole compound from Xenon (series exemplified by 6 XEN402, now known as TV-45070)12 and an aminoheterocycle sulphonamide series from a Pfizer-Icagen collaboration (series exemplified by 7).13 To-date, Xenon have reported efficacy treating pain in patients with congenital erythromelalgia, but no efficacy was seen in a study of postherpatic neuralgia. Also, Convergence has reported positive data from an open-label phase of a trigeminal neuralgia study as well as statistically significant differences in the reduction of pain in a lumbosacral radiculopathy (sciatica) trial.50
In addition to Nav 1.7, a lot of interest has been generated in Nav1.8 as a target for both inflammatory and neuropathic pain. Abbott, in collaboration with Icagen, reported the discovery of a number of biaryl Nav1.8 inhibitors (Fig. 4), exemplified by 8 (A-803467)14 and 9,15 of which A-803467 has been used extensively in preclinical target validation. Pfizer has also disclosed a series of Nav1.8 selective compounds, exemplified by 6,6-biaryl amide 10,16 and progressed an example into the clinic. Unfortunately, in the one result published so far, amide 10 showed no efficacy in a post-surgical dental pain clinical trial.
The TRP channels constitute a family of mammalian cation channels with biological functions that include pain perception and thermosensation.17 TRPV1 is activated by numerous stimuli including heat (>42°C), vanilloids, lipids, and protons/cations. Several highly selective TRPV1 antagonists have advanced into clinical development for the treatment of pain including 11 AMG517,18 12 SB-705498,19 13 ABT-102,20 and 14 MK-229521 (Fig. 5). Unfortunately, reports from many of the clinical studies have shown TRPV1 blockade to have effects on core body temperature, which has halted much of the drug development activity targeting this channel. The use of desensitising TRPV1 agonists has also been shown to reduce pain sensitivity.22,23 This has found clinical application with a topical approach (e.g., Astella's Qutenza® patch, 8% capsaicin) in which a local anesthetic is applied to the painful area prior to topical capsaicin, preventing the initial painful flare that occurs with agonist-induced nociceptor excitation prior to desensitisation.
Figure 5.
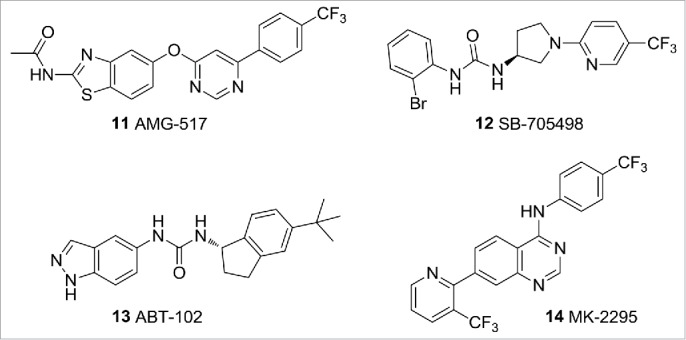
TRPV1 ligands.
TRPM8 is activated in vitro by cool temperatures (10−23°C) and cooling agents such as icilin and menthol. Researchers at Pfizer have recently disclosed that TRPM8 antagonist 15 (PF-05105679), Figure 6, is analgesic in an experimental model of cold pain (the Cold Pressor Test) in humans, without affecting core body temperature.24
Figure 6.
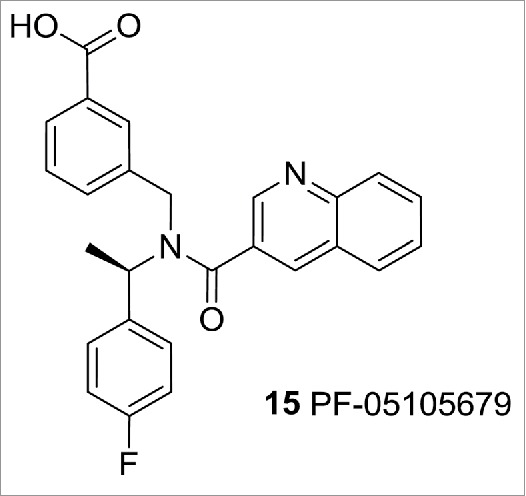
TRPM8 modulator PF-05105679.
TRPA1 is activated by a variety of ligands including exogenous electrophiles such as cinnamaldehyde, allyl isothiocyanate, and the endogenous ligand 4-hydroxynonenal (4-HNE).9 Recombinant TRPA1 is activated by noxious cold (<17°C).17 The TRPA1 channel has been directly linked to pain in humans by a gain-of-function mutation that causes familial episodic pain syndrome.25 The most advanced TRPA1 antagonist is Glenmark's GRC17536, which has shown a statistically significant and clinically relevant response in a Phase 2a clinical trial for treating diabetic peripheral neuropathy. The structure of GRC17536 has not been disclosed, but an exemplar from one of Glenmark Pharmaceutical's patent applications is shown in Figure 7 (compound 16,26 TRPA1 IC50 2.5nM). Another TRPA1 compound comes from a Hydra Biosciences/Cubist Pharmaceuticals partnership. They have taken TRPA1 antagonist CB-625, presumed to be from a series that includes 17 HC-03003127 and 18 HC-068559,28 to the clinic for the potential oral treatment of pain and inflammatory conditions. Hydra/Cubist have reported completion of a single ascending-dose Phase 1 study with CB-625, but efficacy data is not yet available.
Figure 7.

TRPA1 ligands.
TRPV3 is activated by natural products such as camphor, 2-APB, and warm temperatures (>32°C). Several classes of selective TRPV3 antagonists have to-date been disclosed (Fig. 8) that have enabled research into the role of TRPV3 in pain signaling. These include the benzothiazole 1929 and the quinazolin-4-one 20 from Hydra30 and Glenmark's GRC15300 (structure not disclosed), Glenmark reported the completion of a Phase 1 study with GRC 15300 in 2011 and, following a deal with Sanofi-Aventis, opened a Phase 2 trial in neuropathic pain in 2012.51 Unfortunately, Sanofi discontinued the trial in 2014 when the compound failed the Phase 2 proof of concept trial. Glenmark has filed a number TRPV3 patent applications, examples from which include pyridopyrimidine 2131 and benzimidazole 22.32
Figure 8.
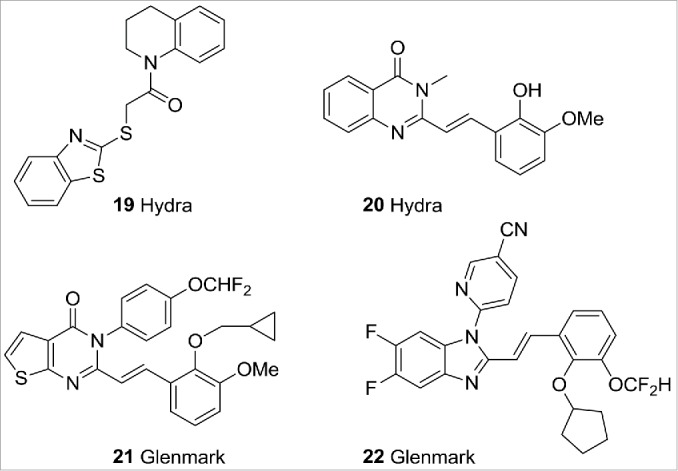
TRPV3 ligands.
TRPV4 is activated by a number of small molecules that include anandamide, 5,6-epoxyeicosatrienoic acid, and GSK1016790A, and by heat (27−34°C). A number of selective TRPV4 antagonists have been disclosed over recent years (Fig. 9). These include RN-173433 from Renovis, Hydra's HC-067047; a compound reported to be efficacious in preclinical models of bladder cystitis,34 and sulphonamide 25 from Pfizer.35 To date, the only TRPV4 ligand to have entered clinical trials is GSK 2798745, a compound of undisclosed structure that has entered a Phase 1 study. This ligand is hoped to be effective in treating pulmonary edema.
Figure 9.

TRPV4 ligands.
There is interest in the pain research field in delivering small molecule N-type Cav2.2 selective compounds. This is driven by the established clinical link of this subtype to neuropathic pain36 and the fact that specific knockdown of Cav2.2 ameliorates pain in chemically induced and neuropathic pain models.37 The only marketed N-type Cav2.2 selective compound is the analgesic peptide Ziconotide, derived from the toxin of the cone snail Conus magus. Ziconotide requires intrathecal administration to be efficacious and is associated with significant side effects. Progress has been made in developing orally acting small molecule N-type inhibitors to overcome these limitations. N-type Cav2.2 chemotypes have now been disclosed from several research groups (Fig. 10) including CNV2197944 from Convergence (likely to be from the same family as compound 26)38 and Z-160 from Zalicus (now Epirus Biopharma),39 both of which have been progressed into Phase 2 studies. The Convergence compound has now completed a pair of Phase 2 studies, in postherpetic neuralgia and diabetic peripheral neuropathy. While the resulting efficacy data is not yet available, it is expected to be revealed at the end of 2015. The Zalicus compound has also finished a pair of Phase 2 studies, in lumbosacral radiculopathy and postherpetic neuralgia. Compound 27 did not meet the primary endpoint for either of these studies.
Figure 10.
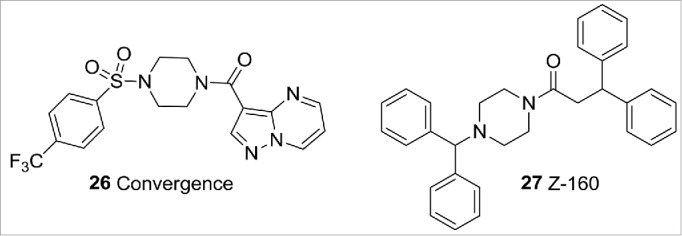
N-type Cav2.2 compounds.
T-type calcium channels are low-voltage activated calcium channels that act as modulators of action potential threshold by increasing the calcium current, leading to further depolarization of the cell and activation of other calcium channels, and ultimately, sodium and potassium channels. They are present within cardiac and smooth muscle, and many neuronal cells within the central nervous system. T-type calcium channel blockade can reduce pain in neuropathic, inflammatory, and visceral pain models. Z944, is a novel, oral, state-dependent, selective T-type calcium channel blocker (structure not disclosed). This blocker has >150-fold selectivity vs. non-T-type voltage-gated ion channels. A Phase 1 study to determine the safety, tolerability, and pharmacokinetics of oral Z944 showed it was safe and well tolerated. A Phase 1b clinical study measuring Laser-Evoked Potentials (LEP) from skin irritated following topical application of capsaicin or exposure to UV light demonstrated that Z944 reduced peak-to-peak amplitudes of LEPs from both capsaicin and UV irritated skin models and reduced subjective Visual Analog Scale (VAS) pain scores in both skin types. On the basis of these findings, Z944 is currently being tcgq.53
Kv7 ion channels are widely expressed in neurons and a genetic association exists between channel mutations in Kv7.2 and Kv7.3 and neuronal hyperexcitability.40 A number of preclinical pain models have been used to examine the effects of activation of Kv7 channels41,42 which support the role of Kv7 channels in pain signaling and the potential opportunities for Kv7 openers in pain therapy. Retigabine was approved in 2011 for use in partial-onset seizures and then later taken on to a Phase 2 proof-of-concept clinical trial for pain from postherpetic neuralgia. Unfortunately Retigabine was non-superior to placebo in this study with respect to reducing pain scores. A structurally similar analog of Retigabine, Flupirtine, is approved for treatment of lower back pain in Europe.
Ionotropic glutamate receptors (iGluRs) are ligand gated ion channels that mediate excitatory neurotransmission.43 Subtype-selective GluN2B receptor antagonists in particular have attracted attention from the pharmaceutical research community for targeting CNS disorders including stroke and Parkinson disease.44 Examples of GluN2B receptor antagonists include Ifenprodil45 and the more selective derivative Traxoprodil46 (Fig. 11). Traxoprodil has progressed into clinical trials for a number of indications including stroke and neuropathic pain.
Figure 11.
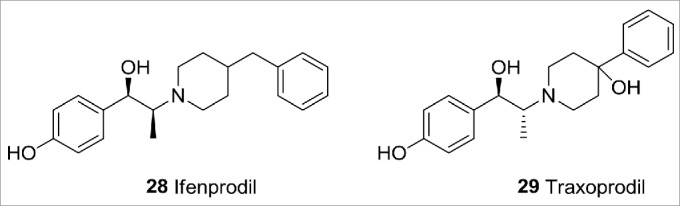
Ionotropic glutamate receptor ligands.
P2X receptors are purinergic cell surface ion channels gated by extracellular ATP.47 P2 × 3, P2 × 2/3, P2 × 4, and P2 × 7 receptors have received much attention over the last few years as potential targets to treat a variety of conditions that include chronic pain and arthritis.48 Evotec, Pfizer, and AstraZeneca have progressed P2 × 7 receptor antagonists to the clinic. Unfortunately, CE-224535 and AZD9056 (Fig. 12) did not demonstrate clinical efficacy in rheumatoid arthritis, although it is not yet known whether compounds such as these may be useful in pain indications.49
Figure 12.
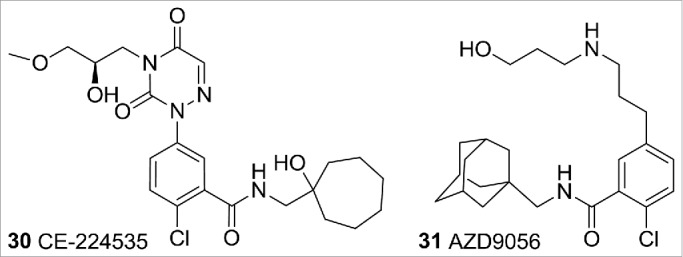
P2 × 7 ligands.
Conclusion and outlook
The modulation of ion channel signaling by small molecules has shown the ability to treat pain. However, current therapies often lack the desired level of efficacy or tolerability to offer optimal pain management. The focus for future research will inevitably be to provide safer and more effective treatments for pain. This future research will include advances in ion channel cloning and screening capabilities, increased knowledge regarding subtype selective molecules, and additional screening against ion channels known to elicit CNS and cardiovascular side effects. Finally, discovery of strong genetic linkages between specific ion channels and their related diseases will help to determine the most beneficial targets. In the end, a greater understanding of ion channel modulation will be the greatest tool we have in developing the next generation of drugs for the modulation of pain in the human disease state.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Lloyd RS, Blythe JO Jr. Clinical experiences with lidocaine as a local anesthetic. J Am Dent Assoc 1949; 39:296-8; PMID:; http://dx.doi.org/ 10.14219/jada.archive.1949.0206. [DOI] [PubMed] [Google Scholar]
- 2.Schindler W, Hafliger F. Derivatives of iminodibenzyl. Helv Chim Acta 1954; 37:472-83; http://dx.doi.org/ 10.1002/hlca.19540370211. [DOI] [Google Scholar]
- 3.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, et al.. An SCN9A channelopathy causes congenital inability to experience pain. Nature 2006; 444 (7121):894-8; PMID:; http://dx.doi.org/ 10.1038/nature05413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fertleman CR, Baker MD, Parker KA, Moffatt S, Elmslie FV, Abrahamsen B, Ostman J, Klugbauer N, Wood JN, Gardiner RM, Rees M. SCN9A mutations in paroxysmal clinical study extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron 2006; 52 (5):767-74; PMID:; http://dx.doi.org/ 10.1016/j.neuron.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 5.Yang Y, Wang Y, Li S, Xu Z, Li H, Ma L, Fan J, Bu D, Liu B, Fan Z, et al.. Mutations in SCN9A, encoding a sodium channel α subunit, in patients with primary erythermalgia. J Med Gen 2004; 41 (3):171-4; http://dx.doi.org/ 10.1136/jmg.2003.012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Data derived from text-mining search of published articles in which high relevance co-occurrences are established between ion channels and disease. Search performed by Dr. Ben Sidders, Pfizer. [Google Scholar]
- 7.Xiong Z, Strichartz G. R. Inhibition by local anesthetics of Ca2+ channels in rat anterior pituitary cells. Eur J Pharmacol 1998; 363:81-90; PMID:; http://dx.doi.org/ 10.1016/S0014-2999(98)00769-9. [DOI] [PubMed] [Google Scholar]
- 8.Bagal SK, Brown AD, Cox PJ, Omoto K, Owen RM, Pryde DC, Sidders B, Skerratt SE, Stevens EB, Storer RI, et al.. Ion Channels as Therapeutic Targets: A Drug Discovery Perspective. J Med Chem 2013; 56 (3):593-624; PMID:; http://dx.doi.org/ 10.1021/jm3011433. [DOI] [PubMed] [Google Scholar]
- 9.Backonja M, Glanzman RL. Gabapentin dosing for neuropathic pain: evidence from randomized, placebo-controlled clinical trials. Clin Ther 2003; 25:81-104; PMID:; http://dx.doi.org/ 10.1016/S0149-2918(03)90011-7. [DOI] [PubMed] [Google Scholar]
- 10.Silverman RB. From basic science to blockbuster drug: the discovery of Lyrica. Angew Chem Int Ed 2008; 47:3500-4; http://dx.doi.org/ 10.1002/anie.200704280. [DOI] [PubMed] [Google Scholar]
- 11.Zajac MA. Process for preparing α-carboxamide pyrrolidine derivatives. WO2011029762A1, 2011. [Google Scholar]
- 12.Goldberg YP, Price N, Namdari R, Cohen CJ, Lamers MH, Winters C, Price J, Young CE, Verschoof H, Sherrington R, et al.. Treatment of Nav1.7-mediated pain in inherited erythromelalgia using a novel sodium channel blocker. Pain 2012; 153:80-5; PMID:; http://dx.doi.org/ 10.1016/j.pain.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 13.Beaudoin S, Laufersweiler MC, Markworth CJ, Marron BE, Millan DS, Rawson DJ, Reister SM, Sasaki K, Storer RI, Stupple PA, et al.. Preparation of heteroarylsulfonamide derivatives for use as analgesics. WO2010079443A1, 2010. [Google Scholar]
- 14.Jarvis MF, Honore P, Shieh C-C, Chapman M, Joshi S, Zhang X-F, Kort M, Carroll W, Marron B, Atkinson R, et al.. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. PNAS 2007; 104:8520-5; PMID:; http://dx.doi.org/ 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scanio MJC, Shi L, Drizin I, Gregg RJ, Atkinson RN, Thomas JB, Johnson MS, Chapman ML, Liu D, Krambis MJ, et al.. Discovery and biological evaluation of potent, selective, orally bioavailable, pyrazine-based blockers of the Na(v)1.8 sodium channel with efficacy in a model of neuropathic pain. Bioorg Med Chem 2010; 18:7816-25; PMID:; http://dx.doi.org/ 10.1016/j.bmc.2010.09.057. [DOI] [PubMed] [Google Scholar]
- 16.Kemp MI. Structural trends among second-generation voltage-gated sodium channel blockers. Progr Med Chem 2010; 49:81-111; http://dx.doi.org/ 10.1016/S0079-6468(10)49003-7. [DOI] [PubMed] [Google Scholar]
- 17.Nilius B, Owsianik G, Voets T, Peters JA. Transient Receptor Potential Cation Channels in Disease. Physiol Rev 2007; 87:165-217; PMID:; http://dx.doi.org/ 10.1152/physrev.00021.2006. [DOI] [PubMed] [Google Scholar]
- 18.Gavva NR, Bannon AW, Hovland DN Jr, Lehto SG, Klionsky L, Surapaneni S, Immke DC, Henley C, Arik L, Bak A, et al.. Repeated administration of vanilloid receptor TRPV1 antagonists attenuates hyperthermia elicited by TRPV1 blockade. J Pharmacol Exp Ther 2007; 323:128-37; PMID:; http://dx.doi.org/ 10.1124/jpet.107.125674. [DOI] [PubMed] [Google Scholar]
- 19.Rami HK, Thompson M, Stemp G, Fell S, Jerman JC, Stevens AJ, Smart D, Sargent B, Sanderson D, Randall AD, et al.. Discovery of SB-705498: A potent, selective and orally bioavailable TRPV1 antagonist suitable for clinical development. Bioorg Med Chem Lett 2006; 16:3287-91; PMID:; http://dx.doi.org/ 10.1016/j.bmcl.2006.03.030. [DOI] [PubMed] [Google Scholar]
- 20.Gomtsyan A, Bayburt EK, Schmidt RG, Surowy CS, Honore P, Marsh KC, Hannick SM, McDonald HA, Wetter JM, Sullivan JP, et al.. Identification of (R)-1-(5-tert-Butyl-2,3-dihydro-1H-inden-1-yl)-3-(1H-indazol-4-yl)urea (ABT-102) as a Potent TRPV1 Antagonist for Pain Management. J Med Chem 2008; 51:392-5; PMID:; http://dx.doi.org/ 10.1021/jm701007g. [DOI] [PubMed] [Google Scholar]
- 21.Bakthavatchatam R, Blum CA, Brielmann HL, Caldwell TM, De LS. Preparation of substituted quinazolin-4-ylamine analogs as VR1 capsaicin receptor antagonists for relieving pain. WO2003062209A2, 2003. [Google Scholar]
- 22.Baamonde A, Lastra A, Juarez L, Hidalgo A, Menendez L. TRPV1 desensitisation and endogenous vanilloid involvement in the enhanced analgesia induced by capsaicin in inflamed tissues. Brain Res Bull 2005; 67:476-81; PMID:; http://dx.doi.org/ 10.1016/j.brainresbull.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 23.Knotkova H, Pappagallo M, Szallasi A. Capsaicin (TRPV1 Agonist) therapy for pain relief: farewell or revival? Clin J Pain 2008; 24:142-54; PMID:; http://dx.doi.org/ 10.1097/AJP.0b013e318158ed9e. [DOI] [PubMed] [Google Scholar]
- 24.(a) Owen R. TRPM8 blockade and its role in cold pain signalling. 17th RSC/SCI Medicinal Chemistry Symposium, 2013. [Google Scholar]; (b) Andrews MD, af Forselles K, Beaumont K, Galan SRG, Glossop PA, Grenie M, Jessiman A, Kenyon AS, Lunn G, Maw G. Discovery of a Selective TRPM8 Antagonist with Clinical Efficacy in Cold-Related Pain. ACS Medicinal Chemistry Letters 2015; 6(4):419-424; PMID:. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kremeyer B, Lopera F, Cox JJ, Momin A, Rugiero F, Marsh S, Woods CG, Jones NG, Paterson KJ, Fricker FR, et al.. A gain-of-function mutation in TRPA1 causes familial episodic pain syndrome. Neuron 2010; 66:671-80; PMID:; http://dx.doi.org/ 10.1016/j.neuron.2010.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar S, Thomas A, Waghmare NT, Margal S, Khairatkar-Joshi N, Mukhopadhyay I. Preparation of thieno-pyrimidinedione derivatives as TRPA1 modulators. WO2010109334A2, 2010. [Google Scholar]
- 27.Eid SR, Crown ED, Moore EL, Liang HA, Choong K-C, Dima S, Henze DA, Kane SA, Urban MO. HC-030031, a TRPA1 selective antagonist, attenuates inflammatory- and neuropathy-induced mechanical hypersensitivity. Mol. Pain 2008; 4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ng H, Weigele M, Moran M, Chong J, Fanger C, Larsen GR, Del CD, Hayward N, Adams S, Ripka A. Preparation of purine nucleobases via coupling reaction for treating disorders related to TRPA1. WO2009002933A1, 2008. [Google Scholar]
- 29.Chong JA, Fanger C, Moran MM, Underwood DJ, Zhen X, Ripka A, Weigele M, Lumma WC Jr, Larsen GR. Benzothiazole derivatives and related compounds for modulating TRPV3 function and their preparation, pharmaceutical compositions and their use for treatment of pain. WO2006122156A2, 2006. [Google Scholar]
- 30.Chong JA, Fanger C, Larsen GR, Lumma WC Jr, Moran MM, Ripka A, Underwood DJ, Weigele M, Zhen X. Dihydroquinazolinone compounds for modulating TRPV3 function and their preparation, pharmaceutical compositions and use in the treatment of pain and related disorders. WO2007056124A2, 2007. [Google Scholar]
- 31.Vs PL, Sachin SC, Abraham T, Khairatkar-Joshi N, Vidya GK. Preparation of fused pyrimidinone compounds as TRPV3 modulators. WO2009130560A1, 2009. [Google Scholar]
- 32.Lingam VSP, Thomas A, Phatangare SK, Mindhe AS, Khatik JY, Khairatkar-Joshi N, Kattige VG. Preparation of fused imidazole derivatives as TRPV3 antagonists useful in treatment and prevention of TRPV3 receptor-mediated diseases. WO2010073128A2, 2010. [Google Scholar]
- 33.Vincent F, Acevedo A, Nguyen MT, Dourado M, DeFalco J, Gustafson A, Spiro P, Emerling DE, Kelly MG, Duncton MAJ. Identification and characterization of novel TRPV4 modulators. Biochem Biophys Res Commun 2009; 389:490-4; PMID:; http://dx.doi.org/ 10.1016/j.bbrc.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 34.Everaerts W, Zhen X, Ghosh D, Vriens J, Gevaert T, Gilbert JP, Hayward NJ, McNamara CR, Xue F, Moran MM, et al.. Inhibition of the cation channel TPRV4 improves bladder function in mice and rats with cyclophosphamide-induced cystitis. PNAS 2010; 107:19084-9, S19084/1-S19084/6; PMID:; http://dx.doi.org/ 10.1073/pnas.1005333107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skerratt SE, Mills JEJ, Mistry J. Identification of false positives in “HTS hits to lead:” The application of Bayesian models in HTS triage to rapidly deliver a series of selective TRPV4 antagonists. MedChemComm 2013; 4:244-51; http://dx.doi.org/ 10.1039/C2MD20259J. [DOI] [Google Scholar]
- 36.Yamamoto T, Takahara A. Recent updates of N-type calcium channel blockers with therapeutic potential for neuropathic pain and stroke. Curr Top Med Chem (Sharjah, United Arab Emirates) 2009; 9:377-95; http://dx.doi.org/ 10.2174/156802609788317838. [DOI] [PubMed] [Google Scholar]
- 37.Altier C, Dale CS, Kisilevsky AE, Chapman K, Castiglioni AJ, Matthews EA, Evans RM, Dickenson AH, Lipscombe D, Vergnolle N, Zamponi GW. Differential role of N-type calcium channel splice isoforms in pain. J Neurosci 2007; 27:6363-73; PMID:; http://dx.doi.org/ 10.1523/JNEUROSCI.0307-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heer JP, Norton D, Ward SE. Preparation of pyrazolo[1,5-a]pyrimidine derivatives for blocking Cav2.2 calcium channels. WO2010007074A1, 2010. [Google Scholar]
- 39.Snutch TP. Synthesis and activity of substituted piperazine and piperidine calcium channel blockers. WO2001045709A1, 2001. [Google Scholar]
- 40.Yang W-P, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, Blanar MA. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem 1998; 273:19419-23; PMID:; http://dx.doi.org/ 10.1074/jbc.273.31.19419. [DOI] [PubMed] [Google Scholar]
- 41.Mruk K, Kobertz WR. Discovery of a novel activator of KCNQ1-KCNE1 K+ channel complexes. PLoS One 2009; 4(1): e4236; PMID:; http://dx.doi.org/ 10.1371/journal.pone.0004236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. Eur J Pharmacol 2003; 460:109-16; PMID:; http://dx.doi.org/ 10.1016/S0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- 43.Bowie D. Ionotropic glutamate receptors & CNS disorders. CNS Neurol Disord: Drug Targets 2008; 7:129-43; PMID:; http://dx.doi.org/ 10.2174/187152708784083821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol 2006; 6:68-74; PMID:; http://dx.doi.org/ 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 45.Reynolds IJ, Miller RJ. Ifenprodil is a novel type of N-methyl-D-aspartate receptor antagonist: interaction with polyamines. Mol Pharmacol 1989; 36:758-65; PMID:. [PubMed] [Google Scholar]
- 46.Chenard BL, Bordner J, Butler TW, Chambers LK, Collins MA, De CDL, Ducat MF, Dumont ML, Fox CB, et al.. (1S,2S)-1-(4-Hydroxyphenyl)-2-(4-hydroxy-4-phenylpiperidino)-1-propanol: A Potent New Neuroprotectant Which Blocks N-Methyl-D-Aspartate Responses. J Med Chem 1995; 38:3138-45; PMID:; http://dx.doi.org/ 10.1021/jm00016a017. [DOI] [PubMed] [Google Scholar]
- 47.Khakh BS, North RA. P2X receptors as cell-surface ATP sensors in health and disease. Nature (London) 2006; 442:527-32; http://dx.doi.org/ 10.1038/nature04886. [DOI] [PubMed] [Google Scholar]
- 48.Guile SD, Ince F, Ingall AH, Kindon ND, Meghani P, Mortimore MP. The medicinal chemistry of the P2 receptor family. Prog Med Chem 2001; 38:115-87; PMID:; http://dx.doi.org/ 10.1016/S0079-6468(08)70093-6. [DOI] [PubMed] [Google Scholar]
- 49.Keystone EC, Wang MM, Layton M, Hollis S, McInnes IB. Clinical evaluation of the efficacy of the P2´7 purinergic receptor antagonist AZD9056 on the signs and symptoms of rheumatoid arthritis in patients with active disease despite treatment with methotrexate or sulphasalazine [Erratum to document cited in CA158:123117]. Ann Rheum Dis 2012; 71:2064; http://dx.doi.org/ 10.1136/annrheumdis-2012-201289corr1. [DOI] [PubMed] [Google Scholar]
- 50.Convergence, http://www.convergencepharma.com/index.asp?page_id=14 Press release.
- 51.Glenmark, http://www.glenmarkpharma.com/GLN_NWS/homepage.aspx?res=P _GLN.
- 52.Zalicus Updates Progress on Z944 Clinical Development http://www.marketwatch.com/story/zalicus-updates-progress-on-z944-clinical-development-2014-05-22.