

Abstract
Next-generation sequencing (NGS) has been rapidly integrated into molecular pathology, dramatically increasing the breadth genomic of information available to oncologists and their patients. This review will explore the ways in which this new technology is currently applied to bolster care for patients with solid tumors and hematological malignancies, focusing on practices and guidelines for assessing the technical validity and clinical utility of DNA variants identified during clinical NGS oncology testing.
Keywords: Cancer genomics, next-generation sequencing, molecular diagnostics
Introduction
Massively parallel sequencing of nucleic acids enables DNA and RNA analysis on a grand scale. A natural implementation of this "next-generation sequencing (NGS)" technology is to assess the unique and complex set of genomic alterations that occur in malignant neoplasms, with the goal of improving patient care through personalized diagnosis, prognosis, and therapy.
The most prevalent current implementation of NGS for oncology is mutation detection via targeted panels1-5. These assays use molecular methods such as multiplex polymerase chain reactions (PCR) to isolate clinically relevant segments of the genome, such as mutation hotspots or coding exons of entire genes. These panels range from a few hundred target loci to many thousands. In these assays, raw sequence reads are first aligned to the reference human genome. Variant calling is then performed to identify small mismatches in these alignments which may represent mutations present in the specimen. Variant analysis and interpretation must then be performed to assess the technical validity and clinical utility of each variant (Figure 1).
Figure1.
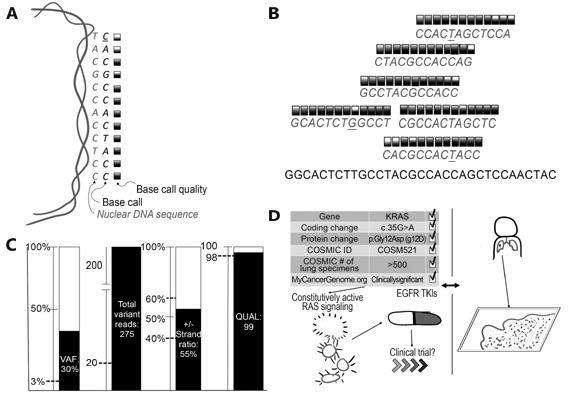
Summary of technical validity and clinical utility assessment for cancer NGS. (A) NGS basecalling, wherein a DNA sequence and corresponding confidence score is generated from a nuclear genomic DNA template. (B) The next step, which compares all available data to the reference and each other. Variant calling is then performed (underlined bases in panel B), comparing base calls across many reads; many false positive variant calls (x'ed out bases) can be filtered, while true positives (circled bases) should generate a strong signal. (C) Multiple quality metrics are generated during variant calling, which can be compared to cutoffs established during assay validation (dashed lines). (D) Detailed review of available databases and literature (left side) and comparison to clinical history and tumor pathology (right side) to assess clinical utility. VAF, variant allele frequency; QUAL, variant call quality; COSMIC, Catalogue of Somatic Mutations in Cancer; TKIs, tyrosine kinase inhibitor therapies.
Before any variant analysis is performed, the data must be checked for overall assay performance and quality. As many surgical pathology specimens have limited tissue amounts and quality, and have been treated with formalin fixation, assay failures occur with some regularity. As there are many different ways to measure assay performance, each assay requires a unique set of parameters established during validation6,7.
Technical validity and clinical utility are the two major issues that must be resolved for every variant identified via NGS somatic variant detection. If a detected variant fails to meet or exceed predetermined validity and utility criteria, it should not be clinically reported as medically relevant. While simple in principal, there exists deep complexity in these areas which merit close consideration.
Terminology
NGS introduces a new suite of vocabulary into the clinical lab, with some specific to oncology testing. The following glossary is provided as reference and to facilitate further discussion in this review.
Genomic analyst (GA)
GA is typically a doctorate or masters level specialist who performs some or all of the initial review of assay quality, technical validity assessment of detected variants, clinical utility assessment of variants, and generating draft reports. Final reports must be signed out by either a board certified MD clinical pathologist or PhD with clinical laboratory board certification.
Base call quality score (Q score)
Q score is a PHRED-scaled probability ranging from 0-20 inversely proportional to the probability that a single sequenced base is correct. For example, a T base call with Q of 20 is considered likely correct with a confidence P-value of 0.01. Any base call with Q<20 should be considered low quality, and any variant identified where a substantial proportion of reads supporting the variant are of low quality should be considered potentially false positive.
Read depth
Read depth (or coverage, conventionally a number followed by "×") is the number of independent reads with overlapping alignment at a locus of interest. This is often expressed as an average or percentage exceeding a cutoff over a set of intervals (such as exons, genes, or panels). For example, a clinical report might say that a panel average coverage is 1,105× with 98% of targeted bases covered >100×.
Mutations
Mutations are events that result in changes of genomic DNA. Variants are deviations from the human genome reference sequence observed in a specimen. The goal of NGS cancer panel testing is to identify variants which are extremely likely to be caused by somatic mutation. The difference is subtle; the two terms are often used interchangeably.
Variant read number (variant reads)
Variant reads is the number of independent sequence reads supporting the presence of a variant. Due to the high error rate of NGS at the per-base call level, calls supported by fewer than 5 variant reads are typically considered to be likely false positive calls.
Variant allele frequency (VAF)
VAF is the percentage of sequence reads observed matching a specific DNA variant divided by the overall coverage at that locus. Because NGS provides a near random sample, VAF is thus a surrogate measure of the proportion of DNA molecules in the original specimen carrying the variant. For constitutional genetic testing, VAF is a measure of diploid zygosity: heterozygous loci should be near 50% VAF, homozygous loci should be near 100%, and reference loci should be near 0%. Deviations from these three expected values should be considered suspicious as potential errors due to incorrect base calls or alignment. For somatic testing, contamination from normal cells and tumor heterogeneity combine to cause unpredictable VAFs. In some clinical scenarios, such as testing a patient for therapeutic resistance mutations, the desired sensitivity dictates that variants with low VAF be included in downstream analysis. This all means that in order to maintain an acceptable level of sensitivity, variant filtering for somatic variant must be highly permissive from a technical validity perspective.
Variant quality scores (QUAL)
QUAL are generated during the variant calling step and a requisite component of the Variant Call File (VCF). Generally speaking, QUAL scores are transformed log-scaled (PHRED) values where, for example, a score of 90 supports the variant call with a P-value of 1×10-9. It is critical to keep in mind that there are significant multiple testing considerations due to the large number of variants that are detected by NGS. The scale for QUAL values vary widely and depend on assay platform, capture method, and variant calling software. As such, it is strongly recommended at this time that each laboratory performing NGS independently assess the performance characteristics of QUAL scores for each assay, preferably by orthogonal testing.
Strand bias (SB)
SB is a measure of how far the observed variant reads deviate from an expectation of equal likelihood of sequencing the plus and minus strands. A high SB score indicates that the variant call may be caused by an artifact of the alignment process rather than a true mutation8. Certain targeting methodologies, including those using Illumina TruSight capture techniques selectively amplify one target strand9; for these assays, SB is not a meaningful measure of quality.
Technical validity
To date, there are no NGS oncology in vitro diagnostics (IVD) cleared by the United States Food and Drug Administration (FDA). As such, all current clinically validated NGS oncology assays are classified as "laboratory developed" tests (LDT) which can vary highly in content and quality from lab to lab10. In the US, LDTs must be performed in laboratories with Clinical Laboratory Improvement Amendments (CLIA) licensure and are subject to state and federal regulations. The New York State Department of Health has issued guidelines for NGS oncology testing, setting a very high bar for clinical validation and ongoing assay performance (Table 1)11.
Table1.
Summary of valuable references and guidelines relevant to clinical NGS oncology testing
| Source | Title | Content summary | Reference | |||
| ACMG: American College of Medical Genetics and Genomics; CDC: United States Centers for Disease Control and Prevention | ||||||
| New York State Board of Health | "Next Generation" Sequencing (NGS) guidelines for somatic genetic variant detection | Detailed standards for technical validity | 11 | |||
| ACMG | Standards and guidelines for the interpretation of sequence Variants | Guidelines for clinical validity assessment, particularly for germline/constitutional variants | 12 | |||
| ACMG | ACMG clinical laboratory standards for next-generation sequencing | Broad summaries of major areas of consideration for clinical validation of all NGS assay | 13 | |||
| CDC | Assuring the quality of next-generation sequencing in clinical laboratory practice | Detailed recommendations for technical validity assessment/validation of all NGS assays | 14 | |||
| CDC | Good laboratory practice for clinical next-generation sequencing informatics pipelines | Detailed recommendations for clinical validity assessment for all NGS assays | 15 | |||
| Quest Diagnostics (reference laboratory) | Annotation of sequence variants in cancer samples processes and pitfalls for routine assays in the clinical laboratory | The framework for a repeatable workflow for clinical validity assessment in use at a high volume testing facility is described | 16 |
For labs without New York approval, there are no matching federal guidelines at this time. As such, it is strongly recommended to prefer laboratories with optional College of American Pathology (CAP) accreditation. The major benefit of CAP accreditation comes from proficiency testing participation, wherein laboratories performing similar assays are given identical reference materials for comparison of results. This proficiency testing gives labs a mechanism and incentives to continually improve their test methodologies by direct comparison with others (Table 1)12-16.
Orthogonal confirmatory testing can validate that detected variants is a direct means of assuring technical validity. This requires a laboratory to have access to duplicate sequencing of entire panels using alternate methodologies (e.g. sequencing using both Life Technologies and Illumina platforms for every specimen), or to have a single-locus test of equivalent sensitivity for every targeted interval. As neither approach is simple or inexpensive to implement, many laboratories eschew such confirmatory testing during routine analysis. While PCR-based dideoxy sequencing ("Sanger" sequencing) is a generally accepted method of confirmatory testing for constitutional clinical NGS17-20, its sensitivity is limited to approximately 10%-20% VAF. As cancer NGS testing routinely identifies clinically relevant mutations from 5%-10% VAF, it Sanger is not an ideal method for confirmation for all variants.
Manual review of aligned reads in a genome browser ("variant inspection") can greatly reduce the risk of false positive or incorrect results (Figure 2). While variant calling software has become increasingly sophisticated, it still cannot yet perform as well as a GA when it comes to pattern recognition. For cancer NGS, it is best to think of the role of variant calling software as to alert the GA to loci of interest. From there, it is the job of the GA to assess first whether any variant is present and if the variant called is the correct change. In some cases, variant inspection will reject dubious variant calls which have received a high QUAL score; this is the necessary cost of setting high sensitivity. In other cases, the variant observed will differ from that called. Complex insertion/deletion events are especially susceptible to this type of error. Given how common this mutation type is in certain tumors, laboratories must be prepared for this issue to be routine rather than exceptional.
Figure2.
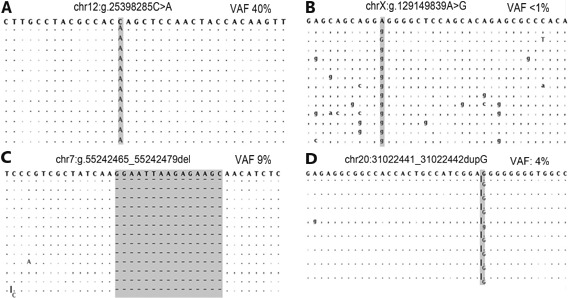
Alignment views of four variants detected by NGS. Each panel depicts a representative set of variant reads for single nucleotide variant (A and B) or insertion/deletion variants (C and D) with either high quality (A and C) or low quality (B and D). The human genome reference sequence is the string of bases along the top of each panel. Aligned basecalls matching the reference are listed as dots (plus strand) or commas (minus strand). High quality variants typically have higher variant allele frequencies (VAF), and variants reads have fewer additional variants. Panel D depicts known false positive locus, likely due to the "homopolymer problem" where certain NGS technologies overcall insertion/deletion variants where the reference sequence has five or more of the same nucleotides in a row (in this case, 8 guanines).
There are many metrics that can be used to assess if a variant call is likely a true positive result. VAF, QUAL, SB, and other metrics may be available depending on the variant callers used (Figure 1). As each LDT NGS assay performs differently, it is necessary to establish filtering criteria anew for all new tests. If hard cutoffs are to be used to filter out low quality variants, a rigorous validation should be performed, including accuracy and limit of detection experiments to determine and test the cutoffs. Alternatively, very inclusive filters can be set to minimize false negative risk. This will increase the burden of confirmatory analysis and/or testing required for each case. Both options can be rate-limiting for small laboratories.
During the validation phase of assay development, it is valuable to test assay performance using reference materials. DNA or false FFPE blocks from positive control cell lines are commercially available for many of the most recurrent mutations. Some reference materials now available contain a variety of mutations with different VAF, genes, and mutation types. These are designed to be run during validation and/or during routine testing to ensure ongoing assay performance. Acrometrix (a division of Thermo Fisher Sciences Inc. Waltham, MA, United States), and Horizon Diagnostics (a division of Horizon Discovery Group plc. Waterbeach, United Kingdom) are two commercial vendors specializing in this area.
Clinical validity
It is tempting to think of tumors as "gene positive" or "gene negative". This is a convenient way to discuss certain classical oncogenic changes such as fusion genes, but it glosses over a significant layer of complexity we are only now beginning to understand. For example, some mutations in EGFR render a tumor more susceptible to targeted tyrosine kinase inhibitor (TKI) therapy; other mutations confer resistance to the same21-24. Certain KRAS mutations are known to be targetable driver mutations while others are commonly observed but seem to have little to no impact on drug response (Figure 1)1,25,26. These now classic examples are the vanguard of a new way of thinking about cancer genes and mutations.
For each detected variant, there are multiple different types of considerations a GA should weigh.
Somatic vs. germline
If normal tissue is available and tested, variant subtraction can be used to directly assess which DNA variants arose in somatically in the tumor. As this is not always feasible (consider hematological malignancies, where blood specimens contain neoplastic cells) or practical, indirect measures should be taken to avoid reporting benign germline variants as relevant for oncology. The front-line tool for indirectly filtering out germline variants is population allele frequency. Any variant that is present on >1% of normal human chromosomes is almost certainly not clinically relevant for cancer. VAF databases such as the Exome Aggregation Consortium (ExAC)27 or dbSNP (which uses 1,000 genome project allele frequencies)28 should be used to perform this filtering. As they match expectations for heterozygous and homozyous frequencies respectively, variants with VAF at nearly 50% or 100% should be considered potentially germline during analysis. Hard filtering for VAF however is not recommended as true mutations can certainly be observed at low VAF.
Tumor type
Many mutations impact different tumor types in different ways, or have only been studied rigorously in a specific set of tumor types but are recurrently observed in other types. This means that variant annotation and interpretation procedures must take tumor type into account prior to rendering a clinical report. Resources such as COSMIC, The Cancer Genome Atlas (TCGA), and MyCancerGenome.org (MCG) have publically available databases with tumor-type specific mutation information and should be referred to when performing variant analysis. Literature review may be necessary for are mutations.
VAF and tumor cellularity
If the VAF and percent tumor cell prediction from pathology are highly discordant, it could indicate an invalid or unreportable variant. If the VAF is far higher than expected, it could indicate that the variant is either germline or in a region of loss of heterozygosity (LoH). If the variant is in a gene where LoH is not anticipated (e.g. an oncogene where activating mutations are the mechanism), germline may be the more likely explanation. A VAF far lower than expected could indicate either false positivity or low level tumor heterogeneity. If the technical validity of a lower than expected VAF variant is very strong, tumor heterogeneity is more likely. If this is the case, the relevance of the mutation may depend on contextual considerations such as mutation type, tumor type, and patient treatment history. For example, the observation of a low relative VAF of a known recurrent acquired resistance mutation is of much stronger clinical validity in a treated patient than the same observation in a treatment naive patient.
It is also important to remember that percent tumor cell prediction, especially surgical pathology/histology, are essentially qualitative assessments never intended to have highly accurate or precise measurements. This is not an indictment of pathologists, rather a statement of fact that the methods used are not high enough resolution to be used in this way. Genomic analysts, who may or may not have any training in anatomic pathology, should be aware that these assessments may be off by as much as 10%-20% when performed by the best practitioners. Flow cytometry assessment of hematological malignancies should be a much more precise and accurate quantitation, though the correlation between flow cytometry results and NGS VAFs is as yet poorly understood.
Highly recurrent mutations
The observation of certain mutations, even at borderline technical quality, represents a "smoking gun" and must be reported. Examples such as BRAF p.V600E and KRAS codon 12/13 mutations should come to mind immediately for experienced cancer genomic analysts. These variants must be reported.
Clinically significant mutation types
In-frame insertions/deletions (indels) and loss of function (LoF) mutations are two mutation types which can present as apparently novel variants at the DNA level despite having well-known implications. Examples include in-frame indels in EGFR exon 19 or LoF mutations in TP53. As practically all genomic analysis software solutions perform comparisons at the DNA (or predicted mRNA) level, this pattern can cause patient variants to be incorrectly classified as of uncertain clinical significance. Until such a times as more sophisticated software tools are available, it is the role of the genomic analyst to determine whether apparently novel variants are in fact of known clinical significance.
This area can become quite complex. For example, acute myeloid leukemia (AML) patients positive for biallelic LoF mutations in CEBPA have a favorable prognosis only if one mutation is in the C terminal region and the other is in the N terminal region29. This means that a specific mutation seen alone should be considered of uncertain significance, whereas that same mutation in conjunction with a second mutation would be clinically relevant to prognosis. This pattern can be challenging to convey in standard databases, requiring either highly specialized software or manual review by experts.
Variants of uncertain clinical significance (VUS)
The number of VUS identified in a given specimen closely correlates with the scale of targets analyzed. As more genes or hotspots are added to tests, more VUS will be observed. Whether and how to report VUS is highly laboratory and test dependent: some labs will always report all VUS, some will never report any, and some will report only when specific conditions are met. The ordering oncologist should be aware of VUS reporting policies and utilize the test that best addresses the clinical needs of their patient. Contacting a lab directly is the best way to ascertain VUS reporting policies; if no such policy exists or cannot be readily ascertained, it may be wise to consider a provider with better clarity. However, due to a lack of evidence supporting the clinical utility of VUS, reporting of VUS should not be considered a necessary component of tumor sequencing analysis.
Integration of cytogenetics information
There are clinically significant cytogenetic abnormalities or nearly all major malignancies routinely detected by karyotype or FISH. Some of these have a significant impact on clinical interpretation of NGS results. For example, there are multiple mechanisms of TP53 loss, including point mutation (detectable by NGS) and gene deletion.
Clinical trials
The unfortunate reality for many patients is that standard of care treatments available are unlikely to result in a positive outcome. A major goal of precision medicine initiatives is to improve and expand on these treatments through the use of targeted therapies, either small molecules or biologics designed to attack cells expressing specific mutations. While there are now several such drugs available, the modest gains achieved so far are hopefully the vanguard of a much larger and more effective suite of targeted therapies.
To assist in managing the complexities of drug development, discovery, and evaluation, the National Cancer Institute (NCI) has created the NCI Match program where patients are connected with clinical trials for which their mutation status makes them eligible. This nationwide collaboration among academic labs, pharmaceutical companies, oncologists, and patients hold great promise. From the molecular pathologist perspective, these trials offer a window into which targets may be clinically utile in the near future, pending their results. In the meantime, labs are developing or even offering testing of targets that remain in the trial phase both to prepare for therapies which may come available in the near future and to enable their patients to enroll in a clinical trial.
Hereditary cancer syndrome variants
There is significant overlap between the genes known to have clinically relevant somatic alterations and hereditary cancer predisposition30,31. As such, panel testing may reveal germline variants of clinical relevance to the patient and the patient’s family. At this time, such variants are considered incidental, as patients are not typically consented or counseled that such information may be identified during testing. As screening for hereditary cancer syndromes is only recommended in high risk patients (e.g. those with family history or atypical presentation), this is a moral grey area with minimal guidance from professional societies or governmental agencies at this time.
Conclusions
Clinical NGS testing in cancer is a new field experiencing rapid change. As a result, the current landscape is extremely varied, with each laboratory deciding how and when to report detected variants. For now, labs must perform extensive assay performance testing and complex validations to ensure a high level of reliability, accuracy, and sensitivity.
It is also important to recognize the current limitations of NGS analysis. Due to the limited number of prospective studies and sample sizes, mutation screening has very limited negative predictive value. This means that at present NGS testing cannot discern between benign and malignant neoplasms from solid or liquid biopsy samples. Rather, as we have seen, it can help guide therapy and management for individuals known to have malignancies.
Genome-wide sequencing, including genome and exome, has enabled major advances in our understanding of the molecular basis of cancer. Some findings, such as the roles of calreticulin (CALR) mutations in myeloproliferative neoplasms32 and IDH1 mutations in gliomas33 have led to rapidly adopted, high-impact clinical tests. Thus the current common practice is to use the results of genome-wide sequencing as a resource to inform clinical test development.
While the benefit of these research efforts have been tremendous, the utility of performing the same tests on clinical specimens is less certain. At present, the total number of genes relevant to targeted therapies, diagnostic and prognostic implications, and clinical trials is likely in the hundreds. For any given cancer type, this number may be far lower: in some cases only half a dozen or so. As such, the majority of variants or mutations detected by genome-wide sequencing of neoplastic specimens cannot be interpreted in a clinical setting and are of limited clinical utility. However, several commercial and academic laboratories offer clinical exome or genome sequencing, and have reported some early success34. So far though, the vast majority of relevant mutations detected by genome-wide sequencing are within genes on standard panels. For rare cancer types or types with unusually broad mutational spectra, genome-wide sequencing may already have clinical use enough above panels to merit semi-routine implementation.
Genome-wide sequencing may confer additional benefits, such as structural rearrangement detection (copy number changes, loss of heretozygosity, ploidy, etc.) and tumor purity and heterogeneity estimation. At this time, the bioinformatics required to perform these additional analyses are not mature enough for clinical diagnostics. Eventually, the benefits of genome-wide sequencing will increase (along with the decrease in the cost of sequencing) to meet a crossing point where panels are no longer sufficient. Thus it is likely a matter of "when?" - not "if?" - genome-wide sequencing replaces panel testing.
Over time, labs will coalesce around similar methods and approaches. The driving forces behind this will be increasingly sophisticated professional guidelines, more powerful reference materials, and availability of in vitro diagnosis (IVD) tests. Professional societies such as the College of American Pathologists (CAP) and the US Centers for Disease Control and Prevention (CDC) have laid the early groundwork for regulations and guidelines that will likely grow more specific and rigorous in years to come. If the pace at which commercial vendors have set continues, the rate-limiting step of mutation-positive control specimen acquisition will be reduced. And it is likely that the US FDA will continue to approve more NGS IVD tests including cancer panels.
All of these trends will combine to make clinical cancer NGS testing less costly and complex to offer, thus making them more accessible to laboratories and patients. Due to mainly economic concerns, DNA testing of solid tumors and hematological malignancies is typically limited to high level tertiary care centers and/or patients with refractory disease at this time. As the cost of testing comes down and as more targeted therapies are available, its net benefit to population health will rise dramatically.
Conflict of interest statement
No potential conflicts of interest are disclosed.
References
- D'Haene N, Le Mercier M, De Nève N, Blanchard O, Delaunoy M, El Housni H, et al. Clinical validation of targeted next generation sequencing for colon and lung cancers. PLoS One. 2015;10: e0138245. doi: 10.1371/journal.pone.0138245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malapelle U, Vigliar E, Sgariglia R, Bellevicine C, Colarossi L, Vitale D, et al. Ion torrent next-generation sequencing for routine identification of clinically relevant mutations in colorectal cancer patients. J Clin Pathol. 2015;68: 64–8. doi: 10.1136/jclinpath-2014-202691. [DOI] [PubMed] [Google Scholar]
- Sie D, Snijders PJ, Meijer GA, Doeleman MW, Van Moorsel MI, Van Essen HF, et al. Performance of amplicon-based next Generation DNA sequencing for diagnostic gene mutation profiling in oncopathology. Cell Oncol (Dordr) 2014;37: 353–61. doi: 10.1007/s13402-014-0196-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh RR, Patel KP, Routbort MJ, Reddy NG, Barkoh BA, Handal B, et al. Clinical validation of a next-generation sequencing screen for mutational hotspots in 46 cancer-related genes. J Mol Diagn. 2013;15: 607–22. doi: 10.1016/j.jmoldx.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Marrone M, Filipski KK, Gillanders EM, Schully SD, Freedman AN. Multi-marker solid tumor panels using next-generation sequencing to direct molecularly targeted therapies. PLoS Curr. 2014; 6.
- Dames S, Chou LS, Xiao Y, Wayman T, Stocks J, Singleton M, et al. The development of next-generation sequencing assays for the mitochondrial genome and 108 nuclear genes associated with mitochondrial disorders. J Mol Diagn. 2013;15: 526–34. doi: 10.1016/j.jmoldx.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Mccourt CM, Mcart DG, Mills K, Catherwood MA, Maxwell P, Waugh DJ, et al. Validation of next generation sequencing technologies in comparison to current diagnostic gold standards for BRAF, EGFR and KRAS mutational analysis. PLoS One. 2013;8: e69604. doi: 10.1371/journal.pone.0069604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Li J, Li CI, Long J, Samuels DC, Shyr Y. The effect of strand bias in illumina short-read sequencing data. BMC Genomics. 2012;13: 666. doi: 10.1186/1471-2164-13-666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato T, Morisada N, Nagase H, Nishiyama M, Toyoshima D, Nakagawa T, et al. Somatic mosaicism of a CDKL5 mutation identified by next-generation sequencing. Brain Dev. 2015;37: 911–5. doi: 10.1016/j.braindev.2015.03.002. [DOI] [PubMed] [Google Scholar]
- Gray PN, Dunlop CL, Elliott AM. Not all next generation sequencing diagnostics are created equal: understanding the nuances of solid tumor assay design for somatic mutation detection. Cancers (Basel) 2015;7: 1313–32. doi: 10.3390/cancers7030837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- "Next Generation" Sequencing (NGS) guidelines for somatic genetic variant detection. Available from http://www.wadsworth.org/labcert/TestApproval/forms/NextGenSeq_ONCO_Guidelines.pdf. [Last accessed on 2015 August 10].
- Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17: 405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Bale SJ, Bayrak-Toydemir P, Berg JS, Brown KK, Deignan JL, et al. ACMG clinical laboratory standards for next-generation sequencing. Genet Med. 2013;15: 733–47. doi: 10.1038/gim.2013.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012;30: 1033–6. doi: 10.1038/nbt.2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gargis AS, Kalman L, Bick DP, Da Silva C, Dimmock DP, Funke BH, et al. Good laboratory practice for clinical next-generation sequencing informatics pipelines. Nat Biotechnol. 2015;33: 689–93. doi: 10.1038/nbt.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee LA, Arvai KJ, Jones D. Annotation of sequence variants in cancer samples: processes and pitfalls for routine assays in the clinical laboratory. J Mol Diagn. 2015;17: 339–51. doi: 10.1016/j.jmoldx.2015.03.003. [DOI] [PubMed] [Google Scholar]
- Strom SP, Lee H, Das K, Vilain E, Nelson SF, Grody WW, Deignan JL, et al. Assessing the necessity of confirmatory testing for exome-sequencing results in a clinical molecular diagnostic laboratory. Genet Med. 2014;16:510–5. doi: 10.1038/gim.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YP, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, et al. Clinical Whole-Exome sequencing for the diagnosis of mendelian disorders. N Engl J Med. 2013;369: 1502–11. doi: 10.1056/NEJMoa1306555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal SM, Yu JH, Chong JX, Dent KM, Conta JH, Tabor HK, et al. Practices and policies of clinical exome sequencing providers: analysis and implications. Am J Med Genet A. 2013; 161A: 935-50.
- Lee H, Deignan JL, Dorrani N, Strom SP, Kantarci S, Quintero-Rivera F, et al. Clinical exome sequencing for genetic identification of rare Mendelian disorders. JAMA. 2014;312: 1880–7. doi: 10.1001/jama.2014.14604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TJ, Bell DW, Sordella R, Gurubhagavatula S, Okimoto RA, Brannigan BW, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350: 2129–39. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304: 1497–500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Pao W, Miller V, Zakowski M, Doherty J, Politi K, Sarkaria I, et al. EGF receptor gene mutations are common in lung cancers from "never smokers" and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101: 13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305: 1163–7. doi: 10.1126/science.1101637. [DOI] [PubMed] [Google Scholar]
- Fiala O, Pesek M, Finek J, Benesova L, Belsanova B, Minarik M. The dominant role of G12C over other KRAS mutation types in the negative prediction of efficacy of epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Cancer Genet. 2013;206: 26–31. doi: 10.1016/j.cancergen.2012.12.003. [DOI] [PubMed] [Google Scholar]
- Riely GJ, Kris MG, Rosenbaum D, Marks J, Li A, Chitale DA, et al. Frequency and distinctive spectrum of KRAS mutations in never smokers with lung adenocarcinoma. Clin Cancer Res. 2008;14: 5731–4. doi: 10.1158/1078-0432.CCR-08-0646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W, Gardner SA, Hovhannisyan H, Natalizio A, Weymouth KS, Chen W, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population database: insights of relevance to variant classification. Genet Med. 2015 Dec 17. doi: 10.1038/gim.2015.180. [Epub ahead of print]
- 1000 Genomes Project Consortium, Abecasis GR, Auton A, Brooks LD, Depristo MA, Durbin RM, et al. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491: 56–65. doi: 10.1038/nature11632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HY, Deng DH, Huang Y, Ye FH, Huang LL, Xiao Q, et al. Favorable prognosis of biallelic CEBPA gene mutations in acute myeloid leukemia patients: a meta-analysis. Eur J Haematol. 2015;94: 439–48. doi: 10.1111/ejh.12450. [DOI] [PubMed] [Google Scholar]
- Laduca H, Stuenkel AJ, Dolinsky JS, Keiles S, Tandy S, Pesaran T, et al. Utilization of multigene panels in hereditary cancer predisposition testing: analysis of more than 2,000 patients. Genet Med. 2014;16: 830–7. doi: 10.1038/gim.2014.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC, Berg JS, Grody WW, Kalia SS, Korf BR, Martin CL, et al. ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet Med. 2013;15: 565–74. doi: 10.1038/gim.2013.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369: 2379–90. doi: 10.1056/NEJMoa1311347. [DOI] [PubMed] [Google Scholar]
- Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321: 1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016 Jan 28. doi: 10.1001/jamaoncol.2015.5699. [Epub ahead of print]