

Abstract
Drugs used to treat cardiovascular disease as well as those used in the treatment of multiple other conditions can occasionally produce exaggerated prolongation of the QT interval on the electrocardiogram and the morphologically distinctive polymorphic ventricular tachycardia (‘torsades de pointes’). This syndrome of drug‐induced long QT syndrome has moved from an interesting academic exercise to become a key element in the development of any new drug entity. The prevailing view, which has driven both clinical care and drug regulation, holds that cardiac repolarization represents a balance between inward currents (primarily through calcium and sodium channels) and outward currents (primarily through rapid and slowed delayed rectifier potassium channels) and that block of the rapid delayed rectifier (I Kr) is the primary mechanism whereby drugs prolong individual action potentials, manifest on the surface electrocardiogram as QT interval prolongation. Such marked action potential prolongation in individual cardiac cells, in turn, is accompanied by arrhythmogenic afterdepolarizations thought to trigger torsades de pointes. This review describes the evidence in support of this construct, and describes the way in which clinical and whole heart experiments have informed molecular mechanisms and vice versa. New data that challenge these views and that may, as a result, lead to new clinical care and drug screening paradigms, are discussed.
A brief history: first associations between long QT and torsades de pointes
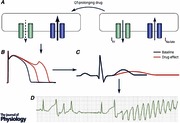
The problem of occasional syncope during therapy with the antiarrhythmic drug quinidine was recognized in the 1920s, and an association with a distinctive polymorphic ventricular tachycardia, subsequently termed ‘torsades de pointes’ (Dessertenne, 1966), was recognized in 1964 (Selzer & Wray, 1964). The 1950s and ’60s also saw the initial descriptions of the autosomal recessive and autosomal dominant forms of the congenital long QT syndrome (cLQTS), a familial disease characterized by QT interval prolongation, and occasionally sudden death with ventricular fibrillation following torsades de pointes as the commonest mechanism. During the 1970s and ’80s, multiple reports appeared of exaggerated QT interval prolongation and torsades de pointes in association with the use of a wide variety of therapeutics, not only those used in cardiac disease but also antibiotics, antipsychotics and other drug classes (see Table 1). A body of basic, translational, and clinical science now links drug actions at the molecular level to the arrhythmia (Abstract Figure), and these are reviewed here.
Table 1.
Therapeutic drug classes associated with torsades de pointes*
| Therapeutic class | Examples of ‘high risk’ drugs |
|---|---|
| Antiarrhythmics | Dofetilide |
| Ibutilide | |
| Quinidine | |
| Sotalol | |
| Antibiotics | Grepafloxacin*, * |
| Pentamidine | |
| Sparfloxacin*, * | |
| Anticancer agents | Arsenic trioxide |
| Vandetanib | |
| Antiemetics | Droperidol |
| Ondansetron | |
| Antihistamines | Astemizole*, * |
| Terfenadine*, * | |
| Antipsychotics | Haloperidol |
| Mesoridazine*, * | |
| Pimozide | |
| Thioridazine | |
| Gastric prokinetic agent | Cisapride*, * |
| Opiate antagonists | Levomethadyl*, * |
| Methadone |
*Not all members of a presented class share equal liability; generally, one or more drugs have especially high liability, and others have no liability. Specific lists are maintained at www.crediblemeds.org. **Withdrawn from the US market because of torsades de pointes liability.
The drug‐induced long QT syndrome (diLQTS) – the association of drug therapy, marked prolongation of the QT interval (generally > 500 ms, rate‐corrected), and torsades de pointes – affected neither clinical practice nor drug development until the early 1990s. At that time, a new, very popular non‐sedating antihistamine, terfenadine, was being considered for over‐the‐counter status when a single case report of torsades de pointes in association with its use was published (Monahan et al. 1990). Studies over the next few years elucidated the mechanism by demonstrating that terfenadine at therapeutic dosages in healthy subjects produces minimal prolongation of the QT interval, but that in overdose, in the presence of liver disease, or with co‐administration of drugs such as ketoconazole inhibiting terfenadine's extensive presystemic metabolism, plasma concentrations of the drug rise dramatically and marked QT prolongation occurs (Honig et al. 1993; Woosley et al. 1993). We now recognize that the antihistamine effects of the drug were attributable to its extensive pre‐systemic biotransformation by CYP3A4 to an active, non‐QT prolonging metabolite, fexofenadine, now marketed as Allegra. Terfenadine was not granted over‐the‐counter status and in fact with the development of fexofenadine as an antihistamine was withdrawn from the market. In the same era, another non‐sedating antihistamine, astemizole, and a gastric prokinetic agent, cisapride, were both withdrawn from the US market because of rare instances of torsades de pointes, often due to co‐administration of drugs that inhibited their metabolism. With these high profile adverse drug events, diLQTS attracted increased attention from both the practice community and drug developers and regulators (Roden, 2004). Over the past two decades, diLQTS and hepatotoxicity have been the most common reasons for drug withdrawal or relabelling.
QT‐prolonging antiarrhythmic drugs such as quinidine, dofetilide and sotalol cause torsades de pointes in 1–5% of exposed subjects (Abraham et al. 2015). By contrast, antibiotics, antipsychotics and other classes of ‘non‐cardiovascular drugs’ associated with diLQTS risk produce torsades de pointes at much lower frequencies (e.g.1/1,000,000 exposures). However, in each of these cases, the decision to prescribe the drug is predicated on the assumption that the risk is outweighed by the benefits. Thus, a drug uniquely effective in certain forms of promyelocytic leukaemia (arsenic trioxide) remains on the market despite a recognized diLQTS risk, whereas a drug used for nasal congestion (a non‐fatal condition for which many other alternatives are available) requires much less of a diLQTS signal for its withdrawal (Roden, 2004).
Clinical case series have identified risk factors for diLQTS that have, in turn, directed studies of basic mechanisms (Roden, 2006): these include underlying bradyarrhythmias or abrupt heart rate slowing (the ‘short–long–short’ cycle length changes; Fig. 1), female sex, hypokalaemia and hypomagnesemia, the presence of advanced underlying structural heart disease, recent conversion from atrial fibrillation, previously unrecognized congenital long QT syndrome, and polypharmacy, which may be especially important if multiple QT‐prolonging drugs are prescribed or if altered drug metabolism (as in the case of the ketoconazole–terfenadine interaction) ensues. The questions, which this review will address, are how to identify patients at risk for diLQTS and how to identify new drug entities, early in the development process, that also carry this risk.
Figure 1. Development of torsades de pointes in a patient with atrial fibrillation treated with sotalol .
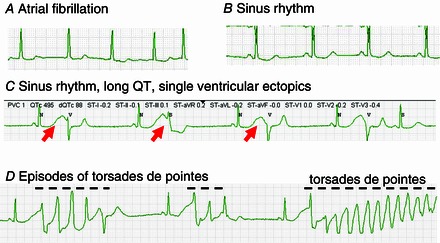
A, atrial fibrillation. B, with sotalol, the rhythm converts to sinus with a borderline prolonged QT interval (∼480 ms). C, several hours later, the QT interval after sinus beats is markedly prolonged (> 600 ms; red arrows), and single ectopic beats, possibly representing early afterdepolarization‐mediated automaticity (Abstract Figure B) are seen. D, this is followed within minutes by bursts of the polymorphic ventricular tachycardia torsades de pointes, indicated by the dashed lines.
Principles of repolarization in cardiomyocytes
The QT interval on the surface electrocardiogram indicates repolarization time in the ventricle, and first electrophysiological principles then dictate that prolongation of the QT interval must reflect prolongation of at least some action potentials within the ventricle. Such action potential prolongation, in turn, can arise either from decreased outward (repolarizing) currents or increased inward (depolarizing) currents. The QT interval is rate dependent so various formulae have been proposed to generate a rate‐corrected QT (QTc) to allow comparisons within individual (e.g. with drug exposure) or across populations. Distinguishing drug‐induced small changes in QT from those due to physiological fluctuation in QT (e.g. a function of posture, meals, exercise, time of day) can be problematic; further, the QT interval (however corrected) is longer in post‐pubertal women than it is in men. QTc values > 460–480 have been used to define long QTc intervals although this is also within the range of normal values seen across large populations. Values > 500 ms are definitely abnormal. In formal evaluations of drug effects on QTc described further below, values > 480, > 500, and drug‐induced changes in QTc > 30 ms or > 60 ms are often reported.
The major repolarizing current in the heart of large mammals (and not rodents) was first identified as a delayed rectifier potassium current, termed I K. Electrophysiological studies in the early 1990s identified two functionally distinct components: (1) a rapidly activating current (I Kr) sensitive to methanesulfonanilide blocking drugs and (2) a very slowly activating component (I Ks) strikingly enhanced by adrenergic activation (Sanguinetti & Jurkiewicz, 1990). It was genetic linkage analysis in large kindreds with cLQTS that identified the genes whose expression leads to I Kr and I Ks (Curran et al. 1995; Wang et al. 1996). The commonest form (type 1) of cLQTS is caused by mutations in KCNQ1, which encodes the pore–potassium channel subunit termed Kv7.1, which underlies I Ks. As with other voltage‐gated potassium channels, four subunits coassemble to form the pore‐forming structure. Further, in the case of KCNQ1, a function‐modifying subunit, encoded by KCNE1, is required to recapitulate the slow activation of I Ks seen in mammalian ventricular cells (Barhanin et al. 1996; Wang et al. 1996). The second commonest cause of cLQTS (type 2) is mutations in KCNH2 (also termed HERG) encoding the Kv11.1 potassium channel. Like Kv7.1, Kv11.1 subunits coassemble as tetramers, and while function‐modifying subunits have been described, it is not clear they are absolutely required to recapitulate I Kr. Together, mutations in genes whose expression leads to I Kr and I Ks account for 80–90% of cLQTS. Mutations in KCNQ1, KCNH2, or KCNE1 cause cLQTS by decreasing outward potassium current either by interfering with gating, or more commonly, decreasing channel cell surface expression. Type 3 cLQTS is caused by mutations in the cardiac sodium channel, encoded by SCN5A (Wang et al. 1995). Ordinarily, sodium channels open rapidly to initiate cardiac action potentials and then very rapidly (within milliseconds) inactivate. LQTS‐associated SCN5A mutations interfere with this fast inactivation process leading to a persistent inward current – sufficient to prolong action potentials – during the plateau of the action potential (Bennett et al. 1995). Mutations in at least 12 other genes have been associated with cLQTS in rare families. In some cases, the mutations decrease I Kr or I Ks or alter SCN5A fast inactivation, while in other cases, the functional defects involve other potassium channels (the inward rectifier channel or the acetylcholine gated potassium channel) or increase inward current through L‐type calcium channels (Schwartz et al. 2013).
The cloning of these channels has not only elucidated mechanisms in congenital and drug‐induced forms of the long QT syndromes, but has literally revolutionized our understanding of the molecular physiology of cardiac repolarization. Examples especially relevant to diLQTS include the following.
Kv11.1 is a major drug target in diLQTS. The vast majority of drugs prolonging the QT interval in humans block I Kr. Indeed, I Kr was initially described as the portion of I K sensitive to block by methanesulfonanilide drugs (Sanguinetti & Jurkiewicz, 1990), such as E4031, d‐sotalol and dofetilide, all potent I Kr blockers (and associated with diLQTS when used in patients). This observation, in turn, led to regulatory recommendations with respect to screening new drug entities for ‘HERG block’ (Food and Drug Administration, HHS, 2005 a). Most drugs block the channel by binding in the pore from within the cell interior. The observation that many drugs across therapeutic classes interact with the Kv11.1 channel has been attributed to two features of the pore structure, an unusually wide interior ‘mouth’ and multiple aromatic groups thought to enable π‐bonding to aromatic groups in methanesulfonanilides and other ‘HERG blockers’ (Mitcheson et al. 2000). In some cases, drug‐induced reduced I Kr has been attributed to reduced cell surface expression of the channel (Kuryshev et al. 2005).
Hypokalaemia prolongs QT and is a risk factor for diLQTS. The Nernst equation indicates that decreased extracellular potassium should increase outward potassium current and thus shorten QT intervals (and prevent diLQTS). However, hypokalaemia in patients prolongs QT and predisposes to diLQTS. An important underlying mechanism for this apparent paradox is that the amplitude of I Kr activated by depolarization to plateau potentials decreases as extracellular potassium is lowered. This unusual behaviour reflects very fast inactivation that open Kv11.1 channels undergo such that, with depolarizing pulses, channels are distributed between open and inactivated states. Decreasing extracellular potassium shifts this balance towards occupancy of inactivated states, and thus decreases overall repolarizing current (Yang et al. 1997). Further, drug block of open channels is enhanced at low extracellular potassium, thereby further enhancing diLQTS risk (Yang & Roden, 1996). As discussed further below, hypokalaemia may also prolong QT by activating CaM kinase and thereby increasing late sodium current (Pezhouman et al. 2015).
Intracellular signalling systems modulate the QT interval and its response to drugs. In human cells, I Ks is very small and enhanced by activators of protein kinase A such as catecholamines or isoproterenol (Marx et al. 2002). In patients with cLQTS due to KCNQ1 mutations, torsades de pointes often develops during adrenergic stimulation (e.g. with emotional or physical stress). With adrenergic stimulation, calcium currents are enhanced and thus the QT interval would ordinarily prolong markedly, especially at high heart rates driven by catecholamines. However, in normal individuals, the QT interval in fact shortens with exercise, thought to reflect enhanced I Ks blunting this increased L‐type calcium current. Patients with cLQTS due to KCNQ1 mutations may display normal or near normal QT intervals at rest, but marked prolongation during and especially after exercise (Schwartz et al. 2013). As described below, recent studies suggest that other signalling pathways may also strikingly modulate the diLQTS in vitro phenotype.
Action potential heterogeneity and afterdepolarizations
There is considerable variability in the shape and duration of action potentials in individual cardiac cells, presumably reflecting the specific repertoire of channels expressed (Antzelevitch, 2007). The extent to which such heterogeneities play a role in the normal ventricle, in which cells are well coupled to each other, is controversial (Opthof et al. 2007). When cardiac cells, notably from the conduction system, are exposed to conditions that produce torsades de pointes in patients (an I Kr‐blocking drug; low extracellular potassium; slow drive rates), action potentials are prolonged and frequently display early afterdepolarizations (EADs), discontinuities in the time course of normal repolarization (Roden & Hoffman, 1985). Triggered activity arising from single EADs likely initiates torsades de pointes and the arrhythmia may be maintained and propagated either by trains of EADs or by heterogeneity of cardiac action potentials leading to unstable reentrant ‘scrolls’ across the ventricular wall. The inward current underlying EADs and triggered upstrokes is carried by reactivated L‐type calcium channels (January & Riddle, 1989), sodium‐calcium exchanger (Wu et al. 1999), or possibly even the sodium channel, depending on their take‐off potential. A conventional view has made a clear distinction between EADs interrupting terminal repolarization at slow drive rates (and inhibited by L‐type calcium channel blockers) contrasting to delayed afterdepolarizations elicited by rapid drive rates in calcium‐overloaded preparations and unaffected by L‐type calcium channel blockers. However, it is probably an oversimplification to invoke such a clean distinction: preparations studied under highly arrhythmogenic conditions may display both delayed and early afterdepolarizations, and several lines of evidence have indicated that intracellular calcium overload predisposes to EADs when action potentials are prolonged.
Identifying patients at risk for diLQTS
Soon after KCNQ1, KCNH2 and SCN5A were identified as cLQTS disease genes, it became apparent that relatives of individuals with severe phenotypes could carry the same mutation but have normal QT intervals and no arrhythmias (Priori et al. 1999). Subsequently, patients were described with such ‘incompletely penetrant’ cLQTS but torsades de pointes on exposure to a QT‐prolonging drug (Donger et al. 1997). These observations, and the increasing recognition of the molecular complexity of normal cardiac repolarization, then led to the ‘reduced repolarization reserve’ concept of diLQTS risk (Roden, 1998). This framework proposes that normal repolarization is determined by multiple, somewhat redundant, mechanisms so lesions in any one of these may remain subclinical, i.e. the QT interval may remain near normal. However, challenge (e.g. by hypokalaemia or with a QT‐prolonging drug) may uncover such subclinical lesions and, in fact, lead to exaggerated action potential prolongation, exaggerated QT interval prolongation, EADs and torsades de pointes. For example, loss‐of‐function variants in KCNQ1 may lead to only very minimal action potential prolongation, because repolarization can still remain near normal because of a normal I Kr. However, administration of an I Kr blocking drug in this setting then leads to marked action potential prolongation because other mechanisms determining repolarization are insufficient to maintain normal action potential durations. This formulation is supported by reports of subclinical KCNQ1 mutations in patients with diLQTS and by in silico action potential reconstructions that demonstrate exposure of reduced repolarization reserve by administration of an I Kr blocker (Donger et al. 1997; Napolitano et al. 1997; Roden & Viswanathan, 2005). Another study compared the effects of challenge with the QT‐prolonging drug sotalol in patients who had experienced diLQTS (after the QT had returned to normal) and controls. In controls, the QTc was prolonged from 422 ± 17 to 450 ± 22 ms in controls while in the diLQTS group, the baseline QTc was slightly longer (434 ± 20 ms) but prolonged markedly to 541 ± 37 ms with drug challenge and 3/20 subjects redeveloped torsades de pointes (Kaab et al. 2003). This result provides strong support for the idea that some subjects remain susceptible to diLQTS even with normal baseline QTc values.
Molecular genetic studies of diLQTS
The question of whether variants predisposing to diLQTS can be identified to prevent exposure of high‐risk subjects has been addressed by a series of studies using contemporary genotyping and sequencing approaches. diLQTS is a rare adverse drug event so accrual of cases has relied on development of international consortia focused on common case definitions (Behr et al. 2013 a). In an initial study of 176 European‐descent subjects with diLQTS, 207 subjects with minimal QTc interval prolongation (< 50 ms) during treatment with a QT‐prolonging antiarrhythmic, and 837 population controls (all of European descent), the KCNE1 single nucleotide polymorphism (SNP) leading to D85N was identified in 8.6% of cases, and 1.8–2.9% of controls (Kaab et al. 2012). This variant is expected to reduce I Ks slightly, and thus, decrease ‘repolarization reserve’, as described above.
The genome‐wide association study (GWAS) paradigm has been applied to study of the QT interval and to diLQTS. Variability in the QT interval itself has been studied in over 100,000 subjects using this approach, and multiple genomic loci have been identified at which variants confer highly statistically significant (e.g. P < 10−200) but very modest influences (odds ratio (OR) < 2; QT variance < 5 ms) on QT (Arking et al. 2014). Interestingly, many of these are in cLQTS disease gene loci, indicating that common variants at these sites modestly modulate the QT interval while rare variants confer the cLQTS phenotype. One of the genes with the strongest signal in GWAS analysis of variability in the QT interval is NOS1AP, encoding an ancillary protein thought to modify function of neuronal nitric oxide synthase. The exact role of NOS1AP in cardiac electrophysiology remains incompletely defined, although NOS1AP variants have been associated with increased risk of diLQTS associated with amiodarone (Jamshidi et al. 2012), with penetrance in the congenital LQTS (Crotti et al. 2009; Tomas et al. 2010), and with risk of sudden cardiac death in the general population (Kao et al. 2009).
A GWAS of 216 diLQTS cases and 771 controls, all drawn from north‐western Europe failed to implicate any SNP as a risk factor at genome‐wide significance (Behr et al. 2013 b). While the sample size is small compared to contemporary GWA studies, there was 80% power to detect a common SNP at minor allele frequency > 10% with OR > 2.7.
Sequencing common cLQTS disease genes has identified potentially causative cLQTS mutations in 5–20% of patients with diLQTS (Yang et al. 2002; Paulussen et al. 2004; Itoh et al. 2009). Most recently, exome sequencing in 65 diLQTS patients and 148 drug‐exposed controls (who did not develop QT interval prolongation > 50 ms) reported significant associations between variants in KCNE1 (as discussed above) and in ACN9, a gene involved in gluconeogenesis not known to play a role in cardiac electrophysiology (Weeke et al. 2014). Rare non‐synonymous variants in a predefined set of seven cLQTS potassium channel disease genes were found in 37% of diLQTS cases compared to 21% of controls. Taken together, these studies continue to provide support for the idea that diLQTS can arise in patients with unrecognized cLQTS, but also highlight the problem that at this point a simple genetic ‘cause’ for diLQTS remains undefined in the majority of individual patients.
Identifying drugs at risk
The identification of conditions under which unusually high terfenadine concentrations could arise, and contribute to torsades de pointes, led to a number of controlled studies of terfenadine's influence on the QT interval. In one study, the effects of terfenadine alone and terfenadine plus ketoconazole (a potent CYP3A4 inhibitor) were examined in six healthy medication free normal volunteers. In the absence of ketoconazole, terfenadine was detectable at the lower limit of assay sensitivity in one subject (Honig et al. 1993). By contrast, terfenadine was readily detectable in five out of six subjects during ketoconazole coadministration. Importantly, baseline QTc was 408 ± 8 ms and prolonged to 416 ± 6 ms during terfenadine alone and to 490 ± 16 ms during coadministration of ketoconazole. In fact, in four out of six subjects, the study was terminated early because of marked QTc interval prolongation so this is an underestimate of the true steady state effect. In another study, 28 normal healthy volunteers and 28 patients with stable heart disease received a normal dose (60 mg) or a high dose (180 mg) of terfenadine (Pratt et al. 1996). In the normal subjects receiving the normal doses, the mean QTc interval prolongation was 6 ms, and was higher (12 ms) in subjects with heart disease. In both groups exposed to the higher dose, the extent of QTc prolongation was greater (19 and 26 ms, respectively). The original intent of the study was also to evaluate the effects of a very high dose (300 mg), but that arm of the study was stopped early when a subject with heart disease died suddenly after 4 days; while the rhythm was not monitored at the time of death, diLQTS was suspected.
These early findings have led to regulatory recommendations to detect a ‘QT signal’ early during drug development and thus prevent the problem of drugs such as terfenadine reaching the market (Food and Drug Administration, HHS, 2005 a,b). The recommendations include characterization of drug effects on individual ion currents (notably I Kr) as well as conduct of a ‘thorough QT (TQT) study’ in which normal volunteers (or patients) are exposed to a therapeutic dose of a drug as well as, if feasible, a supratherapeutic dose or a therapeutic dose with a metabolic inhibitor. In such studies, it is standard practice to include a ‘positive control’ arm, generally the antibiotic moxifloxacin, which produces a predictable 10–15 ms QTc prolongation and thus can be used to establish that the QT reading method is accurate (Bloomfield et al. 2008). Interestingly, moxifloxacin is an I Kr blocker and has been associated with torsades de pointes (Altin et al. 2007), but only very rarely, probably less than 1 in 100,000 (Frothingham, 2001; Haverkamp et al. 2012). The TQT study is a relatively expensive enterprise and can only be undertaken after a therapeutic dose of a drug has been defined, i.e. relatively late during the drug development process. A change in QTc as a function of time compared to change in QTc after administration of a placebo (the ΔΔQTc) of < 10 ms is generally taken to indicate a ‘negative’ TQT study, i.e. one that suggests the drug is low risk for producing diLQTS.
Challenging current paradigms
The awkwardness of the timing of the TQT study as well as its reliance on a relatively crude metric of cardiac repolarization (the QTc) has led to proposals for other methods, or combinations of novel methods (Darpo et al. 2014, 2015), to screen for high risk drug candidate molecules earlier in the development process. One is the use of cardiomyocytes derived from induced pluripotent stem cells (iPSCs), in which exaggerated responses to challenge with I Kr blocking drugs have been seen in the presence of congenital LQTS mutations (Liang et al. 2013). A second is to incorporate increasingly sophisticated in silico models to predict individual current changes that may or may not be especially arrhythmogenic (Sarkar & Sobie, 2011). A third method is to examine not simply the QTc, but multiple other electrocardiographic parameters to distinguish signals produced by highly arrhythmogenic drugs versus drugs that may prolong the QT but have less arrhythmogenic potential (Johannesen et al. 2014). For example, the antianginal ranolazine is known to block late sodium current and to block I Kr, and to produce modest QTc prolongation in some patients. However, torsades de pointes has not been reported with this drug, and in fact, some preclinical and clinical evidence suggest it may be useful as a treatment for some cases of torsades de pointes (Antzelevitch et al. 2011). Initial reports suggest that ranolazine may generate qualitatively different ECG changes beyond simple QTc prolongation, such as T morphological changes, from more arrhythmogenic agents such as dofetilide. The TQT study is often performed late during drug development, after the doses of drug to be used clinically have been established (Vicente et al. 2015).
A new signalling pathway in QT regulation?
In 2012, Lu and colleagues reported that acute exposure to the epidermal growth factor receptor (EGFR) blocker nilotinib, which is known to prolong the QT interval, did not affect canine ventricular cardiomyocyte action potentials (Lu et al. 2012). However, with prolonged (hours) exposure to the drug, marked action potential prolongation was seen and multiple electrophysiological effects documented; these included decreased cell surface expression of ion channels conducting both inward and outward current, as well as a striking increase in late sodium current. The increase in late sodium current was rapidly reversed when phosphatidylinositol‐3,4,5‐trisphosphate (PIP3), a downstream effector of phosphatidylinositol‐4,5‐bisphosphate 3‐kinase (PI3K), was included in the pipette (i.e. the intracellular solution perfusing the cells). This finding suggested that nilotinib block of EGFR led to PI3K inhibition with decreased PIP3, and that this decrease, in turn, generated a large inward sodium current, prolonging action potentials. We have gone on to examine the effects of drugs previously thought to be ‘pure’ I Kr blockers, such as dofetilide, moxifloxacin and others, that had all been associated with diLQTS but at very different frequencies across the population (Yang et al. 2014). We found that dofetilide markedly prolongs action potentials in adult mouse cardiomyocytes (a preparation in which I Kr is absent) after hours of exposure, and this was reversed by pipette PIP3. By contrast, no such effect was seen with moxifloxacin. These effects on action potential were accompanied by generation of early afterdepolarizations (EADs) and delayed afterdepolarizations (DADs), which were also reversed by pipette PIP3, and these effects were also seen in human iPSC‐derived cardiomyocytes. In other work, decreased PI3 kinase signalling has also been documented in mouse models of diabetes and may, therefore, explain the clinical observation that diabetics have longer QT intervals than normal subjects (Lu et al. 2013); it appears that the α‐isoform of PI3K is responsible (Lu et al. 2012; Yang et al. 2015), but the details of how this wide range of drugs interact with the PI3K pathway remain to be worked out. The complexities of QT regulation under pathological conditions is further illustrated by a recent study that showed that hypokalaemia activates CaM kinase, which in turns increases late sodium current to prolong QT and to generate EADs and ventricular fibrillation (Pezhouman et al. 2015). The identification of new signalling pathways modulating cardiac electrical behaviour holds the promise of leading to new paradigms in both drug screening and identification of individual patients at risk for this adverse drug effect. One of the interesting outstanding questions in studies of drug‐induced (and of congenital) long QT syndromes is the failure of the paradigm illustrated in the Abstract Figure to explain why most heartbeats in affected patients do not elicit torsades de pointes; it is conceivable that variable activity of these or other signalling pathways (e.g. autonomic tone) confers greater or lesser risk for each heartbeat.
Summary
This review has emphasized the way in which clinical science can pose important mechanistic questions at the fundamental molecular and genetic levels and how this information can ultimately feed back to the clinic. Many clinical observations still remain incompletely explained, and challenges remain, such as explaining the female preponderance of diLQTS, understanding the interesting period of increased susceptibility in the hours following conversion of atrial fibrillation to sinus rhythm, and the way to identify subjects at increased risk. Further, mechanistic work has also raised new questions: what are the causes of the distinct morphology in torsades de pointes? How do diverse structures such as nilotinib and dofetilide interact with PI3K signalling to generate late current? How can iPSC‐derived cardiomyocytes be deployed efficiently to screen new drug entities? Ongoing studies in these areas should improve prediction of diLQTS in individual patients and in new drug candidates.
Additional information
Competing interests
None.
Funding
The author's work was supported by grants from the US National Institutes of Health (GM115305 and HL49989).
Biography
Dan Roden is Professor of Medicine and Pharmacology, and Biomedical Informatics, Director of the Oates Institute for Experimental Therapeutics, and Assistant Vice‐Chancellor for Personalized Medicine at Vanderbilt University School of Medicine. He received his medical degree and training in Internal Medicine from McGill University. He then went to Vanderbilt where he trained in Clinical Pharmacology and Cardiology, and has been a faculty member there since. His initial career focus – which he has maintained – was studies of the clinical, genetic, cellular and molecular basis of arrhythmia susceptibility and variability responses to arrhythmia therapies. In particular, he proposed the term ‘reduced repolarization reserve’ to describe a framework for analysing the way in which multiple components of the complex repolarization process interact to culminate in long QT‐related arrhythmia. He has created and led international consortia analysing genomic risk for arrhythmias. In addition to studies of mechanisms underlying QT interval prolongation and the functional consequences of ion channel mutations in the whole heart, his laboratory is using large electronic health records as tools for discovering new genotype–phenotype associations across the spectrum of human traits, and for implementing genomic variants to personalize care.

This review was presented at the symposium “Cardiac Arrhythmias: Challenges for Diagnosis and Treatment. A symposium in honour of George Ralph Mines (1886‐1914)”, which took place at McGill University, Montreal, QC, Canada, between 6–7 November 2014.
References
- Abraham JM, Saliba WI, Vekstein C, Lawrence D, Bhargava M, Bassiouny M, Janiszewski D, Lindsay BD, Militello M, Nissen SE, Poe S, Tanaka‐Esposito C, Wolski K & Wilkoff BL (2015). Safety of oral dofetilide for rhythm control of atrial fibrillation and atrial flutter. Circ Arrhythm Electrophysiol 8, 772–776. [DOI] [PubMed] [Google Scholar]
- Altin T, Ozcan O, Turhan S, Ongun OA, Akyurek O, Karaoguz R & Guldal M (2007). Torsade de pointes associated with moxifloxacin: a rare but potentially fatal adverse event. Can J Cardiol 23, 907–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C (2007). Role of spatial dispersion of repolarization in inherited and acquired sudden cardiac death syndromes. Am J Physiol Heart Circ Physiol 293, H2024–H2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C, Burashnikov A, Sicouri S & Belardinelli L (2011). Electrophysiologic basis for the antiarrhythmic actions of ranolazine. Heart Rhythm 8, 1281–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, Johnson AD, Lundby A, Gudbjartsson DF, Noseworthy PA, Eijgelsheim M, Bradford Y, Tarasov KV, Dorr M, Muller‐Nurasyid M, Lahtinen AM, Nolte IM, Smith AV, Bis JC, Isaacs A, Newhouse SJ, Evans DS, Post WS, Waggott D, Lyytikainen LP, Hicks AA, Eisele L, Ellinghaus D, Hayward C, Navarro P, Ulivi S, Tanaka T, Tester DJ, Chatel S, Gustafsson S, Kumari M, Morris RW, Naluai AT, Padmanabhan S, Kluttig A, Strohmer B, Panayiotou AG, Torres M, Knoflach M, Hubacek JA, Slowikowski K, Raychaudhuri S, Kumar RD, Harris TB, Launer LJ, Shuldiner AR, Alonso A, Bader JS, Ehret G, Huang H, Kao WH, Strait JB, Macfarlane PW, Brown M, Caulfield MJ, Samani NJ, Kronenberg F, Willeit J, Smith JG, Greiser KH, Meyer Zu Schwabedissen H, Werdan K, Carella M, Zelante L, Heckbert SR, Psaty BM, Rotter JI, Kolcic I, Polasek O, Wright AF, Griffin M, Daly MJ, Arnar DO, Holm H, Thorsteinsdottir U, Denny JC, Roden DM, Zuvich RL, Emilsson V, Plump AS, Larson MG, O'Donnell CJ, Yin X, Bobbo M, D'Adamo AP, Iorio A, Sinagra G, Carracedo A, Cummings SR, Nalls MA, Jula A, Kontula KK, Marjamaa A, Oikarinen L, Perola M, Porthan K, Erbel R, Hoffmann P, Jockel KH, Kalsch H, Nothen MM, den Hoed M, Loos RJ, Thelle DS, Gieger C, Meitinger T, Perz S, Peters A, Prucha H, Sinner MF, Waldenberger M, de Boer RA, Franke L, van der Vleuten PA, Beckmann BM, Martens E, Bardai A, Hofman N, Wilde AA, Behr ER, Dalageorgou C, Giudicessi JR, Medeiros‐Domingo A, Barc J, Kyndt F, Probst V, Ghidoni A, Insolia R, Hamilton RM, Scherer SW, Brandimarto J, Margulies K, Moravec CE, Greco MF, Fuchsberger C, O'Connell JR, Lee WK, Watt GC, Campbell H, Wild SH, El Mokhtari NE, Frey N, Asselbergs FW, Mateo Leach I, Navis G, van den Berg MP, van Veldhuisen DJ, Kellis M, Krijthe BP, Franco OH, Hofman A, Kors JA, Uitterlinden AG, Witteman JC, Kedenko L, Lamina C, Oostra BA, Abecasis GR, Lakatta EG, Mulas A, Orru M, Schlessinger D, Uda M, Markus MR, Volker U, Snieder H, Spector TD, Arnlov J, Lind L, Sundstrom J, Syvanen AC, Kivimaki M, Kahonen M, Mononen N, Raitakari OT, Viikari JS, Adamkova V, Kiechl S, Brion M, Nicolaides AN, Paulweber B, Haerting J, Dominiczak AF, Nyberg F, Whincup PH, Hingorani AD, Schott JJ, Bezzina CR, Ingelsson E, Ferrucci L, Gasparini P, Wilson JF, Rudan I, Franke A, Muhleisen TW, Pramstaller PP, Lehtimaki TJ, Paterson AD, Parsa A, Liu Y, van Duijn CM, Siscovick DS, Gudnason V, Jamshidi Y, Salomaa V, Felix SB, Sanna S, Ritchie MD, Stricker BH, Stefansson K, Boyer LA, Cappola TP, Olsen JV, Lage K, Schwartz PJ, Kaab S, Chakravarti A, Ackerman MJ, Pfeufer A, de Bakker PI & Newton‐Cheh C (2014). Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nat Genet 46, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M & Romey G (1996). KvLQT1 and IsK (minK) proteins associate to form the I Ks cardiac potassium current. Nature 384, 78–80. [DOI] [PubMed] [Google Scholar]
- Behr ER, January C, Schulze‐Bahr E, Grace AA, Kaab S, Fiszman M, Gathers S, Buckman S, Youssef A, Pirmohamed M & Roden D (2013. a). The International Serious Adverse Events Consortium (iSAEC) phenotype standardization project for drug‐induced torsades de pointes. Eur Heart J 34, 1958–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behr ER, Ritchie MD, Tanaka T, Kääb S, Crawford DC, Nicoletti P, Floratos A, Sinner MF, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze‐Bahr E, Zumhagen S, Guicheney P, Bishopric NH, Marshall V, Shakir S, Dalageorgou C, Bevan S, Jamshidi Y, Bastiaenen R, Myerburg RJ, Schott JJ, Camm AJ, Steinbeck G, Norris K, Altman RB, Tatonetti N, Jeffery S, Kubo M, Nakamura Y, Shen Y, George AL Jr & Roden DM (2013. b). Genome wide analysis of drug‐induced Torsades de Pointes: lack of common variants with large effect sizes. PloS One 8, e78511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett PB, Yazawa K, Makita N & George AL Jr (1995). Molecular mechanism for an inherited cardiac arrhythmia. Nature 376, 683–685. [DOI] [PubMed] [Google Scholar]
- Bloomfield DM, Kost JT, Ghosh K, Hreniuk D, Hickey LA, Guitierrez MJ, Gottesdiener K & Wagner JA (2008). The effect of moxifloxacin on QTc and implications for the design of thorough QT studies. Clin Pharmacol Ther 84, 475–480. [DOI] [PubMed] [Google Scholar]
- Crotti L, Monti MC, Insolia R, Peljto A, Goosen A, Brink PA, Greenberg DA, Schwartz PJ & George AL Jr (2009). NOS1AP is a genetic modifier of the long‐QT syndrome. Circulation 120, 1657–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED & Keating MT (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80, 795–803. [DOI] [PubMed] [Google Scholar]
- Darpo B, Garnett C, Benson CT, Keirns J, Leishman D, Malik M, Mehrotra N, Prasad K, Riley S, Rodriguez I, Sager P, Sarapa N & Wallis R (2014). Cardiac Safety Research Consortium: Can the thorough QT/QTc study be replaced by early QT assessment in routine clinical pharmacology studies? Scientific update and a research proposal for a path forward. Am Heart J 168, 262–272. [DOI] [PubMed] [Google Scholar]
- Darpo B, Garnett C, Keirns J & Stockbridge N (2015). Implications of the IQ‐CSRC prospective study: time to revise ICH E14. Drug Saf 38, 773–780. [DOI] [PubMed] [Google Scholar]
- Dessertenne F (1966). La tachycardie ventriculaire à deux foyers opposés variables. [Ventricular tachycardia with 2 variable opposing foci]. Arch Mal Coeur 59, 263–272. [PubMed] [Google Scholar]
- Donger C, Denjoy I, Berthet M, Neyroud N, Cruaud C, Bennaceur M, Chivoret G, Schwartz K, Coumel P & Guicheney P (1997). KVLQT1 C‐terminal missense mutation causes a forme fruste long‐QT syndrome. Circulation 96, 2778–2781. [DOI] [PubMed] [Google Scholar]
- Food and Drug Administration, HHS (2005. a). International Conference on Harmonisation: guidance on S7B Nonclinical Evaluation of the Potential for Delayed Ventricular Repolarization (QT Interval Prolongation) by Human Pharmaceuticals; availability. Notice. Fed Regist 70, 61133–61134. [PubMed] [Google Scholar]
- Food and Drug Administration, HHS (2005. b). International Conference on Harmonisation: guidance on E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs; availability. Notice. Fed Regist 70, 61134–61135. [PubMed] [Google Scholar]
- Frothingham R (2001). Rates of torsades de pointes associated with ciprofloxacin, ofloxacin, levofloxacin, gatifloxacin, and moxifloxacin. Pharmacotherapy 21, 1468–1472. [DOI] [PubMed] [Google Scholar]
- Haverkamp W, Kruesmann F, Fritsch A, van Veenhuyzen D & Arvis P (2012). Update on the cardiac safety of moxifloxacin. Curr Drug Saf 7, 149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig PK, Wortham DC, Zamani K, Conner DP, Mullin JC & Cantilena LR (1993). Terfenadine‐ketoconazole interaction. Pharmacokinetic and electrocardiographic consequences. JAMA 269, 1513–1518. [PubMed] [Google Scholar]
- Itoh H, Sakaguchi T, Ding WG, Watanabe E, Watanabe I, Nishio Y, Makiyama T, Ohno S, Akao M, Higashi Y, Zenda N, Kubota T, Mori C, Okajima K, Haruna T, Miyamoto A, Kawamura M, Ishida K, Nagaoka I, Oka Y, Nakazawa Y, Yao T, Jo H, Sugimoto Y, Ashihara T, Hayashi H, Ito M, Imoto K, Matsuura H & Horie M (2009). Latent genetic backgrounds and molecular pathogenesis in drug‐induced long QT syndrome. Circ Arrhythm Electrophysiol 2, 511–523. [DOI] [PubMed] [Google Scholar]
- Jamshidi Y, Nolte IM, Dalageorgou C, Zheng D, Johnson T, Bastiaenen R, Ruddy S, Talbott D, Norris KP, Snieder H, George AL Jr, Marshall V, Shakir S, Kannankeril PJ, Munroe PB, Camm AJ, Jeffery S, Roden DM & Behr ER (2012). Common variation in the NOS1AP gene is associated with drug‐induced QT prolongation and ventricular arrhythmia. J Am Coll Cardiol 60, 841–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- January CT & Riddle JM (1989). Early afterdepolarizations: Mechanism of induction and block: A role for L‐type Ca2+ current. Circ Res 64, 977–990. [DOI] [PubMed] [Google Scholar]
- Johannesen L, Vicente J, Gray RA, Galeotti L, Loring Z, Garnett CE, Florian J, Ugander M, Stockbridge N & Strauss DG (2014). Improving the assessment of heart toxicity for all new drugs through translational regulatory science. Clin Pharmacol Ther 95, 501–508. [DOI] [PubMed] [Google Scholar]
- Kaab S, Crawford DC, Sinner MF, Behr ER, Kannankeril PJ, Wilde AA, Bezzina CR, Schulze‐Bahr E, Guicheney P, Bishopric NH, Myerburg RJ, Schott JJ, Pfeufer A, Beckmann BM, Martens E, Zhang T, Stallmeyer B, Zumhagen S, Denjoy I, Bardai A, Van Gelder IC, Jamshidi Y, Dalageorgou C, Marshall V, Jeffery S, Shakir S, Camm AJ, Steinbeck G, Perz S, Lichtner P, Meitinger T, Peters A, Wichmann HE, Ingram C, Bradford Y, Carter S, Norris K, Ritchie MD, George AL Jr & Roden DM (2012). A large candidate gene survey identifies the KCNE1 D85N polymorphism as a possible modulator of drug‐induced torsades de pointes. Circ Cardiovasc Genet 5, 91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaab S, Hinterseer M, Nabauer M & Steinbeck G (2003). Sotalol testing unmasks altered repolarization in patients with suspected acquired long‐QT‐syndrome–a case‐control pilot study using i.v. sotalol. Eur Heart J 24, 649–657. [DOI] [PubMed] [Google Scholar]
- Kao WH, Arking DE, Post W, Rea TD, Sotoodehnia N, Prineas RJ, Bishe B, Doan BQ, Boerwinkle E, Psaty BM, Tomaselli GF, Coresh J, Siscovick DS, Marban E, Spooner PM, Burke GL & Chakravarti A (2009). Genetic variations in nitric oxide synthase 1 adaptor protein are associated with sudden cardiac death in US white community‐based populations. Circulation 119, 940–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryshev YA, Ficker E, Wang L, Hawryluk P, Dennis AT, Wible BA, Brown AM, Kang J, Chen XL, Sawamura K, Reynolds W & Rampe D (2005). Pentamidine‐induced long QT syndrome and block of hERG trafficking. J Pharmacol Exp Ther 312, 316–323. [DOI] [PubMed] [Google Scholar]
- Liang P, Lan F, Lee AS, Gong T, Sanchez‐Freire V, Wang Y, Diecke S, Sallam K, Knowles JW, Wang PJ, Nguyen PK, Bers DM, Robbins RC & Wu JC (2013). Drug screening using a library of human induced pluripotent stem cell–derived cardiomyocytes reveals disease‐specific patterns of cardiotoxicity. Circulation 127, 1677–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Jiang YP, Wu CY, Ballou LM, Liu S, Carpenter ES, Rosen MR, Cohen IS & Lin RZ (2013). Increased persistent sodium current due to decreased PI3K signaling contributes to QT prolongation in the diabetic heart. Diabetes 62, 4257–4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Wu CY, Jiang YP, Ballou LM, Clausen C, Cohen IS & Lin RZ (2012). Suppression of phosphoinositide 3‐kinase signaling and alteration of multiple ion currents in drug‐induced long QT syndrome. Sci Transl Med 4, 131ra150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR & Kass RS (2002). Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1‐KCNE1 potassium channel. Science 295, 496–499. [DOI] [PubMed] [Google Scholar]
- Mitcheson JS, Chen J, Lin M, Culberson C & Sanguinetti MC (2000). A structural basis for drug‐induced long QT syndrome. Proc Natl Acad Sci USA 97, 12329–12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monahan BP, Ferguson CL, Killeavy ES, Lloyd BK, Troy J & Cantilena LR Jr (1990). Torsades de pointes occurring in association with terfenadine use. JAMA 264, 2788–2790. [PubMed] [Google Scholar]
- Napolitano C, Priori SG, Schwartz PJ, Cantu F, Paganini V, Matteo PS, de Fusco M, Pinnavaia A, Aquaro G & Casari G (1997). Identification of a long QT syndrome molecular defect in drug‐induced torsades de pointes. Circulation 96, I‐211. [Google Scholar]
- Opthof T, Coronel R, Wilms‐Schopman FJ, Plotnikov AN, Shlapakova IN, Danilo P Jr, Rosen MR & Janse MJ (2007). Dispersion of repolarization in canine ventricle and the electrocardiographic T wave: T(p‐e) interval does not reflect transmural dispersion. Heart Rhythm 4, 341–348. [DOI] [PubMed] [Google Scholar]
- Paulussen AD, Gilissen RA, Armstrong M, Doevendans PA, Verhasselt P, Smeets HJ, Schulze‐Bahr E, Haverkamp W, Breithardt G, Cohen N & Aerssens J (2004). Genetic variations of KCNQ1, KCNH2, SCN5A, KCNE1, and KCNE2 in drug‐induced long QT syndrome patients. J Mol Med 82, 182–188. [DOI] [PubMed] [Google Scholar]
- Pezhouman A, Singh N, Song Z, Nivala M, Eskandari A, Cao H, Bapat A, Ko CY, Nguyen TP, Qu Z, Karagueuzian HS & Weiss JN (2015). Molecular basis of hypokalemia‐induced ventricular fibrillation. Circulation 132, 1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt CM, Ruberg S, Morganroth J, McNutt B, Woodward J, Harris S, Ruskin J & Moye L (1996). Dose‐response relation between terfenadine (Seldane) and the QTc interval on the scalar electrocardiogram: distinguishing a drug effect from spontaneous variability. Am Heart J 131, 472–480. [DOI] [PubMed] [Google Scholar]
- Priori SG, Napolitano C & Schwartz PJ (1999). Low penetrance in the long‐QT syndrome: clinical impact. Circulation 99, 529–533. [DOI] [PubMed] [Google Scholar]
- Roden DM (1998). Taking the idio out of idiosyncratic – predicting torsades de pointes. Pacing Clin Electrophysiol 21, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Roden DM (2004). Drug‐induced prolongation of the QT interval. N Engl J Med 350, 1013–1022. [DOI] [PubMed] [Google Scholar]
- Roden DM (2006). Long QT syndrome: reduced repolarization reserve and the genetic link. J Intern Med 259, 59–69. [DOI] [PubMed] [Google Scholar]
- Roden DM & Hoffman BF (1985). Action potential prolongation and induction of abnormal automaticity by low quinidine concentrations in canine Purkinje fibers. Relationship to potassium and cycle length. Circ Res 56, 857–867. [DOI] [PubMed] [Google Scholar]
- Roden DM & Viswanathan PC (2005). Genetics of acquired long QT syndrome. J Clin Invest 115, 2025–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC & Jurkiewicz NK (1990). Two components of cardiac delayed rectifier K+ current: Differential sensitivity to block by class III antiarrhythmic agents. J Gen Physiol 96, 195–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar AX & Sobie EA (2011). Quantification of repolarization reserve to understand interpatient variability in the response to proarrhythmic drugs: A computational analysis. Heart Rhythm 8, 1749–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz PJ, Ackerman MJ, George AL Jr & Wilde AAM (2013). Impact of genetics on the clinical management of channelopathies. J Am Coll Cardiol 62, 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer A & Wray HW (1964). Quinidine syncope, paroxysmal ventricular fibrillations occurring during treatment of chronic atrial arrhythmias. Circulation 30, 17–26. [DOI] [PubMed] [Google Scholar]
- Tomas M, Napolitano C, De Giuli L, Bloise R, Subirana I, Malovini A, Bellazzi R, Arking DE, Marban E, Chakravarti A, Spooner PM & Priori SG (2010). Polymorphisms in the NOS1AP gene modulate QT interval duration and risk of arrhythmias in the long QT syndrome. J Am Coll Cardiol 55, 2745–2752. [DOI] [PubMed] [Google Scholar]
- Vicente J, Johannesen L, Mason JW, Crumb WJ, Pueyo E, Stockbridge N & Strauss DG (2015). Comprehensive T wave morphology assessment in a randomized clinical study of dofetilide, quinidine, ranolazine, and verapamil. J Am Heart Assoc 4, e001615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD & Keating MT (1996). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 12, 17–23. [DOI] [PubMed] [Google Scholar]
- Wang Q, Shen J, Splawski I, Atkinson D, Li Z, Robinson JL, Moss AJ, Towbin JA & Keating MT (1995). SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80, 805–811. [DOI] [PubMed] [Google Scholar]
- Weeke P, Mosley JD, Hanna D, Delaney JT, Shaffer C, Wells QS, Van Driest S, Karnes JH, Ingram C, Guo Y, Shyr Y, Norris K, Kannankeril PJ, Ramirez AH, Smith JD, Mardis ER, Nickerson D, George AL Jr & Roden DM (2014). Exome sequencing implicates an increased burden of rare potassium channel variants in the risk of drug‐induced long QT interval syndrome. J Am Coll Cardiol 63, 1430–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woosley RL, Chen Y, Freiman JP & Gillis RA (1993). Mechanism of the cardiotoxic actions of terfenadine. JAMA 269, 1532–1536. [PubMed] [Google Scholar]
- Wu Y, Roden DM & Anderson ME (1999). Calmodulin kinase inhibition prevents development of the arrhythmogenic transient inward current. Circ Res 84, 906–912. [DOI] [PubMed] [Google Scholar]
- Yang P, Kanki H, Drolet B, Yang T, Wei J, Viswanathan PC, Hohnloser SH, Shimizu W, Schwartz PJ, Stanton MS, Murray KT, Norris K, George AL Jr & Roden DM (2002). Allelic variants in Long QT disease genes in patients with drug‐associated torsades de pointes. Circulation 105, 1943–1948. [DOI] [PubMed] [Google Scholar]
- Yang T, Chun YW, Stroud DM, Mosley JD, Knollmann BC, Hong C & Roden DM (2014). Screening for acute IKr block is insufficient to detect torsades de pointes liability: role of late sodium current. Circulation 130, 224–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Hong CC & Roden DM (2015). Inhibition of the α‐subunit of PI3 kinase increases late sodium current (INa‐L) and generates arrhythmias. Heart Rhythm 12, S150. [Google Scholar]
- Yang T & Roden DM (1996). Extracellular potassium modulation of drug block of IKr: Implications for torsades de pointes and reverse use‐dependence. Circulation 93, 407–411. [DOI] [PubMed] [Google Scholar]
- Yang T, Snyders DJ & Roden DM (1997). Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circ Res 80, 782–789. [DOI] [PubMed] [Google Scholar]