

Abstract
Long‐QT syndrome type‐2 (LQT2) is characterized by reduced functional expression of the human ether‐à‐go‐go related (hERG) gene product, resulting in impaired cardiac repolarization and predisposition to fatal arrhythmia. Previous studies have implicated abnormal trafficking of misfolded hERG as the primary mechanism of LQT2, with misfolding being caused by mutations in the hERG gene (inherited) or drug treatment (acquired). More generally, environmental and metabolic stresses present a constant challenge to the folding of proteins, including hERG, and must be countered by robust protein quality control (QC) systems. Disposal of partially unfolded yet functional plasma membrane (PM) proteins by protein QC contributes to the loss‐of‐function phenotype in various conformational diseases including cystic fibrosis (CF) and long‐QT syndrome type‐2 (LQT2). The prevalent view has been that the loss of PM expression of hERG is attributed to biosynthetic block by endoplasmic reticulum (ER) QC pathways. However, there is a growing appreciation for protein QC pathways acting at post‐ER cellular compartments, which may contribute to conformational disease pathogenesis. This article will provide a background on the structure and cellular trafficking of hERG as well as inherited and acquired LQT2. We will review previous work on hERG ER QC and introduce the more novel view that there is a significant peripheral QC at the PM and peripheral cellular compartments. Particular attention is drawn to the unique role of the peripheral QC system in acquired LQT2. Understanding the QC process and players may provide targets for therapeutic intervention in dealing with LQT2.
Abbreviations
- CF
cystic fibrosis (CF)
- CFTR
cystic fibrosis transmembrane conductance regulator
- CHIP
C‐terminus of Hsc70‐interacting protein
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum‐associated degradation
- hERG
Human ether‐à‐go‐go related gene
- HSP
heat shock protein
- LQT2
long‐QT syndrome type‐2
- NEF
nucleotide exchange factor
- PM
plasma membrane
- QC
quality control
- WT
wild‐type
Introduction
The human ether‐a‐go‐go related gene (hERG1, gene name KCNH2) encodes the Kv11.1 inwardly rectifying K+ channel commonly referred to as hERG. In cardiac myocytes hERG is expressed as the rapidly activating delayed rectifier potassium current (I Kr) that is involved in cardiac repolarization (Trudeau et al. 1995). Loss of hERG function is associated with long‐QT syndrome type‐2 (LQT2), characterized by impaired ventricular repolarization, extended action potential duration and increased risk of potentially fatal torsades de pointes arrhythmia (Keating & Sanguinetti, 2001; Sanguinetti & Tristani‐Firouzi, 2006). The loss of hERG function as seen in LQT2 can be caused by mutations in the hERG gene (inherited LQT2) or as a result of off‐target drug effects (acquired LQT2). As with other proteins, the normal maturation and function of the hERG channel requires proper protein folding and conformational integrity, which is constantly challenged by physical, environmental and metabolic stresses. Consequently, cells require robust protein quality control (QC) systems to monitor folding status and to direct unfolded molecules within each cellular compartment to either refolding or degrading pathways. Owing to the fact that a growing number of diseases are related to protein misfolding, the study of protein QC mechanisms as potential therapeutic targets has received increasing attention. A large subset of acquired and inherited LQT2 involves degradation of non‐native hERG channels by the protein QC machinery, resulting in a reduction in cell‐surface expression and contributing to the overall loss of functional phenotype. In many cases, misfolded channels targeted for degradation are at least partially functional, making protein QC systems a significant player in LQT2 pathogenesis (Anderson et al. 2006; Ke et al. 2013). For many years, the hERG loss‐of‐expression phenotype was attributed to impaired biosynthetic maturation and plasma membrane (PM) insertion. Recent work has uncovered an extensive QC network targeting misfolded plasma membrane proteins, including hERG, for degradation. Removal of misfolded hERG from the cell periphery is likely to act in parallel with the established biosynthetic maturation defect and almost certainly plays a significant role in LQT2 pathogenesis. This review will highlight recent progress on the mechanisms underlying hERG quality control and their contribution to LQT2 pathogenesis.
Background
Structure and maturation of hERG
The hERG channel is a tetrameric (Vandenberg et al. 2012) structure containing a transmembrane core and two cytosolic structures: an N‐terminal Per‐Arnt‐Sim (PAS) domain and a C‐terminal cyclic nucleotide‐binding domain (CNBD) (Fig. 1 A). The transmembrane region forms the voltage sensor, the selectivity filter and the ion‐conducting pore. Interactions between the hERG cytosolic domains and the transmembrane region regulate channel‐gating kinetics (Vandenberg et al. 2004).
Figure 1. hERG channel structure .
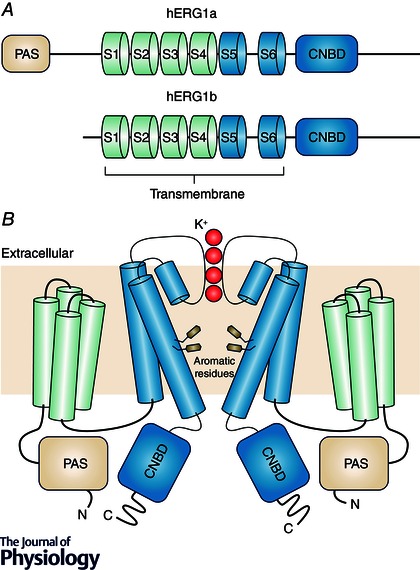
A, domain structure of hERG1a (top) and hERG1b (bottom). hERG1a contains a cytosolic Per‐Arnt‐Sim (PAS) and cyclic nucleotide binding domain (CNBD), as well as 6 transmembrane helices S1–S6. The hERG1b N‐terminus lacks a PAS domain and instead contains a unique 35‐amino‐acid sequence. Domains are not drawn to scale. B, putative topology of the hERG K+ channel tetramer. For clarity, only one pair of opposing subunits comprising the hERG tetramer are shown. K+ ions passing through the channel selectivity filter are shown in red. Also shown are aromatic amino acids Y652 and F656 involved in drug‐induced hERG block.
In cardiac cells, hERG is expressed as two isoforms: hERG1a, which constitutes the full‐length channel that conducts a current at the cell surface, and hERG1b, which lacks most of the cytosolic N‐terminus including the PAS domain and instead has a short 35‐residue N‐terminal cytosolic sequence (Phartiyal et al. 2008) (Fig. 1 A). Both hERG1a and ‐1b homotetramers form functional ion channels; however, whilst hERG1a is expressed at the cell surface, hERG1b is confined to the ER in the absence of hERG1a co‐expression, presumably due to the presence of an ‘RXR’ ER retention signal on the hERG1b N‐terminus (Phartiyal et al. 2008). Consequently, most studies investigating hERG cellular processing and quality control rely upon exogenous overexpression of hERG1a alone. In cardiac myocytes, incorporation of hERG1b regulates the kinetics of channel gating and is essential for normal cardiac repolarization (Larsen et al. 2008; Jones et al. 2014); whether the addition of hERG1b subunits influences hERG folding/misfolding remains relatively unexplored.
As with other polytypic plasma membrane proteins, nascent hERG channels are synthesized and N‐linked glycosylated in the endoplasmic reticulum (ER) (Fig. 2). Channels are then trafficked to the Golgi and subjected to additional glycosylation before being exported to the plasma membrane (PM) (Zhou et al. 1998; Petrecca et al. 1999; Gong et al. 2002). This additional glycosylation increases the apparent molecular mass from 135 kDa (core‐glycosylated form) to 155 kDa (mature complex glycosylated form) (Fig. 2 inset). At the cell periphery, mature hERG channels cycle between the PM and endocytic compartments (Apaja et al. 2013).
Figure 2. hERG cellular processing .
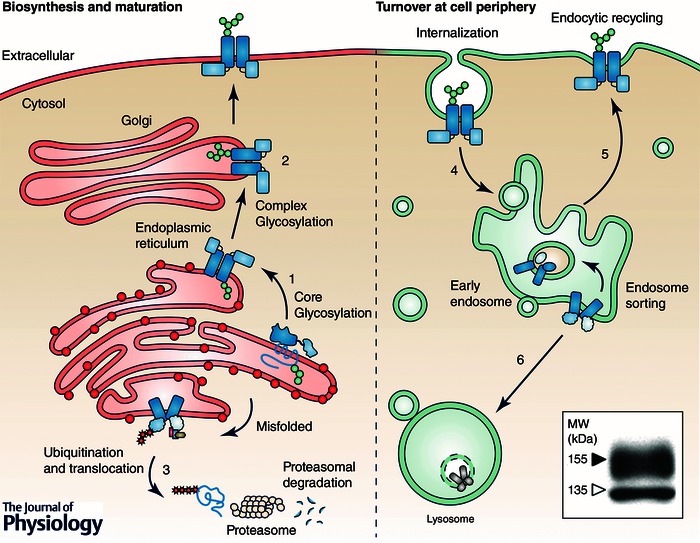
Overview of hERG cellular processing. Nascent channels are cotranslationally inserted into the ER membrane (1) where they undergo N‐linked glycosylation to an apparent molecular mass of 135 kDa. Upon proper folding and assembly, hERG is exported to the Golgi where it is subject to additional glycosylation, increasing the apparent molecular mass to 155 kDa prior to export to the plasma membrane (2). Nascent channels, which fail to fold at the ER, are targeted for proteasomal degradation (3). PM‐resident hERG is internalized (4) and either recycled back to the PM (5) or targeted for lysosomal degradation (6). Inset, typical immunoblot of WT hERG. Indicated are the immature 135 kDa core‐glycosylated and mature 155 kDa complex glycosylated forms (open and filled arrow respectively). hERG is detected in stably transfected HeLa cells by immunoblotting against an engineered HA‐epitope in the S1–S2 extracellular loop (Ficker et al. 2003).
Inherited LQT2
The congenital form of LQT2 is dominantly inherited, with a prevalence estimated to be 1:6000. It is linked to nearly 500 hERG mutations, with no single mutation being predominant (Schwartz et al. 2009). It is noteworthy that over 30% of these LQT2 mutations can cause nonsense mutations, resulting in a truncated, non‐functional form of hERG mRNA that is likely to result in nonsense mediated mRNA decay (Gong et al. 2007). Studies have shown that deletion/truncation at the hERG CNBD is poorly tolerated (Akhavan et al. 2005). However, mRNA truncated downstream of the CNBD can produce functional channels if nonsense‐mediated mRNA decay is inhibited (Gong et al. 2011). The dominant mechanisms for inherited LQT2 pathogenesis identified to date are the presence of missense mutations in hERG, which result in altered channel function and reduced channel expression due to impaired ER exit and accelerated degradation. The latter has been proposed as the predominant mechanism, accounting for almost 90% of the mutants tested (Anderson et al. 2006, 2014; Ke et al. 2013) (see Fig. 2 for example). There is significant variation in the expression defect between mutations; some completely abolish expression of mature hERG while others exhibit a milder phenotype (Ke et al. 2013; Anderson et al. 2014). In some cases, rescue of hERG expression by incubation at low temperature restored PM channel activity (Anderson et al. 2006, 2014; Ke et al. 2013). Since low temperature rescue is not a clinical treatment option, there is more emphasis placed on folding and trafficking pathways that could restore abrasive inherited mutations. Though it has not yet been explored, it is also interesting to speculate on the presence of modest hERG mutations that produce a negligible clinical phenotype in isolation, but possibly act synergistically with pharmacological or environmental factors to generate a more severe defect under certain conditions.
Acquired LQT2
The hERG channel is the target for the vast majority of drug‐induced arrhythmias and represents a major cause of withdrawal of drugs from the market (Roden, 2004; Waring et al. 2015). A recent in silico study suggests that up to 5% of approved drugs bind hERG as an off‐target effect (Lounkine et al. 2012). In general, drugs can impact hERG either directly by binding to the channel or indirectly through modifying cellular processes.
The majority of compounds affecting hERG function block channel conductance by directly binding two non‐conserved aromatic amino acid residues exposed within the channel pore (Y652, F656, Fig. 1 B) (Lees‐Miller et al. 2000; Mitcheson et al. 2000; Sanchez‐Chapula et al. 2002; Fernandez et al. 2004). A subset of hERG blockers has additional effects on channel trafficking. Some hERG blockers have been shown to reduce hERG PM expression by impairing ER exit and accelerating degradation from the cell surface (Dennis et al. 2011). These effects are likely to synergize with the functional block to produce the acquired LQT2 phenotype. Interestingly, other hERG blockers have the opposite effect on expression and trafficking, being able to increase the expression of mutant hERG channels (Zhou et al. 1998; Ficker et al. 2002; Kuryshev et al. 2005; Rajamani et al. 2006; Takemasa et al. 2008). The structural basis determining whether a hERG blocker promotes or impairs hERG expression remains unknown. Pharmacological rescue of mutant hERG expression presents a promising therapeutic strategy for inherited LQT2; however, current hERG ‘pharmacochaperones’ are limited by concomitant functional block of the rescued channels. A number of hERG ‘activators’ bind to sites outside the channel pore and modulate hERG gating (Kang et al. 2005; Casis et al. 2006; Perry et al. 2007; Grunnet et al. 2011). Whether binding to these non‐pore sites can rescue mutant hERG trafficking remains to be established.
Pharmacological agents can also impact hERG indirectly in the absence of binding. This was found to be the case for acquired LQT2 associated with cardiac glycosides and diuretic‐induced hypokalaemia. Depletion of intracellular K+ via cardiac glycosides and depletion of extracellular K+ have both been shown to destabilize the hERG conformation or defect maturation (Wang et al. 2007; Guo et al. 2009).
hERG domains
Mutations affecting hERG conformation have been identified in both the PAS and CNBD cytosolic domains, as well as within the membrane‐spanning helices and extracellular/intracellular loops (Anderson et al. 2006, 2014). Deletion of the entire CNBD or any conserved structural motif within the CNBD has a severe impact on channel expression, suggesting that an intact and properly folded CNBD is necessary for stable channel conformation and trafficking (Akhavan et al. 2005). Indeed, mutations in this region tend to be more severe in their trafficking defect, and poorly correctible by low‐temperature incubation and/or pharmacochaperones (Anderson et al. 2014).
The role of the N‐terminal PAS domain in hERG folding is less clear‐cut. Truncated hERG1a lacking the entire N‐terminal region appears to traffic normally (Phartiyal et al. 2008; Ke et al. 2014); however, a significant number of LQT2‐associated mutations reducing channel expression have been identified in this region (Harley et al. 2012). Recent studies have investigated the impact of these mutations on both the thermal stability of the isolated domain and hERG gating kinetics (Harley et al. 2012; Ke et al. 2013). Most of the mutations examined reduced the thermal stability of the isolated domain. However, in some cases the extent of thermal destabilization did not correspond with the severity of trafficking defect. In many cases, channel destabilization was associated with abnormal gating kinetics, suggesting altered domain–domain interactions. The hERG PAS domain contains a hydrophobic patch that is thought to be involved in domain interactions (Morais Cabral et al. 1998). It is possible that such interactions are required to conceal this hydrophobic area, which, if exposed, could be recognized by protein QC systems. Alternatively, it is possible that while complete absence of a PAS domain interaction is tolerated, an abnormal interaction negatively affects the co‐operative folding of other domains. The relative importance of local destabilization of the PAS domain vs. loss of domain–domain interactions remains to be directly examined.
Acquired LQT2 by either direct drug binding or K+ depletion is thought to act at the channel pore. In a recent study, we found that perturbations at the channel pore (via K+ depletion) or by a mutation in an extracellular loop induced unfolding of the hERG cytosolic domains (Apaja et al. 2013). This suggests that local perturbations within the hERG transmembrane region trigger co‐operative unfolding of additional domains. A similar mechanism whereby misfolding of one domain triggered co‐operative misfolding of others was shown to contribute to the conformational destabilization caused by several disease‐associated cystic fibrosis transmembrane conductance regulator (CFTR) mutations, including ΔF508 (Du & Lukacs, 2009; Rabeh et al. 2012).
ER quality control
Nascent hERG at the ER must successfully undergo several stages of protein folding prior to Golgi export, including folding of the polypeptide chain, post‐translational folding after release from the ribosome, and oligomer assembly. To guide hERG and other proteins through these stages, the ER has its own quality control system (ERQC). There has been much emphasis on understanding the key players of the ERQC and their role in different protein misfolding diseases. Impaired hERG biosynthetic secretion and maturation, presumably due to the action of ERQC machinery, remains one of the most established mechanism for LQT2 pathogenesis (Kuryshev et al. 2005; Anderson et al. 2006; Rajamani et al. 2006; Takemasa et al. 2008).
The mediators of the ERQC system are the molecular chaperones. These proteins both promote the proper folding of nascent polypeptides but also send terminally misfolded substrates for degradation. Degradation of misfolded substrates is mediated by a process called ER associated degradation (ERAD), involving binding of molecular chaperones and co‐chaperones, recruitment of ubiquitination machinery, dislocation of transmembrane segments from the ER membrane into the cytosol, and proteasomal degradation. We will discuss the ERQC in two parts: the chaperone/co‐chaperone system and the ubiquitination/degradation machinery associated with ERAD.
HSP70/90 chaperone systems
The predominant molecular chaperones involved in ERQC are the heat‐shock protein (Hsp) families Hsp70 and Hsp90, along with associated co‐chaperones (Fig. 3) (Hartl et al. 2011). The function of these proteins has been covered in detail elsewhere (Mayer, 2013; Young, 2014). Briefly, Hsp70 cycles between a high substrate affinity (ADP‐bound) state and a low substrate affinity (ATP‐bound) state. The Hsp40/DNAJ co‐chaperone family assists in Hsp70 substrate binding by stimulating the hydrolysis of ATP to ADP. Conversely, nucleotide exchange factors (NEFs) stimulate the release of substrate through the exchange of bound ADP for ATP. Hsp70‐bound substrates can be passed to Hsp90, which further promotes substrate folding (Fig. 3). The Hsp90 chaperone family also functions via an ATPase cycle that is mechanistically unrelated to Hsp70, and is assisted and modulated by a complicated co‐chaperone network (Pearl & Prodromou, 2006; Li et al. 2012). The Hsp70 and Hsp90 networks have both been implicated in the folding and trafficking mechanisms of hERG (Ficker et al. 2003).
Figure 3. Quality control of misfolded hERG at the ER .
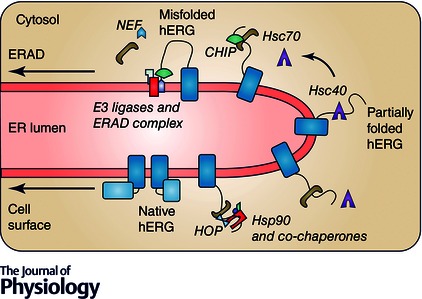
Partially folded hERG is recognized by Hsp40s such as DNAJA1 and DNAJA2. This binding stimulates the hydrolysis of ATP to ADP, initiating Hsp70/Hsc70 substrate binding and Hsp40 dissociation. DNAJA1 assists folding by subsequent binding of Hsp70, Hsp90 and co‐chaperones, resulting in a native form of hERG, which is trafficked to the cell surface. DNAJA2 promotes degradation by initiating CHIP E3 ubiquitin ligase binding. NEFs promote the release of ADP and re‐binding of ATP and release of Hsc70 from the substrate, possibly regulating hERG folding or degradation. Chaperone‐independent E3 ubiquitin ligases and other proteins may be involved in ER associated degradation (ERAD) of hERG.
The chaperone systems need to strike a balance between refolding of salvageable substrates and degradation of terminally misfolded molecules. The two primary members of the Hsp70 family, the constitutively expressed heat‐shock cognate (Hsc)‐70 and the stress inducible Hsp70, appear to play opposing roles although they are regulated by the same co‐chaperones. Overexpression of Hsp70 increases wild‐type (WT)‐hERG expression levels and I Kr, whereas Hsc70 overexpression does not (Ficker et al. 2003; Li et al. 2011). Hsp70 and Hsc70 were previously proposed to play opposite roles in the ER processing of ENaC channels, suggesting that Hsp70 and Hsc70 may promote folding and degradation, respectively (Goldfarb et al. 2006). The reason for this difference between Hsp70 and Hsc70 is unknown but may involve the co‐chaperone C‐terminus of Hsc70‐interacting protein (CHIP), an E3 ubiquitin ligase (see below). Furthermore, trafficking‐deficient hERG channels with cytosolic‐domain mutations tend to preferentially recruit Hsc70 over Hsp70 compared to the WT channel. However, a subset of trafficking‐defective mutations in the channel core reduced Hsc70 binding, suggesting a more complicated interplay between specific hERG conformational defects and chaperone/co‐chaperone recognition (Li et al. 2011).
Hsp90 was initially shown to interact with immature hERG at the ER and to be required for the maturation of hERG and I Kr (Fig. 3) (Ficker et al. 2003). More recently, hERG was shown to be preferentially bound by Hsp90α over Hsp90β to promote trafficking, highlighting a more precise relationship for Hsp90 in the folding of hERG (Peterson et al. 2012). The specificity of chaperone roles in hERG folding is further complicated by the many co‐chaperones that regulate or modify chaperone activity.
hERG co‐chaperones
Co‐chaperones from the Hsp40 family (DNAJs) and NEFs contribute to the Hsp70/Hsc70 reaction cycle and can regulate chaperone function. DNAJs promote the hydrolysis of ATP into ADP, changing the Hsp70/Hsc70 from an open, substrate‐accepting state to a closed, substrate‐bound state, and NEFs promote the reverse process by stimulating the exchange of ADP with ATP (Hartl et al. 2011).
The impact of DNAJs in substrate folding/degradation seems to be specific to the type of DNAJ and misfolded substrate. It is apparent that while DNAJA1 (Hdj2) and DNAJA2 are highly homologous and are the most abundant DNAJs, they are functionally distinct (Tzankov et al. 2008; Baaklini et al. 2012). Previous work has shown that DNAJA1 and Hsc70 were both required for CHIP E3 ubiquitin ligase binding to promote degradation of CFTR (Younger et al. 2004). Similarly, both DNAJs are thought to promote degradation of hERG through the binding of the pro‐degradative Hsc70 chaperone (Walker et al. 2010). However, there appear to be functional differences between individual DNAJs. Specifically, knockdown of DNAJA1 and to a lesser extent DNAJA2 also impaired hERG trafficking, suggesting that normal levels of DNAJA1 in particular are needed for Hsp70‐assisted folding. (Fig. 3) (Walker et al. 2010).
The NEFs consist of three structural families: BAG (Bcl2‐associated athanogene 1 related) proteins, the Hsp110 family and HspBP1 (Hsp70 binding protein 1). The NEFs induce the release of the protein substrate from the Hsp70 complex, and under appropriate conditions, can enhance Hsp70‐mediated folding (Tzankov et al. 2008; Rauch & Gestwicki, 2014). Hsp110s are distinct from the other NEFs because they are thought to bind substrate directly and are distant homologues of Hsp70 (Xu et al. 2008; Saxena et al. 2012). Although the roles of NEFs in the folding of other membrane spanning channels have been studied (Young, 2014), their function on hERG is much less explored. One NEF, Bag2, is involved in the Hsp70 cycle of CFTR folding and was also found to associate with hERG (Arndt et al. 2005; Walker et al. 2007). The exact role of Bag2 on hERG ERQC is not established.
Additional interactions with the substrate protein occur outside the Hsp70 folding cycle, which further complicates the identification of key regulators in hERG protein quality control. One group of co‐chaperones are able to bind to Hsc/Hsp70 through tetratricopeptide‐repeat (TPR) adaptor domains (Carrigan et al. 2006). These proteins can attach various activities to Hsc/Hsp70 or Hsp90 (Young et al. 2003, 2004). For example, CHIP is a soluble E3 ubiquitin ligase that promotes degradation of hERG and other substrates bound to Hsc/Hsp70 or Hsp90 in the cytosol and in ERAD (Connell et al. 2001; Meacham et al. 2001). Another well‐characterized TPR co‐chaperone is HSP‐organizing protein (HOP), which connects Hsc/Hsp70 with Hsp90 so that Hsp90‐dependent folding can proceed (Scheufler et al. 2000; Pearl & Prodromou, 2006; Li & Buchner, 2013). HOP was found in complex with hERG and may aid the release of hERG from Hsc70 and the recruitment of Hsp90 (Walker et al. 2007). Since CHIP and HOP both bind to the same C‐terminal motif of Hsc70, the competition between these co‐chaperones will result in either degradation or Hsp90 substrate transfer, respectively (Fig. 3) (Scheufler et al. 2000; Meacham et al. 2001).
It seems logical to use the model of the much more characterized folding cycle of CFTR as an example to target potential co‐chaperones involved in hERG folding (Wang et al. 2006; Okiyoneda et al. 2010), though it is noteworthy to point out that we do not see comparable co‐chaperone interactions with hERG to what has been so far described with CFTR (unpublished results). Through enhancing pro‐folding co‐chaperones and manipulating pro‐degradative co‐chaperones, it is reasonable to speculate that combined manipulation of Hsc/Hsp70 and co‐chaperones could promote the proper folding and trafficking of hERG.
Endoplasmic reticulum associated proteasomal degradation
The chaperone system provides the means to identify misfolded proteins directed for degradation (Hartl et al. 2011). However in ERAD, misfolded substrate is polyubiquitinated by an E3 ubiquitin ligase, sometimes independent of chaperones, causing dislocation from the ER followed by unfolding of the substrate and degradation by the 26S proteasome (Hampton & Sommer, 2012). Identification of E3 ligases involved in protein substrate degradation has, therefore, become an interesting area of exploration. In fact, one such cytosolic E3 ligase, CHIP, has been identified in degrading misfolded proteins including those found at the ER such as CFTR and hERG (Meacham, 2001; Murata et al. 2001). The presence of certain members in the Hsp70 folding cycle seems to influence CHIP binding, as seen with some of the DNAJs increasing substrate interaction with CHIP, while Hsp90 prevents it, thus indicating that CHIP predominantly acts through Hsp70 (Walker et al. 2010; Iwai et al. 2013). While CHIP plays a prominent role in hERG degradation, it is recognized that several other ER‐resident E3 ligases and ERAD proteins may be involved in hERG degradation (Fig. 3). Indeed, for CFTR, the transmembrane E3 ligases gp78 and RMA1 have been shown to promote degradation of ΔF508‐CFTR (Sun et al. 2006; Younger et al. 2006; Morito et al. 2008). A number of proteins including gp78, its homologue E3 ligase HRD1, the derlin family and associated factors are involved in quality control ERAD of other transmembrane proteins as well (Smith et al. 2011; Hampton & Sommer, 2012). It seems likely that these E3 ligases and ERAD proteins may also be involved in hERG degradation. The exploration of such ERAD components for hERG have yet to be reported, though they may prove to be an additional mechanism for regulating hERG degradation that could be manipulated for clinical use.
Peripheral quality control
For many years, it was believed that disposal of partially misfolded hERG by the ERQC represents the primary, if not the sole, mechanism for LQT2 pathogenesis (Kuryshev et al. 2005; Anderson et al. 2006; Rajamani et al. 2006; Takemasa et al. 2008). However, there is growing evidence that misfolded channels may be trafficked to the cell membrane and their regulation would require proteostasis mechanisms acting in peripheral compartments (Anderson et al. 2006; Apaja et al. 2013). Consistent with this notion, we recently reported that misfolded hERG channels at the PM are polyubiquitinated and rapidly internalized into the endocytic pathway and targeted for lysosomal delivery (Fig. 4). A similar paradigm was previously described for other misfolded membrane proteins including disease‐associated mutant CFTR and G‐protein‐coupled receptors (Okiyoneda et al. 2010; Apaja et al. 2013).
Figure 4. Quality control of misfolded hERG at peripheral cellular compartments .
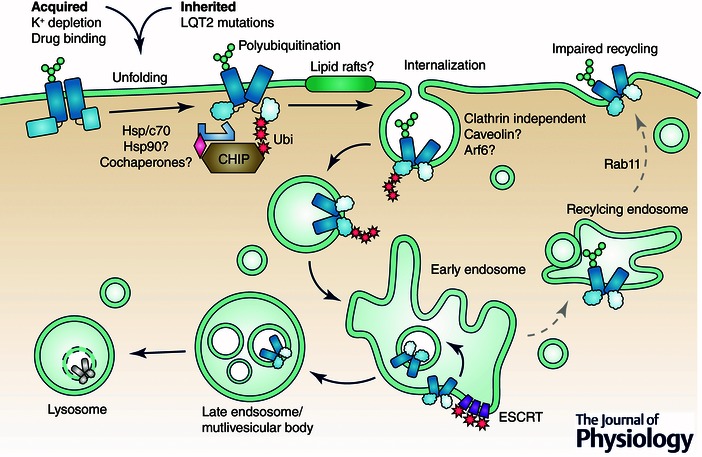
Misfolded hERG cytosolic domains are recognized by cytosolic chaperone proteins such as Hsp/Hsc70 and Hsp90. Chaperone binding results in recruitment of CHIP E3 ubiquitin ligase and subsequent ubiquitination. Polyubiquitin serves as a signal for rapid internalization via a clathrin‐independent mechanism. Possible pathways include recruitment to lipid rafts and subsequent caveolin‐mediated internalization or an Arf6‐dependent mechanism. Internalized proteins may be prevented from recycling back to the plasma membrane and are instead sorted to intraluminal vesicles and targeted for lysosomal degradation by the ESCRT machinery.
Molecular mechanisms responsible for ubiquitination of misfolded hERG at the PM
For several years, the CHIP E3 ubiquitin ligase has been known to ubiquitinate misfolded cytosolic and PM proteins (Okiyoneda et al. 2010; Apaja et al. 2013). We recently demonstrated that CHIP also promotes the removal of misfolded hERG from the PM via addition of K48‐ and K63‐linked polyubiquitin chains (Apaja et al. 2013). Disruption of CHIP binding to Hsc/p70 and Hsp90 prevented the removal of misfolded channels from the PM, suggesting a role of cytosolic chaperones in recognition of misfolded hERG. In contrast to hERG QC at the ER, the role of cytosolic chaperones/co‐chaperones in hERG refolding and/or degradation at the PM remains to be explored. It has been reported that ablation of Hsc70, Hsp90, or co‐chaperones Bag1, DNAJB2 (HSJ1), DNAJA1 (Hdj2) and DNAJC7 (Tpr2) prevented removal of misfolded CFTR from the PM (Okiyoneda et al. 2010). Interestingly, Hsp70 upregulation did not affect turnover of misfolded CFTR, yet is involved in degradation of other misfolded PM‐protein substrates (Apaja et al. 2010; Okiyoneda et al. 2010). Furthermore, Hsp90 is involved in the degradation of misfolded CFTR, but it conversely stabilizes a large number of conformationally unstable protein kinases at the PM (Taipale et al. 2010; Taipale et al. 2012). While there are several reports of the roles of chaperone/co‐chaperones in other protein folding and stability at the PM, the key modulators for hERG folding have yet to be established. Interestingly, Hsp90 inhibition did not inhibit the removal of misfolded hERG from the PM (unpublished data). These observations suggest that the role of individual chaperones/co‐chaperones in peripheral protein QC can be substrate specific.
Interestingly, CHIP knockdown only partially abolished polyubiquitination and turnover of misfolded PM hERG, suggesting the involvement of other E3 ligases. Previous studies have implicated Nedd4‐2‐mediated monoubiquitination in the regulation of hERG PM expression under steady‐state conditions and in response to kinase serine/threonine‐protein kinase (SGK) signalling (Lamothe & Zhang, 2013). However, Nedd4 and Nedd4‐2 ablation had no effect on the turnover of misfolded hERG, suggesting that Nedd4‐2 is primarily involved in regulating PM hERG in response to second‐messenger signalling rather than quality control (Apaja et al. 2013; Lamothe & Zhang, 2013). This is supported by the observation that Nedd4‐2‐mediated ubiquitination is dependent on binding to a C‐terminal PY motif on the channel, whereas an E3 ligase involved in quality control would be expected to recruit via hydrophobic regions or cytosolic chaperones (Guo et al. 2012).
Internalization and recycling of misfolded hERG
Most PM proteins are targeted for clathrin‐mediated endocytosis upon ubiquitination (Piper et al. 2014). Interestingly, there is evidence suggesting that hERG is internalized via a clathrin‐independent pathway; however, some controversy exists over the exact mechanism. Caveolin was found to be involved in the degradation of PM hERG in response to unfolding via extracellular K+ depletion and Nedd4‐2‐mediated ubiquitination (Guo et al. 2012), suggesting that misfolded hERG is internalized via caveolae and lipid rafts. In contrast, a recent study found hERG plasma membrane levels were regulated under steady‐state conditions via an Arf6‐dependent internalization pathway that is independent of caveolin and dynamin (Karnik et al. 2013). Arf6 was shown to be involved in a rapid redistribution of hERG from the PM into endocytic compartments following a short chase (1 h). In contrast, caveolin has only been shown to be involved in degradation of PM hERG over a relatively long time scale (more than 4 h). One interpretation of these data is that the initial internalization step is Arf6 dependent whereas caveolin regulates subsequent endosomal sorting and trafficking processes (Sandvig et al. 2011). If this scenario proves to be correct, one could postulate that the internalization of misfolded and ubiquitinated hERG also occurs via an Arf6‐dependent mechanism. Another possibility is that internalization is caveolin dependent and Arf6 regulates hERG PM expression through its role in endocytic recycling (Grant & Donaldson, 2009). Further work is required to clarify the hERG internalization machinery, which could represent a novel ubiquitin‐dependent clathrin‐independent endocytosis pathway. In addition to accelerating internalization, ubiquitination of misfolded hERG was found to impair endocytic recycling. It was recently shown that internalized hERG is recycled by a Rab11‐dependent mechanism (Lamothe & Zhang, 2013; Chen et al. 2015). It is possible that ubiquitination impairs this process, although the involvement of other recycling pathways for internalized hERG channels cannot be ruled out.
Endosomal sorting for lysosomal delivery
At the endosomal membrane, ubiquitinated cargoes are generally sorted into intraluminal vesicles destined for lysosomal delivery by the endosomal sorting complex for transport (ESCRT) machinery. Lysosomal delivery of misfolded hERG was found to be ESCRT dependent (Sun et al. 2011; Apaja et al. 2013). Intriguingly monoubiquitination was implicated in the downregulation of PM hERG in response to extracellular K+ depletion, whereas intracellular K+ depletion and mutations were found to induce K48‐ and K68‐linked polyubiquitination (Sun et al. 2011; Apaja et al. 2013). Due to the tetrameric structure of hERG, it is possible that mono/multimono‐ubiquitination of several hERG subunits would generate a sufficient sorting signal (Barriere et al. 2007). On the other hand, the role of monoubiquitination was established primarily on measurements in cells overexpressing a Lys‐free ubiquitin to suppress formation of ubiquitin‐linked chains. It is possible that exogenous ubiquitin overexpression artificially promoted substrate monoubiquitination. Additionally, even limited incorporation of endogenous ubiquitin would permit formation of linked chains. In contrast, the observation of hERG polyubiquitination was based on direct immunodetection of endogenously ubiquitinated hERG using validated poly‐ubiquitin‐specific antibodies.
Clinical relevance of hERG QC at the cell periphery
Degradation of hERG by peripheral QC machinery is likely to complement the established ER‐exit defect and presents a barrier to the development of therapeutic interventions; however, the relative contribution of these systems remains open to speculation. It is likely that the peripheral QC systems play an important role in acquired LQT2. The plasma concentration of most orally administered drugs peaks shortly after dosage, then declines over time as a result of metabolism and excretion. The turnover of WT‐hERG upon biosynthetic inhibition is relatively slow (T 1/2 ∼8–12 h (Staudacher et al. 2011; Apaja et al. 2013). Consequently, it is unlikely that the ERQC machinery alone (which only blocks hERG maturation) will be able to appreciably reduce PM hERG expression before drug plasma concentrations drop to safe levels. On the other hand, removal of misfolded channels from the PM occurs much faster (T 1/2 ∼2 h), and thus is likely to make a larger contribution to the acquired LQT2 phenotype during the transient period of peak drug concentration (Staudacher et al. 2011; Apaja et al. 2013). Another intriguing prospect is the possibility that the ER and peripheral QC systems possess a different degree of stringency, with some misfolded hERG conformers recognized by one system but not the other.
Conclusion
The study of hERG in recent years has yielded valuable insights into LQT2 pathogenesis and protein QC pathways in general. The models for protein quality control considered for hERG are based upon recent work on other membrane proteins, in particular CFTR. Although diverse clients may be subject to these same QC pathways, there is a growing appreciation that the underlying molecular machinery may involve a large degree of substrate specificity. In particular, the function of molecular chaperones/co‐chaperones to promote either folding or degradation appears to be at least partially dependent on substrate identity and expression levels. It is also likely that additional substrate specificity may exist within the ubiquitination and endocytosis machinery.
For many years, research has been focused on ER QC pathways. Recently, there has been a growing appreciation that the conformation of mature proteins at the PM is policed by an overlapping yet distinct mechanism. This peripheral QC pathway is likely to contribute to the loss‐of‐expression phenotype in many conformational diseases, including LQT2 and CFTR.
hERG is unique in that misfolding is often induced by pharmacological agents in addition to inherited mutations. Acquired LQT2 is noteworthy in that peripheral QC may represent one of the important mechanisms that contributes to LQT2 due to pharmacokinetic considerations.
Complex interactions may also arise between hERG polymorphisms and approved drugs. This may be an area where personalized medicine may be of use to prevent synergistic interactions between acquired and inherited LQT2 factors. Indeed, adverse interactions have been shown to occur between CFTR, pharmacological correctors and potentiators in a mutation‐ and patient‐specific manner (Cholon et al. 2014; Veit et al. 2014). Understanding the structural basis of hERG misfolding may provide insights into the interaction between hERG polymorphisms and clinically relevant drugs.
Additional information
Competing interests
None declared.
Acknowledgements
B.F. and B.W. share first authorship. This work was supported by operating grants from the Canadian Institutes of Health Research (CIHR) to A.S., J.C.Y. and G.L., from Cystic Fibrosis Canada to J.C.Y. and G.L. and from the Heart and Stroke Foundation Canada to A.S. G.L. holds a Canada Research Chair in Molecular and Cellular Biology of CF and Other Conformational Diseases (Tier 1) and J.C.Y. holds a Canada Research Chair in Molecular Chaperones (Tier 2). B.F. is funded by a CIHR Banting and Best Doctoral Scholarship.
Biography
Jason Young is Associate Professor in the Department of Biochemistry and Canada Research Chair in Molecular Chaperones (Tier 2). His research investigates the biochemical mechanisms of chaperone and co‐chaperone activity, and the consequences of this essential activity at a cellular level. Brittany Williamson is an MSc student in the Department of Biochemistry. Gergely Lukacs is Professor and Canada Research Chair in Molecular and Cellular Biology of CF and Other Conformational Diseases (Tier 1). His research elucidates the molecular and cellular basis of cystic fibrosis and other folding related diseases with a focus on peripheral quality control of membrane proteins. Brian Foo is a PhD student in the Department of Physiology and the recipient of a Banting and Best CIHR Doctoral Scholarship. Alvin Shrier is Professor and Hosmer Chair of Physiology. His research activity focuses on hERG potassium channel quality control and cardiac dynamics.

References
- Akhavan A, Atanasiu R, Noguchi T, Han W, Holder N & Shrier A (2005). Identification of the cyclic‐nucleotide‐binding domain as a conserved determinant of ion‐channel cell‐surface localization. J Cell Sci 118, 2803–2812. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Delisle BP, Anson BD, Kilby JA, Will ML, Tester DJ, Gong Q, Zhou Z, Ackerman MJ & January CT (2006). Most LQT2 mutations reduce Kv11.1 (hERG) current by a class 2 (trafficking‐deficient) mechanism. Circulation 113, 365–373. [DOI] [PubMed] [Google Scholar]
- Anderson CL, Kuzmicki CE, Childs RR, Hintz CJ, Delisle BP & January CT (2014). Large‐scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat Commun 5, 5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaja PM, Foo B, Okiyoneda T, Valinsky WC, Barriere H, Atanasiu R, Ficker E, Lukacs GL & Shrier A (2013). Ubiquitination‐dependent quality control of hERG K+ channel with acquired and inherited conformational defect at the plasma membrane. Mol Biol Cell 24, 3787–3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaja PM, Xu H & Lukacs GL (2010). Quality control for unfolded proteins at the plasma membrane. J Cell Biol 191, 553–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt V, Daniel C, Nastainczyk W, Alberti S & Hohfeld J (2005). BAG‐2 acts as an inhibitor of the chaperone‐associated ubiquitin ligase CHIP. Mol Biol Cell 16, 5891–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baaklini I, Wong MJ, Hantouche C, Patel Y, Shrier A & Young JC (2012). The DNAJA2 substrate release mechanism is essential for chaperone‐mediated folding. J Biol Chem 287, 41939–41954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriere H, Nemes C, Du K & Lukacs GL (2007). Plasticity of polyubiquitin recognition as lysosomal targeting signals by the endosomal sorting machinery. Mol Biol Cell 18, 3952–3965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrigan PE, Sikkink LA, Smith DF & Ramirez‐Alvarado M (2006). Domain:domain interactions within Hop, the Hsp70/Hsp90 organizing protein, are required for protein stability and structure. Protein Sci 15, 522–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casis O, Olesen SP & Sanguinetti MC (2006). Mechanism of action of a novel human ether‐a‐go‐go‐related gene channel activator. Mol Pharmacol 69, 658–665. [DOI] [PubMed] [Google Scholar]
- Chen J, Guo J, Yang T, Li W, Lamothe SM, Kang Y, Szendrey JA & Zhang S (2015). Rab11‐dependent recycling of the human ether‐a‐go‐go‐related gene (hERG) channel. J Biol Chem 290, 21101–21113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholon DM, Quinney NL, Fulcher ML, CR Esther Jr, Das J, Dokholyan NV, Randell SH, Boucher RC & Gentzsch M (2014). Potentiator ivacaftor abrogates pharmacological correction of ΔF508 CFTR in cystic fibrosis. Sci Transl Med 6, 246ra296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J & Patterson C (2001). The co‐chaperone CHIP regulates protein triage decisions mediated by heat‐shock proteins. Nat Cell Biol 3, 93–96. [DOI] [PubMed] [Google Scholar]
- Dennis AT, Nassal D, Deschenes I, Thomas D & Ficker E (2011). Antidepressant‐induced ubiquitination and degradation of the cardiac potassium channel hERG. J Biol Chem 286, 34413–34425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du K & Lukacs GL (2009). Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell 20, 1903–1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez D, Ghanta A, Kauffman GW & Sanguinetti MC (2004). Physicochemical features of the HERG channel drug binding site. J Biol Chem 279, 10120–10127. [DOI] [PubMed] [Google Scholar]
- Ficker E, Dennis AT, Wang L & Brown AM (2003). Role of the cytosolic chaperones Hsp70 and Hsp90 in maturation of the cardiac potassium channel HERG. Circ Res 92, e87–e100. [DOI] [PubMed] [Google Scholar]
- Ficker E, Obejero‐Paz CA, Zhao S & Brown AM (2002). The binding site for channel blockers that rescue misprocessed human long QT syndrome type 2 ether‐a‐gogo‐related gene (HERG) mutations. J Biol Chem 277, 4989–4998. [DOI] [PubMed] [Google Scholar]
- Goldfarb SB, Kashlan OB, Watkins JN, Suaud L, Yan W, Kleyman TR & Rubenstein RC (2006). Differential effects of Hsc70 and Hsp70 on the intracellular trafficking and functional expression of epithelial sodium channels. Proc Natl Acad Sci USA 103, 5817–5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Anderson CL, January CT & Zhou Z (2002). Role of glycosylation in cell surface expression and stability of HERG potassium channels. Am J Physiol Heart Circ Physiol 283, H77–H84. [DOI] [PubMed] [Google Scholar]
- Gong Q, Stump MR & Zhou Z (2011). Inhibition of nonsense‐mediated mRNA decay by antisense morpholino oligonucleotides restores functional expression of hERG nonsense and frameshift mutations in long‐QT syndrome. J Mol Cell Cardiol 50, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Q, Zhang L, Vincent GM, Horne BD & Zhou Z (2007). Nonsense mutations in hERG cause a decrease in mutant mRNA transcripts by nonsense‐mediated mRNA decay in human long‐QT syndrome. Circulation 116, 17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant BD & Donaldson JG (2009). Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol 10, 597–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunnet M, Abbruzzese J, Sachse FB & Sanguinetti MC (2011). Molecular determinants of human ether‐a‐go‐go‐related gene 1 (hERG1) K+ channel activation by NS1643. Mol Pharmacol 79, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Massaeli H, Xu J, Jia Z, Wigle JT, Mesaeli N & Zhang S (2009). Extracellular K+ concentration controls cell surface density of IKr in rabbit hearts and of the HERG channel in human cell lines. J Clin Invest 119, 2745–2757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, Wang T, Li X, Shallow H, Yang T, Li W, Xu J, Fridman MD, Yang X & Zhang S (2012). Cell surface expression of human ether‐a‐go‐go‐related gene (hERG) channels is regulated by caveolin‐3 protein via the ubiquitin ligase Nedd4‐2. J Biol Chem 287, 33132–33141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY & Sommer T (2012). Finding the will and the way of ERAD substrate retrotranslocation. Curr Opin Cell Biol 24, 460–466. [DOI] [PubMed] [Google Scholar]
- Harley CA, Jesus CS, Carvalho R, Brito RM & Morais‐Cabral JH (2012). Changes in channel trafficking and protein stability caused by LQT2 mutations in the PAS domain of the HERG channel. PloS One 7, e32654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A & Hayer‐Hartl M (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. [DOI] [PubMed] [Google Scholar]
- Iwai C, Li P, Kurata Y, Hoshikawa Y, Morikawa K, Maharani N, Higaki K, Sasano T, Notsu T, Ishido Y, Miake J, Yamamoto Y, Shirayoshi Y, Ninomiya H, Nakai A, Murata S, Yoshida A, Yamamoto K, Hiraoka M & Hisatome I (2013). Hsp90 prevents interaction between CHIP and HERG proteins to facilitate maturation of wild‐type and mutant HERG proteins. Cardiovasc Res 100, 520–528. [DOI] [PubMed] [Google Scholar]
- Jones DK, Liu F, Vaidyanathan R, Eckhardt LL, Trudeau MC & Robertson GA. (2014). hERG 1b is critical for human cardiac repolarization. Proc Natl Acad Sci USA 111, 18073–18077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J, Chen XL, Wang H, Ji J, Cheng H, Incardona J, Reynolds W, Viviani F, Tabart M & Rampe D (2005). Discovery of a small molecule activator of the human ether‐a‐go‐go‐related gene (HERG) cardiac K+ channel. Mol Pharmacol 67, 827–836. [DOI] [PubMed] [Google Scholar]
- Karnik R, Ludlow MJ, Abuarab N, Smith AJ, Hardy ME, Elliott DJ & Sivaprasadarao A (2013). Endocytosis of HERG is clathrin‐independent and involves arf6. PloS One 8, e85630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Hunter MJ, Ng CA, Perry MD & Vandenberg JI (2014). Role of the cytoplasmic N‐terminal Cap and Per‐Arnt‐Sim (PAS) domain in trafficking and stabilization of Kv11.1 channels. J Biol Chem 289, 13782–13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Ng CA, Hunter MJ, Mann SA, Heide J, Hill AP & Vandenberg JI (2013). Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain‐domain interactions. Biochem J 454, 69–77. [DOI] [PubMed] [Google Scholar]
- Keating MT & Sanguinetti MC (2001). Molecular and cellular mechanisms of cardiac arrhythmias. Cell 104, 569–580. [DOI] [PubMed] [Google Scholar]
- Kuryshev YA, Ficker E, Wang L, Hawryluk P, Dennis AT, Wible BA, Brown AM, Kang J, Chen XL, Sawamura K, Reynolds W & Rampe D (2005). Pentamidine‐induced long QT syndrome and block of hERG trafficking. J Pharmacol Exp Therap 312, 316–323. [DOI] [PubMed] [Google Scholar]
- Lamothe SM & Zhang S (2013). The serum‐ and glucocorticoid‐inducible kinases SGK1 and SGK3 regulate hERG channel expression via ubiquitin ligase Nedd4‐2 and GTPase Rab11. J Biol Chem 288, 15075–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen AP, Olesen SP, Grunnet M & Jespersen T (2008). Characterization of hERG1a and hERG1b potassium channels—a possible role for hERG1b in the IKr current. Pflugers Arch 456, 1137–1148. [DOI] [PubMed] [Google Scholar]
- Lees‐Miller JP, Duan Y, Teng GQ & Duff HJ (2000). Molecular determinant of high‐affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol Pharmacol 57, 367–374. [PubMed] [Google Scholar]
- Li J & Buchner J (2013). Structure, function and regulation of the hsp90 machinery. Biomed J 36, 106–117. [DOI] [PubMed] [Google Scholar]
- Li J, Soroka J & Buchner J. (2012). The Hsp90 chaperone machinery: conformational dynamics and regulation by co‐chaperones. Biochim Biophys Acta 1823, 624–635. [DOI] [PubMed] [Google Scholar]
- Li P, Ninomiya H, Kurata Y, Kato M, Miake J, Yamamoto Y, Igawa O, Nakai A, Higaki K, Toyoda F, Wu J, Horie M, Matsuura H, Yoshida A, Shirayoshi Y, Hiraoka M & Hisatome I (2011). Reciprocal control of hERG stability by Hsp70 and Hsc70 with implication for restoration of LQT2 mutant stability. Circ Res 108, 458–468. [DOI] [PubMed] [Google Scholar]
- Lounkine E, Keiser MJ, Whitebread S, Mikhailov D, Hamon J, Jenkins JL, Lavan P, Weber E, Doak AK, Cote S, Shoichet BK & Urban L (2012). Large‐scale prediction and testing of drug activity on side‐effect targets. Nature 486, 361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP (2013). Hsp70 chaperone dynamics and molecular mechanism. Trends Biochem Sci 38, 507–514. [DOI] [PubMed] [Google Scholar]
- Meacham GC, Patterson C, Zhang W, Younger JM & Cyr DM (2001). The Hsc70 co‐chaperone CHIP targets immature CFTR for proteasomal degradation. Nat Cell Biol 3, 100–105. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C & Sanguinetti MC (2000). A structural basis for drug‐induced long QT syndrome. Proc Natl Acad Sci USA 97, 12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais Cabral JH, Lee A, Cohen SL, Chait BT, Li M & Mackinnon R (1998). Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95, 649–655. [DOI] [PubMed] [Google Scholar]
- Morito D, Hirao K, Oda Y, Hosokawa N, Tokunaga F, Cyr DM, Tanaka K, Iwai K & Nagata K (2008). Gp78 cooperates with RMA1 in endoplasmic reticulum‐associated degradation of CFTRΔF508. Mol Biol Cell 19, 1328–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata S, Minami Y, Minami M, Chiba T & Tanaka K (2001). CHIP is a chaperone‐dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep 2, 1133–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC & Lukacs GL (2010). Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science 329, 805–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl LH & Prodromou C (2006). Structure and mechanism of the Hsp90 molecular chaperone machinery. Annu Rev Biochem 75, 271–294. [DOI] [PubMed] [Google Scholar]
- Perry M, Sachse FB & Sanguinetti MC (2007). Structural basis of action for a human ether‐a‐go‐go‐related gene 1 potassium channel activator. Proc Natl Acad Sci USA 104, 13827–13832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LB, Eskew JD, Vielhauer GA & Blagg BS (2012). The hERG channel is dependent upon the Hsp90α isoform for maturation and trafficking. Mol Pharm 9, 1841–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrecca K, Atanasiu R, Akhavan A & Shrier A (1999). N‐linked glycosylation sites determine HERG channel surface membrane expression. J Physiol 515, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phartiyal P, Sale H, Jones EM & Robertson GA (2008). Endoplasmic reticulum retention and rescue by heteromeric assembly regulate human ERG 1a/1b surface channel composition. J Biol Chem 283, 3702–3707. [DOI] [PubMed] [Google Scholar]
- Piper RC, Dikic I & Lukacs GL (2014). Ubiquitin‐dependent sorting in endocytosis. Cold Spring Harb Perspect Biol 6, a016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabeh WM, Bossard F, Xu H, Okiyoneda T, Bagdany M, Mulvihill CM, Du K, di Bernardo S, Liu Y, Konermann L, Roldan A & Lukacs GL (2012). Correction of both NBD1 energetics and domain interface is required to restore ΔF508 CFTR folding and function. Cell 148, 150–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajamani S, Eckhardt LL, Valdivia CR, Klemens CA, Gillman BM, Anderson CL, Holzem KM, Delisle BP, Anson BD, Makielski JC & January CT (2006). Drug‐induced long QT syndrome: hERG K+ channel block and disruption of protein trafficking by fluoxetine and norfluoxetine. Br J Pharmacol 149, 481–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch JN & Gestwicki JE (2014). Binding of human nucleotide exchange factors to heat shock protein 70 (Hsp70) generates functionally distinct complexes in vitro. J Biol Chem 289, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM (2004). Drug‐induced prolongation of the QT interval. N Engl J Med 350, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Chapula JA, Navarro‐Polanco RA, Culberson C, Chen J & Sanguinetti MC (2002). Molecular determinants of voltage‐dependent human ether‐a‐go‐go related gene (HERG) K+ channel block. J Biol Chem 277, 23587–23595. [DOI] [PubMed] [Google Scholar]
- Sandvig K, Pust S, Skotland T & van Deurs B (2011). Clathrin‐independent endocytosis: mechanisms and function. Curr Opin Cell Biol 23, 413–420. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC & Tristani‐Firouzi M (2006). hERG potassium channels and cardiac arrhythmia. Nature 440, 463–469. [DOI] [PubMed] [Google Scholar]
- Saxena A, Banasavadi‐Siddegowda YK, Fan Y, Bhattacharya S, Roy G, Giovannucci DR, Frizzell RA & Wang X (2012). Human heat shock protein 105/110 kDa (Hsp105/110) regulates biogenesis and quality control of misfolded cystic fibrosis transmembrane conductance regulator at multiple levels. J Biol Chem 287, 19158–19170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheufler C, Brinker A, Bourenkov G, Pegoraro S, Moroder L, Bartunik H, Hartl FU & Moarefi I (2000). Structure of TPR domain‐peptide complexes: critical elements in the assembly of the Hsp70‐Hsp90 multichaperone machine. Cell 101, 199–210. [DOI] [PubMed] [Google Scholar]
- Schwartz PJ, Stramba‐Badiale M, Crotti L, Pedrazzini M, Besana A, Bosi G, Gabbarini F, Goulene K, Insolia R, Mannarino S, Mosca F, Nespoli L, Rimini A, Rosati E, Salice P & Spazzolini C (2009). Prevalence of the congenital long‐QT syndrome. Circulation 120, 1761–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL & Weissman JS (2011). Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staudacher I, Wang L, Wan X, Obers S, Wenzel W, Tristram F, Koschny R, Staudacher K, Kisselbach J, Koelsch P, Schweizer PA, Katus HA, Ficker E & Thomas D (2011). hERG K+ channel‐associated cardiac effects of the antidepressant drug desipramine. Naunyn Schmiedebergs Arch Pharmacol 383, 119–139. [DOI] [PubMed] [Google Scholar]
- Sun F, Zhang R, Gong X, Geng X, Drain PF & Frizzell RA (2006). Derlin‐1 promotes the efficient degradation of the cystic fibrosis transmembrane conductance regulator (CFTR) and CFTR folding mutants. J Biol Chem 281, 36856–36863. [DOI] [PubMed] [Google Scholar]
- Sun T, Guo J, Shallow H, Yang T, Xu J, Li W, Hanson C, Wu JG, Li X, Massaeli H & Zhang S (2011). The role of monoubiquitination in endocytic degradation of human ether‐a‐go‐go‐related gene (hERG) channels under low K+ conditions. J Biol Chem 286, 6751–6759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taipale M, Jarosz DF & Lindquist S (2010). HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11, 515–528. [DOI] [PubMed] [Google Scholar]
- Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI & Lindquist S (2012). Quantitative analysis of HSP90‐client interactions reveals principles of substrate recognition. Cell 150, 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemasa H, Nagatomo T, Abe H, Kawakami K, Igarashi T, Tsurugi T, Kabashima N, Tamura M, Okazaki M, Delisle BP, January CT & Otsuji Y (2008). Coexistence of hERG current block and disruption of protein trafficking in ketoconazole‐induced long QT syndrome. Br J Pharmacol 153, 439–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B & Robertson GA (1995). HERG, a human inward rectifier in the voltage‐gated potassium channel family. Science 269, 92–95. [DOI] [PubMed] [Google Scholar]
- Tzankov S, Wong MJ, Shi K, Nassif C & Young JC (2008). Functional divergence between co‐chaperones of Hsc70. J Biol Chem 283, 27100–27109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenberg JI, Perry MD, Perrin MJ, Mann SA, Ke Y & Hill AP. (2012). hERG K+ channels: structure, function, and clinical significance. Physiol Rev 92, 1393–1478. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Torres AM, Campbell TJ & Kuchel PW (2004). The HERG K+ channel: progress in understanding the molecular basis of its unusual gating kinetics. Eur Biophys J 33, 89–97. [DOI] [PubMed] [Google Scholar]
- Veit G, Avramescu RG, Perdomo D, Phuan PW, Bagdany M, Apaja PM, Borot F, Szollosi D, Wu YS, Finkbeiner WE, Hegedus T, Verkman AS & Lukacs GL (2014). Some gating potentiators, including VX‐770, diminish ΔF508‐CFTR functional expression. Sci Transl Med 6, 246ra297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker VE, Atanasiu R, Lam H & Shrier A (2007). Co‐chaperone FKBP38 promotes HERG trafficking. J Biol Chem 282, 23509–23516. [DOI] [PubMed] [Google Scholar]
- Walker VE, Wong MJ, Atanasiu R, Hantouche C, Young JC & Shrier A (2010). Hsp40 chaperones promote degradation of the HERG potassium channel. J Biol Chem 285, 3319–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wible BA, Wan X & Ficker E (2007). Cardiac glycosides as novel inhibitors of human ether‐a‐go‐go‐related gene channel trafficking. J Pharmacol Exp Ther 320, 525–534. [DOI] [PubMed] [Google Scholar]
- Wang X, Venable J, LaPointe P, Hutt DM, Koulov AV, Coppinger J, Gurkan C, Kellner W, Matteson J, Plutner H, Riordan JR, Kelly JW, Yates JR 3rd & Balch WE (2006). Hsp90 cochaperone Aha1 downregulation rescues misfolding of CFTR in cystic fibrosis. Cell 127, 803–815. [DOI] [PubMed] [Google Scholar]
- Waring MJ, Arrowsmith J, Leach AR, Leeson PD, Mandrell S, Owen RM, Pairaudeau G, Pennie WD, Pickett SD, Wang J, Wallace O & Weir A (2015). An analysis of the attrition of drug candidates from four major pharmaceutical companies. Nat Rev Drug Discov 14, 475–486. [DOI] [PubMed] [Google Scholar]
- Xu Z, Page RC, Gomes MM, Kohli E, Nix JC, Herr AB, Patterson C & Misra S (2008). Structural basis of nucleotide exchange and client binding by the Hsp70 cochaperone Bag2. Nat Struct Mol Biol 15, 1309–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC (2014). The role of the cytosolic HSP70 chaperone system in diseases caused by misfolding and aberrant trafficking of ion channels. Disease Models Mech 7, 319–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Agashe VR, Siegers K & Hartl FU (2004). Pathways of chaperone‐mediated protein folding in the cytosol. Nat Rev Mol Cell Biol 5, 781–791. [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ & Hartl FU (2003). Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112, 41–50. [DOI] [PubMed] [Google Scholar]
- Younger JM, Chen L, Ren HY, Rosser MF, Turnbull EL, Fan CY, Patterson C & Cyr DM (2006). Sequential quality‐control checkpoints triage misfolded cystic fibrosis transmembrane conductance regulator. Cell 126, 571–582. [DOI] [PubMed] [Google Scholar]
- Younger JM, Ren HY, Chen L, Fan CY, Fields A, Patterson C & Cyr DM (2004). A foldable CFTRΔF508 biogenic intermediate accumulates upon inhibition of the Hsc70‐CHIP E3 ubiquitin ligase. J Cell Biol 167, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Ye B, Fan Z, Makielski JC, Robertson GA & January CT (1998). Properties of HERG channels stably expressed in HEK 293 cells studied at physiological temperature. Biophys J 74, 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]