

Abstract
The p38/MAPK-activated kinase 2 (MK2) pathway is involved in a series of pathological conditions (inflammation diseases and metastasis) and in the resistance mechanism to antitumor agents. None of the p38 inhibitors entered advanced clinical trials because of their unwanted systemic side effects. For this reason, MK2 was identified as an alternative target to block the pathway, but avoiding the side effects of p38 inhibition. However, ATP-competitive MK2 inhibitors suffered from low solubility, poor cell permeability, and scarce kinase selectivity. Fortunately, non-ATP-competitive inhibitors of MK2 have been already discovered that allowed circumventing the selectivity issue. These compounds showed the additional advantage to be effective at lower concentrations in comparison to the ATP-competitive inhibitors. Therefore, although the significant difficulties encountered during the development of these inhibitors, MK2 is still considered as an attractive target to treat inflammation and related diseases, to prevent tumor metastasis, and to increase tumor sensitivity to chemotherapeutics.
Introduction
The pharmacological treatment of inflammatory diseases, including rheumatoid arthritis, was based for many years on prostaglandin synthesis inhibitors and NSAIDs, such as COX 2 inhibitors.1 A very important step forward in the treatment of these diseases was allowed by the disease modifying anti-rheumatic drugs (DMARD)2 that interfere with molecular and cellular steps crucial for the propagation of inflammatory disease. An example is represented by the anti-cytokine drugs, such as the monoclonal antibody adalimumab or the genetically engineered fusion protein etanercept, constituted by two recombinant human TNF-receptor p75 monomers fused with the Fc domain of human immunoglobulin G1. On the other hand, the p38 MAPK/MAPK-activated kinase 2 (MK2) signaling pathway has been studied for many years for its involvement in inflammation, cell migration, and cell cycle regulation.2-5 Experimental evidence clearly showed that production of pro-inflammatory cytokines (such as TNFα and interleukins), induction of enzymes such as COX-2, and emergence of related inflammatory diseases mainly depended on activation of the p38α MAPK/MK2 signaling pathway. On this basis, many small molecules have been described as p38α inhibitors, several of them entered clinical trials, but none progressed to phase III6 mainly because of their systemic side effects (hepatotoxicity, cardiac toxicity, central nervous system disorders). Another reason why p38 inhibitors are not suitable drugs for chronic anti-inflammatory diseases derives from the original observation that C-reactive protein levels (a biomarker of inflammation) undergo to an initial reduction just after administration of the p38 inhibitors, to return to baseline values after few week treatments.7 This phenomenon was initially attributed to a physiological escape that involved other inflammatory pathways. Further studies demonstrated that inhibition of p38 activity also suppressed a feedback control by which p38 blocked upstream kinases, such as the transforming growth factor-β activated kinase 1 (TAK1) [via TAK-binding protein 1 (TAB1) phosphorylation)]8. Consequent activation of TAK1 in turn induced downstream kinases (such as the c-Jun N-terminal kinase JNK, involved in inflammatory signals and liver dysfunctions) to hyperactivation.9 An additional consequence of p38 inhibition is the loss of mixed-lineage kinase (MLK) activation that is responsible for the production of anti-inflammatory agents, such as IL 10 and the dual specificity phosphatase 1 (DUSP1).10 These results suggested that inhibition of p38 kinase activity was not the optimal approach for treating chronic inflammatory diseases, such as rheumatoid arthritis. However, additional experimental results showed that a dual inhibitor able to block both the p38 kinase activity and its activation in cells did not lead to resistance mechanisms occurred after administration of classical p38 kinase inhibitor.11 Although this compound entered phase II clinical trials for the treatment of rheumatoid arthritis in combination with methotrexate, it suffered from a solubility/exposure issue.12
Moreover, the fact that p38 is involved in the regulation of more than 60 substrates with various physiological roles13 may contribute to account for the problems consequent to administration of p38 inhibitors.9
The current exception to the disappointing clinical results obtained for the treatment of inflammatory diseases with p38 inhibitors is represented by the chronic obstructive pulmonary disease, for which the imminent results of the phase II studies with RV-568 (structure not disclosed) could be of pivotal importance to understand if p38 inhibition is as effective as phosphodiesterase-4 inhibition and corticosteroid inhalation in the treatment of the disease.6,14
In the last years, the significant unwanted side-effects caused by direct inhibitors of p38 MAPK and their failure in clinical trials prompted the researchers to point toward downstream targets of the signaling pathway, such as MK2. Following this approach, the feedback control of p38 on the TAB1-TAK1 system is maintained, as well as the activation of downstream anti-inflammatory targets (MLK).
MK2 is the first substrate of p38 which was identified. It is involved in regulation of TNFα and interleukin-6 (IL-6) production, by increasing the stability and translation of the corresponding mRNA. In particular, MK2 phosphorylates tristetraprolin (TTP), inhibits TTP destabilizing activity toward TNF mRNA,15-16 thus permitting its translation. MK2 is currently considered as a novel DMARD ligand and a possible alternative to p38 for the treatment of inflammatory diseases. In fact, differently from p38α knockout mice that suffer from embryonic lethality and compromised fertility, health of MK2-null mice is not affected. Moreover, MK2 knockout mice show levels of cytokines significantly reduced in the serum and in the brain, as well as fewer or no symptoms in arthritis or lung sensitization models. In addition, neuroprotective effects (also in the case of ischemic injury) found after MK2 depletion suggest that chronic neuroinflammation associated with neurodegenerative diseases such as Parkinson’s disease, Alzheimer’s disease, and multiple sclerosis could be in part modulated by MK2 activity.
MK2 is also involved in the process of actin regulation. Among various actin-modulating proteins that are targets of MK2 [examples are represented by the Lin-11 Isl-1 Mec-3 kinase (LIMK), filamentous actin capping protein Z-interacting protein (CAP-zip), p16 subunit of the seven-member actin-related protein-2/3 complex (p16-Arc)], Hsp27 plays a pivotal role in actin remodeling and cell migration. Hsp27 in its unphosphorylated form is able to act as an actin filament cap-binding protein, thus inhibiting polymerization of globular actin into filamentous actin (F-actin). MK2-mediated Hsp27 phosphorylation5 blocks capping activity, thus promoting actin polymerization and remodeling.17 In addition, phosphorylated Hsp27 is unable to undergo to multimeric self-aggregation, with the consequent loss of its chaperoning activity.18 Involvement of the MK2/Hsp27 system in actin remodeling and cell migration is also of pivotal importance for cancer cell invasion and metastasis. Finally, the p38 MAPK/MK2 pathway also activates the checkpoint signaling to G2/M arrest and cell survival after DNA damage caused by chemotherapy, thus leading to resistance to therapeutical protocols.
Very recent evidence demonstrated that MK2 is also involved in cardiac hypertrophy and fibrosis. In fact, MK2 inhibitors are able to alleviate cardiac fibrosis in myocardial infarction.19 MK2 inactivation significantly reduced cardiomyocyte hypertrophy in mice by reducing COX-2 protein synthesis, but not affecting mRNA levels and protein stability.20 Contrasting results have been obtained in the case of pulmonary fibrosis in mice, where disruption of MK2 prevented myofibroblast formation and might contribute to fibrosis instead of preventing or reducing it.21 On the contrary, a peptide inhibitor of MK2 decreased the fibrotic responses in idiopathic pulmonary fibrosis.22
These findings clearly suggest that targeting MK2 to block downstream events could be equivalent to a direct inhibition of p38α, with the advantage of lacking p38-dependent side effects. Therefore, although the significant difficulties encountered during the development of these inhibitors, MK2 is still considered as an attractive target because the inhibitors of the MK2 activity could serve as therapeutic agents for the treatment of diseases associated with inflammation and neuroinflammation. They may be also used to reduce migration of cancer cells and metastasis formation. Moreover, given the ability of MK2 to activate a cell cycle checkpoint, MK2 inhibitors are also considered as effective tools to bypass DNA reparation induced by chemotherapy, and thus to increase tumor cell sensitivity to chemotherapy. Finally, MK2 inhibitors may represent an alternative and innovative approach to the treatment of fibrosis-related diseases.
Almost all of disclosed MK2 inhibitors belong to the type I class of inhibitors that bind to the ATP binding site of the kinase and thus compete with intracellular ATP to inhibit phosphorylation and activation of the substrates. After discovering a plethora of compounds with minimal to modest in vitro activity toward MK2,2,23 significant improvements in efficacy and safety have been made in comparison to previous generation compounds. In fact, compounds with in vivo efficacy have been already reported.24 However, one of the major limitations of the MK2 inhibitors discovered so far is their low biochemical efficiency (BE), expressed as the ratio between Ki (the binding affinity to the kinase) and EC50 (cellular activity). In fact, ability to bind MK2 is usually 10-100-fold higher than functional response (i.e., drug concentration that inhibits TNFα production in cells), resulting in BE values in the range between 0.1 and 0.01. Among all of the MK2 inhibitors disclosed, only very few exceptions showed optimal BE values (see compounds 61 and 81, with BE of about 0.5 and 1, respectively). Studies on the biochemical mechanisms of drug action demonstrate that two-thirds of marketed drugs show BE values higher than 0.4,25 thus suggesting that MK2 inhibitors are unlikely to become a drug. In fact, whether high concentrations of compounds are required to have a cellular efficacy, their cytotoxicity and off-target effects could be exacerbated, thus increasing attrition probability. On the contrary, compounds that do not compete with ATP could be active at lower concentrations and have higher probability to be optimized to become a drug. Unfortunately, the currently available non-ATP competitive and uncompetitive MK2 inhibitors do not give any experimental support to this hypothesis, being their BE far from the optimal values (see below).
The high affinity of the inactive MK2 to ATP has been proposed as the major determinant of low BE values for MK2 inhibitors. In fact, many other kinases have low ATP affinity in their inactive form and high ATP affinity in their active form. As a consequence, their known inhibitors have been selected among compounds that bind the inactive form of the kinase, do not compete with the high intracellular ATP concentration, and, consequently, are required at low concentrations to give cellular effects. Differently, given the high affinity of the inactive MK2 to ATP, putative ligands of the ATP binding site of either the active or inactive form of MK2 are required to compete with the high levels of ATP. For this purpose, higher concentrations of ATP-competitive inhibitors are required for cellular activity, thus decreasing BE values. On the basis of these considerations and taking into account the importance of MK2 in modulating inflammation, cell cycle, and cell motility, non-ATP-competitive and allosteric inhibitors of MK2 are under investigation as modulators of the p38 MAPK/MK2 signaling pathway.
In recent years, excellent surveys of the studies on the biology of p38/MK2 pathway have been reported,2-5 also describing the efforts made to find new and effective MK2 inhibitors.2,4,23 In this review, the most important classes of small molecules able to inhibit MK2 by an ATP-competitive and a non-ATP-competitive mechanism are reviewed. In particular, the rational design planned by medicinal chemists is described, as well as the structure-affinity and structure-activity relationships that could be elaborated by comparison of molecular structures and affinity/activity data of the disclosed compounds.
Structural and functional features of MK2
The human MK2 is a Ser/Thr protein kinase with a primary sequence comprised of 400 amino acids (UniProt entry P49137, Figure 1),26 divided into a proline-rich N-terminal region (the sequence 10-40, a unique feature among MAP kinases, mainly arranged in β-strands), a protein kinase catalytic domain (residues 64-325), and a regulatory domain at the C-terminal portion, the latter consisting in α-helices. In particular, the region ranging from positions 328 and 364 is constituted by an autoinhibitory α-helix that is arranged in such a way to block the access to the catalytic machinery. Deletion of the regulatory domain unveils the active site, allows substrate to access, and causes an increase of the catalytic activity. In a similar way, phosphorylation of Thr222 and Ser272 of the catalytic domain, as well as Thr334 by p38α (also reported as MAPK14, which is one of the four p38 MAP kinases, UniProt entry Q16539) leads to a conformational rearrangement of the autoinhibitory α helix with a consequent kinase activation. Ser9, Thr25, and Ser328 are additional phosphorylation sites, with the latter processed by autocatalysis. Moreover, the C-terminal region 366-390 represents the p38 MAPK-binding site, also referred to as the docking region. A a bipartite nuclear localization signal (NLS, 371-374 and 385-389, respectively) and a nuclear export signal (NES, a motif with the sequence 356-365) are of crucial importance for interaction with p38 and its migration to the cytoplasm.
Figure 1.

MK2 structure. A Pro-rich N-terminal region is connected to the kinase catalytic domain that contains two major phosphorylation sites (Thr222 and Ser272). A third phosphorylation site (Thr334) precedes the nuclear export signal (NES, responsible for MK2 translocation from the nucleus to the cytoplasm) and a bipartite nuclear localization signal (NLS, responsible for accumulation of the p38/MK2 complex within the nucleus of resting cells). A C-terminal domain (p38 binding site) guarantees for direct interactions between MK2 and p38.
In resting cells, the constitutively active NLS maintains the p38/MK2 complex within the nucleus. Early studies27 based on fluorescence resonance energy transfer (FRET) allowed the identification of an inactive closed conformation of MK2 with part of the autoinhibitory domain (the amino acid sequence 339-353 of the C-terminal region)28 masking the binding site. Upon cellular stress-based activation of p38, a p38-dependent phosphorylation of MK2 Thr334 induces a conformational rearrangement of the autoinhibitory domain of MK2 to an open form. The catalytic domain is unmasked and the NLS is revealed, with consequent kinase activation and translocation of the MK2-p38 complex from the nucleus to the cytoplasm. In a similar way, phosphomimetic mutations, such as T334D or T334E, increase the levels of MK2 within the cytoplasm. On the other hand, phosphorylation of Thr222 is responsible for activation of downstream targets involved in cell motility, cell cycle and apoptosis, as well as in mRNA stabilization.
Early kinetic and thermodynamic studies on the catalysis and function of the MK2-p38 complex showed direct interactions between the C-terminal sequence 370-400 of MK2 and p38 to form a tight complex (Kd = 20 nM).29 The importance of this sequence was also suggested by the fact that its deletion was able alone to abrogate p38-dependent MK2 phosphorylation and activation. A 2.7 Å resolution crystal structure of the heterodimeric complex between unphosphorylated MK2 and unphosphorylated p38α (PDB entry 2oza, Table 1)30 showed a direct contact between the N-terminal portions of both proteins, as well as additional interactions between their C-termini. The Gly-rich portion of the phosphate binding loop (P loop, constituted by the 71-76 GXGXXG sequence belonging to the ATP binding site) was in a β-sheet conformation, as already found in constitutively active MK2 enzymes (see below). Among the five major interface regions of the complex, the C-terminal docking region of MK2 was crucial for the MK2-p38 interactions. Another crystal structure of the p38-MK2 heterodimeric complex was solved at 4.0 Å resolution (PDB entry 2onl), that represented the first example of full length kinase crystallized in a complex.31
Table 1. Structures of the MK2 kinase and its complexes stored in the PDB.
| PDB entry Release date |
Chain | Solved sequence/ Protein Constructa |
Resolutionb | Ligand | Binding modec | Reference |
|---|---|---|---|---|---|---|
| 1kwp 2002-09-18 |
A, B | 46-385 47-400 |
2.80 Å | apoenzyme | 36 | |
| no PDB entry 2003 |
45-371 | 3.0 Å |
|
|
37 | |
| 1ny3 2003-10-14 |
A | 46-345 41-364 |
3.00 Å | ADP | see Figure 2 | 35 |
| 1nxk 2003-10-14 |
A, B, C, D | 42-345 41-364 |
2.70 Å |
|
|
35 |
| 2okr 2007-02-06 |
C, F (MK2) A, D (p38α) |
370-393 370-400 |
2.0 Å | p38α-MK2 heterodimer | 31 | |
| 2onl 2007-02-06 |
C, D (MK2) A, B (p38α) |
46-393 Full length |
4.0 Å | p38α-MK2 heterodimer | 31 | |
| 2jbod 2007-03-20 |
A | 44-347 41-364 |
3.10 Å |
|
|
63 |
| 2jbpe 2007-03-20 |
A, B, C, D, E, F, G, H, I, J, K, L |
46-350 41-364 |
3.31 Å |
|
|
63 |
| 2oza 2007-04-03 |
A (MK2) B (p38α) |
51-390 47-400 |
2.70 Å | p38α-MK2 heterodimer | 30 | |
| 2p3g 2007-06-12 |
X | 45-364 45-371 |
3.80 Å |
|
|
46 |
| 2pzy 2007-07-31 |
A, B, C, D | 42-346 41-364 |
2.90 Å |
|
|
60 |
| 3fpm 2009-04-07 |
A | 44-345 41-364 |
3.30 Å |
|
|
77 |
| 3fyk 2009-04-07 |
X | 45-364 45-371 |
3.50 Å |
|
|
51 |
| 3fyj 2009-04-07 |
X | 45-364 45-371 |
3.80 Å |
|
|
52 |
| 3kc3 2010-01-12 |
A, B, C, D, E, F, G, H, I, J, K, L |
43-347 41-364 |
2.90 Å |
|
|
42 |
| 3ka0 2010-01-12 |
A | 47-364 47-366f |
2.90 Å |
|
|
42 |
| 3kga 2010-01-26 |
A | 47-364 47-364 |
2.55 Å |
|
|
43 |
| 3gok 2010-03-02 |
A, B, C, D, E, F, G, H, I, J, K, L |
46-350 47-400 |
3.20 Å |
|
unpublished resultsg |
|
| 3a2c 2010-05-12 |
A, B, C, D, E, F, G, H, I, J, K, L |
47-344 41-364 |
2.90 Å |
|
|
38 |
| 3m2w 2010-07-28 |
A | 47-364 47-364 |
2.41 Å |
|
|
73 |
| 3m42 2011-03-23 |
A | 47-364 47-364 |
2.68 Å |
|
|
72 |
| 3r2y 2011-05-25 |
A | 46-365 41-364 |
3.00 Å |
|
|
64 |
| 3r30 2011-05-25 |
A | 46-365 41-364 |
3.20 Å |
|
|
64 |
| 3r2b 2011-05-25 |
A, B, C, D, E, F, G, H, I, J, K, L |
47-345 41-364 |
2.90 Å |
|
|
64 |
| 3wi6 2013-12-18 |
A, B, C, D, E, F |
46-347 41-364 |
3.00 Å |
|
|
40 |
| 4tyh 2015-07-22 |
A (MK2) B (p38α) |
51-400 | 3.00 Å | p38-MK2 heterodimer with a p38 inhibitor |
101 |
When more than one MK2 chain has been crystallized, the solved sequence and protein construct are referred to the first chain listed in the previous column. Solved sequence: the amino acid sequence listed in the PDB structure. Protein construct: the MK2 protein construct used in the study.
All the structures have been obtained by X-ray diffraction.
Graphical representations of the binding mode of MK2 inhibitors have been elaborated with Discovery Studio 3.0 Visualizer software (Accelrys Software, Inc.) by the corresponding PDB files. The coordinates of the complex between MK2 and AMP-PNP have been taken from the patent text (https://www.google.com.ar/patents/EP1578687A2?cl=en, accessed August 7, 2015), edited to correct typos probably due to a previous scanning from original patent documents, and then converted to a PDB file by means of an in house script.
Bipyramidal crystals, by soaking.
Orthorhombic crystals, by co-crystallyzation
T222E mutant.
Scheich, C.; Smith, M. A.; Barker, J. D.; Kahmann, J.; Hesterkamp, T.; Schade, M. Unpublished results.
A series of point mutations were found to differently affect MK2 activity. In particular, K93R and D207A are defective mutations that abrogate kinase activity; T222A and S272A strongly decrease kinase activity; T334A slightly decreases kinase activity; T222D or S272D are mimic of phosphorylated states and result in a slight increase of basal kinase activity; T334D or T334E are mimic of phosphorylated states and result in a significant increase of basal kinase activity; T222E associated with T334E are mimic of phosphorylated states and lead to constitutive protein kinase activity; K353R abrogates MK2 SUMOylation (that is a post-translational reversible modification affecting MK2 activity) thus leading to an increase of its protein kinase activity. A summary of mutagenesis data and the corresponding literature references can be found at the UniProt web site.32
A shorter splicing variant of MK2 (the isoform 2) does also exist, bearing the alternative GCLHDKNSDQATWLTRL sequence instead of the canonical 354-400 amino acid sequence, thus lacking the NES, NLS, and p38 docking domain.
Crystallization studies
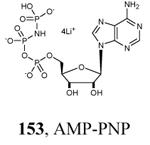
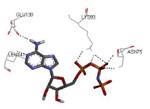
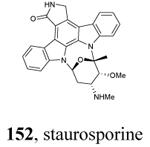
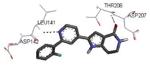
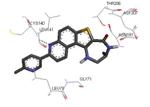
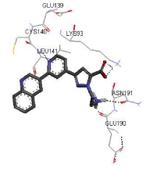
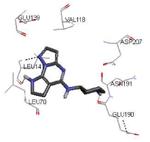
Crystallization of the kinase domain of MK2 is a difficult task to be performed, and the structures of MK2 alone or in complex with small molecule ligands and inhibitors have been solved only to low/medium resolution. A survey of the three dimensional structures of MK2 alone or in complex with small molecules is reported in Table 1. Despite the low/medium resolution gained for most of the MK2 crystal structures, MK2 can be efficiently expressed by Escherichia coli, thus allowing for high-throughput design and production of protein constructs. X-ray crystallography studies reveal that MK2 can be arranged in trimer structures that are however unable to give interactions with p38. Seven different crystal forms of MK2 (referred to as rods, plates, cubes, bipyramids, hexagonal bullets, hexagonal bullets collapsed from the latter form, and sharp blocks) have been identified and described by different research groups.33 An analysis of the MK2 crystal structures deposited within the PDB34 reveals that the construct 41-364 (a constitutively active form of the enzyme) has been most often used to obtain complexes between the protein and various small molecule inhibitors (Table 1). Moreover, a construct design strategy based on a rational mutagenesis approach was set up to optimize crystallization conditions and to define the construct(s) to be most efficiently expressed. Truncations at both the C- and N-terminus, mutations of solvent-exposed charged amino acids to Ala, deletion of the activation loop, as well as changes at the phosphorylation sites were the structural variations applied to the MK2 sequence. The 47-366 MK2 construct bearing the T222E activating mutation was the sole structure that diffracted well and was solved to 2.9 Å as a bipyramidal crystal structure (PDB entry 3ka0). The pH range and the precipitating reagents were crucial keys for a more effective crystallization. Deletion of the activation loop was not tolerated, while the overall crystallization process was more sensitive to N-truncation in comparison to C-truncation.33 Another study showed that deletion of the 1-40 sequence of the Pro-rich domain resulted in higher expression level and enhanced solubility of MK2, while best crystals were built upon removing part of the C terminal domain (namely, the sequence 365-400).35 The constitutively active form 41-364 of MK2 was thus crystallized with both ADP and the broad-spectrum kinase inhibitor staurosporine 152 (PDB entries 1ny3 and 1nxk, solved at 3.2 and 2.7 Å, respectively, Table 1). The complex with ADP allowed the identification of the pockets that accommodated the molecular portions of ATP and ADP (Figure 2). In particular, the “phosphate binding region” (a cavity delimited by Lys93, Asn191, Asp207, and capped by Ile74) was filled by the diphosphate moiety of ADP. Glu145, Glu190, Leu70, Gly71, and Leu72 constituted the “sugar pocket” and surrounded the ribose moiety of ADP. The adenine residue was accommodated within the hinge region, delimited by Glu139, Cys140, Leu141, and Asp142. Finally, a rather small hydrophobic area between the adenine binding region and the solvent, not occupied by ADP, constituted the “front pocket”. On the other hand, the complex with 152 (Table 1) showed a binding mode of the inhibitor within the ATP binding site very similar to that found in the complexes with CDK2, Src, Lck, and, in particular, with PKA. The ATP binding site was characterized by a narrow and deep groove, resulting from a closed conformation. As a Met was the gatekeeper amino acid (Met138), the ATP binding pocket had a reduced size and a narrow shape in comparison to other kinases. As a consequence, planar compounds were preferably accommodated within the pocket and their structure was difficult to be decorated to improve affinity and kinase selectivity. This finding anticipated that identification of selective kinase inhibitors could result a challenging exercise, given the high similarity of the kinase binding sites.
Figure 2.

Stereographical representation of the major interactions between ADP and the ATP binding site of MK2, as taken from the PDB entry 1ny3. The adenine ring is involved in two hydrogen bonds (represented as black dotted lines) with Glu139 and Leu141. An additional hydrogen bond is found between the pyrophosphate moiety and the charged terminal group of Lys93. Amino acids of the phosphate binding region, the sugar pocket, the hinge region are shown, as well as the gatekeeper Met138.
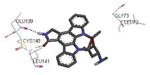
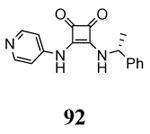
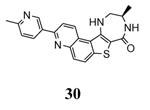
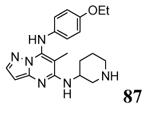
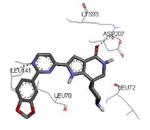
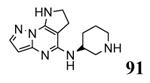
The unbound MK2 (the apo form, PDB entry 1kwp, 2.8 Å resolution) was the first crystallized structure, characterized by an inactive kinase conformation with the Gly-rich loop in an α-helical form, the substrate binding site locked by the autoinhibitory domain (even if the ATP binding pocket was in an open form), and the Lys93-Glu104 salt bridge disrupted.36 Differently, the complex between MK2 and 153 (AMP-PNP, a non-hydrolyzable ATP analogue, namely the γ-imino-ATP, Table 1) showed a different inactive conformation with the Gly-rich loop in a β-sheet form, and the Lys93-Glu104 salt bridge disrupted.37 All the remaining complexes between MK2 and small molecules (including ADP) were in an active conformation with the Gly-rich loop in a β-sheet form, and the Lys93-Glu104 salt bridge present. Exceptionally, the complex with the pyrazolo[1,5-a]pyrimidine 87 (TEI-I01800, Table 12) was the first structure in the active form, with the Lys93-Glu104 salt bridge, but with the Gly-rich loop in an α-helical form (PDB entry 3a2c, 2.9 Å resolution).38-39 It was hypothesized that the conformational shift of MK2 from β-sheet to α-helical was induced by the presence of the large p-ethoxyanilino side chain at the position 7 that forced the structure of the inhibitor to a non-planar conformation to avoid intramolecular clashes with the methyl group at position 6. To support this hypothesis, an analogue compound was designed without the aryl moiety at position 7 and with a pyrroline ring bridging the positions 6 and 7 of the pyrazolo-pyrimidine scaffold, thus leading to a pyrazolo[1,5-a]pyrrolo[3,2-e]pyrimidine compound. The new tricyclic compound 91 (also referred to as TEI-L03090, Table 12) showed micromolar inhibitory activity toward both MK2 and CDK2 (4700 and 630 nM, respectively), without appreciable selectivity. It was cocrystallized with MK2 that showed a Gly-rich loop in an β-sheet form (PDB entry 3wi6, 2.99 Å resolution).40 The N1 and 8-NH of the inhibitor showed two hydrogen bonds with both the NH and carbonyl groups of Leu141. Moreover, the piperidino basic nitrogen was involved in an additional hydrogen bonds with the backbone carbonyl group of Glu190. Similar interactions were also found in the complex with 87, although they showed better geometrical parameters (distance and angles) in comparison to the complex with 91.38 The inhibitor was able to be accommodated within the ATP binding site of MK2 in its β form.
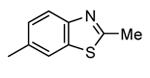
Table 12. Pyrazolopyrimidine derivatives from Teijin Pharma and BioFocus.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | R1 | R2 | IC50a (nM) |
| 84 |
|
H |
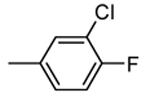
|
1300 |
| 85 |
|
Me |
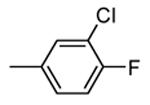
|
400 |
| rac-86 |
|
H |
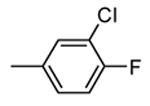
|
40 |
| 87, TEI-I01800b |
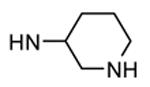
|
Me |
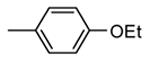
|
130 |
| 88b |
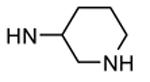
|
Me |
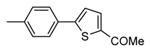
|
57 |
| 89b |
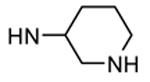
|
Me |
|
76 |
| 90b |
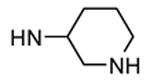
|
Me |
|
54 |
| 91, TEI-L03090 | 4700 | |||
Expressed as the inhibition of MK2 activity toward a peptide substrate.
Compounds 87-90 were in their (S)-configuration.
In the attempt to rationalize the kinase selectivity profile of 87, analysis of its complex with CDK2 showed a strained conformation of the inhibitor that led to the lack of interactions between its substituent at the position 7 and the ATP binding site of CDK2, that assumed a β-sheet form. This unprofitable interaction pattern may be responsible for the lower affinity of 87 toward CDK2 in comparison to MK2 and consequently for its 177-fold kinase selectivity.41
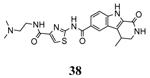
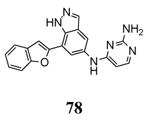
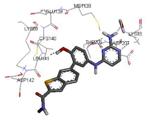
Another study demonstrated that, similarly to the complex between MK2 and 87,38 2,4-diaminopyrimidine 80 induced the Gly-rich loop to assume an α helical conformation (PDB entry 3ka0),42 even if the Lys93-Glu104 salt bridge was maintained. On the contrary, the 2,4-diaminopyrimidine analogue 78 was cocrystallyzed with MK2 in the β-form (PDB entry 3kc3).42
1. ATP-competitive MK2 inhibitors
The need to discover new therapeutics for inflammatory diseases led to hypothesize the downstream MK2 as a druggable target. High throughput screening (HTS) campaigns and the release of the three-dimensional structure of MK2 in its apo-form and in complex with ADP or with various small molecule inhibitors gave impetus to the discovery of many ATP-competitive MK2 inhibitors. However, the classical approach to block this kinase by competing with ATP for its binding site generated two challenging issues to be solved in the case of MK2 inhibitors. First, the ATP binding site of MK2 is structurally similar to that of other kinases (MK3, MK5, PKA, CDK2, etc.), thus strongly affecting selectivity. Second, either the high cellular levels of ATP or the high affinity of ATP for its binding site on MK2 caused very low BE for small molecule ATP-competitive inhibitors. Finally, solubility and permeability profiles appropriate for in vivo administration were a very difficult task to be addressed.
Routine hit-to-lead and lead optimization studies led to the identification of compounds with subnanomolar affinity toward MK2. Disasppointingly, the corresponding activity in cell-based assays was significantly lower in most cases. On the other hand, selected compounds showed oral anti-inflammatory efficacy in models of inflammation, but none have entered clinical trials yet.
The large number of small molecule ATP-competitive MK2 inhibitors identified and disclosed in the recent years belong to different chemical classes, but share common pharmacophoric portions. An analysis of the complexes between MK2 and its inhibitors deposited within the PDB showed a conserved hydrogen bond acceptor motif able to interact with the backbone NH group of Leu141. Moreover, a hydrogen acceptor group (often represented by a carbonyl oxygen atom) was an additional anchor point that interacted with the terminal ammonium group of Lys93 of the phosphate binding region. Hydrophobic contacts also contributed to the stabilization of the complexes, together with hydrogen bond interactions specific for each inhibitor. The ability of a few compounds to induce a conformational rearrangement of the Phe90 side chain and to open up an additional binding pocket could be a very important step in the process to discover MK2 selective compounds.43
In the next sections, a survery of the most important ATP-competitive MK2 inhibitors is reported, together with biological data from in vitro and in vivo assays, as well as structure-activity relationship considerations.
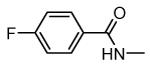
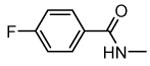
1.1. Compounds from Pfizer Global Research and Development
1.1.1. Benzopyranopyridines
Benzopyranopyridine derivatives were identified by HTS as moderate inhibitors of MK2 with an ATP-competitive mechanism.44 The prototypical compound 1 (Figure 3), that showed an IC50 value of about 2 μM (expressed as the inhibition of MK2 ability to phosphorylate a peptide susbtrate), was modified by deleting the malononitrile group and by decoration of the condensed phenyl ring. Resulting derivatives showed that different substituents and substitution patterns were tolerated, increasing MK2 inhibition to submicromolar values. The same compounds were also able to reduce TNFα expression in U937 cells treated with LPS, but with a different trend in comparison to the results of the enzymatic assay. The best compound 2 (IC50 = 130 nM) also affected TNFα production in an acute rat model of inflammation (EC50 = 920 nM), after LPS administration. Given its MK2 inhibitory activity, the ability of 2 to inhibit Hsp27 phosphorylation consequent to the p38/MK2 pathway activation, was also assayed in Werner syndrome (WS) cells.45 In fact, this pathway could be involved in F-actin stress fiber formation in WS cells, leading them to resemble aged cells. Thus, inhibition of MK2 activity was expected to reactivate the Hsp27 capping activity on actin, resulting in reduced actin polymerization. However, although 2 blocked Hsp27 phosphorylation in WS cells at 25 μM, higher amounts of stress fibers were found with respect to control. This result led to the suggestion that F-actin stress fiber production may derive from off-target effects possibly caused by LIMK activation and consequent inactivation of cofilin, a protein with depolymerizing activity toward actin.17
Figure 3.

Benzopyranopyridine derivatives from Pfizer Global Research and Development.
1.1.2. Pyrrolopyridinones
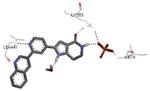
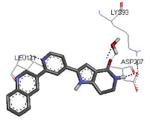
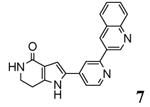
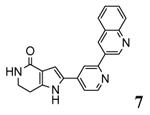
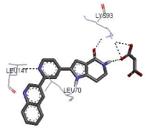
A new class of MK2 inhibitors was obtained by hit optimization of a pyrrolopyridinone compound identified by HTS.46 Preliminary SAR considerations were obtained by decoration of the pyridine appendage (Table 2). Inhibition of MK2 activity in a cell-free assay significantly increased from unsubstituted pyridine (3, IC50 = 171 nM) to the corresponding quinoline-substituted analogue (7, 8.5 nM). The presence of an aromatic substituent was profitable for activity (compare 3 and 4), and the nitrogen atom of the quinoline seemed to have a crucial role in defining the inhibitory activity toward MK2 (compare 7 and 6). Moreover, activity was maintained in the low micromolar range by a variety of substituents at the phenyl ring of 4, even if meta and para substituents were preferred in comparison to groups at the ortho positions. This suggested that a planar or quasi-planar conformation of the arylpyridine side chain could be important for activity. The new compounds showed an ATP-competitive mechanism of action and were selective toward a large panel of kinases, apart MK3 and MK5 that shared a high degree of sequence identity with MK2 and possessed a larger hinge region and front pocket that could accommodate the arylpyridine substituent. X-ray crystallography on the complex between MK2 and 5 (PDB entry 2p3g, Table 1) confirmed the expected binding mode of the inhibitor, with the arylpyridine moiety pointing toward the protein surface and partially exposed to the solvent. The pyridine nitrogen and the lactam moiety were responsible for the hydrogen bond interactions with protein residues.
Table 2. Pyrrolopyridinone derivatives from Pfizer.

| |||
|---|---|---|---|
|
| |||
| Compd | R | IC50a (nM) | EC50b (nM) |
| 3 | H | 171 | 19000 |
| 4 | Ph | 66 | 1400 |
| 5 | 2-F-Ph | 126 | 4800 |
| 6 | 3-naphthyl | 52 | 4200 |
| 7 | 3-quinoline | 8.5 | 4400 |
Expressed as the inhibition of MK2 ability to phosphorylate the KKKALSRQLSVAA peptide.
Expressed as the ability to inhibit TNFα production in LPS-stimulated U937 cells.
Improvement of MK2 inhibition in cell-free assays (with IC50 spanning from micromolar to nanomolar concentrations) did not correlate with increased potency in the cell-based assays. In fact, reduction of cellular production of TNFα, resulting from MK2 inhibition, was obtained with micromolar concentrations of the inhibitors (Table 2). The weak efficacy of the new compounds in U937 cell-based assay could be attributed to poor physico-chemical properties. However, even if efforts have been made to improve solubility and enhance oral bioavailability of these compounds,47 the sole solubility can not explain the gap found between activity in cell-free and cell-based assays. As an example, 5 showed a 160 μM solubility, significantly higher than that of the quinoline analogue 7 (< 0.4 μM). These compounds had comparable activity in U937 cell assay (EC50 of about 4-5 μM), while the inhibitory activity of the less soluble compound in the cell free assay was 15-fold better than that of 5. However, one of the most reliable explanation for the lack of correlation between the two sets of data could be based on the ATP competitive mechanism of action of these compounds. In fact, their interaction with the MK2 target was disfavored by either the high cellular concentration of ATP (1-5 mM) or its affinity value for MK2 (Km = 30 μM). The mechanism of action of these compounds were confirmed in U937 cells stimulated with LPS, where 5 inhibited both phosphorylation of Hsp27 at Ser78 and TNFα production. Moreover, the inability of 5 to inhibit kinases involved in the production of TNFα, as well as to block p38 pathway components that were upstream of MK2, strongly suggested that this compound acted on U937 cells by interfering with MK2. TNFα production was also strongly reduced (> 80%, when 5 was administered 2 h prior to LPS stimulation) by oral administration in a rat model of acute inflammation. In this case, compound concentration in the plasma samples was about 3 μg/mL. The same compound also improved cell viability in a nephrotic syndrome-related podocyte injury model by inhibition of MK2 activity and consequent reduction of COX-2 mRNA expression and Hsp27 phosphorylation.48 On the other hand, 7 was used as MK2 inhibitor to study how senescence underwent an acceleration in fibroblasts from human WS, without reaching conclusive and unambiguous results.49
1.1.3. Carbolines from Pfizer Global Research and Development
A commercially available β-carboline (8, Table 3) was also identified as a weak ATP competitive inhibitor of MK2 (2500 nM in the enzymatic assay).50 Attempts to improve its biological profile have been made by changes at the methoxy substituents. Replacement with a hydroxy (9) and a carboxymethyl group (10) retained a single-digit micromolar activity (4500 and 6600 nM, respectively), while deletion (unsubstitution of the phenyl ring), replacement with different substituents, and variation of the substitution pattern caused a significant loss in activity. Moreover, aromatization of the piperidine ring was not tolerated, as well as replacement of the nitrogen atom with an oxygen (pyrane analogue) or a methylene group (cyclohexyl analogue), thus suggesting that basicity and non-planar arrangement of the condensed ring played an important role for activity. The pendant carboxylic group appeared to be as a mandatory substituent for activity. While N-alkylation of the basic nitrogen was tolerated only for small groups such as Me (11) and Et (12), alkylation of the pyrrole nitrogen was detrimental for activity. SAR analysis at the positions 3 and 4 of the piperidine ring showed that a Me and Et group at position 4 as in 13 and 14 maintained a micromolar activity (2200 nM), similarly to hydrogen bond acceptor/donor groups (such as the hydroxymethyl and aminomethyl substituents of 15 and 16, respectively) at position 3 (3000 and 5900 nM, respectively). Considering that small alkyl groups were tolerated at both the positions 2 (corresponding to the nitrogen atom) and 4 of the condensed piperidine ring, two- and three-carbon bridges between N2 and C4 have beeb built. While 18 maintained a micromolar activity (5100 nM), its homologue 17 had a better profile, with a 18-fold increased activity toward MK2 (290 nM) and selectivity against a panel of structurally related kinases, including MK3 and MK5. In a cell-based assay on U937 cells, 17 showed a dramatic loss of activity (81 μM) that was attributed to weak cellular permeability. Disruption of the zwitterionic form by esterification of the carboxy group led to a decreased activity in cell-free assay and a significant enhancement in activity in cell-based assay. The most active compounds was the isopropyl derivative 19 that showed a two-log unit improvement in activity toward U937 cells (0.83 μM), probably consequent to an intracellular conversion of the ester prodrug into the corresponding free acidic and biologically active form. When 19 was administered in rats 1 h before LPS stimulation, it was able to inhibit TNFα production by more than 84%.
Table 3. β-Carboline derivatives from Pfizer.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | R1 | X | IC50a (nM) |
| 8 | 6-OMe | 1-COOH | NH | 2500 |
| 9 | 6-OH | 1-COOH | NH | 4500 |
| 10 | 6-COOMe | 1-COOH | NH | 6600 |
| 11 | 6-OMe | 1-COOH | NMe | 1000 |
| 12 | 6-OMe | 1-COOH | NEt | 5800 |
| 13 | 6-OMe | 1-COOH, 4-Me | NH | 2200 |
| 14 | 6-OMe | 1-COOH, 4-Et | NH | 2200 |
| 15 | 6-OMe | 1-COOH, 3-CH2OH | NH | 3000 |
| 16 | 6-OMe | 1-COOH, 3-CH2NH | NH | 5900 |
| 17 | H | CH2 | 290 | |
| 18 | H | CH2CH2 | 5100 | |
| 19 | n-Pr | CH2 | 10100 | |
Expressed as the inhibition of MK2 ability to phosphorylate the KKKALSRQLSVAA peptide.
1.1.4. Polycondensed benzothiophene derivatives
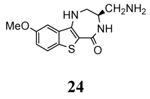
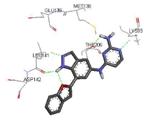
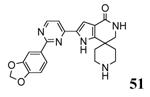
A benzothiophene condensed with a seven membered lactam ring constituted the scaffold of a new series of MK2 inhibitors (Table 4).51 These compounds however showed structural features common to known MK2 inhibitors: a methoxy substituent that interacted with the hinge region and a lactam moiety condensed to a five-membered heterocyclic ring that was at hydrogen bond distance from conserved catalytic residues of the MK2 active site. In the attempt to find compounds with significant inhibitory activity toward MK2 and selectivity against similar kinases, changes to the original scaffold (20) suggested that the nitrogen at position 5 was crucial for activity (its replacement with sulphur or a methylene spacer was detrimental), although it was unable to make hydrogen bond interactions with MK2, as evidenced in the co-crystallized complex structure between MK2 and the amino derivative of 20 (24, PDB entry 3fyk). The importance of this nitrogen atom could reside in its electronic properties as electron-donating group, and in its ability to induce a different conformation and shape of the seven-membered ring in comparison to S and to a CH2 group. Moreover, configuration of the stereogenic center at position 3 affected activity. In fact, 21 had significantly better inhibitory activity in cell-free and cell-based assay (40 and 700 nM, respectively) in comparison to its enantiomer 22 (300 and 3800 nM, respectively). Unfortunately, 21 was not selective (less than 10-fold selectivity) toward 18 out of a panel of 90 kinases. Transformation of the C3 methyl group of 21 into the corresponding primary alcohol (23) and amine (24) susbtituents led to an enhancement of the activity in the enzymatic assay (14 and 5 nM, respectively), while the results from a cell-based assay on U937 cells were not encouraging in particular for 24 (2600 nM), probably because the protonatable amine nitrogen affected cell permeability. Introduction of bulkier linear and cyclic amino groups at C3, as well as replacement of the C7 methoxy group with various alkoxy and aryloxy substituents afforded compounds with weaker activity in both assays.
Table 4. Polycondensed benzothiophene derivatives from Pfizer.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R1 | R2 | IC50a (nM) | EC50b (nM) |
| 20 | H | H | 180 | 1400 |
| 21 | Me | H | 40 | 700 |
| 22 | H | Me | 300 | 3800 |
| 23 | CH2OH | H | 14 | 400 |
| 24 | CH2NH | H | 5 | 2600 |
| 25 | 16 | 90 | ||
| 26 | H | 1 | 50 | |
| 27 | Cl | 2 | 130 | |
| 28 | Ph | 9 | 180 | |
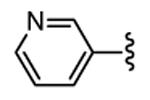
| 29 |
|
5 | 220 | |
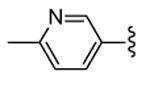
| 30, PF022 |
|
5 | 150 | |
| 31, PF318 |
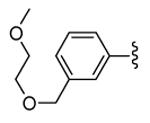
|
25 | 255 | |
| 32, PF029 |
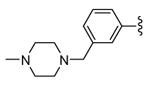
|
2 | 150 | |
Expressed as the inhibition of MK2 ability to phosphorylate the KKKALSRQLSVAA peptide.
Expressed as the ability to inhibit TNFα production in LPS-stimulated U937 cells.
Previous SAR analysis suggested that pyrrolopyridinone compounds, bearing a conformationally constrained (ridig) moiety (such as the arylpyridine group found in 3 and its congeners, Table 2) able to occupy the hinge region of MK2, could gain good selectivity toward a panel of kinases, also similar to MK2.46 On this basis, with the aim of enhancing kinase selectivity of the benzothiophene derivatives, their molecular portion bearing the methoxy substituent was rigidified with an additional condensed ring.52 A furan (25) and a pyridine ring (26) caused an increase in activity (16 and 1 nM in the enzymatic assay, 90 and 50 nM in the cell-based assay, respectively), without selectivity toward CDK2 and other 26 kinases out of a panel of 109 kinases (26 showed a 0.8 nM activity toward CDK2). In the next step, the position adjacent to the heteroatom was decorated by an aryl moiety reminiscent of the aromatic ring found in pyrrolopyridinone derivatives, such as 4-7 (Table 2). The 3-Cl (27), 3-Ph (28), 3-pyrid-3-yl (29), and 3-(4-methylpyrid-3-yl) (30) derivatives of 26 retained a single-digit nanomolar activity in the enzymatic assay (2, 9, 5, and 5 nM, respectively) with a slight reduction of cellular potency (130, 180, 220, and 150 nM, respectively). They however showed a selectivity for MK2 versus CDK2 ranging from 10 to >1000-fold. The most active compounds 29 and 30 (also referred to as PF-3644022 or PF022) were demonstrated to reduce TNFα production by a mechanism of action that selectively inhibited MK2. In fact, only 4 out of 50 kinases assayed were inhibited by more than 70% by 30 at a 1 μM concentration. The crystallographic structure of the complex between MK2 and 30 (PDB entry 3fyj) clearly described the interactions between the inhibitor and the MK2 catalytic site. Although the lactam moiety was reported to interact with the conserved Lys93 and Asp207, dihedral angles of the crystallographic structure assumed sub-optimal values. The sole hydrogen bond contact involved the selectivity-element (the bipyridyl moiety) that was in part accommodated within the hinge region and pointed toward the solvent. In particular, the nitrogen atom of the condensed pyridine ring made a hydrogen bond with the NH backbone group of Leu141, as the methoxy oxygen atom of 24 did.
An in deep biological profiling of 30 confirmed an ATP-competitive mechanism of action and a good selectivity toward a panel of 200 human kinases.24 Interestingly, only 16 of these kinase were inhibited by more than 50%, and 11 of them belonged to Ca++/calmodulin-dependent kinase group that also comprised MK2. Expectedly, inhibitory activity of 30 toward MK2 and MK5 was similar, while a 10-fold selectivity was found for MK3. In functional assays, TNFα production stimulated by LPS administration was inhibited at submicromolar concentrations (150 nM) in U937 cells, and a comparable IC50 value (201 nM) accounted for the inhibition of MK2 ability to phosphorylate Hsp27. Taken together, these data strongly supported the hypothesis that a decrease of TNFα production was consequent to selective inhibition of MK2. In addition, pharmacokinetic parameters of 30 was considered appropriate for proceeding to in vivo evaluation. In fact, this compound showed rapid absorption (Tmax of about 10 min in a 3 mg/kg suspension), oral bioavailability, good half-life (about 9 h), and low clearance. Oral administration showed efficacy of 30 in the rat model of acute inflammation, with a dose-dependent response for inhibition of TNFα production (inhibition was higher than 90%). Incouraging results were also obtained in the rat model of chronic inflammation, where arthritis was induced by administration of peptidoglycan-polysaccharide complexes from group A streptococcal cell wall (an animal models of systemic inflammation induced by bacterial endotoxins). A significant reduction of chronic paw swelling was found (about 80%), even if lower than that induced by p38 inhibitors (90-95%).
Unfortunately, 30 showed acute hepatotoxicity in beagle dogs and monkeys. An impressive increase (up to 25000%) of bile salts and bilirubin total amount was found that caused reversible liver injury.53 To understand the mechanism leading to increased levels of the serum biomarkers of hepatotoxicity, hepatic metabolism of benzothiophene derivatives was studied. In addition to various hydroxylated metabolites, 30 was transformed in a pair of diastereomeric glutathione conjugates at position 11.54 Moreover, an additional glutathione metabolite was generated by metabolic attack to the pendant pyridine ring. Incorporation of a polyether side chain as a metabolism shunt led to 31 (PF318) where the O-dealkylation occurred as the principal biotransformation way, while the diazepinone-conjugate disappeared. This compounds also retained a 25 nM inhibitory activity toward MK2, although its cellular potency was significantly reduced to 255 nM. Surprisingly, enhanced exposure and plasma concentrations upon oral administration caused hepatic tissue damage of increased severity in comparison to that found for 30. Considering that the level of bilirubin and bile salts are affected by hepatobiliary transporters (the multi-drug resistance-associated protein 2, MRP2, and the bile salt export pump, BSEP, respectively), MK2 inhibitors were evaluated for their activity toward these transporters. Both MRP2 and BSEP, as well as P-gp were strongly inhibited by 30 and its derivative 31, with IC50 values ranging from 7 to 38 μM, thus suggesting off-target effects toward these transporters. Previous SAR considerations showing that the cationic form of basic compounds negatively affected their affinity for MRP2, prompted the researcher to introduce a piperazine ring instead of the polyether chain. The resulting compound 32 (PF029) did not inhibit the activity of MRP2, BSEP, and P-gp at the maximum test dose (70-80 μM), and, consequently, did not induce liver toxicity in dogs (5-300 mg/kg oral dose).
In a very recent work, 30 has been used as a probe to confirm the link between both p38/MK2 and Sonic Hedgehog (Shh) pathways in breast cancer cells. In particular, experimental evidence demonstrated that Shh served as activator of p38 and MK2 by inducing their phosphorylation. Phospho-p38 and phospho MK2 in turn acted as activators of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), thus resulting in enhanced glycolityc activity and cell proliferation in MCF-7 and MDA-MB-231 cells. Significantly decreased levels of phospho-PFKFB3 have been found upon treatment with the MK2 inhibitor 30, thus demonstrating a direct involvement of MK2 in the phosphorylation and activation of PFKFB3.55 This very intriguing result suggested that the combined administration of MK2 inhibitors and small molecules able to block the Hh pathway56-59 could be a useful strategy for the treatment of specific tumors, such as the breast cancer.
1.2. Compounds from Boehringer-Ingelheim Pharmaceuticals
1.2.1. Carbolines
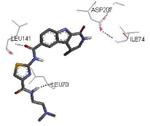
A dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) was used to evaluate the ability of small molecules to inhibit MK2 catalysis.60 As a result, a new class of carboline derivatives with micromolar activity was discovered (one of the prototypical entries of this class was the tetrahydro-β-carbolinone 33 with an IC50 of 5400 nM, Table 5). Improved potency was obtained by substitution of the amide group with aromatic and heteroaromatic amide moieties. While substituted phenyl ring resulted in slightly enhanced activity, a significant improvement was gained with pyridine (34, 520 nM) and thiazole (35, 820 nM) rings. Further decoration of the thiazole with a 4-amide (36) or 4-methylamide (37) substituents ameliorated the activity by about one log unit (89 and 72 nM, respectively). Lengthening the amide chain by polar group (as in 38) led to compounds with IC50 in the range between 20 and 35 nM. The three-dimensional structure of the complex between MK2 and 38 (PDB entry 2pzy) showed the ligand accommodated within the ATP binding site and interacting with the hinge region. The two amide moieties made hydrogen bonds with the NH of Leu141 and the carbonyl group of Leu70. The terminal amine substituent, as well as the lactam moiety that seemed to affect activity, were not involved in hydrogen bonds with MK2.
Table 5. Carboline analogues from Boehringer-Ingelheim Pharmaceuticals.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R1 | R1, R2 | IC50a (nM) | EC50b (nM) |
| 33 | NH2 | 5400 | ||
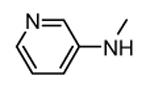
| 34 |
|
520 | ||
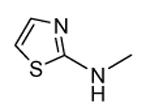
| 35 |
|
820 | ||
| 36 |
|
89 | ||
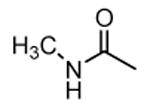
| 37 |
|
72 | ||
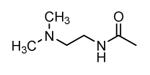
| 38 |
|
34 | ||
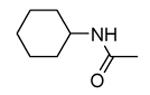
| 39 |
|
44 | 1600 | |
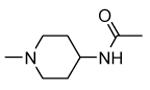
| 40 |
|
10 | >5000 | |
| 41 |
|
20 | 1400 | |
| 42 |
|
5 | >5000 | |
| 43 |
|
H, Me | 7 | 8900 |
| 44 |
|
H, Me | 4 | 1670 |
| 45 |
|
H, Me | 3 | 735 |
| 46 |
|
Me, Me | 2 | 430 |
| 47 |
|
-(CH2)3- | 2 | 300 |
Expressed as the inhibition of MK2 in a DELFI assay.
Expressed as the ability to inhibit TNFα production in LPS-stimulated THP-1 cells.
Evaluation of TNFα production in THP-1 cells stimulated by LPS showed that the new compounds were inactive (IC50 > 10 μM) with the sole exception of 39 (44 nM in the cell-free assay, 1600 nM in the cell-based assay) that lacked a terminal polar group. This result suggested that polar groups could affect membrane permeability and result in the lack of cellular activity.
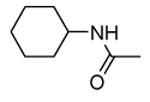
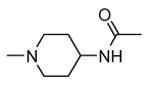
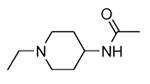
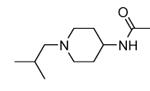
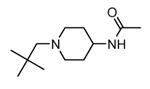
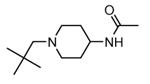
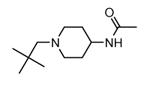
Transformation of the indolopyridinone scaffold into the corresponding dihydropyrazino[1,2-a]indolone61 resulted in a slight improvement in cell-free assay, although cellular potency was maintained at micromolar concentrations (compare activity data for 39 and 41, as well as and 40 and 42). However, the basic nitrogen atom of the piperidine analogues led to better solubility (about 100 mg/L for 42 at pH = 7.4, in comparison to cyclohexyl compounds, such as 39 and 41, that showed a solubility lower than 1.5 mg/L). In the attempt to combine the positive effect of the piperazine ring on cell-free assay with an improvement of cellular activity, basicity of the nitrogen atom was attenuated by the synthesis of less polar derivatives. Exploration of the N-alkyl series led to compounds that maintained both a nanomolar activity in cell-free assay and a solubility of about 30 mg/L, while showing a measurable submicromolar activity in the cell based assay (43-45). The neo-pentyl group at the piperidine ring of 45 was the most profitable substituent for activity and was kept fixed when a SAR analysis was attempted at the opposite pyrazinone ring. The gem-dimethyl (46) and the spiro cyclobutyl analogue 47 showed a further increase in cellular potency (430 and 300 nM, respectively), with a significant kinase selectivity. Evaluation of the effects of 47 on the phosphorylation of upstream (p38) and downstream (Hsp27) components le to the suggestion that cellular activity of such a compound is mediated by MK2 inhibition. However, 47 did not affected TNFα production in the LPS assay, probably because of its high binding to plasma proteins.61
Introduction of polar groups (hydroxymethyl and aminomethyl) at the position adjacent to the lactam moiety of both the carboline and pyrazinoindolone derivatives led to compounds with weak activity in the cell-free assay (the best values were in the two-digit nanomolar concentrations), and with cellular potency higher than 10000 nM.62
1.3. Compounds from Merck Research Laboratories
1.3.1. Spiropiperidines
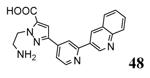
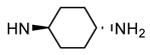
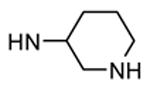
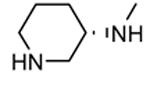
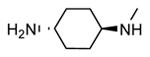
Compounds bearing a common quinolinylpyridine system (Figure 4) were used in a hybridization approach to find new entries able to block MK2 activity via an ATP-competitive mechanism of action. The three-dimensional coordinates of the complexes between MK2 and 7 (PDB entry 2jbo and 2jbp,63 as well as 3r2y)64 or 48 (PDB entry 3r30),64 two ATP-competitive MK2 inhibitors previously described by Pfizer,46,65 showed that the common cycles of the two ligands were accommodated with the same orientation and conformation within the binding site of MK2. The quinoline ring pointed toward the solvent, and occupied the front pocket of the ATP binding site without any specific interaction with the protein. The pyridine nitrogen made a hydrogen bond with the NH group of the Leu141 backbone. Moreover, the carbonyl group of both ligands interacted by hydrogen bond with the ammonium terminal group of Lys93. The distinctive molecular portions of the inhibitors also showed contacts with MK2. In particular, the NH lactam group of 7 interacted by a hydrogen bond with the carboxyl terminus of Asp207, while the terminal amino group of 48 made a bifurcated hydrogen bond with both the carbonyl groups of Glu190 backbone and Asn191 side chain. In the attempt to maintain the interactions with MK2 found for the common molecular portions and to gain the specific interactions of the NH lactam of 7 and the amine group of 48, a molecular hybridization approach has been performed to synthesize new hybrid putative MK2 inhibitors. The first hybrid inhibitor 49 was designed by changing the pyrrolopyridinone system into a pyrazolopiperazine nucleus on the basis of the easier synthetic feasibility of the pyrazole scaffold in comparison to the pyrrole analogue. Moreover, a methyleneamino side chain was added, reminiscent of compound 48. Unfortunately, affinity of 49 toward MK2 was significantly decreased in a cell-free immobilized metal ion affinity-based fluorescence polarization (IMAP) assay (151 nM) in comparison to both parent compounds (49 and 28 nM, respectively).64 Details on the binding mode of 49 into the MK2 ATP binding site were obtained by the X-ray crystallographic structure (not disclosed) that showed a suboptimal arrangement of the amino group that was unable to make hydrogen bonds with Asn191. Given the initial failure of this approach, a small library of additional compounds was designed to explore the chemical space around the overall molecular structure, and in particular to restore the interactions with amino acid residues of the ribose pocket. To have a good affinity and cellular activity profile, the most challenging issue to be solved for a compound was to set up the right balance between solubility and membrane permeability. Satisfactory results were obtained by combination of the pyrrolopyridinone scaffold with a piperidine ring in aza-spiro compounds, such as 50 and 51 (Table 6). They showed nanomolar affinity toward MK2 (6.3 and 4.3 nM, respectively), and micromolar inhibitory activity (4800 and 910 nM, respectively) for the production of TNFα in THP-1 cells. Compound 51 also affected Hsp27 phosphorylation (620 nM) and was selective toward a large panel of kinases. Moreover, the presence of the spiropiperidine basic moiety improved several pharmacokinetic parameters, such as lipophilicity and solubility, as well as plasma protein binding and microsomial stability. Unexpectedly, absorption was very poor following oral administration (rat AUC 0.016 μM·h at a 10 μmol/kg dose), while a moderate bioavailability was detected upon intravenous and sub-cutaneous administration (rat AUC of 1.26 and 2.71 μM·h at a 4 μmol/kg and 10 μmol/kg dose, respectively). Bioavailability consequent to sub-cutaneous adminstration led 51 to be efficacious in the in vivo inhibition of LPS-induced acute TNFα production in rats. Based on these results, 51 was considered as a good starting point for a lead optimization process focused on the enhancement of oral bioavailability. Replacement of the methylenedioxophenyl moiety of 51 with aryl (variously substituted phenyl moieties) and heteroaryl (naphthyl, quinolyl, pyridyl, and pyrimidyl) substituents was tolerated and resulted in compounds with nanomolar affinity.66 This suggested that the aromatic portion accommodated within the front region of the ATP binding site was not of crucial importance in determining affinity toward MK2. Cellular activity of these compounds was in the micromolar range in most cases, and seemed to be correlated with lipophilicity values higher than 1 (expressed as calculated logP). However, an increased lipophilicity did not result in enhanced membrane permeability and oral bioavailability. The basicity of the unsubstituted 4-piperidine, that was introduced to ameliorate the solubility profile of these compounds, was suspected to be in part responsible of the lack of permeability and oral bioavailability.66 The most simple attempt made to reduce pKa values of these compounds was to change the substitution pattern of the piperazine ring, by moving the basic nitrogen toward the electron-deficient pyrrole ring, resulting in a net electron-withdrawing balance. Accordingly with this hypothesis, 4- and 3-piperidyl analogues showed a difference of more than 0.4 log unit in their clogP. As an example, the 3-piperidyl analogue of 51 (52) showed an experimental pKa = 8.3, one log unit lower than that found for 51 (9.3). Moreover, pharmacokinetic parameters were in general improved: Caco-2 permeability was 34 nm/s, AUC in rats after oral administration was 3.2 μM·h, with reduced microsomial stability both in human and rat (>60 and 48 min half-life, respectively). The oral exposure (bioavailability after oral administration) significantly improved in comparison to 4-piperidine analogues was in part counterbalanced by a 6-fold reduced affinity toward MK2 (28 nM) with respect to 51 (4.3 nM). Similar results in terms of affinity and pharmacokinetic parameters were found for compounds bearing an alkyloxy-substituted pyridine nucleus instead of the methylenedioxophenyl moiety, even if the oral bioavailability decreased to lower values (0.88 μM·h). Given the enhanced pharmacokinetic profile of 3-piperidyl spiro analogues, they were synthesized as enantiomerically pure compounds (ee > 99%). As a general trend, the (S)-enantiomer showed affinity and cellular activity higher than that of the (R)-stereoisomer. As an example, (S)-52 showed an affinity 4- and 12-fold better in comparison to the racemic compound and the (R)-stereoisomer, respectively. A similar trend was found for inhibitory activity in cell-based assay, that occurred at sub-micromolar concentrations. Oral availability of (S)-52 was not improved with respect to the racemic mixture (F = 48%, AUC = 3.5 μM·h).
Figure 4.

Hybridization approach to design pyrazolo-pyperazines.
Table 6. Spiro-piperidines from Merck Research Laboratories.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | Y, X | IC50a (nM) | EC50b (nM) |
| 50 |
|
N, 4-NH | 6.3 | 4800, 1600 |
| 51 |
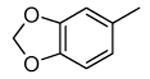
|
N, 4-NH | 4.3 | 910, 620 |
| 52 |
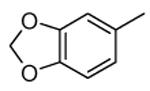
|
N, 3-NH | 28 | 1400, 930 |
| (S)-52 |
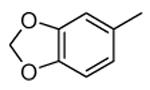
|
N, 3-NH | 7.4 | 850, 500 |
| (R)-52 |
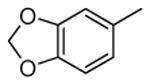
|
N, 3-NH | 91 | 8300, 7700 |
| 53 |
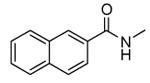
|
CH, 4-NH | 0.7 | 280, 320, 230 |
| 54 |
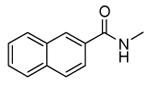
|
CH, 4-NMe | 2.6 | 1100, 730, 70 |
| 55 |
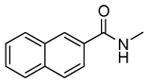
|
CH, 3-NH | 7.6 | 780, 1100, 70 |
| (S)-55 |
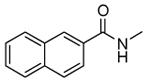
|
CH, 3-NH | 2.5 | 510, 530, 170 |
| 56 |
|
CH, 3-NH | 8.9 | 320, 870, 70 |
| (S)-56 |
|
CH, 3-NH | 2.4 | 310, 320, 70 |
Expressed as the inhibition of MK2 in a IMAP assay.
Expressed as the ability to inhibit TNFα production and Hsp27 phosphorylation in LPS-stimulated THP-1 cells, as well as to inhibit TNFα production in hPBMC cells, respectively.
The lack of any specific interaction between the terminal aryl moiety (i.e., the methylenedioxophenyl system of 51) and the front pocket of the ATP binding site prompted the researchers to introduce an amide spacer able in principle to make hydrogen bond contacts.67 Replacement of the original moiety with a benzamido and naphthylamido terminal group, in combination with a pyridine instead of the central pyrimidine ring gave very active compounds. Better activity found for pyridine-containing compounds was attributed to a conformational preference caused by electrostatic favorable interactions between the pyridine N and the amide NH, as well as between the amide CO and a pyridine CH moiety.
A few compounds taken as representative examples of three sub-series of 4-piperidine, 3-piperidine, and 3-pyrrolidine derivatives showed impressive enhancement of both MK2 affinity (with values up to sub-nanomolar concentrations) and cellular activity (up to two-digit nanomolar concentrations).67 A SAR analysis of biological data allowed to rule out several considerations: i) unsubstituted piperidine derivatives had better affinity in comparison to the corresponding alkylated analogues (compare 53 and 54); ii) values of MK2 affinity and cellular activity in the Hsp27 and TNFα assays showed a similar trend (compound with better affinity were also more active in cells); iii) unexpectedly, biological data for the assay on human peripheral blood mononuclear cells (hPBMC) showed very high potency, but their trend was opposite in comparison to that of affinity and other cell-based assay: among congeneric compounds, those with lower affinity and lower cellular activity in the TNFα and Hsp27 assays had very high activity in the hPBMC assay (compare 54 and 53, 55 and (S)-55), probably due to off-target effects; iv) among 3-piperidine derivatives 52, 55, and 56, the (S)-enantiomers were confirmed to be more active than the corresponding (R)-enantiomers and racemic mixtures; v) although logP and pKa values for these compounds could suggest acceptable membrane permeability, PAMPA values were in general very low (around 0 nm/s), apart for several fluorinated derivatives, such as 56.
1.3.2. Aminopyrazinylthiourea derivatives
A HTS led to the identification of a micromolar inhibitor of MK2 (57, IC50 = 2.0 μM, EC50 = 7.9 μM in reducing LPS-stimulated TNFα production in THP-1 cells) with an aminopyrazinylthiourea scaffold (Figure 5).68 An intensive SAR study showed that the thiourea spacer can not be replaced by an urea system, the aminopyrazine moiety was a mandatory feature, while replacement of the chlorine with small alkyl groups was tolerated. In particular, the corresponding methyl, ethyl, propyl, and cyclopropyl analogues showed IC50 values of 4.0, 1.5, 2.3, and 0.47 μM, respectively, in the MK2 enzyme assay. In alternative, decoration of the phenyl ring led to identify a series of carbamates with improved activity. As examples, methyl, ethyl, n-butyl, i-butyl, t-butyl, and benzyl carbamate side chains at position 4 of the parent compound showed IC50 values of 0.94, 0.31, 0.23, 0.15, 0.19, and 0.46 μM, respectively. A cyclic carbamate was also found to have a 0.21 μM inhibition of MK2. The best chemical features found for the right part (small alkyl groups such as n-Pr and c-Pr) and for the left part (t-Bu and cyclic carbamates) of the molecular scaffold were combined and led to compounds with a two-digit nanomolar inhibition of MK2 (from 15 to 45 nM) and micromolar activity (from 0.75 to 3.0 μM) in cells. Because they were also effective in reducing TNFα production in an acute inflammation mouse model, further modifications were attempted. Though thiourea was found to be a fundamental structural feature for activity, it was replaced with other groups in the attempt to avoid its potential cytotoxicity. However, compounds bearing an amide, sulfonamide, and guanidine moiety, as well as a five- or six-membered heterocycle instead of the thiourea group showed a weak activity (in the micromolar range) in the enzyme and cell-based assays.69
Figure 5.

Aminopyrazinylthiourea from Merck Research Laboratories.
1.3.3. Imidazo[1,2-a]pyrazines
In addition to play a critical role in the signal transduction pathways that regulate the production of pro-inflammatory cytokines, MK2 was recently identified as a cell cycle checkpoint kinase acting at the cytoplasm during the late G2/M phase. In a similar way, also CHK1 is a serine/threonine protein kinase that is the effector of the G2 phase checkpoint. As a consequence, both MK2 and CHK1 inhibitors are considered as possible anticancer agents able to block cell cycle checkpoints, cell cycle arrest, and DNA repair induced by chemotherapeutics. In this context, a HTS campaign for the identification of new ATP-competitive CHK1 inhibitors led to the discovery of an imidazo[1,2-a]pyrazine (58, Table 7) with submicromolar in vitro potency (IC50 = 650 nM) toward MK2.70 Attempts to improve the potency and selectivity of this compound led to diaminocyclohexyl derivatives that showed a significantly better profile. As an example, 59 had a IC50 = 60 nM and a 18-fold selectivity toward CHK1. Its mechanism of action at the molecular level was not investigated, although a similar compound was co-crystallized within the ATP binding site of CHK1. Any further change to the structural features of 59 led to loss in MK2 potency.
Table 7. Imidazopyrazines Merck Research Laboratories.

| ||
|---|---|---|
|
| ||
| Compd | R | IC50a (nM) |
| 58 |
|
650 |
| 59 |
|
60 |
Expressed as the inhibition of MK2 ability to phosphorylate a peptide substrate.
1.4. Compounds from Novartis Institutes for BioMedical Research
1.4.1. Pyrrolopyrimidinones and pyrazoles
Another class of MK2 inhibitors71 was derived directly by the pyrrolopyridinones previously reported.46 The new pyrrolopyrimidinones were obtained by transformation of the pyridone moiety into a pyrimidinone ring (Table 8), keeping fixed the pyridino substituent at the position 2 of the pyrrole. The two hydrogen bond donors (NH) groups at both the pyrrole and lactam moiety appeared as crucial keys for activity, while a p-F styryl chain at the position 2 of the pyridine led to 60 with submicromolar activity (200 and 350 nM, respectively) in both the cell-free and hPBMC cellular assay. In an attempt to ameliorate the poor solubility of 60 (3 mg/L), the fluorine was replaced with larger polar groups. Although the most active compound 61 showed a solubility enhanced to 41 mg/L, its activity (51 and 110 nM) was significantly lower than that of the pyrrolopyridinones described by Pfizer (single-digit nanomolar inhibitory activity in cell-free assay and a single-digit micromolar activity in U937 cells).46 It also showed a modest selectivity toward a panel of about 30 kinases, with a 24 nM activity toward the closely related MK5. MK2 was identified as the target of this compound, although targeting of kinases upstream of p38 can not be excluded.
Table 8. Pyrrolopyrimidinones and pyrazoles from Novartis.

| |||
|---|---|---|---|
|
| |||
| Compd | R | IC50a (nM) | EC50b (nM) |
| 60 | F | 200 | 350 |
| 61 |
|
51 | 110 |
| 62 | 2000 | ||
| 63 | p-OMe | 84 | |
| 64c |
|
82 | 5300 (4100) |
| 65c |
|
61 | 2500 (2000) |
Expressed as the inhibition of MK2 activity toward a peptide substrate.
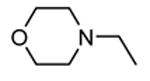
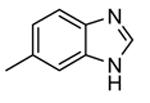
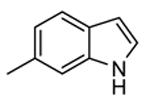
Expressed as the ability to inhibit TNFα production in hPBMC cells stimulated with both LPS and IFNγ, and to inhibit Hsp27 phosphorylation in anisomycin-stimulated THP-1 cells (in parentheses).
R group is in meta position.
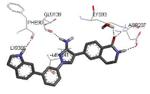
Following a scaffold-hopping strategy, the pyrrolopyrimidinone core was simplified to give a benzamide scaffold bearing a five-membered heterocycle at the para position, instead of the pyridine substituent (Table 8).43 A 3-aminopyrazole was identified as the heterocycle the most profitable for activity. As an example, 62 showed a micromolar activity (2000 nM) in a cell-free assay. Not surprisingly, further analysis of the benzamide portion revealed that rigidification into the corresponding lactam moiety (as in 63) resulted in a significant improvement in affinity (84 nM), but without cellular activity even if this compound showed appreciable permeability. Moreover, in the search for better substituents than the methoxy group, a benzimidazole (64) and an indole nucleus (65) maintained a 82 and 61 nM activity, respectively, and gained, at the same time, a single-digit micromolar activity in both cellular assays (inhibition of TNFα production in hPBMC stimulated by LPS, and inhibition of Hsp27 phosphorylation in THP-1 cells stimulated by anisomycin). X-ray crystallographic studies on the complex between MK2 and 65 (PDB entry 3kga) showed that the aminopyrazole moiety was accommodated within the hinge region (as the arylpyridine group of the parent compounds) and made hydrogen bonds with both Glu139 and Leu141. On the other hand, the lactam carbonyl gave the usual hydrogen bonds with the conserved residues Lys93 and Asp207. Very interestingly, the indole portion of the inhibitors forced the structure of MK2 to an induced fit of Phe90. As a result, a hydrophobic pocket was opened behind the hinge region, that was probably responsible for the high kinase selectivity found for 65. A 68% inhibition of the TNFα production in LPS-treated mice was found at a 100 mg/kg oral dosage, while no inhibition was measured at a 30 mg/kg oral dose. This was probably due to the low solubility of 65 (< 2 mg/L) that, at the 30 mg/kg dose, only guaranteed a 1700 nM blood concentration, lower than that required to exert an effect in cells (in fact, in hPBMC, TNFα production was inhibited with an EC50 = 2500 nM).
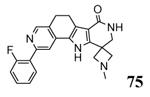
1.4.2. Pyrrolo-isoquinolines and tetracyclic derivatives
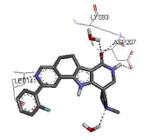
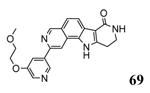
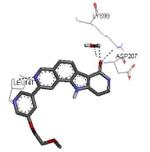
A HTS campaign led to the identification of a micromolar inhibitor of MK2 (3800 nM) bearing a pyrrolo[2,3-f]isoquinoline amide scaffold 66 (Table 9)72 reminiscent of the pyrrolopyridine compounds previously reported by others.46 A combination of the structural features of both classes of compounds resulted in tetracyclic derivatives with five-, six-, and seven-membered lactam rings. SAR analysis of the MK2 inhibitory properties showed that the six-membered lactam ring was better for activity, as already found for pyrazinoindolone derivatives previously described.62 Moreover, the NH lactam group was important for hydrogen bond interactions, and thus did not tolerate alkyl substituents. One of the most active compounds of this series was 67, bearing a styryl chain already found as a profitable substituent for pyrrolopyrimidinones,71 had a 1 and 9 nM activity in cell-free assay and as inhibition of TNFα production in hPBMC, respectively. A very interesting selectivity (about 200-fold) was also found toward c-Jun N-terminal kinase 2 (JNK2), whose inhibition may affect TNFα production in cells. Unfortunately, the overall selectivity toward a panel of 26 kinases was significantly low (13 kinases were inhibited by 67 with IC50 < 1000 nM). The corresponding 3-F,4-OMe-phenyl derivative 68, although underwent a reduction in activity (15 and 97 nM in the cell-free and in the hPBMC assay, respectively), also inhibited Hsp27 phosphorylation at 500 nM concentration and was very selective toward kinases (JNK2 was inhibited with a 1420 nM IC50, while none of the remaining 25 kinases was affected). A similar activity/selectivity profile was found for 69. These findings suggested that phenyl and pyridyl substituents conferred kinase selectivity, while styryl groups broadened activity toward a wide range of kinases. None of the tetracyclic compounds showed activity after oral administration in LPS-treated mice, with the sole exception of the ketone derivative 70 that reduced TNFα release by 73% (13 μM plasma level after 2 h).
Table 9. Tricyclic and tetracyclic pyrrole derivatives from Novartis.

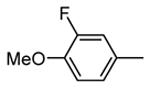
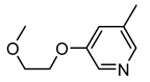
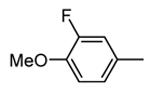
Expressed as the inhibition of MK2 activity toward a peptide substrate.
Expressed as the ability to inhibit TNFα production in hPBMC cells stimulated with both LPS and IFNγ, and to inhibit Hsp27 phosphorylation in anisomycin-stimulated THP-1 cells (in parentheses).
Lack of solubility and oral bioavailability were the major limitation of the tetracyclic compounds described above. To address this issue, a systematic approach to modify each of the four rings was applied (Table 10). In particular, de-aromatization of the central phenyl ring of 69 into a cyclohexadiene moiety (71) maintained an activity comparable to that of the parent compound, while ring expansion or contraction led to significantly less active compounds.73 Next, three- and four-membered spiroalkanes were attached to the lactam moiety with different substitution patterns. In particular, spirocyclopropanes 72 and 73 showed an appreciable activity profile as MK2 inhibitors and in cell assays, even if they still suffered from low solubility (9 and <4 μM, respectively, in comparison to 8 μM found for 71). A dramatic enhancement of MK2 inhibitory activity was found with spiroazetidines at the lactam moiety. The unsubstituted derivative 74 had a 17 nM activity toward MK2 and submicromolar ability to inhibit TNFα production and Hsp27 phosphorylation in cell-based assays (100 nM). Its solubility was considerably improved to 337 μM. The corresponding N-methyl and N-ethyl analogues (75 and 76, respectively) retained submicromolar cellular activity, while their MK2 inhibition occurred at concentrations lower than 3 nM. In the case of the N-methyl azetidine derivatives (such as 75), inhibition of MK2 occurred at the same concentration, irrespective of the aryl substituent at the pyridine ring. Importantly, the sole derivatives bearing a o-F phenyl substituent at the pyridine ring showed solubility values from 337 (74) to >1000 μM (77), probably consequent to an out-of-plane rotation of the pendant phenyl ring in comparison to the tetracyclic core. Finally, changing the pyridine into a pyrimidine, as well as the central pyrrole into a N-methyl pyrrole or furan led to compounds with lower activity and solubility. Compounds 73 and 75 were further tested for their ability to inhibit LPS-induced TNFα production in vivo in mice models. A good oral activity was found after a 100 mg/kg administration: TNFα release was inhibited by 96 and 90%, respectively, at 30 and 6 μM plasma concentration. Encouraging pharmacokinetic parameters for 72 in rats at 1 mg/kg po (in terms of maximal plasma concentration, clearance, and half-life time) allowed its evaluation in the chronic models of rheumatoid arthritis. A significant reduction in swelling was observed, without body weight reduction. However, considering that 1 μM 72 showed a > 95% inhibition of 14 out of 262 human kinases, as well as its high plasma concentration in rats and mice, the overall effects observed in chronic model may in part derive from off-target activity toward kinases different from MK2. Differently from the spirocyclopropanes as 72, that showed appreciable absorption and acceptable clearance in vivo, the spiroazetidines as 75 had the best activity toward MK2 and in cell-based assays. Details of the binding mode of 75 into the ATP-binding pocket of MK2 (PDB entry 3m2w) revealed the important role of the nitrogen atom of the azetidine ring that made a water-bridged hydrogen bond interaction with the carboxy terminus of Glu145, in addition to the usual hydrogen bond between the nitrogen atom of the condensed pyridine and Lys141.
Table 10. Tetracyclic pyrrole derivatives from Novartis.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | X | IC50a (nM) | EC50b (nM) |
| 71 |
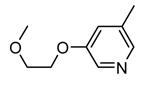
|
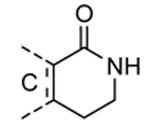
|
40 | 400 (1700) |
| 72 |
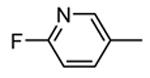
|
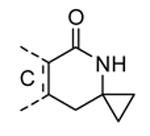
|
50 | 300 (1100) |
| 73 |
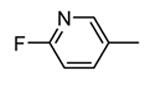
|
|
27 | 700 (1400) |
| 74 |
|
|
17 | 100 (100) |
| 75 |
|
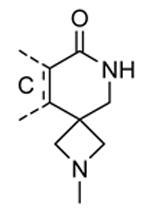
|
<3 | 100 (300) |
| 76 |
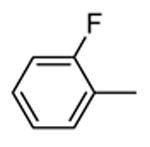
|
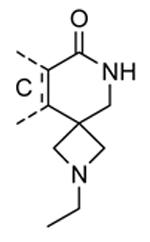
|
<3 | 700 (1500) |
| 77 |
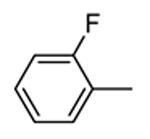
|
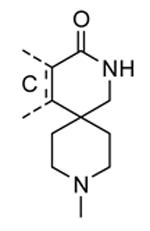
|
4 | 200 (700) |
Expressed as the inhibition of MK2 activity toward a peptide substrate.
Expressed as the ability to inhibit TNFα production in hPBMC cells stimulated with both LPS and IFNγ, and to inhibit Hsp27 phosphorylation in anisomycin-stimulated THP-1 cells (in parentheses).
1.5. Compounds from Abbott Laboratories
1.5.1. Aminopyrimidines
The X-ray three-dimensional structure of the complex between MK2 and 2,4-diaminopyrimidine derivatives 78 and 80 (PDB entries 3kc3 and 3ka0) showed an unexpected binding mode of the inhibitors within the ATP binding site.42 In fact, the diaminopyrimidine system was known to interact with the hinge region of various kinases and expected to form hydrogen bonds with Glu139 and Leu141 of the MK2 hinge region. On the contrary, the indazolepyrimidine 78 and the phenolpyrimidine 80 (Table 11) showed their aminopyrimidine moiety at hydrogen bond distance from the catalytic Lys93 and Thr206 near the DFG region, while the opposite benzofuran and benzothiophene ring were accommodated within the hinge region and pointed to the solvent. Given this binding mode, four different molecular portions were modified to optimize potency and selectivity of the small molecule inhibitors:74 i) to better interact with the extended hinge region, the 2-benzofuran ring of 78 was replaced with the 2-benzothiophene found in 80, thus resulting in 79 that maintained a MK2 inhibitory activity of 160 nM. The phenol moiety (see 80) was not further considered because of its metabolic liability; ii) an aromatic and aliphatic amide introduced in place of the primary amine group led to inactive compounds; iii) a significant enhancement in activity to 56 nM was gained by rigidification of the NH bridge into a bicyclic pyrrolo[2,3-d]pyrimidine moiety as in 81; iv) in turn, the NH group of the newly generated pyrrole nucleus was substituted with various polar groups to gain interactions with the Glu145 and Glu190 residues of the ribose pocket by hydrogen bonds and salt bridges. The most active compounds (82 and 83) had better MK2 inhibitory activity (19 and 35 nM, respectively) in comparison to 81 (56 nM). These compounds also showed a submicromolar inhibition of TNFα production in LPS-stimulated peripheral human monocytes, but their oral bioavailability at a 10 mg/kg dose was negligible. Given the weak enzymatic and cellular activity of the compounds, as well as a very low oral exposure, further development of the series was abandoned.
Table 11. Aminopyrimidine derivatives from Abbott Laboratories.

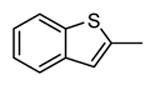
Expressed as the inhibition of MK2 activity toward a peptide substrate by using a homogeneous time-resolved fluorescence method.
Expressed as the ability to inhibit TNFα production in LPS-stimulated peripheral human monocytes (PHM).
1.6. Compounds from Teijin Pharma and BioFocus
1.6.1. Pyrazolo[1,5-a]pyrimidines
A HTS campaign on a focused kinase library comprised of 5075 compounds led to the identification of a micromolar MK2 inhibitor belonging to the class of pyrazolo[1,5-a]pyrimidine derivatives (84, Table 12).75 This compound acted as an ATP-competitive MK2 inhibitor and showed an IC50 = 1300 nM in a cell-free assay, as well as a significantly better affinity toward CDK2 (55 nM). In the attempt to improve MK2 affinity and to enhance selectivity, a large series of derivatives was designed and synthesized. Among them, C2-substituted analogues of 84 were inactive, while insertion of a halogen substituent at the position C3 led to compounds that retained a single-digit micromolar activity, while gaining a single-digit nanomolar activity toward CDK2. As a consequence, both the positions C2 and C3 were left unsubstituted in the next derivatives. A three-fold better cell-free activity (400 nM) was found when the diaminocyclohexyl side chain at C5 was replaced by a racemic 3-piperidylamino moiety, as in 86. A similar trend was observed by introduction of small alkyl groups (Me and Et) at the position C6. As an example, 85 showed a 400 nM activity and improved selectivity toward CDK2. Interesting results were also obtained by changing the C7 side chain to a p-ethoxyaniline moiety, as in 87. In particular, the (S)-enantiomer of 87 had a 130 nM activity, about a log unit better than that of the corresponding (R)-enantiomer (1200 nM), and a 177-fold selectivity. Although several structural features of the complex between MK2 and the pyrazolo[1,5-a]pyrimidine (and pyridine) derivatives was predicted by docking simulations,76 the availability of the three-dimensional coordinates of the complex between 87 and MK2 (PDB entry 3a2c)38-39 shed light into their direct interactions. In particular, a non-planar conformation that minimized steric clashes between the C6 and C7 substituents of the inhibitor was found. Moreover, the pendant aryl moiety was accommodated within a pocket originated by a β-sheet-to-α-helix conformational rearrangement occurred at the Gly-rich loop of MK2. The analysis of the complex suggested further variations at the position C7. While larger alkoxy groups instead of the p-methoxy substituent of 87 led to compounds that retained similar activity (ranging from 130 to 160 nM) and gained some CDK2 selectivity, biaryl groups and bicyclic heteroaryl moieties increased activity by about 20-fold (as in 88-90). Both 87 and 90 were able to reduce TNFα production in LPS-stimulated THP-1 cells (EC50 of 7.2 and 7.6 μM, respectively) and in a murine model of endotoxin shock (C57BL/6 mice treated with a 100 mg/kg oral dose showed 14% of serum TNFα). Pharmacokinetic parameters (bioavailability, total clearance, and half-life time) were also satisfactory.
1.7. Compounds from Wyeth Research
1.7.1. Squarate derivatives
A squarate derivative (92, Table 13) was identified by means of a HTS campaign and co-crystallyzed with the 41-364 truncated form of MK2 at 3.3 Å resolution (PDB entry 3fpm).77 Hydrogen bonds between the terminal ammonium group of Lys93 and one of the carbonyl oxygen atoms of 92, as well as between the pyridine nitrogen and the NH group of Leu141 (hinge region) were the major interactions found in the crystal structure. The micromolar MK2 inhibitory activity of 92 in a cell-free assay (8.9 μM), its poor kinase selectivity, and the ability to significantly inhibit members of the CYP450 family (in the micromolar and submicromolar range), suggested the need to improve both potency and selectivity. Changes at the benzylamino portion showed that the presence of a methyl group with the appropriate configuration was a crucial key for activity. In fact, the (S)-enantiomer (93) and the unsubstituted derivative (94) underwent a deep decrease of activity. Moreover, decoration of the phenyl ring led to the identification of various hydrogen donor groups at the meta position as profitable substituents, with the 3-hydroxyl group of 95 and 96 being the most effective (0.38 and 0.39 μM, respectively). They both gained a 20-fold higher activity in comparison to the parent compound 92, and 96 also showed an inhibition of CYP450 isozymes higher than 10 μM. On the other hand, variations at the pyridine ring resulted in micromolar inhibition of MK2, with the sole exception of 97 that showed an IC50 = 0.67 μM. The most active compounds were also evaluated for their ability to inhibit the production of TNFα in THP-1 cells pretreated with LPS. The increased polarity of 95 and 96 seemed to be related to a lower activity in the cell-based assay in comparison to 92 (34 and >40 vs 11 μM found for 92). On the contrary, 97, obtained by keeping fixed the core structure of 92, showed a 0.67 and a 1.1 μM activity in cell-free and cell-based assays, even if off-target effects have not been investigated. Kinase selectivity was also ameliorated.
Table 13. Squarate derivatives from Wyeth Research.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | R1 | R2 | IC50a (μM) |
| 92 | H |
 Me Me |
Ph | 8.9 |
| 93 | H |
 Me Me |
Ph | >100 |
| 94 | H | H | Ph | 88 |
| 95 | H | Me | m-OH-Ph | 0.38 |
| 96 | H | CONH2 | m-OH-Ph | 0.39 |
| 97 |
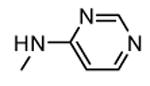
|
 Me Me |
Ph | 0.67 |
Expressed as the inhibition of MK2 activity toward a peptide substrate.
1.8. MK2 inhibitors from marine sponges
Crude extracts of marine sponges were evaluated for their ability to inhibit MK2 activity. Two meroterpenoids were found in the metanolic portion obtained from Acanthodendrilla sp.78 that was collected in 1996 on reefs near Makassar (Indonesia). Repeated extractions and fractionations, followed by reverse-phase HPLC yielded (+)-makassaric acid 98 and the structurally related (+)-subersic acid 99 (Figure 6). Their MK2 inhibition, evaluated by means of an ELISA based assay on the 41-353 human recombinant MK2, was in the micromolar range (9.6 and 20 μM, respectively). Later, a short and very versatile synthesis of 98 was proposed as a tool to obtain small libraries of analogue compounds, in the attempt to figure out SAR considerations, and finally to improve MK2 inhibitory activity.79
Figure 6.

MK2 inhibitors extracted from marine sponges.
1.9. MK2 inhibitors designed by computer-aided drug discovery approaches
In the attempt to find new inhibitors of MK2, a series of theoretical studies based on both structure- and ligand-based drug design approaches have been performed starting from the structure of MK2 and already known MK2 inhibitors. 3D QSAR,80-82 pharmacophoric models,83 and docking studies81 were generated for pyrrolopyridinones disclosed by Anderson,46 for carbolines described by Wu,60 and for thiourea derivatives described by Lin.68 Disappointingly, although all of these models were described as useful tools for designing novel MK2 inhibitors, none of them was effectively used for this purpose. A more elaborated computational protocol based on compounds reported by Novartis,43,71-72 comprised also of database search within commercially available collections of compounds, led to hypothesize several original scaffolds as putative MK2 inhibitors that, however, have not been tested.84
Recently, a computational protocol for the study of tautomerization and ionization of the previously described pyrrolopyridinone46 and benzothiophene derivatives51-52 showed that inclusion of all tautomers and isomers was a mandatory condition to obtain reliable and robust models to be used in the hit-to-lead and lead optimization steps of the ligand design process.85
1.10. Peptides as inhibitors of MK2 activity
A 23-mer cell-permeant peptide able to inhibit MK2 activity was built by linking a modified analogue of the selective, not cell-permeant, peptidic inhibitor of MK2 (KKKALNRQLGVAA) with a cell-penetrating peptide (CPP).86 The resulting sequence 100 (WLRRIKAWLRRIKALNRQLGVAA) was evaluated for its ability to reduce MK2-dependent Hsp27 phosphorylation. A 80% decrease of Hsp27 phosphorylation was obtained with 200 μM 100 in an in vitro kinase assay that evaluated recombinant human Hsp27 phosphorylation by purified MK2. As a comparison, the original non-penetrating peptide reduced Hsp27 phosphorylation by only 64%. Hsp27 phosphorylation was also reduced in keloid fibroblasts treated with the inhibitor peptide (10 μM). At the same concentration, expression of connective tissue growth factor and collagen was also decreased by 68 and 76%, respectively. A reduced concentration of the inhibitor (5 μM) did not affect these parameters. Unfortunately, the new peptidic inhibitor showed a kinase selectivity lower than that found for the original MK2 inhibitor. In the attempt to design more specific kinase inhibitors with a peptidic scaffold, additional CPP were added to the MK2 inhibitor peptide KALNRQLGVAA.87 As a result, although the peptide 101 bearing a YARAAARQARA CPP (YARAAARQARAKALNRQLGVAA) showed a significant decrease of MK2 inhibitory activity in comparison to 100 (5.8 versus 0.74 μM), its selectivity toward a panel of 43 kinases was ameliorated. The most relevant examples were represented by p38α, interleukin-1 receptor-associated kinase 4, myosin light chain kinase, PKBβ, PKCδ, and rho-associated coiled-coil-containing kinase 1, whose percent kinase activity were 61, 12, 4, 18, 11, and 0 after treatment with 30 μM 100, in comparison to 100, 68, 66, 96, 105, and 95 after treatment with 300 μM 101. Moreover, while 100 induced cell death at 40 μM, the YARAAARQARA-containing peptide 101 could be used at concentrations up to 3000 μM without toxicity toward pleural mesothelial cells. As expected, these peptides were also able to reduce pro-inflammatory cytokine (TNFα and IL-6) production in LPS-induced THP-1 monocytes, as well as to decrease Hsp27 phosphorylation in the same cells.88 Further studies on 101 led to the identification of the analogue 102 (YARAAARQARAKALARQLGVAA, also referred to as MMI-0100, bearing a Asn to Ala substitution at the position 15) that showed kinase selectivity and a MK2 inhibitory activity corresponding to IC50 = 22 μM.87 This peptide was used in various in vivo animal models. As examples, its anti-inflammatory activity was used to reduce formation of scars and adhesions without affecting intestinal healing in cases of abdominal surgery.89 Moreover, treatment with 102 resulted in a significant reduction of intimal hyperplasia in ex vivo and in vivo mouse vein graft models,90 as also previously found for 100 in a human saphenous vein model.91 It also inhibited cardiac fibrosis in myocardial infarction,19 and affected MK2-induced inflammatory and fibrotic responses in idiopathic pulmonary fibrosis.22
Recent studies on the role of matrix stiffness on the uptake of MK2 inhibitory peptides shed light on an unusual effect found upon treatment with 102 that was required in millimolar concentrations for efficacy in cells, while it was active in the range between 10-100 μM in animal models. This evidence resulted from the fact that matrix stiffness (of polystyrene used for tissue culture) and cell density affected uptake.92 These results suggested that studies on possible changes of drug uptake consequent to diseases-induced tissue rigidity should be included in the drug discovery processes and, in principle, also in the personalized therapies.
2. Non-ATP-competitive MK2 inhibitors
Although the significant number of small molecules able to inhibit MK2 by an ATP-competitive mechanism of action, they in general showed poor BE, expressed as the ratio between binding affinity (Ki toward MK2) and cellular activity (EC50). This is mainly due to the high binding affinity of ATP toward MK2 (2 μM) and the high cellular concentration of ATP (about 2-5 mM). As a consequence, a mechanism of action different from ATP-competitive could enhance BE of MK2 inhibitors. In fact, a non-ATP-competitive inhibitor is expected to achieve inhibitory efficacy easier than compounds that do compete with the high concentration of cellular ATP and its high affinity for MK2. Moreover, non-ATP-competitive inhibitors should guarantee higher selectivity profiles as a consequence of the fact that they do not bind the ATP binding site that is similar among kinases.
Several attempts have been made to identify and optimize small molecules acting as non-ATP-competitive and uncompetitive inhibitors of MK2. As an example, Merck researchers applied a particular HTS to find allosteric, non-ATP-competitive MK2 inhibitors by application of the Automated Ligand Identification System (ALIS) technique. ALIS is an integrated affinity selection–mass spectrometry protocol comprised of sequential rapid size-exclusion chromatography, reverse-phase chromatography, and electrospray ionization mass spectrometry, efficiently used for rapid and direct identification of non-covalent small molecule ligands from combinatorial libraries and mixtures. Three steps are included in the overall process: the soluble target biomolecule is exposed to a mass-encoded small molecule library to allow the formation of any suitable ligand-protein complex (affinity-based ligand selection); the resulting complexes are separated from non-binding protein and ligands by size-exclusion chromatography during a rapid and low temperature step that also allows identification of complex with weak affinity ligands; finally, a reverse-phase chromatography column induces complex dissociation, while the eluted ligand is analyzed by a high-resolution mass spectrometer and identified on the basis of its molecular weight. Further details on the ALIS method and its advantages in comparison to other affinity selection–mass spectrometry techniques are reported in the original literature.93
ALIS was included in a HTS procedure with recombinant purified MK2 consisting of the minimal kinase domain, spanning from residue 41 to 338. Truncation of both the amino and carboxy termini was mainly chosen to reduce termal stability of the kinase domain. In fact, an α-helical motif (sequence 334-364) of the carboxy terminus, folded on the kinase domain surface, interacts with the substrate binding cavity, thus stabilizing the kinase domain and limiting its accessibility by ligands. Its removal could increase the probability of the ALIS screening to find small molecules that are able to bind both the classical and allosteric sites on MK2.
2.1. Compounds from Merck Research Laboratories
2.1.1. Furan-2-carboxamide derivatives
Application of ALIS protocol to Merck mixture-based combinatorial libraries led to the identification of a diaryl 2-furyl amide 103 (Table 14) that showed a novel binding mode in comparison to the classical ATP-competitive small molecules.94 Different NMR studies and biochemical (enzymatic analysis of MK2 inhibition) experiments were set up to demonstrate that 103 bound MK2 without competing with ATP (non-ATP-competitive inhibition) and that its binding was site-specific. This compound showed a single-digit micromolar activity in a DELFIA immunoassay (5500 nM) and toward SW1353 chondrosarcoma cells (about 2000 nM) without appreciable cytotoxicity, as well as a significant kinase selectivity in an in house profiling panel assay. Interestingly, its activity was not affected by variations of ATP concentration.
Table 14. Furan-2-carboxamides from Merck Research Laboratories.

| |||||
|---|---|---|---|---|---|
|
| |||||
| Compd | R | R1 | X | IC50 (nM) | EC50b (nM) |
| 103 | Cl | H |
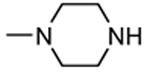
|
5500 | 2000 |
| 104 | Cl | Et |
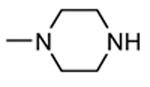
|
130 | 800 |
| 105 | Cl |
|
|
110 | 350 (5000) |
| 106 | CN |
|
|
115 | |
| 107 | CN |
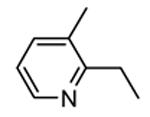
|
|
79 | |
| 108c |
|
10 | |||
| 109c |
|
10 | |||
| 110c |
|
5.4 | |||
| 111 | CN |
|
|
9.0 | 3460 |
| 112 | CN |
|
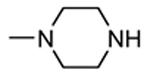
|
12 | |
| 113 | CN |
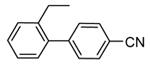
|
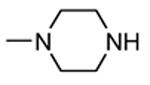
|
8.0 | 2950 |
| 114 | CN |
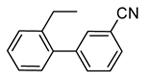
|
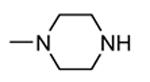
|
19 | 1590 |
| 115 | CN |
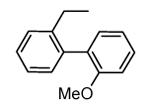
|
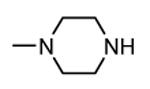
|
20 | 1960 |
| 116 | CN |
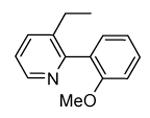
|
|
12 | 734 |
| 117 | CN |
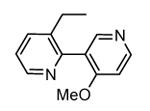
|
|
9 | 159 |
| 118 | CN |
|
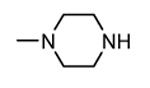
|
12 | |
| 119 | CN |
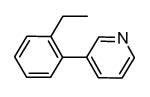
|
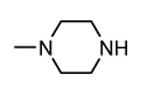
|
9 | 1240 |
| 120 | CN |
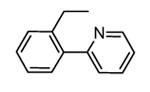
|
|
14 | 2540 |
| 121 | CN |
|
|
8 | 310 |
| 122 | CN |
|
|
27 | 880 |
Cell-free activity was determined in a DELFIA immunoassay
Cellular activity is expressed as inhibition of Hsp27 phosphorylation in SW1353 chondrosarcoma cells (103-105) or TNFα production in THP-1 cells (106-122). Both values are reported for 105.
Substituents R and R1 were not specified for these compounds.
A classical medicinal chemistry approach was planned in the attempt to improve biochemical (in a cell DELFIA immunoassay) and cell-based activity (inhibition of phosphorylation of the Hsp27 natural substrate in SW1353 cells), as well as ADME-Tox parameters. Changes at the p-Cl phenyl ring in terms of substitution pattern and substituents (Me, OMe, CF3) did not result in better compounds. Similarly, small variations at the piperazinephenyl moiety and replacement of the central furan core with five- and six-membered heterocycles resulted in compounds with activity comparable to or lower than that of the parent compound. A significant improvement in activity resulted from N-substitution of the carboxamide group, probably due to a conformational rearrangement of the amide bond to a cis-isomer that was reported as more stable by about 3 kcal/mol. Tertiary amides bearing homologous alkyl chains showed submicromolar activity in both assays, without cytotoxicity. The ethyl chain led to the best compound (104), with IC50 and EC50 values of 130 and 800 nM, respectively. Even better results were obtained with heterocyclic substituents. Among them, the 2-pyridyl derivative 105, with IC50 and EC50 values of 110 and 350 μM, was further profiled: apart a >50% inhibition toward CK1γ3, it did not affect activity of a panel of 150 kinases; no inhibition was found toward a panel of CYP450 enzymes. Its pharmacokinetic properties showed appreciable oral bioavailability in rat, with inhibition of pro-inflammatory cytokine secretion in human THP-1 acute monocytic leukemia cells consequent to MK2 inhibition. LPS-stimulated THP-1 cells treated with 105 suffered from inhibition of TNFα and IL-6 production. MMP13 secretion was inhibited in both SW1353 chondrosarcoma and human primary chondrocyte cells. Although the promising biological profile shown by 105, its inhibitory activity of TNFα production in THP-1 was about 5 μM, thus suggesting a significant difference between binding in cell-free assay and cellular activity.
As previously found, changes at the p-Cl phenyl moiety did not result in more active compounds, with the sole exception of the p-CN analogue 106 that retained the same affinity found for 105 (about 110 nM).95 Moreover, the corresponding methylated analogue 107 showed a further improvement of affinity (79 nM).
Given that the para-substitution was the best substitution pattern for the central phenyl ring C, diverse nitrogenated heterocyclic rings were attached instead of the terminal piperazine portion. Although the 3- and 4-piperidine, as well as the 1,4-diazepane analogues only had a submicromolar affinity, the azetidine (108), pyrrolidine (109), and 2-piperidine (110) derivatives showed an affinity significantly enhanced (10, 10, and 5.4 nM, respectively) in comparison to the piperazine analogue 105. Unexpectedly, compounds bearing these cycloalkylamino side chain were not further studied and discussed, while the piperazine ring was considered the best group to be kept within the structure of these MK2 inhibitors.95 Finally, replacement of the pyridine ring with a biaryl system as in 111 suggested that only the ortho-substitution pattern of the central aromatic ring D was optimal to give active compounds. In fact, ortho-biphenyl derivatives bearing one polar substituent at the terminal phenyl ring E (112-115) showed IC50 ranging from 8 to 20 nM and EC50 (expressed as inhibition of TNFα production in THP-1 cells) about three orders of magnitude higher (from 1590 to 3460 nM). Substituents and substitution pattern on the terminal phenyl ring did not show a significant influence in determining affinity and cellular activity. Similar activity values were found for compounds bearing a biaryl system that contains a vicinal or distal pyridine ring (116-121) as well as a terminal pyrimidyl group (122).
2.1.2. Dihydrooxadiazoles and imidazoles
In a next step, the amide bond of the furan-2-carboxamide derivatives was rigidified by replacement with a five- and six-membered heterocyclic ring (Table 15).96 Only a dihydrooxadiazole core (123) was appropriate for affinity (50 nM, measured in a IMAP assay), although cellular activity was more than two orders of magnitude higher (6700 nM, measured as inhibition of Hsp27 phosphorylation in THP-1 cells). Affinity and activity were tolerant to the transformation of the 5-phenyl ring into pyridine (124-127), pyrimidine (128-129), and imidazole (130) analogues. Also a pyrimidylphenyl system as in 131 was profitable for both affinity and cellular activity (40 and 730 nM, respectively). Modification of 131 gave some SAR suggestions. In particular, para position for Cl is optimal for activity and affinity: in fact, m-Cl or m,p-dihalo derivatives showed a more than 5-fold reduction in affinity. On the other hand, replacement of Cl with a F as in 132 maintained the same biological profile (50 and 760 nM for affinity and cellular activity, respectively), while a p-CN group resulted in a 8 nM affinity (133). The two enantiomers of 133 were resolved (although their absolute configuration was not determined): only one of them retained a 6 nM enzymatic activity and a 170 nM inhibitory activity toward phosphorylation of Ser78 of the natural substrate Hsp27. The mechanism of action of this compound was confirmed to be as non-ATP-competitive, as already found for its analogue and parent compounds. Unfortunately, a significant gap between cell-free enzymatic data and cellular activity was maintained, probably due to low solubility and high plasma protein binding (about 97% in both rat and human). Moreover, moderate absorption and rat hepatocyte clearance were also found for 133, thus resulting in a poor pharmacokinetic profile.
Table 15. Rigidification of the cis-amide into five-membered heterocyclic rings.

| ||||
|---|---|---|---|---|
|
| ||||
| Compd | R | X | IC50a (nM) | EC50b (nM) |
| 123 | Cl |
|
50 | 6700 |
| 124 | Cl |
|
30 | 3000 |
| 125 | Cl |
|
50 | |
| 126 | Cl |
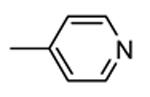
|
30 | 2700 |
| 127 | Cl |
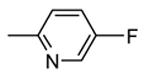
|
60 | 3700 |
| 128 | Cl |
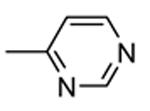
|
60 | |
| 129 | Cl |
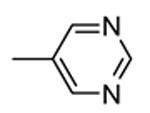
|
70 | |
| 130 | Cl |
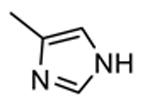
|
70 | 2400 |
| 131 | Cl |
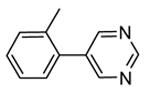
|
40 | 730 |
| 132 | F |
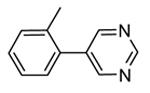
|
50 | 760 |
| 133 | CN |
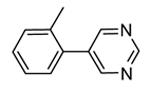
|
8 | 1200 |
| 134 | 7.7 | |||
Measured in a IMAP assay.
Cellular activity is expressed as inhibition of LPS-induced Hsp27 Ser78 phosphorylation in THP-1 cells.
In addition, keeping fixed the p-Cl or p-CN phenyl moiety and a 1,2-disubstitution pattern on the central five-membered core ring, it was changed to triazole and tetrazole moiety, as isosteric groups of the cis-amide moiety. The new compounds showed an affinity decrease to two-digit nanomolar concentrations.97 Better results were obtained with the imidazole ring that allowed restoration of a single-digit nanomolar affinity, as in 134 (7.7 nM). Further attempts to modify the piperazine ring to mitigate the oxidative metabolism on the attached phenyl ring and to fine tune pKa values were unsuccessful and resulted in less active compounds.
2.1.3. Tricyclic azepinone derivatives
Following the rigidification approach to block the anilide system into the conformationally preferred E-conformer, a methylene bridge was inserted between the furan ring and the central phenyl ring of linear amide compounds (such as 103) to yield conformationally-constrained azepinone derivatives.98 The most simple derivatives of this series (namely, the unsubstituted lactam 135 and its methyl derivative 136, Table 16) showed a comparable weak affinity (96 and 79 nM, respectively), although significantly improved in comparison to 103 (5500 nM). This finding further supported the hypothesis that a conformational change from Z- to E-conformation occurred in N-alkylated tertiary anilides (such as 104) in comparison to secondary amides (103) could be responsible for the 5-40-fold improvement in affinity. Based on previous suggestions showing that a biaryl moiety at the amide nitrogen atom was optimal for both affinity and cellular activity, pyridinylphenyl and pyrimidinylphenyl moieties with an ortho-substitution pattern were added to the nitrogen atom of the lactam moiety. Among pyridinyl derivatives, only methoxy-substituted compounds 137-139 with a different substitution pattern showed a single-digit nanomolar affinity (4.9, 8.4, and 6.8 nM, respectively). Unsubstituted pyridinyl derivatives and their monofluoro analogues underwent a 2-5-fold drop in affinity. Differently, pyrimidinyl compounds 140-141 showed a 5.7 and 1.9 nM affinity, respectively. The cellular activity of the best compound 141 in inhibiting Hsp27 phosphorylation corresponded to an EC50 of 138 nM, while inhibition of TNFα secretion from THP-1 cells was 150 nM. Unfortunately, tricyclic lactams showed not negligible cytotoxicity toward SW1353 cells, with CC50 values in the range of two-digit micromolar concentrations. Finally, 141 was confirmed to have a non-ATP-competitive mechanism of action, and retained a high selectivity toward a panel of 21 kinases.
Table 16. Further rigidification to azepinones (Merck Research Laboratories).

| |||
|---|---|---|---|
|
| |||
| Compd | R | X | IC50a (nM) |
| 135 | Cl | H | 96 |
| 136 | Cl | Me | 79 |
| 137 | CN |
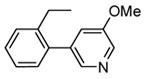
|
4.9 |
| 138 | CN |
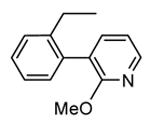
|
8.4 |
| 139 | CN |
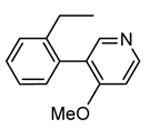
|
6.8 |
| 140 | Cl |
|
5.7 |
| 141 | CN |
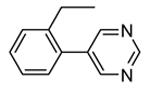
|
1.9 |
Measured in a IMAP assay.
2.1.4. Tetracyclic compounds
Further restriction of conformational flexibility of the azepinone derivatives by condensation of a fourth five-membered heterocyclic ring resulted in compounds with single-digit nanomolar affinity and cellular activity in the range between 260 and 2500 nM (Table 17).99 Among chloro analogues, an imidazole, a methyl-imidazole, and a triazole moiety (as found in 142, 146, and 144) resulted in comparable affinity (7.0, 9.2, and 9.3 nM, respectively) and activity (ranging from 1500 and 2500 nM). On the other hand, the corresponding ciano derivatives 143 and 145 had similar affinity (2.9 and 3.8 nM, respectively), but 6-8-fold better activity (260 nM) in comparison to their corresponding chloro analogues. Importantly, solubility of ciano compounds was slightly lower than that of chloro derivatives, thus suggesting that cellular activity may depend on different molecular parameters, in addition to solubility. Finally, transformation of 142 into the azocine analogue 147 resulted in a drop of affinity (18 nM), although a 2-fold enhanced solubility.
Table 17. Condensation of a fourth condensed ring to azepinones.

| |||||
|---|---|---|---|---|---|
|
| |||||
| Compd | R | R1 | X | IC50a (nM) | EC50b (nM) |
| 142 | Cl | H | CH | 7.0 | 1500 |
| 143 | CN | H | CH | 2.9 | 260 |
| 144 | Cl | H | N | 9.3 | 2100 |
| 145 | CN | H | N | 3.8 | 260 |
| 146 | Cl | Me | CH | 9.2 | 2500 |
| 147 | 18 | ||||
Measured in a IMAP assay.
Cellular activity is expressed as inhibition of LPS-induced Hsp27 Ser78 phosphorylation in THP-1 cells.
3. Uncompetitive MK2 inhibitors
3.1. Compounds from AstraZeneca
3.1.1. Anilinoquinolines with an uncompetitive mechanism of action
Screening the AstraZeneca core compound collection by HTS led to the discovery of the 4-anilino-6-phenyl-quinoline 148 (Table 18) as micromolar inhibitors of MK2 activity (IC50 = 5200 nM in a cell-free assay).100 Attempts to rule out SAR considerations suggested the position 6 of the quinoline ring as the most profitable to be changed. In particular, a series of p-substituted halo-derivatives (149-151) showed enhanced activity up to 400 nM (149). Unfortunately, due to very low permeability (evaluated in human Caco-2 cells), these compounds had only a micromolar ability to inhibit TNFα production in LPS-treated THP-1 cells. As an example, 151 showed an EC50 = 4300 nM. Kinetic studies of reaction velocity (MK2 activity) in the presence of various ATP concentrations and 2 μM inhibitor strongly suggested an uncompetitive mechanism of action, further supported by the fact that inhibition of MK2 occurred better at higher ATP concentrations. Given the low stability in human microsomes, compound cytotoxicity, and low permeability, these compounds were not further studied.
Table 18. ATP uncompetitive quinoline derivatives from AstraZeneca.

| ||
|---|---|---|
|
| ||
| Compd | R | IC50a (nM) |
| 148 | H | 5200 |
| 149 | Br | 400 |
| 150 | Cl | 600 |
| 151 | F | 700 |
Expressed as the inhibition of MK2 activity toward a peptide substrate.
Conclusions and future perspective
The p38 MAPK/MK2 signaling pathway is involved in a series of pathological conditions, such as inflammation diseases and metastasis, as well as in the resistance mechanism to antitumor agents. Inhibition of this pathway by blocking p38 was unsuccessful and none of the p38 inhibitors was able to enter phase III clinical trials because of the unwanted and important systemic side effects. For this reason, in recent years, MK2 was selected as a druggable target in alternative to p38, in the attempt to reduce or abrogate the systemic side effects emerged by treatment with p38 inhibitors. In the field of anti-inflammatory therapy, MK2 showed the very promising profile of a disease-modifying anti-rheumatic drug target. In fact, differently from COX-2 inhibitors and other compounds classified as palliative drugs (i.e., NSAIDs), MK2 is able to interfere with key steps in the propagation of inflammation. As a consequence, compounds able to modulate MK2 activity are expected to be more efficacious and safe in comparison to NSAIDs and p38 inhibitors, respectively. Early studies led to discover ATP-competitive inhibitors. Unexpectedly, their optimization to lead-like and drug-like compounds suffered from major limitations in terms of scarce solubility, cell permeation, as well as kinase selectivity. A suboptimal cellular permeability led to disappointing results in both cell-based assays and in vivo models. As a consequence, enhancement of this pharmacokinetic parameter still remains a challenging issue to be addressed.
In the last years, promising MK2 inhibitors were identified among compounds able to block MK2 activity by a non-ATP-competitive or ATP-uncompetitive mechanism of action. These compounds have the peculiarity of interacting with a binding site different from that of ATP, thus avoiding selectivity issues among kinases. They also show the additional advantage to be effective at lower concentrations in comparison to the ATP-competitive inhibitors. In fact, non-ATP-competitive inhibitors, by definition, are not required to compete with the high ATP concentrations found within the cells and with the high affinity of ATP for the inactive and active forms of MK2. Lower but effective concentrations of MK2 inhibitors should guarantee less pronounced side effects.
Although the difficulties encountered in the process of developing drug-like MK2 inhibitors, a renewed interest derives from the important results on the pathological roles of this kinase in several diseases. The role of p38/MK2 as activators of a cell cycle checkpoint also suggested that MK2 could be a target for compounds acting as chemotherapeutic enhancers able to improve the effectiveness of already known chemotherapeutic drugs. Moreover, this enzyme is also involved in tumor metastasis and other dysfunctions linked to actin remodeling. Recent studies hypothesized that MK2 inhibitors could be effective in reducing or inhibiting cardiac fibrosis in myocardial infarction in humans and in decreasing inflammatory and fibrotic responses in idiopathic pulmonary fibrosis.
Given the pivotal importance of the p38-MK2 signaling pathway in inflammation, cell cycle, actin remodeling and metastasis, MK2 still remains a very promising target and the identification of drug-like MK2 inhibitors with appropriate pharmacokinetics and pharmacodynamics is an appealing challenge for medicinal chemists. Efforts to be made in the very near future should be focused on the identification of new compounds with a profile of significant kinase selectivity, appropriate solubility and cell permeability, to be submitted to clinical trials. To this aim, non-ATP-competitive or ATP-uncompetitive inhibitors of MK2 appear to be the most promising compounds to be studied and optimized.
Acknowledgments
This work was partially supported by grant R01 GM069832 from the National Institutes of Health to S.F..
Abbreviations used
- MAPKAPK2
mitogen-activated protein kinase-activated protein kinase 2
- IL-6
interleukin-6
- DMARD
disease-modifying anti-rheumatic drugs
- BE
biochemical efficiency
- TAK1
transforming growth factor-β activated kinase 1
- TAB1
TAK-binding protein 1
- JNK
c-Jun N-terminal kinase
- MLK
mixed-lineage kinases
- DUSP1
dual specificity phosphatase 1
- TTP
tristetraprolin
- CAP-zip
filamentous actin capping protein Z-interacting protein
- p16-Arc
p16 subunit of the seven-member actin-related protein-2/3 complex
- NLS
nuclear localization signal
- NES
nuclear export signal
- WS
Werner syndrome
- MRP2
multi-drug resistance-associated protein 2
- BSEP
bile salt export pump
- DELFIA
dissociation-enhanced lanthanide fluorescent immunoassay
- ALIS
Automated Ligand Identification System
- IMAP
immobilized metal ion affinity-based fluorescence polarization
- F-actin
filamentous actin
- LIMK
Lin-11 Isl-1 Mec-3 kinase
- hPBMC
human peripheral blood mononuclear cells
- JNK2
c-Jun N-terminal kinase 2
- CPP
cell-penetrating peptide
- PFKFB3
6-phosphofructo-2-kinase/fructose 2,6-bisphosphatase 3
- Shh
Sonic Hedgehog
Biographies
Biography
Mario Fiore graduated in 2012 in Pharmacy at the University of Siena (Italy) working on small molecule inhibitors of MK2. In 2014 has awarded a Master’s degree in Nutrition at Funiber (Fondazione Universitaria Iberoamericana), and in 2015 a Master’s degree in Cosmetic Tecniques and Chemistry at the University of Siena. Currently, he has a collaboration with Inuvance.
Stefano Forli obtained his Ph.D. from the Università di Siena in 2006 in molecular modeling applied to medicinal chemistry. He is currently Staff Scientist at the Molecular Graphics Laboratory, Department of Integrative Structural and Computational Biology at The Scripps Research Institute. He has been involved in numerous drug design projects, including the study of compounds that interfere with the polymerization cascade of α/β tubulin and actin, dual inhibitors of c-Src/Abl kinases, and COX-1/2 selective inhibitors. He is currently involved in the study of inhibitors of several HIV-1 therapeutic targets and in the development of software for drug design, notably AutoDock docking software.
Fabrizio Manetti received a Ph.D. in Pharmaceutical Sciences at the Department of Pharmaceutical and Applied Chemistry at the University of Siena in 1996. From 2002, he is a researcher in Medicinal Chemistry at the Department of Biotechnology, Chemistry and Pharmacy of the same University. His research activity of the last few years was focused on small molecule inhibitors of COX-2 and on modulators of kinases involved in cytoskeleton reorganization, actin remodeling, and cancer (i.e., 14-3-3, LIMK, and ERK-8). Additional research projects are based on ligand-based drug design approaches to find new modulators of the Hedgehog signaling pathway. He is co-founder of Lead Discovery Siena, a spin off operating in the field of drug design and lead optimization.
Footnotes
The authors declare no competing financial interest.
References
- (1).Biava M, Porretta GC, Poce G, Battilocchio C, Botta M, Manetti F, Rovini M, Cappelli A, Sautebin L, Rossi A, Pergola C, Ghelardini C, Galeotti N, Makovec F, Giordani A, Anzellotti P, Tacconelli S, Patrignani P, Anzini M. Enlarging the NSAIDs family: ether, ester and acid derivatives of the 1,5-diarylpyrrole scaffold as novel anti-inflammatory and analgesic agents. Curr. Med. Chem. 2011;18:1540–1554. doi: 10.2174/092986711795328364. [DOI] [PubMed] [Google Scholar]
- (2).Edmunds JJ, Talanian RV. MAPKAP kinase 2 as a target for anti-inflammatory drug discovery. In: Levi JI, Laufer S, editors. Anti-Inflammatory Drug Discovery. The Royal Society of Chemistry; 2012. pp. 158–180. RSC Drug Discovery Series No. 26. Chapter 6. [Google Scholar]
- (3).Moens U, Kostenko S, Sveinbiornsson B. The role of mitogen-activated protein kinase-activated protein kinases (MAPKAPKs) in inflammation. Genes. 2013;4:101–133. doi: 10.3390/genes4020101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gaestel M. What goes up must come down: molecular basis of MAPKAP kinase 2/3-dependent regulation of the inflammatory response and its inhibition. Biol. Chem. 2013;394:1301–1315. doi: 10.1515/hsz-2013-0197. [DOI] [PubMed] [Google Scholar]
- (5).Gurgis FMS, Ziaziaris W, Munoz L. Mitogen-activated protein kinase-activated protein kinase 2 in neuroinflammation, heat shock protein 27 phosphorylation, and cell cycle: role and targeting. Mol. Pharmacol. 2014;85:345–356. doi: 10.1124/mol.113.090365. [DOI] [PubMed] [Google Scholar]
- (6).Norman P. Investigational p38 inhibitors for the treatment of chronic obstructive pulmonary disease. Expert Opin. Investig. Drugs. 2015;24:383–392. doi: 10.1517/13543784.2015.1006358. [DOI] [PubMed] [Google Scholar]
- (7).Genovese MC. Inhibition of p38: has the fat lady sung? Arthritis Rheum. 2009;60:317–320. doi: 10.1002/art.24264. [DOI] [PubMed] [Google Scholar]
- (8).Cheung PC, Campbell DG, Nebreda AR, Cohen P. Feedback control of the protein kinase TAK1 by SAPK2a/p38α. EMBO J. 2003;22:5793–5805. doi: 10.1093/emboj/cdg552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cohen P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell. Biol. 2009;21:317–324. doi: 10.1016/j.ceb.2009.01.015. [DOI] [PubMed] [Google Scholar]
- (10).Gaestel M, Kotlyarov A, Kracht M. Targeting innate immunity protein kinase signalling in inflammation. Nat. Rev. Drug Discov. 2009;8:480–499. doi: 10.1038/nrd2829. [DOI] [PubMed] [Google Scholar]
- (11).Liu C, Lin J, Wrobleski ST, Lin S, Hynes J, Wu H, Dyckman AJ, Li T, Wityak J, Gillooly KM, Pitt S, Shen DR, Zhang RF, McIntyre KW, Salter-Cid L, Shuster DJ, Zhang H, Marathe PH, Doweyko AM, Sack JS, Kiefer SE, Kish KF, Newitt JA, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K. Discovery of 4-(5-(cyclopropylcarbamoyl)-2-methylphenylamino)-5-methyl-N-propylpyrrolo[1,2-f][1,2,4]triazine-6-carboxamide (BMS-582949), a clinical p38α MAP kinase inhibitor for the treatment of inflammatory diseases. J. Med. Chem. 2010;53:6629–6639. doi: 10.1021/jm100540x. [DOI] [PubMed] [Google Scholar]
- (12).Liu C, Lin J, Hynes J, Wu H, Wrobleski ST, Lin S, Dhar TG, Vrudhula VM, Sun JH, Chao S, Zhao R, Wang B, Chen BC, Everlof G, Gesenberg C, Zhang H, Marathe PH, McIntyre KW, Taylor TL, Gillooly K, Shuster DJ, McKinnon M, Dodd JH, Barrish JC, Schieven GL, Leftheris K. Discovery of ((4-(5-(Cyclopropylcarbamoyl)-2-methylphenylamino)-5-methylpyrrolo[1,2-f][1,2,4]triazine-6-carbonyl)(propyl)carbamoyloxy)methyl-2-(4-(phosphonooxy)phenyl)acetate (BMS-751324), a clinical prodrug of p38α MAP kinase inhibitor. J. Med. Chem. 2015;58:7775–7784. doi: 10.1021/acs.jmedchem.5b00839. [DOI] [PubMed] [Google Scholar]
- (13).Trempolec N, Dave Coll N, Nebreda AR. SnapShot: p38 MAPK substrates. Cell. 2013;152:924–924.e1. doi: 10.1016/j.cell.2013.01.047. [DOI] [PubMed] [Google Scholar]
- (14). [accessed October 19, 2015]; https://clinicaltrials.gov/ct2/show/NCT01867762.
- (15).Hitti E, Iakovleva T, Brook M, Deppenmeier S, Gruber AD, Radzioch D, Clark AR, Blackshear PJ, Kotlyarov A, Gaestel M. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine rich element. Mol. Cell Biol. 2006;26:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ronkina N, Menon MB, Schwermann J, Tiedje C, Hitti E, Kotlyarov A, Gaestel M. MAPKAP kinases MK2 and MK3 in inflammation: complex regulation of TNF biosynthesis via expression and phosphorylation of tristetraprolin. Biochem. Pharmacol. 2010;80:1915–1920. doi: 10.1016/j.bcp.2010.06.021. [DOI] [PubMed] [Google Scholar]
- (17).Manetti F. LIM kinases are attractive targets with many macromolecular partners and only a few small molecule regulators. Med. Res. Rev. 2012;32:968–998. doi: 10.1002/med.20230. [DOI] [PubMed] [Google Scholar]
- (18).Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo A-P, Buchner J, Gaestel M. Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor α by phosphorylation. J. Biol. Chem. 1999;274:18947. doi: 10.1074/jbc.274.27.18947. [DOI] [PubMed] [Google Scholar]
- (19).Xu L, Yates CC, Lockyer P, Xie L, Bevilacqua A, He J, Lander C, Patterson C, Willis M. MMI-0100 inhibits cardiac fibrosis in myocardial infarction by direct actions on cardiomyocytes and fibroblasts via MK2 inhibition. J. Mol. Cell Cardiol. 2014;77:86–101. doi: 10.1016/j.yjmcc.2014.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Streicher JM, Ren S, Herschman H, Wang Y. MAPK-activated protein kinase-2 in cardiac hypertrophy and cyclooxygenase-2 regulation in heart. Circ. Res. 2010;106:1434–1443. doi: 10.1161/CIRCRESAHA.109.213199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu T, Warburton RR, Guevara OE, Hill NS, Fanburg BL, Gaestel M, Kayyali US. Lack of MK2 inhibits myofibroblast formation and exacerbates pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2007;37:507–517. doi: 10.1165/rcmb.2007-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Vittal R, Fisher A, Gu H, Mickler EA, Panitch A, Lander C, Cummings OW, Sandusky GE, Wilkes DS. Peptide-mediated inhibition of MK2 ameliorates bleomycin-induced pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 2013;49:47–57. doi: 10.1165/rcmb.2012-0389OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Schlapbach A, Huppertz C. Low-molecular-weight MK2 inhibitors: a tough nut to crack! Future Med. Chem. 2009;1:1243–1257. doi: 10.4155/fmc.09.98. [DOI] [PubMed] [Google Scholar]
- (24).Mourey RJ, Burnette BL, Brustkern SJ, Scott Daniels J, Hirsch JL, Hood WF, Meyers MJ, Mnich SJ, Pierce BS, Saabye MJ, Schindler JF, South SA, Webb EG, Zhang J, Anderson DR. A benzothiophene inhibitor of mitogen-activated protein kinase-activated protein kinase 2 inhibits tumor necrosis factor α production and has oral anti-inflammatory efficacy in acute and chronic models of inflammation. J. Pharmacol. Exp. Ther. 2010;333:797–807. doi: 10.1124/jpet.110.166173. [DOI] [PubMed] [Google Scholar]
- (25).Swinney DC. Biochemical mechanisms of drug action: what does it take for success? Nature Rev. Drug Discov. 2004;3:801–808. doi: 10.1038/nrd1500. [DOI] [PubMed] [Google Scholar]
- (26).The Universal Protein Resource (UniProt) [accessed July 8, 2015]; http://www.uniprot.org/uniprot/P49137.
- (27).Neininger A, Thielemann H, Gaestel M. FRET-based detection of different conformations of MK2. EMBO Rep. 2001;2:703–708. doi: 10.1093/embo-reports/kve157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Zu Y-L, Ai T, Huang C-K. Characterization of an autoinhibitory domain in human mitogen-activated protein kinase-activated protein kinase 2. J. Biol. Chem. 1995;270:202–206. doi: 10.1074/jbc.270.1.202. [DOI] [PubMed] [Google Scholar]
- (29).Lukas SM, Kroe RR, Wildeson J, Peet GW, Frego L, Davidson W, Ingraham RH, Pargellis CA, Labadia ME, Werneburg BG. Catalysis and function of the p38αTMK2a signaling complex. Biochemistry. 2004;43:9950–9960. doi: 10.1021/bi049508v. [DOI] [PubMed] [Google Scholar]
- (30).White A, Pargellis CA, Studts JM, Werneburg BG, Farmer BT., II Molecular basis of MAPK-activated protein kinase 2:p38 assembly. Proc. Natl. Acad. Sci. USA. 2007;104:6353–6358. doi: 10.1073/pnas.0701679104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).ter Haar E, Prabakhar P, Liu X, Lepre C. Crystal structure of the p38α-MAPKAP kinase 2 heterodimer. J. Biol. Chem. 2007;282:9733–9739. doi: 10.1074/jbc.M611165200. [DOI] [PubMed] [Google Scholar]
- (32). [accessed August 4, 2015]; http://www.uniprot.org/uniprot/P49137#showFeatures.
- (33).Argiriadi MA, Sousa S, Banach D, Marcotte D, Xiang T, Tomlinson MJ, Demers M, Harris C, Kwak S, Hardman J, Pietras M, Quinn L, DiMauro J, Ni B, Mankovich J, Borhani DW, Talanian RV, Sadhukhan R. Rational mutagenesis to support structure-based drug design: MAPKAP kinase 2 as a case study. BMC Struct. Biol. 2009;9:16. doi: 10.1186/1472-6807-9-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, Bourne PE. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. www.rcsb.org. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Underwood KW, Parris KD, Federico E, Mosyak L, Czerwinski RM, Shane T, Taylor M, Svenson K, Liu Y, Hsiao C-L, Wolfrom S, Maguire M, Malakian K, Telliez J-B, Lin L-L:, Kriz RW, Seehra J, Somers WS, Stahl ML. Catalytically active MAP KAP kinase 2 structures in complex with staurosporine and ADP reveal differences with the autoinhibited enzyme. Structure. 2002;11:627–636. doi: 10.1016/s0969-2126(03)00092-3. [DOI] [PubMed] [Google Scholar]
- (36).Meng W, Swenson LL, Fitzgibbon MJ, Hayakawa K, ter Haar E, Behrens AE, Fulghum JR, Lippke JA. Structure of mitogen-activated protein kinase-activated protein (MAPKAP) kinase 2 suggests a bifunctional switch that couples kinase activation with nuclear export. J. Biol. Chem. 2002;277:37401–37405. doi: 10.1074/jbc.C200418200. [DOI] [PubMed] [Google Scholar]
- (37).Kurumbail RG, Pawlitz JL, Stegeman RA, Stallings WC, Shieh H-S, Mourey RJ, Bolten SL, Broadus RM. Crystalline structure of human MPAKAP kinase-2. 2003 Sep 18; WO2003/076333, A2.
- (38).Fujino A, Fukushima K, Namiki N, Kosugi T, Takimoto-Kamimura M. Structural analysis of an MK2 inhibitor complex: insight into the regulation of the secondary structure of the Gly-rich loop by TEI-I01800. Acta Crystallogr. 2010;66:80–87. doi: 10.1107/S0907444909046411. [DOI] [PubMed] [Google Scholar]
- (39).Fujino-Nakajima A. Structural studies of ligand-protein complex: the importance of structural change for drug design. Hokkaido University; [accessed July 31, 2015]. Mar 25, 2014. Doctoral thesis. http://eprints.lib.hokudai.ac.jp/dspace/handle/2115/56257. [Google Scholar]
- (40).Fujino A, Fukushima K, Kubota T, Matsumoto Y, Takimoto-Kamimura M. Structure of the β-form of human MK2 in complex with the non-selective kinase inhibitor TEI-L03090. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2013;69:1344–1348. doi: 10.1107/S1744309113030534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Fujino A, Fukushima K, Kubota T, Kosugi T, Takimoto-Kamimura M. Crystal structure of human cyclin-dependent kinase-2 complex with MK2 inhibitor TI-I01800: insight into the selectivity. J. Synchrotron Rad. 2013;20:905–909. doi: 10.1107/S0909049513020736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Argiriadi MA, Ericsson AM, Harris CM, Banach DL, Borhani DW, Calderwood DJ, Demers MD, DiMauro J, Dixon RW, Hardman J, Kwak S, Li B, Mankovich JA, Marcotte D, Mullen KD, Ni B, Pietras M, Sadhukhan R, Sousa S, Tomlinson MJ, Wang L, Xiang T, Talanian RV. 2,4-Diaminopyrimidine MK2 inhibitors. Part I: observation of an unexpected inhibitor binding mode. Bioorg. Med. Chem. Lett. 2010;20:330–333. doi: 10.1016/j.bmcl.2009.10.102. [DOI] [PubMed] [Google Scholar]
- (43).Velcicky J, Feifel R, Hawtin S, Heng R, Huppertz C, Koch G, Kroemer M, Moebitz H, Revesz L, Scheufler C, Schlapbach A. Novel 3-aminopyrazole inhibitors of MK-2 discovered by scaffold hopping strategy. Bioorg. Med. Chem. Lett. 2010;20:1293–1297. doi: 10.1016/j.bmcl.2009.10.138. [DOI] [PubMed] [Google Scholar]
- (44).Anderson DR, Hegde S, Reinhard E, Gomez L, Vernier WF, Lee L, Liu S, Sambandam A, Snider PA, Masih L. Aminocyanopyridine inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2) Bioorg. Med. Chem. Lett. 2005;15:1587–1590. doi: 10.1016/j.bmcl.2005.01.067. [DOI] [PubMed] [Google Scholar]
- (45).Davis T, Bagley MC, Dix MC, Murziani PGS, Rokicki MJ, Widdowson CS, Zayed JM, Bachler MA, Kipling D. Synthesis and in vivo activity of MK2 and MK2 substrate-selective p38αMAPK inhibitors in Werner syndrome cells. Bioorg. Med. Chem. Lett. 2007;17:6832–6835. doi: 10.1016/j.bmcl.2007.10.036. [DOI] [PubMed] [Google Scholar]
- (46).Anderson DR, Meyers MJ, Vernier WF, Mahoney MW, Kurumbail RG, Caspers N, Poda GI, Schindler JF, Reitz DB, Mourey RJ. Pyrrolopyridine inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2) J. Med. Chem. 2007;50:2647–2654. doi: 10.1021/jm0611004. [DOI] [PubMed] [Google Scholar]
- (47).Chiang P-C, South SA, Daniels JS, Anderson DR, Wene SP, Albin LA, Mourey RJ, Selbo JG. Aqueous versus non-aqueous salt delivery strategies to enhance oral bioavailability of a mitogen-activated protein kinase-activated protein kinase (MK-2) inhibitor in rats. J. Pharm. Sci. 2009;98:248–256. doi: 10.1002/jps.21425. [DOI] [PubMed] [Google Scholar]
- (48).Pengal R, Guess AJ, Agrawal S, Manley J, Ransom RF, Mourey RJ, Benndorf R, Smoyer WE. Inhibition of the protein kinase MK-2 promotes podocytes from nephrotic syndrome-related injury. Am. J. Physiol. Renal Physiol. 2011;301:F509–F519. doi: 10.1152/ajprenal.00661.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Davis T, Rokicki MJ, Bagley MC, Kipling D. The effect of small-molecule inhibition of MAPKAPK2 on cell ageing phenotypes of fibroblasts from human Werner syndrome. Chem. Cent. J. 2013;7:18. doi: 10.1186/1752-153X-7-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Trujillo JI, Meyers MJ, Anderson DR, Hegde S, Mahoney MW, Vernier WF, Buchler IP, Wu KK, Yang S, Hartmann SJ, Reitz DB. Novel tetrahydro-β-carboline-1-carboxylic acids as inhibitors of mitogen activated protein kinase activated protein kinase 2 (MK-2) Bioorg. Med. Chem. Lett. 2007;17:4657–4663. doi: 10.1016/j.bmcl.2007.05.070. [DOI] [PubMed] [Google Scholar]
- (51).Anderson DR, Meyers MJ, Kurumbail RG, Caspers N, Poda GI, Long SA, Pierce BS, Mahoney MW, Mourey RJ. Benzothiophene inhibitors of MK2. Part 1: structure-activity relationships, assessment of selectivity and cellular potency. Bioorg. Med. Chem. Lett. 2009;19:4878–4881. doi: 10.1016/j.bmcl.2009.02.015. [DOI] [PubMed] [Google Scholar]
- (52).Anderson DR, Meyers MJ, Kurumbail RG, Caspers N, Poda GI, Long SA, Pierce BS, Mahoney MW, Mourey RJ, Parikh MD. Benzothiophene inhibitors of MK2. Part 1: improvements in kinase selectivity and cell potency. Bioorg. Med. Chem. Lett. 2009;19:4882–4884. doi: 10.1016/j.bmcl.2009.02.017. [DOI] [PubMed] [Google Scholar]
- (53).Daniels JS, Lai Y, South SA, Chiang P-C, Walker D, Feng B, Mireles R, Whiteley LO, Mckenzie JW, Stevens J, Mourey R, Anderson D, Davis JW., II Inhibition of hepatobiliary transporters by a novel kinase inhibitor contributes to hepatotoxicity in beagle dogs. Drug. Metab. Lett. 2013;7:15–22. doi: 10.2174/18723128112066660018. [DOI] [PubMed] [Google Scholar]
- (54).South SA, Stevens J, Walker G, Parikh M, Walker D, Rock D, Thompson D, Mourey R, Anderson D, Daniels JS. Hepatic metabolism of a novel kinase inhibitor reveals a dual mechanism of bioactivation. Drug Metab. Rev. 2008;40:224–225. [Google Scholar]
- (55).Ge X, Lyu P, Gu Y, Li L, Li J, Wang Y, Zhang L, Fu C, Cao Z. Sonic Hedgehog stimulates glycolysis and proliferation of breast cancer cells: modulation of PFKFB3 activation. Biochem. Biophys. Res. Commun. 2015;464:862–868. doi: 10.1016/j.bbrc.2015.07.052. [DOI] [PubMed] [Google Scholar]
- (56).Manetti F, Taddei M, Petricci E. Structure-activity relationships and mechanism of action of small molecule Smoothened modulators discovered by high-throughput screening and rational design. Top. Med. Chem. 2015;16:43–108. [Google Scholar]
- (57).Solinas A, Faure H, Roudaut H, Traiffort E, Schoenfelder A, Mann A, Manetti F, Taddei M, Ruat M. Acylthiourea, acylurea, and acylguanidine derivatives with potent hedgehog inhibiting activity. J. Med. Chem. 2012;55:1559–1571. doi: 10.1021/jm2013369. [DOI] [PubMed] [Google Scholar]
- (58).Roudaut H, Traiffort E, Gorojankina T, Vincent L, Faure H, Schoenfelder A, Mann A, Manetti F, Solinas A, Taddei M, Ruat M. Identification and mechanism of action of the acylguanidine MRT-83, a novel potent Smoothened antagonist. Mol. Pharmacol. 2011;79:453–460. doi: 10.1124/mol.110.069708. [DOI] [PubMed] [Google Scholar]
- (59).Manetti F, Faure H, Roudaut H, Gorojankina T, Traiffort E, Schoenfelder A, Mann A, Solinas A, Taddei M, Ruat M. Virtual screening-based discovery and mechanistic characterization of the acylthiourea MRT-10 family as Smoothened antagonists. Mol. Pharmacol. 2010;78:658–665. doi: 10.1124/mol.110.065102. [DOI] [PubMed] [Google Scholar]
- (60).Wu J-P, Wang J, Abeywardane A, Andersen D, Emmanuel M, Gautschi E, Goldberg DR, Kashem MA, Lukas S, Mao W, Martin L, Morwick T, Moss N, Pargellis C, Patel UR, Patnaude L, Peet GW, Skow D, Snow RJ, Ward Y, Werneburg B, White A. The discovery of carboline analogs as potent MAPKAP-K2 inhibitors. Bioorg. Med. Chem. Lett. 2007;17:4664–4669. doi: 10.1016/j.bmcl.2007.05.101. [DOI] [PubMed] [Google Scholar]
- (61).Goldberg DR, Choi Y, Cogan D, Corson M, DeLeon R, Gao A, Gruenbaum L, Hao MH, Joseph D, Kashem MA, Miller C, Moss N, Netherton MR, Pargellis CP, Pelletier J, Sellati R, Skow D, Torcellini C, Tseng Y-C, Wang J, Wasti R, Werneburg B, Wu JP, Xiong Z. Pyrazinoindolone inhibitors of MAPKAP-K2. Bioorg. Med. Chem. Lett. 2008;18:938–941. doi: 10.1016/j.bmcl.2007.12.037. [DOI] [PubMed] [Google Scholar]
- (62).Xiong Z, Gao DA, Cogan DA, Goldberg DR, Hao M-H, Moss N, Pack E, Pargellis C, Skow D, Trieselmann T, Werneburg B, White A. Synthesis and SAR studies of indole-based MK2 inhibitors. Bioorg. Med. Chem. Lett. 2008;18:1994–1999. doi: 10.1016/j.bmcl.2008.01.119. [DOI] [PubMed] [Google Scholar]
- (63).Hillig RC, Eberspaecher U, Monteclaro F, Huber M, Nguyen D, Mengel A, Muller-Tiemann B, Egner U. Structural basis for a high affinity inhibitor bound to protein kinase MK2. J. Mol. Biol. 2007;369:735–745. doi: 10.1016/j.jmb.2007.03.004. [DOI] [PubMed] [Google Scholar]
- (64).Barf T, Kaptein A, de Wilde S, van der Heijden R, van Someren R, Demont D, Schultz-Fademrecht C, Versteegh J, van Zeeland M, Seegers N, Kazemier B, van de Kar B, van Hoek M, de Roos J, Klop H, Smeets R, Hofstra C, Hornberg J, Oubrie A. Structure-based lead identification of ATP-competitive MK2 inhibitors. Bioorg. Med. Chem. Lett. 2011;21:3818–3822. doi: 10.1016/j.bmcl.2011.04.018. [DOI] [PubMed] [Google Scholar]
- (65).Hanau CE, Mershon SM, Graneto MJ, Meyers MJ, Hegde SG, Buchler IP, Wu KK, Liu S, Nacro K. Acyclic pyrazole compounds for the inhibition of mitogen-activated protein kinase-activated protein kinase-2. 2004 Jul 15; WO2004/058176 A2.
- (66).Kaptein A, Oubrie A, de Zwart E, Hoogenboom N, de Wit J, van de Kar B, van Hoek M, Vogel G, de Kimpe V, Schultz-Fademrecht C, Borsboom J, van Zeeland M, Versteegh J, Kazemier B, de Roos J, Wijnands F, Dulos J, Jaeger M, Leandro-Garcia P, Barf T. Discovery of selective and orally available spiro-3-piperidyl ATP-competitive MK2 inhibitors. Bioorg. Med. Chem. Lett. 2011;21:3823–3827. doi: 10.1016/j.bmcl.2011.04.016. [DOI] [PubMed] [Google Scholar]
- (67).Oubrie A, Kaptein A, de Zwart E, Hoogenboom N, Goorden R, van de Kar B, van Hoek M, de Kimpe V, van der Heijden R, Borsboom J, Kazemier B, de Roos J, Scheffers M, Lommerse J, Schultz-Fademrecht C, Barf T. Novel ATP competitive MK2 inhibitors with potent biochemical and cell-based activity throughout the series. Bioorg. Med. Chem. Lett. 2012;22:613–618. doi: 10.1016/j.bmcl.2011.10.071. [DOI] [PubMed] [Google Scholar]
- (68).Lin S, Lombardo M, Malkani S, Hale JJ, Mills SG, Chapman K, Thompson JE, Zhang WX, Wang R, Cubbon RM, O’Neill EA, Luell S, Carballo Jane E, Yang L. Novel 1-(2-aminopyrazin-3-yl)methyl-2-thioureas as potent inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2) Bioorg. Med. Chem. Lett. 2009;19:3238–3242. doi: 10.1016/j.bmcl.2009.04.088. [DOI] [PubMed] [Google Scholar]
- (69).Lin S, Malkani S, Lombardo M, Yang L, Mills SG, Chapman K, Thompson JE, Zhang WX, Wang R, Cubbon RM, O’Neill EA, Hale JJ. Design, synthesis, and biological evaluation of aminopyrazine derivatives as inhibitors of mitogen-activated protein kinase-activated protein kinase 2 (MK-2) Bioorg. Med. Chem. Lett. 2015 doi: 10.1016/j.bmcl.2015.09.016. doi:10.1016/j.bmcl.2015.09.016. [DOI] [PubMed] [Google Scholar]
- (70).Meng Z, Ciavarri JP, McRiner A, Zhao Y, Zhao L, Reddy PA, Zhang X, Fischmann TO, Whitehurst C, Siddiqui MA. Potency switch between CHK1 and MK2: discovery of imidazo[1,2-a]pyrazine- and imidazo[1,2-c]pyrimidine-based kinase inhibitors. Bioorg. Med. Chem. Lett. 2013;23:2863–2867. doi: 10.1016/j.bmcl.2013.03.100. [DOI] [PubMed] [Google Scholar]
- (71).Schlapbach A, Feifel R, Hawtin S, Heng R, Koch G, Moebitz H, Revesz L, Scheufler C, Velcicky J, Waelchli R, Huppertz C. Pyrrolo-pyrimidones: a novel class of MK2 inhibitors with potent cellular activity. Bioorg. Med. Chem. Lett. 2008;18:6142–6146. doi: 10.1016/j.bmcl.2008.10.039. [DOI] [PubMed] [Google Scholar]
- (72).Revesz L, Schlapbach A, Aichholz R, Feifel R, Hawtin S, Heng R, Hiestand P, Jahnke W, Koch G, Kroemer M, Moebitz H, Scheufler C, Velcicky J, Huppertz C. In vivo and in vitro SAR of tetracyclic MAPKAP-K2 (MK2) inhibitors. Part I. Bioorg. Med. Chem. Lett. 2010;20:4715–4718. doi: 10.1016/j.bmcl.2010.04.024. [DOI] [PubMed] [Google Scholar]
- (73).Revesz L, Schlapbach A, Aichholz R, Dawson J, Feifel R, Hawtin S, Littlewood-Evans A, Koch G, Kroemer M, Moebitz H, Scheufler C, Velcicky J, Huppertz C. In vivo and in vitro SAR of tetracyclic MAPKAP-K2 (MK2) inhibitors. Part II. Bioorg. Med. Chem. Lett. 2010;20:4719–4723. doi: 10.1016/j.bmcl.2010.04.023. [DOI] [PubMed] [Google Scholar]
- (74).Harris CM, Ericsson AM, Argiriadi MA, Barberis C, Borhani DW, Burchart A, Calderwood DJ, Cunha GA, Dixon RW, Frank KE, Johnson EF, Kamens J, Kwak S, Li B, Mullen KD, Perron DC, Wang L, Wishart N, Wu X, Zhang X, Zmetra TR, Talanian RV. 2,4-Diaminopyrimidine MK2 inhibitors. Part I: structure-based inhibitor optimization. Bioorg. Med. Chem. Lett. 2010;20:334–337. doi: 10.1016/j.bmcl.2009.10.103. [DOI] [PubMed] [Google Scholar]
- (75).Kosugi T, Mitchell DR, Fujino A, Imai M, Kambe M, Kobayashi S, Makino H, Matsueda Y, Oue Y, Komatsu K, Imaizumi K, Sakai Y, Sugiura S, Takenouchi O, Unoki G, Yamakoshi Y, Cunliffe V, Frearson J, Gordon R, Harris CJ, Kalloo-Hosein H, Le J, Patel G, Simpson DJ, Sherborne B, Thomas PS, Suzuki N, Takimoto-Kamimura M, Kataoka K. Mitogen-activated protein kinase-activated protein kinase 2 (MAPKAP-K2) as an antiinflammatory target: discovery and in vivo activity of selective pyrazolo[1,5-a]pyrimidine inhibitors using a focused library and structure-based optimization approach. J. Med. Chem. 2012;55:6700–6715. doi: 10.1021/jm300411k. [DOI] [PubMed] [Google Scholar]
- (76).Miglani R, Cliffe IA, Voleti SR. Assessment of the putative binding conformation of a pyrazolopyridine class of inhibitors of MAPKAPK2 using computational studies. Eur. J. Med. Chem. 2010;45:98–105. doi: 10.1016/j.ejmech.2009.09.030. [DOI] [PubMed] [Google Scholar]
- (77).Lovering F, Kirincich S, Wang W, Combs K, Resnick L, Sabalski JE, Butera J, Liu J, Parris K, Telliez JB. Identification and SAR of squarate inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK-2) Bioorg. Med. Chem. 2009;17:3342–3351. doi: 10.1016/j.bmc.2009.03.041. [DOI] [PubMed] [Google Scholar]
- (78).Williams DE, Telliez J-B, Liu J, Tahir A, van Soest R, Andersen RJ. Meroterpenoid MAPKAP (MK2) inhibitors isolated from the Indonesian marine sponge Acanthodendrilla sp. J. Nat. Prod. 2004;67:2127–2129. doi: 10.1021/np049808d. [DOI] [PubMed] [Google Scholar]
- (79).Basabe P, Martin M, Bodero O, Blanco A, Marcos IS, Diez D, Urones JG. Synthesis of (+)-makassaric acid, a protein kinase MK2 inhibitor. Tetrahedron. 2010;66:6008–6012. [Google Scholar]
- (80).Yang ZQ, Sun PH, Chen WM. 3D QSAR analysis on pyrrolopyridine analogs as mitogen-activated protein kinase-activated protein kinase 2 (MK-2) inhibitors. Lett. Drug Des. Discov. 2007;4:557–561. [Google Scholar]
- (81).Nayana RS, Bommisetty SK, Singh K, Bairy SK, Nunna S, Pramod A, Muttineni R. Structural analysis of carboline derivatives as inhibitors of MAPKAP K2 using 3D QSAR and docking studies. J. Chem. Inf. Model. 2009;49:53–67. doi: 10.1021/ci800294y. [DOI] [PubMed] [Google Scholar]
- (82).Hao M, Ren H, Luo F, Zhang S, Qiu J, Ji M, Si H, Li G. A computational study on thiourea analogs as potent MK-2 inhibitors. Int. J. Mol. Sci. 2012;13:7057–7079. doi: 10.3390/ijms13067057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Kaushik U, Sharma V, Kumar V. Computation of pharmacophore models for the prediction of mitogen-activated protein kinase activated protein kinase-2 inhibitory activity of pyrrolopyridines. Med. Chem. Res. 2012;21:3777–3784. [Google Scholar]
- (84).Wang T-J, Zhou L, Fei J, Li Z-C, He L. Applications of 3D-QSAR and structure-based pharmacophore modeling, virtual screening, ADMET, and molecular docking of putative MAPKAP-K2 (MK2) inhibitors. Med. Chem. Res. 2013;22:4418–4429. [Google Scholar]
- (85).Natesan S, Subramaniam R, Bergeron C, Balaz S. Binding affinity prediction for ligands and receptors forming tautomers and ionization species: inhibition of mitogen-activated protein kinase-activated protein kinase 2 (MK2) J. Med. Chem. 2012;55:2035–2047. doi: 10.1021/jm201217q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Lopes LB, Flynn C, Komalavilas P, Panitch A, Brophy CM, Seal BL. Inhibition of Hsp27 phosphorylation by a cell-permeant MAPKAP kinase 2 inhibitor. Biochem. Biophys. Res. Commun. 2009;382:535–539. doi: 10.1016/j.bbrc.2009.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Ward B, Seal BL, Brophy CM, Panitch A. Design of a bioactive cell-penetrating peptide: when a transduction domain does more than transduce. J. Pept. Sci. 2009;15:668–674. doi: 10.1002/psc.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Brugnano JL, Chan BK, Seal BL, Panitch A. Cell-penetrating peptides can confer biological function: regulation of inflammatory cytokines in human monocytes by MK2 inhibitor peptides. J. Control. Rel. 2011;155:128–133. doi: 10.1016/j.jconrel.2011.05.007. [DOI] [PubMed] [Google Scholar]
- (89).Ward BC, Kavalukas S, Brugnano J, Barbul A, Panitch A. Peptide inhibitors of MK2 show promise for inhibition of abdominal adhesions. J. Surg. Res. 2011;169:e27–e36. doi: 10.1016/j.jss.2011.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Muto A, Panitch A, Kim N, Park K, Komalavilas P, Brophy CM, Dardik A. Inhibitor of mitogen activated protein kinase activated protein kinase II with MMI-0100 reduces intimal hyperplasia ex vivo and in vivo. Vasc. Pharmacol. 2012;56:47–55. doi: 10.1016/j.vph.2011.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Lopes LB, Brophy CM, Flynn CR, Yi z., Bowen BP, Smoke C, Seal B, Panitch A, Komalavilas P. A novel cell permeant peptide inhibitor of MAPKAP kinase II inhibits intimal hyperplasia in a human saphenous vein organ culture model. J. Vasc. Surg. 2010;52:1596–1607. doi: 10.1016/j.jvs.2010.06.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Brugnano JL, Panitch A. Matrix stiffness affects endocytic uptake of MK2-inhibitor peptides. PloS ONE. 2014;9:e84821. doi: 10.1371/journal.pone.0084821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Annis DA, Athanasopoulos J, Curran PJ, Felsch JS, Kalghatgi K, Lee WH, Nash HM, Orminati J-PA, Rosner KE, Shipps JW, Jr., Thaddupathy RRA, Tyler AN, Vilenchik L, Wagner CR, Wintner EA. An affinity selection-mass spectrometry method for the identification of small molecule ligands from self-encoded combinatorial libraries. Discovery of a novel antagonist of E. coli dihydrofolate reductase. Int. J. Mass. Spectrometry. 2004;238:77–83. [Google Scholar]
- (94).Huang X, Shipps GW, Jr., Cheng CC, Spacciapoli P, Zhang X, McCoy MA, Wyss DF, Yang X, Achab A, Soucy K, Montavon DK, Murphy DM, Whitehurst CE. Discovery and hit-to-lead optimization of non-ATP competitive MK2 (MAPKAPK2) inhibitors. ACS Med. Chem. Lett. 2011;2:632–637. doi: 10.1021/ml200113y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Huang X, Zhu X, Chen X, Zhou W, Xiao D, Degrado S, Aslanian R, Fossetta J, Lundell D, Tian F, Trivedi P, Palani A. A three-step protocol for lead optimization: quick identification of key conformational features and functional groups in the SAR studies of non-ATP competitive MK2 (MAPKAPK2) inhibitors. Bioorg. Med. Chem. Lett. 2012;22:65–70. doi: 10.1016/j.bmcl.2011.11.074. [DOI] [PubMed] [Google Scholar]
- (96).Qin J, Dhondi P, Huang X, Aslanian R, Fossetta J, Tian F, Lundell D, Palani A. Discovery of a potent dihydrooxadiazole series of non-ATP competitive MK2 (MAPKAPK2) inhibitors. ACS Med. Chem. Lett. 2012;3:100–105. doi: 10.1021/ml200238g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Xiao D, Zhu X, Sofolarides M, Degrado S, Shao N, Rao A, Chen X, Aslanian R, Fossetta J, Tian F, Trivedi P, Lundell D, Palani A. Discovery of a novel series of potent MK2 non-ATP competitive inhibitors usign 1,2-substituted azoles as cis-amide isosteres. Bioorg. Med. Chem. Lett. 2014;24:3609–3613. doi: 10.1016/j.bmcl.2014.05.024. [DOI] [PubMed] [Google Scholar]
- (98).Xiao D, Palani A, Huang X, Sofolarides M, Zhou W, Chen X, Aslanian R, Guo Z, Fossetta J, Tian F, Trivedi P, Spacciapoli P, Whitehurst CE, Lundell D. Conformation constraint of anilides enabling the discovery of tricyclic lactams as potent MK2 non-ATP competitive inhibitors. Bioorg. Med. Chem. Lett. 2013;23:3262–3266. doi: 10.1016/j.bmcl.2013.03.109. [DOI] [PubMed] [Google Scholar]
- (99).Rao AU, Xiao D, Huang X, Zhou W, Fossetta J, Lundell D, Tian F, Trivedi P, Aslanian R, Palani A. Facile synthesis of tetracyclic azepine and oxazocine derivatives and their potential as MAPKAP-K2 (MK2) kinase. Bioorg. Med. Chem. Lett. 2012;22:1068–1072. doi: 10.1016/j.bmcl.2011.11.113. [DOI] [PubMed] [Google Scholar]
- (100).Olsson H, Sjo P, Ersoy O, Kristoffersson A, Larsson J, Norden B. 4-Anilino-6-phenyl-quinoline inhibitors of mitogen activated protein kinase-activated protein kinase 2 (MK2) Bioorg. Med. Chem. Lett. 2010;20:4738–4740. doi: 10.1016/j.bmcl.2010.06.107. [DOI] [PubMed] [Google Scholar]
- (101).Cumming JG, Debreczeni JE, Edfeldt F, Evertsson F, Harrison M, Holdgate GA, James MJ, Lamont SG, Oldham K, Sullivan JE, Wells SL. Discovery and characterization of MAPK-activated protein kinase-2 prevention of activation inhibitors. J. Med. Chem. 2005;58:278–293. doi: 10.1021/jm501038s. [DOI] [PubMed] [Google Scholar]