

Abstract
Precision medicine aims to identify the right drug, for the right patient, at the right dose, at the right time, which is particularly important in cancer therapy. Problems such as the variability of treatment response and resistance to medication have been long-standing challenges in oncology, especially for development of new medications. Solid tumors, unlike hematologic malignancies or brain tumors, are remarkably diverse in their cellular origins and developmental timing. The ability of next-generation sequencing (NGS) to analyze the comprehensive landscape of genetic alterations brings promises to diseases that have a highly complex and heterogeneous genetic composition such as cancer. Here we provide an overview of how NGS is able to facilitate precision medicine and change the paradigm of cancer therapy, especially for solid tumors, through technical advancements, molecular diagnosis, response monitoring and clinical trials.
Keywords: Precision medicine, cancer therapy, next-generation sequencing, solid tumor
Introduction
Precision medicine, also often called personalized medicine, has been defined as identifying the right drug, for the right patient, at the right dose, at the right time1. This concept relies heavily on access to information on an individual’s unique genetic characteristics to tailor therapy. Today, about 10% of labels for Food and Drug Administration (FDA)-approved drugs contain pharmacogenomic information1. Precision medicine is not a new concept but the availability of large-scale human genome databases, the advent of powerful methods such as next-generation sequencing (NGS) and advancement of computational tools have created an opportunity for significant progress. Precision medicine is particularly important in oncology because along-standing problem is the variability of treatment response, especially in early stage clinical trials. Drugs that fail to induce disease regression in most patients or prolong median progression-free survival are deemed inactive and often abandoned, even when the drug exhibits profound activity in a small number of patients. Other challenges include unexplained drug resistance, genomic heterogeneity of tumors, insufficient means for monitoring responses and tumor recurrence, and limited knowledge about the use of drug combinations.
NGS has the ability to characterize genetic variations simultaneously in a much more cost-effective fashion than traditional Sanger sequencing2. The rapid growth of NGS in the last decade brings promise to diseases that have a highly complex and heterogeneous genetic composition such as cancer. This article will provide an overview of how NGS technology is able to change cancer therapy, especially for solid tumors (Figure 1).
Figure1.
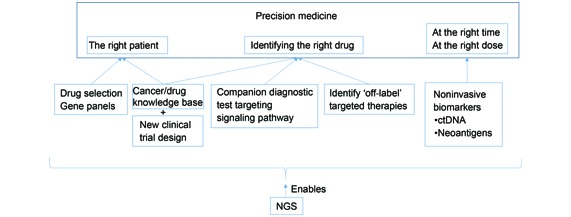
How NGS technology enables precision medicine in cancer.
Technical perspectives of NGS in solid tumor tissue
Solid tumors are remarkably diverse in their cellular origins and developmental timing. Unlike hematologic malignancies or brain tumors, solid tumors can arise in any of the three germ cell lineages, which provides a unique opportunity to study the effect of cellular origin and developmental stage3. The features of solid tumors also brings challenges for analytical tools. Solid tumor specimens have cellular heterogeneity (i.e. mixtures of tumor and stroma) and/or molecular heterogeneity (i.e. subclones of varying genotypes). It's critical to have a technology to simultaneously detect (have a comprehensive view of) the whole molecular picture. NGS technologies must balance the breadth of content with sequencing depth to reveal low-abundance mutations that may be clinically relevant in a cost-effective way.
NGS also has the advantage of detecting low level mosaic mutations that are common in certain solid tumors. High-depth sequencing (i.e. 1,000 fold coverage) may be able to detect such low-level variants. In one study4, high-coverage NGS of the APC gene found a high prevalence of pathogenic mosaic APC mutations below the detection thresholds of routine diagnostics in adenomatous polyposis. Another recent study on tuberous sclerosis (TSC) patients showed that amongst 45 subjects with identified mutations in either TSC1 or TSC2, mosaicism was observed in the majority (26 of 45, 58%)5. Both studies demonstrated how NGS can detect low level mosaicism that can help molecular diagnosis, genetic counseling and guide therapy.
Another challenge is that most solid tumor specimens that are examined by anatomical pathologists are fixed in formalin and embedded in paraffin (FFPE). In order to perform large-scale retrospective studies, FFPE specimens are inevitable. Formalin introduces crosslinks that can both fragment DNA and cause chemical alterations that may alter sequencing results6. Therefore, NGS technologies must be compatible with small quantities of potentially fragmented and cross-linked DNA. Modification of DNA extraction methods, two-step PCR enrichment and optimized bioinformatics algorithms have improved the background noise of variant detection in FFPE samples7.
The impact of NGS on precision cancer therapy
Precision medicine-the right patient
The identification of patients with oncogenic driver mutations provides the opportunity to use the genomic information of individual tumors to guide the selection of rational therapeutics in an attempt to improve the outcome of patients with advanced cancers. Early methods to explore the genomic foundations of cancers involved targeted exploration of specific variants and genes in a low-throughput fashion. The discovery of genomic aberrations like EGFR mutations and ALK rearrangements, and the subsequent targeted therapies leading to improved outcomes in a subset of patients with advanced non–small cell lung cancer (NSCLC), is an example of such a successful biomarker-driven drug development8,9. The examination of chronic myelogenous leukemia cells for BCR-ABL mutations before using an ABL inhibitor is another classic example that clinical benefit could be predicted by the presence of certain molecular characteristics in the tumor that are highly correlated with a favorable pharmacologic effect10. This targeted approach is highly specific but provides limited information and assumes that each type of cancer progresses through a similar, if not identical, process of genetic hits. For example, the output for FDA-approved EGFR and BRAF is limited to either one mutational hotspot (BRAF V600) or mutations in a handful of exons (EGFR exons 18, 19, 20 and 21) and are only approved for specific cancer types.
NGS technology allows multiple genes to be analyzed simultaneously in one run and can provide enough depth of coverage to detect minor allele frequencies in a cost-effective manner. Targeted gene panels are widely implemented and currently the best option for tumor characterization in clinical cancer practice. In a pilot study in 10 patients, all of the tumor samples showed biologically or clinically meaningful genomic alterations in the 137 genes sequenced, including several that might predict sensitivity or resistance to therapeutic agents or provide useful prognostic information11. A more recent study validated the utilization of NGS analysis of 25 cancer-associated genes in 78 tumor specimens to detect mutations at actionable loci12. Clinical laboratories have developed NGS-based cancer gene panels to guide patient management. By mainly using a 50-gene cancer panel, Tafe et al.13 group was able to provide treatment recommendations based on targetable genetic alterations for over 56% of cases. There are several commercially available panels that can be validated and implemented in diagnostic laboratories, ranging from 15 genes to over 400 genes to even the entire coding region (Table 1 for examples of commercially available cancer panels). These panels are intended to test germline and/or somatic mutations and contain genes with high and low penetrance. When designing a panel it is important to consider the clinical importance of the genes to be included and the targeted area in the genes to be sequenced. Pathogenic variants in low penetrance cancer genes and variants of unknown significance can bring tremendous challenges in genetic counseling and clinical decision making.
Table1.
Examples of commercially available cancer panels
| Company | Panel name | Number of genes | Coverage | Note |
| Life sciences | Ion ampliSeq hotspot cancer panel | 50 | None indicated | Compatible with FFPE sample |
| Life sciences | Ion ampliSeq comprehensive cancer panel | 409 | None indicated | Detect CNV; compatible with FFPE sample |
| Illumina | TruSight cancer panel | 94 | 20× | Germline only |
| Illumina | TruSight tumor 15 panel | 15 | ≥500× | Solid tumor somatic variants |
| Illumina | TruSight amplicon cancer panel | 48 | ~1000× | Somatic mutational hotspots |
| Illumina | TruSight RNA pan-cancer panel | 21,043 exonic region | 3 million reads per sample | Gene expression, variant and fusion detection in 1,385 cancer associated genes |
| IDT | xGen® pan-cancer panel | 127 | >97% of targeted covered at 30× | Can be expanded by adding custom probes |
In the research setting, NGS has now made it possible to characterize genomic alterations and allowed examination of cancer exomes (i.e. the combined protein coding exons) and even whole cancer genomes in unprecedented detail. Recent studies support the emerging concept of the "mutation signature"14, which postulates that the combination of mutations present is more predictive of the response to treatment than individual gene mutation status. A study conducted by multiple centers found that of 5 patients with unstable genomes and/or a high BRCAmutational signature burden identified through whole genome sequencing (WGS) on pancreatic ductal adenocarcinomas, 2 had exceptional responses and 2 had robust partial responses, while 3 patients who did not have any of these molecular characteristics did not respond15. Studies on malignant melanoma and small cell lung carcinoma also demonstrated the value of WGS for evaluating the signature of somatic mutations by providing greater resolution and mechanistic insight into mutational signatures due to known carcinogens16,17. By analyzing WGS of 30 Asian lung cancers, mutational signatures separated the patients into two categories of either the never-smokers or all the smokers or ex-smokers18. In the next decade, more cancer genomes will be generated to reveal additional mutation signatures that will improve disease management.
Another emerging approach to identify the right patients is reflected in clinical trial designs. Sometimes, the subpopulation of patients with a particular molecular profile who are predicted to respond to a given therapeutic can be quite small. For example, Only about 5% of lung cancer patients have the ALK gene rearrangement19, which means a great deal of screening was required to identify and enroll patients in studies. In the new clinical "basket trial" design, patients are screened simultaneously for a large number of genetic aberrations using a NGS-based multi-gene panel to determine their eligibility for a large number of clinical trials involving different therapeutic interventions. This sort of approach is particularly useful when the cancer type or the mutation is rare. However an oncology research network system is required for the success of this approach. Some forward-looking models propose a multi-institutional collaboration that employs a multi-gene panel assay in which the cost of the screening assay is shared by different drug development entities. While this approach would significantly reduce the cost of screening patients for rare subpopulations of patients in phase 2 and phase 3 trials for each individual company, it presents the equally interesting question of whether drug developers will collaborate with competitors in such basket trials. Enrolling participants with similar mutations instead of the same clinical cancer types can permit enough patients to be studied since regulatory agencies such as the US FDA are not likely to approve a drug on the basis of data from only a couple of individuals20. Drug developers, diagnostic companies, and regulatory agencies will have to work together to navigate this paradigm shift. In addition, methods of a data mapper and loader and queries for rapid retrieval of data related to clinical efficacy to inform clinical interpretation of molecular aberrations have been developed. The Patterson et al.21 group demonstrated the structured and organized design of the JAX Clinical Knowledgebase which can be queried readily to access comprehensive data for clinical reporting via customized reporting queries.
Precision medicine-identifying the right drug
A companion diagnostic is defined as a diagnostic test that is linked to a specific therapeutic and/or is required for the safe and effective use of the drug. Current testing for precision medicine links a specific drug to a specific gene and can be summarized as "one-drug/one-gene diagnosis." One-gene tests that are FDA-approved for mutations in EGFR, KRAS, and BRAF are perfect examples. However, most cancers are genetically complex, and are better defined by the dysregulation of signaling pathways rather than a defined set of mutations, making the one-gene/one-drug model unsustainable. The poor long-term treatment response of many cancers is best explained by therapy-resistant subclones present within the primary tumor22. The major pathways implicated in colorectal carcinogenesis are a good example, ranging from PI3K/mTOR, mitogen-activated protein kinases (MAPK), and Wnt pathways. These pathways are controlled via complex crosstalk, negative feedback, and other compensatory mechanisms23. Briffa et al.24 demonstrated through colorectal cancer cell lines that single gene mutational analysis is insufficient for stratification of tumors with respect to therapy. Targeted NGS analysis of tumor samples of patients with refractory advanced/metastatic hepatocellular carcinoma detected molecular aberrations leading to putative activation of the PI3K/AKT/mTOR pathway. Patients received therapies including an mTOR inhibitor and all demonstrated therapeutic benefit25. Additional systematically organized clinical trials are needed to investigate the impact of signaling pathway guided therapy.
Deeper characterization of patients' malignant tumors by NGS will also open new strategies to apply 'off-label' targeted therapies, e.g. for rare tumors, otherwise resistant tumors etc. In a study of how NGS altered clinical practice on advanced NSCLC, one patient with an ERBB2 -mutated adenocarcinoma received off-label afatinib and another patient with a MET exon 14 splicing mutation was offered off-label crizotinib at first progression26. The molecular alterations would not be detected through the standard practice in advanced NSCLC, namely single gene assays. These tyrosine kinase inhibitors are acknowledged as targeted therapies for consideration as per the National Comprehensive Cancer Network (NCCN) guidelines for NSCLC, even though they are not FDA-approved for the indications27.
Precision medicine-the right dose and at the right time
Monitoring treatment response is important in all phases of cancer management to avoid continuing ineffective therapies, to prevent unnecessary side-effects and to determine the benefit of new therapeutics. In the absence of metastasis, there is no good method to accurately predict resistance to therapy and recurrence. Moreover, the search for biologic markers that predict response to treatment has been very difficult. Especially for patients with solid tumors, a major obstacle has been the availability and quantity of tissue that can be studied in the period immediately before the initiation of treatment, especially for individuals with recurrent disease. Serial sampling of tumor material through repeat biopsies is usually not feasible, which limits our understanding of genomic evolution during disease progression and treatment. The potential of circulating cell-free DNA (cfDNA) to monitor treatment response has been actively explored in recent years. cfDNA contains tumor-specific sequences that harbor the somatic genomic alterations found in a patient's tumor (circulating tumor DNA: ctDNA), which can be detected through blood by NGS. ctDNA enters the circulation following apoptosis and/or necrosis of tumor cells and is typically fragmented to around 160-180 bp, reflecting the degradation of DNA into nucleosomal units that is characteristic of the apoptotic process. Before, during and after treatment, ctDNA analysis by NGS could potentially predict resistance and recurrence. The biggest advantage of ctDNA is the analysis can be done repeatedly without invasion. Studies have shown that somatic mutations identified in ctDNA are widely representative of the underlying tumor genome and can provide an alternative noninvasive method of tumor sampling28-30.
Plasma samples collected immediately before the administration of each treatment cycle may reflect overall tumor burden, whereas the ability to measure increases in ctDNA release immediately after treatment may prove to be an early indicator of tumor cell death and treatment response. This noninvasive biomarker also has potential clinical utility to monitor the delivery of targeted therapies. Moreover, quantitative assessment of ctDNA levels may also prove to be an important indicator of prognosis. An association between ctDNA levels and prognosis in several malignancies has been demonstrated in small patient cohorts31,32. Furthermore, another use of ctDNA analysis is to be used as a biomarker after potentially curative treatment to identify individuals at risk of relapse. Future long-term studies in larger patient populations are needed to validate the role of ctDNA as a surrogate biomarker for disease-free and overall survival.
The analysis of ctDNA is challenging and requires highly sensitive techniques due to the small fraction of tumor-specific DNA present within the background levels of normal cfDNA. A study by Bettegowda et al.32 compared many different types of cancer to determine in which cancers ctDNA level could be detected and represent a useful clinical tool. They found that less than 50% of patients with medulloblastomas, metastatic kidney, prostate or thyroid cancers and less than 10% of patients with gliomas, harbored detectable ctDNA levels. In addition, the amount of ctDNA from patients with the same cancer varied.
Other emerging biomarkers to measure drug response are neoantigens that arise as a consequence of tumor-specific mutations (mutations only seen in tumor tissue when comparing with normal sources)33. Exome sequencing can be paired with protein mass spectrometry to determine the presentation of neoantigens that are created by somatic mutations and correlated with the overall rate of somatic mutation and clinical response34. Through genome wide somatic neoepitope analysis and HLA typing, candidate tumor neoantigens can be identified for each patient. A neoantigen landscape has been defined that is specifically present in tumors with a strong response to CTLA-4 blockade35. Neoantigen assays could become very important for predicting responses to immunotherapy, a form of therapy that is rapidly being employed, especially for use in melanoma35.
Limitations and concerns of NGS
WGS of tumors is an unbiased approach that provides extensive genomic information about a tumor and can provide information at the single nucleotide level as well as detect structural variations such as large rearrangements, gross deletions and duplications16,36. However, the cost of sequencing and complexity of interpretation are still not feasible for routine clinical WGS of tumor specimens. Exome sequencing maybe an alternative, but it is still not cost-effective due to the minimum requirement of sequencing coverage to detect mosaicism. Copy number variants (CNVs), an important type of mutation in cancer, can be determined using NGS data from WGS, but cannot be readily detected using exome or gene panels as the data only represent a fraction of the genome. Today, microarray analysis is still the most reliable way to analyze CNVs. The analysis can be accurate when analyzing alleles from germline samples, but is limited when analyzing heterogeneous somatic samples, as they cannot confidently call low-level amplifications37. In addition, other important cancer mutation mechanisms that lead to altered gene expression such as methylation profile, changes in non-coding regions, rearrangements, and miRNA expression cannot be detected by NGS on DNA. Other techniques, such as RNA seq, can be helpful to profile tumors.
As with any genetic test, there is a risk of detecting secondary or incidental findings, which are defined as findings unrelated to the indication for obtaining the test, but of medical value for patient care. It is possible that a DNA sequence analysis of tumor tissue may identify variants relevant to familial cancer or other syndromes in addition to somatically acquired mutations. Although germline mutations can be distinguished from somatic changes by paired analysis of germline and tumor tissue, current workflows and economic realities limit NGS to the tumor specimen itself16 in most laboratories. Moreover, because there is overlap between mutations described as somatic changes and those linked to familial cancer syndromes, correct classification of these variants can be difficult. In a cohort of patients undergoing tumor genomic testing for gastrointestinal malignancies, three BRCA2 mutations carriers were incidentally identified, which underscores the need to develop a framework for communication of risks to patients undergoing routine tumor-only sequencing38. Laboratories should have policy in place on how to handle secondary findings before performing the test.
Conclusions
The success of precision medicine depends on having accurate diagnostic tests that identify patients who can benefit from targeted therapies. NGS technologies have revealed a more detailed molecular characterization of cancers helping to realize the great promise of precision medicine. NGS-based cancer gene panels analyze the molecular features of tumor tissues simultaneously and can provide enough depth of coverage to detect minor allele frequencies in a cost-effective manner. New clinical "basket trial" design with multiple centers involved has achieved some success to help guide the right patients with rare cancers to the right drug. Signaling pathway guided cancer therapy has gained success and off-label drug use based on NGS results has been successful. The use of cfDNA has brought new hopes to deliver the drug to patients at the right dose and the right time.
However, the overwhelming complexity of the cancer genome suggests that we are in the earliest phases of interpreting molecular results and translating that data into knowledge that is useful to clinicians and to treat cancer patients. Many more cancer genomes need to be analyzed in order to achieve a deeper understanding of cancers and develop additional tools for molecular analysis. Additional clinical trials with molecular criteria conducted in adult and pediatric patients are needed. Clinical applications of ctDNA are likely to expand in coming years with the development of future genotype-based targeted therapies. Finally, information about cancer generated through NGS technology such as transcriptomes by RNA seq and high-resolution cancer methylomes, orthogonal technologies such as functional studies are needed together with NGS to hasten the era of precision medicine in cancer. Optimal exploitation of all these data through integrated analyses across the different cancer types will lead to a comprehensive understanding of the genetic events that lie at the basis of tumor development and evolution. As a result, a comprehensive map of cellular alterations will benefit all cancer patients.
Conflict of interest statement
Yuan Xue works for a for-profit molecular diagnostic company. There is no conflict for William R. Wilcox.
References
- Mendoza MC. HIM and the path to personalized medicine. J AHIMA. 2010;81: 38–42; quiz 43. [PubMed] [Google Scholar]
- Metzker ML. Sequencing technologies - the next generation. Nat Rev Genet. 2010;11: 31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- Chen X, Pappo A, Dyer MA. Pediatric solid tumor genomics and developmental pliancy. Oncogene. 2015;34: 5207–15. doi: 10.1038/onc.2014.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spier I, Drichel D, Kerick M, Kirfel J, Horpaopan S, Laner A, et al. Low-level APC mutational mosaicism is the underlying cause in a substantial fraction of unexplained colorectal adenomatous polyposis cases. J Med Genet. 2016;53: 172–9. doi: 10.1136/jmedgenet-2015-103468. [DOI] [PubMed] [Google Scholar]
- Tyburczy ME, Dies KA, Glass J, Camposano S, Chekaluk Y, Thorner AR, et al. Mosaic and Intronic Mutations in TSC1/TSC2 Explain the Majority of TSC Patients with No Mutation Identified by Conventional Testing. PLoS Genet. 2015;11: e1005637. doi: 10.1371/journal.pgen.1005637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Do H, Dobrovic A. Sequence artifacts in DNA from formalin-fixed tissues: causes and strategies for minimization. Clin Chem. 2015;61: 64–71. doi: 10.1373/clinchem.2014.223040. [DOI] [PubMed] [Google Scholar]
- Hadd AG, Houghton J, Choudhary A, Sah S, Chen L, Marko AC, et al. Targeted, high-depth, next-generation sequencing of cancer genes in formalin-fixed, paraffin-embedded and fine-needle aspiration tumor specimens. J Mol Diagn. 2013;15: 234–47. doi: 10.1016/j.jmoldx.2012.11.006. [DOI] [PubMed] [Google Scholar]
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304: 1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in Non-Small-Cell lung cancer. N Engl J Med. 2010;363: 1693–703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An X, Tiwari AK, Sun Y, Ding PR, Ashby CR, Chen ZS. BCR-ABL tyrosine kinase inhibitors in the treatment of Philadelphia chromosome positive chronic myeloid leukemia: a review. Leuk Res. 2010;34: 1255–68. doi: 10.1016/j.leukres.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2: 82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell CE, Al-Kateb H, Bredemeyer AJ, Duncavage EJ, Spencer DH, Abel HJ, et al. Validation of a next-generation sequencing assay for clinical molecular oncology. J Mol Diagn. 2014;16: 89–105. doi: 10.1016/j.jmoldx.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tafe LJ, Gorlov IP, De Abreu FB, Lefferts JA, Liu X, Pettus JR, et al. Implementation of a molecular tumor board: the impact on treatment decisions for 35 patients evaluated at Dartmouth-Hitchcock medical center. Oncologist. 2015;20: 1011–8. doi: 10.1634/theoncologist.2015-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500: 415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch AM, Chang DK, Kassahn KS, Bailey P, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 2015;518: 495–501. doi: 10.1038/nature14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, Mcbride DJ, Humphray SJ, Greenman CD, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463: 191–6. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance ED, Stephens PJ, O'meara S, Mcbride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463: 184–90. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan VG, Ebert PJ, Ting JC, Lim E, Wong SS, Teo AS, et al. Whole-genome sequencing of Asian lung cancers: second-hand smoke unlikely to be responsible for higher incidence of lung cancer among Asian never-smokers. Cancer Res. 2014;74: 6071–81. doi: 10.1158/0008-5472.CAN-13-3195. [DOI] [PubMed] [Google Scholar]
- Butrynski JE, D'adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med. 2010;363: 1727–33. doi: 10.1056/NEJMoa1007056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willyard C. 'Basket studies' will hold intricate data for cancer drug approvals. Nat Med. 2013;19: 655. doi: 10.1038/nm0613-655. [DOI] [PubMed] [Google Scholar]
- Patterson SE, Liu R, Statz CM, Durkin D, Lakshminarayana A, Mockus SM. The clinical trial landscape in oncology and connectivity of somatic mutational profiles to targeted therapies. Hum Genomics. 2016;10: 4. doi: 10.1186/s40246-016-0061-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366: 883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318: 1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- Briffa R, Um I, Faratian D, Zhou Y, Turnbull AK, Langdon SP, et al. Multi-Scale genomic, transcriptomic and proteomic analysis of colorectal cancer cell lines to identify novel biomarkers. PLoS One. 2015;10: e0144708. doi: 10.1371/journal.pone.0144708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F, Kaseb AO, Tsimberidou AM, Wolff RA, Kurzrock R. Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next Generation sequencing. Oncotarget. 2014;5: 3012–22. doi: 10.18632/oncotarget.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangachari D, Vanderlaan PA, Le X, Folch E, Kent MS, Gangadharan SP, et al. Experience with targeted next generation sequencing for the care of lung cancer: insights into promises and limitations of genomic oncology in day-to-day practice. Cancer Treat Commun. 2015;4: 174–81. doi: 10.1016/j.ctrc.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettinger DS, Akerley W, Borghaei H, Chang AC, Cheney RT, Chirieac LR, et al. Non-small cell lung cancer, version 2. Non-small cell lung cancer, version 2. 2013. J Natl Compr Canc Netw. 2013;11: 645–53; quiz 653. doi: 10.6004/jnccn.2013.0084. [DOI] [PubMed] [Google Scholar]
- De Mattos-Arruda L, Weigelt B, Cortes J, Won HH, Ng CK, Nuciforo P, et al. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann Oncol. 2014;25: 1729–35. doi: 10.1093/annonc/mdu239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry AR, Mouliere F, El Messaoudi S, Mollevi C, Lopez-Crapez E, Rolet F, et al. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat Med. 2014;20: 430–5. doi: 10.1038/nm.3511. [DOI] [PubMed] [Google Scholar]
- Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013;368: 1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- Nygaard AD, Garm Spindler KL, Pallisgaard N, Andersen RF, Jakobsen A. The prognostic value of KRAS mutated plasma DNA in advanced non-small cell lung cancer. Lung Cancer. 2013;79: 312–7. doi: 10.1016/j.lungcan.2012.11.016. [DOI] [PubMed] [Google Scholar]
- Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6: 224ra24. doi: 10.1126/scitranslmed.3007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348: 69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014;371: 2189–99. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318: 420–6. doi: 10.1126/science.1149504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013;31: 1023–31. doi: 10.1038/nbt.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catenacci DV, Amico AL, Nielsen SM, Geynisman DM, Rambo B, Carey GB, et al. Tumor genome analysis includes germline genome: are we ready for surprises. Int J Cancer. 2015;36: 1559–67. doi: 10.1002/ijc.29128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15: 747–56. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]