

Abstract
Genetic analyses have revealed an association of the gene encoding the Rab3A-interacting molecule (RIM1) with an autosomal dominant cone-rod dystrophy CORD7. However, the pathogenesis of CORD7 has remained unclear. Recently, we have revealed that RIM1 exerts functional impacts on voltage-dependent Ca2+ channel (VDCC) currents and anchors neurotransmitter-containing vesicles to VDCCs, controlling neurotransmitter release. On the basis of this study, we demonstrate here that the mouse RIM1 arginine-to-histidine substitution (R655H), which corresponds to the human CORD7 mutation, modifies RIM1 function in regulating VDCC currents elicited by the P/Q-type Cav2.1 and L-type Cav1.4 channels. Thus, we can raise an interesting possibility that CORD7 phenotypes including retinal deficits and enhanced cognition are at least partly underlaid by altered regulation of presynaptic VDCC currents.
Keywords: Calcium Channels, L-Type; Phenotype; Presynaptic Terminals; Retinitis Pigmentosa; Transfection; Animals; Calcium Channels, N-Type; Cell Line; GTP-Binding Proteins; Genotype; Humans; Membrane Potentials; Mice; Mutation
Originally identified as a putative effector for the synaptic vesicle protein Rab3, RIM1 is expressed in the brain and retinal photoreceptors where it is localized to the presynaptic ribbons in ribbon synapses.1 RIM1 interacts with other presynaptic active zone protein components, including Munc13, ELKS (also known as CAST), RIM-BP and liprins, to form a protein scaffold in the presynaptic nerve terminal.2–6 Studies of mouse knockouts revealed that, in different types of synapses, RIM1 is essential for Ca2+-triggered neurotransmitter release as well as different forms of synaptic plasticity.5,7,8
A mutation has been identified for an autosomal dominant cone-rod dystrophy CORD7 in the RIM1 gene that is localized to chromosome 6q14.9 A four-generation British family with CORD7 first became aware of reduced color vision and visual acuity between the ages of 20 and 40 years. As the disorder progressed, they showed difficulty seeing in bright light, and one individual reported visual problems in dim light. At the onset of symptoms, retinal pigmentary changes were already present around the fovea, simulating a bull’s eye dystrophy, which developed to macular atrophy.10 Interestingly, the affected individuals also showed significantly enhanced cognitive abilities across a range of domains.11 Thus, the family with the CORD7 RIM1 mutation is characterized by retinal dystrophy as well as by enhanced brain function. However, mechanisms underlying these two phenotypes are yet to be elucidated.
Recently, we have revealed a previously unknown interaction between two components of the presynaptic active zone, RIM1 and VDCCs12, that are essential for Ca2+-triggering of neurotransmitter release.13 RIM1 associates with VDCC β-subunits via its C terminus to markedly suppress voltage-dependent inactivation among different neuronal VDCCs. In addition, membrane docking of vesicles is enhanced also by RIM1. Consistently, in pheochromocytoma neuroendocrine PC12 cells and in cultured cerebellar neurons, neurotransmitter release is significantly potentiated by RIM1. Thus, RIM1 association with VDCC β in the presynaptic active zone supports release via two mechanisms: sustaining Ca2+ influx through inhibition of channel inactivation, and anchoring neurotransmitter-containing vesicles in the vicinity of VDCCs. On the basis of this study, we specifically test here whether the RIM1 mutation associated with CORD7 affects regulatory effects of RIM1 on inactivation properties of VDCC currents, to gain insights in molecular mechanisms that underly the two phonotypes in CORD7 patients.
The mouse RIM1 mutation which corresponds to human CORD7 mutation R844H9, is a nucleotide (G to A) substitution that replaces Arg-655 with His in the middle C2A domain (Fig. 1A), reported previously for its ability to bind to VDCC α1-subunit.6 The mouse clone differs from the human clone in having a deletion in the region between the Zn2+-finger-like and PDZ domains. In this region, any specific functional domains have not been identified so far. Coimmunoprecipitation experiments suggest an intact interaction between RIM1 mutant R655H and the VDCC β4b-subunit (Fig. 1B). To elucidate the functional effect of the RIM1 mutant, we characterized whole-cell Ba2+ currents through recombinant VDCCs expressed as α1α2/δβ4b complexes containing neuronal α1-subunits Cav2.1 of P/Q-type VDCCs or Cav1.4 of L-type VDCCs. These VDCCs were selected, because P/Q-type Cav2.1 plays an important role in neurotransmitter release from central neuron13, while L-type Cav1.4 is found at high densities in photoreceptor terminals14,15 and known for its association with X-linked congenital stationary night blindness.16 Compared to wild-type RIM1 (WT), R655H elicited significantly increased non-inactivating component of P/Q-type Cav2.1 currents (from 0.30 ± 0.04 (n = 9) to 0.43 ± 0.03 (n = 6); P < 0.01) (Fig. 1C). Inactivation parameters of L-type Cav1.4 current were unaffected by WT or by R655H (Fig. 1D) (see Table 1 for the estimated half-inactivation potential and the slope factor). In terms of activation properties, activation curve was shifted toward negative potentials (see Table 1 for the voltages for half-maximal activation and slope factors) and activation speed was increased by R655H in P/Q-type (Cav2.1) currents (Fig. 2A). Furthermore, RIM1-mediated augmentation of the current density in Cav2.1 currents was significantly enhanced by R655H (Fig. 2B) (see Table 1 the average of current densities). In contrast to Cav2.1 currents, Cav1.4 currents showed RIM1-mediated hyperpolarizing shift of activation curve abolished by the mutation (Fig. 2C). Effects of R655H on activation speed (Fig. 2C) and current densities were indistinguishable from those of WT in Cav1.4 channels (Fig. 2D).
Figure 1. CORD7 mutation R655H affects regulation of Cav2.1 channel inactivation by RIM1 via β association.
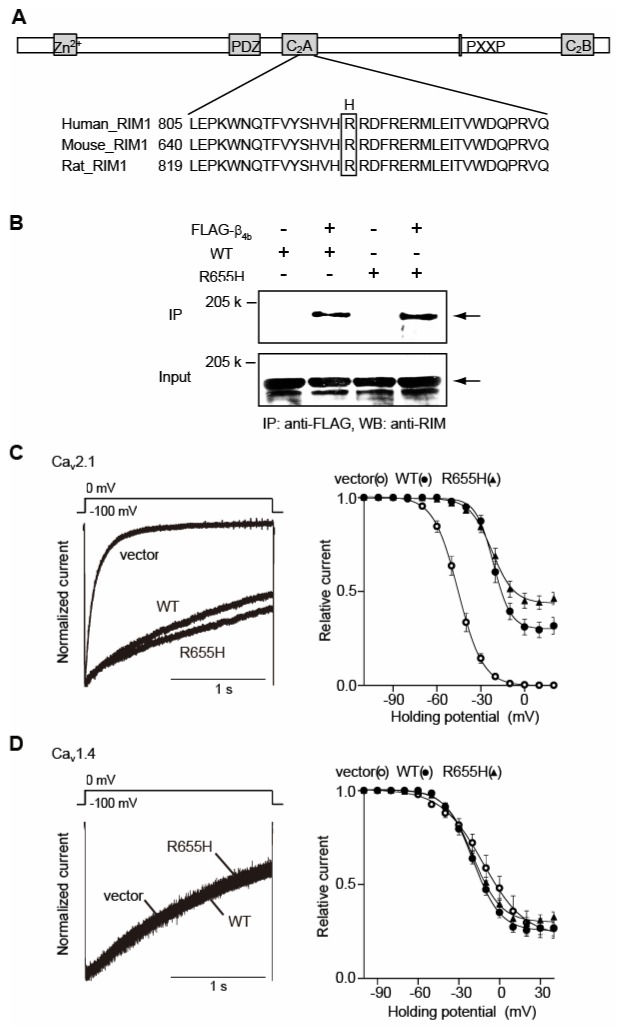
(A) Amino acid sequence alignment of C2A domains of human, mouse and rat RIM1. The position of the CORD7 substitution (H) is indicated. (B) Physical association of recombinant β4b and RIM1 mutant in HEK293 cells. The interaction is evaluated by immunoprecipitation (IP) with anti-FLAG antibody, followed by western blotting (WB) with anti-RIM antibody. Left panel: co-immunoprecipitation of wild-type RIM1 (WT) with FLAG- β4b. Right panel: co-immunoprecipitation of R655H with FLAG-β4b. (C) Effects of WT and R655H on the inactivation properties of P/Q-type Cav2.1 currents in BHK cells co-expressing α2/δ and β4b. Left panel: inactivation of P/Q-type Cav2.1 currents. The peak amplitudes are normalized for Ba2+ currents elicited by 2-s pulses to 0 mV from a holding potential (Vh) of −100 mV before and after expression of WT or R655H. Right panel: inactivation curves of P/Q-type Cav2.1 currents in BHK cells co-expressing α2/δ and β4b. The voltage dependence of inactivation, determined by measuring the amplitude of the peak currents evoked by 20-ms test pulses to 0 mV following 2-s prepulses to potentials from −100 to 20 with increments of 10 mV from a Vh of −100 mV, was fitted with the Boltzmann’s equation. (D) Effects of WT and R655H on the inactivation properties of L-type Cav1.4 currents in BHK cells co-expressing α2/δ and β4b. Left panel: inactivation of L-type Cav1.4 currents. Right panel: inactivation curves of L-type Cav1.4 currents in BHK cells co-expressing α2/δ and β4b. The voltage dependence of inactivation, determined as in (C) above.
Table 1.
Effects of mouse RIM1R655H constructs on current density, activation, and inactivation of Cav2.1, or Cav1.4 channel 1)2).
| Cav2.1 | Current density 3) | Activation parameters 4)
|
Inactivation parameters 5)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (pA/pF) | S.E. | V 0.5 | S.E. | k | S.E. | a | S.E. | V 0.5 | S.E. | k | S.E. | |
| vector | −11.43 | 1.48 | −7.19 | 1.24 | 4.47 | 1.26 | 1.00 | 0.00 | −45.90 | 1.77 | −7.53 | 0.27 |
| RIM1 | −30.45 | 5.34 * | −9.11 | 1.55 | 5.57 | 0.23 | 0.70 | 0.04 *** | −21.26 | 1.24 *** | −5.63 | 0.65 |
| RIM1R655H | −47.73 | 6.71 ***# | −13.35 | 0.89 **# | 5.07 | 0.16 | 0.57 | 0.03 ***## | −22.00 | 1.18 *** | −7.97 | 0.97 # |
| Cav1.4 | Current density 3) | Activation parameters 4)
|
Inactivation parameters 5)
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (pA/pF) | S.E. | V 0.5 | S.E. | k | S.E. | a | S.E. | V 0.5 | S.E. | k | S.E. | |
| vector | −14.71 | 1.71 | −6.08 | 1.77 | 9.69 | 0.83 | 0.79 | 0.06 | −10.50 | 3.40 | −15.12 | 1.00 |
| RIM1 | −31.48 | 7.29 * | −16.16 | 1.02 *** | 8.32 | 0.53 | 0.75 | 0.03 | −19.54 | 1.78 * | −9.69 | 0.33 *** |
| RIM1R655H | −28.13 | 4.70 * | −5.41 | 0.87 ### | 9.92 | 0.59 | 0.73 | 0.02 | −18.00 | 1.56 * | −10.84 | 0.74 *** |
*P < 0.05, **P < 0.01, ***P < 0.001 versus vector (ANOVA, Fisher’s test).
#P < 0.05, ##P < 0.01, ###P < 0.001 versus RIM1 (ANOVA, Fisher’s test).
Amplitude of Ba2+ currents evoked by depolarizing pulse to 0 mV from V h of −100 mV are divided by capacitance.
V 0.5 is the half-maximal activation voltage, and k is the slope factor.
a is the rate of inactivating component, V 0.5 is the half-inactivation potential and k is the slope factor.
Figure 2. CORD7 mutation R655H affects regulation of Cav1.4 channel activation by RIM1 via β association.
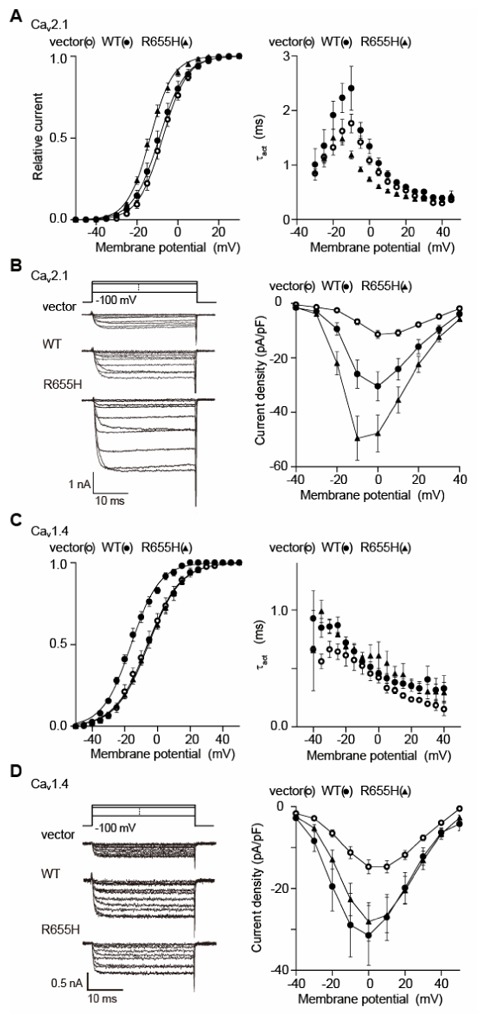
(A) Effects of WT and R655H on activation properties of Cav2.1 currents in BHK cells co-expressing α2/δ and β4b. Left panel: effects of WT and R655H on activation curves of Cav2.1 currents. Tail currents elicited by repolarization to −60 mV after 5-ms test pulse from −50 to 50 mV are used to determine activation curves. Right panel: effects on activation speed of Cav2.1 channels. Time constants are obtained by fitting the activation phase of currents elicited by 5-ms test pulse from −30 mV to 45 mV with a single exponential function. (B) Effects of WT and R655H on P/Q-type Cav2.1 current amplitude. Left panel: representative traces for Ba2+ currents evoked by test pluses from −40 mV to 50 mV with 10-mV increments in BHK cells co-expressing α2/δ and β4b. Right panel: current density-voltage (I–V) relationships of Cav2.1. The Vh is −100 mV. (C) Effects of WT and R655H on activation properties of Cav1.4 currents in BHK cells co-expressing α2/δ and β4b. Left panel: effects of WT and R655H on activation curves of Cav1.4 currents. Right panel: effects on activation speed of Cav1.4 channels. (D) Effects of WT and R655H on L-type Cav1.4 current amplitude. Left panel: representative traces for Ba2+ currents on application of test pluses from −40 mV to 50 mV with 10-mV increments in BHK cells co-expressing α2/δ and β4b. Right panel: I–V relationships of Cav1.4. The Vh is −100 mV.
Here, we demonstrate that the mouse RIM1 mutant R655H, which carries the counterpart mutation for human CORD7, alters RIM1 function in regulating VDCC currents. In the P/Q-type Cav2.1 VDCC known for its important role in neurotransmitter release in central synapses, CORD7 mutation elicited acceleration of activation, and enhanced RIM1-mediated suppression of inactivation and augmentation of current density, likely leading to an enhancement of neurotransmitter release and synaptic transmission. By contrast, in the L-type Cav1.4 VDCC known for its predominant role in glutamate release from photoreceptor terminals, the CORD7 mutation abolished the RIM1-mediated hyperpolarizing shift of activation curve, likely resulting in impaired synaptic transmission in ribbon synapses of the visual system. These different effects of CORD7 mutation on different presynaptic VDCCs may eventually lead to the two reported phenotypes, retinal dystrophy and enhanced cognitive abilities, of distinct nervous tissues.
References
- 1.Wang Y, Okamoto M, Schmitz F, Hofmann K, Südhof TC. Rim is a putative Rab3 effector in regulating synaptic-vesicle fusion. Nature. 1997;388:593–598. doi: 10.1038/41580. [DOI] [PubMed] [Google Scholar]
- 2.Wang Y, Sugita S, Südhof TC. The RIM/NIM family of neuronal C2 domain proteins. J Biol Chem. 2000;275:20033–20044. doi: 10.1074/jbc.M909008199. [DOI] [PubMed] [Google Scholar]
- 3.Betz A, Thakur P, Junge HJ, Ashery U, Rhee J-S, Scheuss V, Rosenmund C, Rettig V, Brose N. Functional interaction of the active zone proteins Munc13-1 and RIM1 in synaptic vesicle priming. Neuron. 2001;30:183–196. doi: 10.1016/s0896-6273(01)00272-0. [DOI] [PubMed] [Google Scholar]
- 4.Ohtsuka T, Takao-Rikitsu E, Inoue E, Inoue M, Takeuchi M, Matsubara K, Deguchi-Tawarada M, Satoh K, Morimoto K, Nakanishi H, Takai Y. CAST: a novel protein of the cytomatrix at the active zone of synapses that forms a ternary complex with RIM1 and Munc13-1. J Cell Biol. 2002;158:577–590. doi: 10.1083/jcb.200202083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schoch S, Castillo PE, Jo T, Mukherjee K, Geppert M, Wang Y, Schmitz F, Malenka RC, Südhof TC. RIM1α forms a protein scaffold for regulating neurotransmitter release at the active zone. Nature. 2002;415:321–326. doi: 10.1038/415321a. [DOI] [PubMed] [Google Scholar]
- 6.Coppola T, Magnin-Lüthi S, Perret-Menoud V, Gattesco S, Schiavo G, Regazzi R. Direct interaction of the Rab3 effector RIM with Ca2+ channels, SNAP-25, and Synaptotagmin. J Biol Chem. 2001;276:32756–32762. doi: 10.1074/jbc.M100929200. [DOI] [PubMed] [Google Scholar]
- 7.Castillo PE, Schoch S, Schmitz F, Südhof TC, Malenka RC. RIM1α is required for presynaptic long-term potentiation. Nature. 2002;415:327–330. doi: 10.1038/415327a. [DOI] [PubMed] [Google Scholar]
- 8.Schoch S, Mittelstaedt T, Kaeser PS, Padgett D, Feldmann N, Chevaleyre V, Castillo PE, Hammer RH, Han W, Schmitz F, Lin W, Südhof TC. Redundant functions of RIM1α and RIM2α in Ca2+-triggered neurotransmitter release. EMBO J. 2006;25:5852–5863. doi: 10.1038/sj.emboj.7601425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson S, Halford S, Morris AG, Patel RJ, Wilkie SE, Hardcastle AJ, Moore AT, Zhang K, Hunt DM. Genomic organization and alternative splicing of human RIM1, a gene implicated in autosomal dominant cone-rod dystrophy (CORD7) Genomics. 2003;81:304–314. doi: 10.1016/s0888-7543(03)00010-7. [DOI] [PubMed] [Google Scholar]
- 10.Kelsell RE, Gregory-Evans K, Gregory-Evans CY, Holder GE, Jay MR, Weber BHF, Moore AT, Biro AC, Hunt DM. Localization of a Gene (CORD7) for a dominant cone-rod dystrophy to chromosome 6q. Am J Hum Genet. 1998;63:274–279. doi: 10.1086/301905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sisodiya S, Thompson P, Need A, Harris S, Weale M, Wilkie S, Michaelides M, Free S, Walley N, Gumb C, Gerrelli D, Ruddle P, Whalley L, Starr J, Hunt D, David G, Deary I, Moore A. Genetic enhancement of cognition in a kindred with cone-rod dystrophy due to RIMS1 mutation. J Med Genet. 2007;44:373–380. doi: 10.1136/jmg.2006.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kiyonaka S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, Beedle AM, Mori E, Hara Y, De Waard M, Kanagawa M, Itakura M, Takahashi M, Campbell KP, Mori Y. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tsien RW, Ellinor PT, Horne WA. Molecular diversity of voltage-dependent Ca2+ channels. Trens Pharmacol Sci. 1991;12:349–354. doi: 10.1016/0165-6147(91)90595-j. [DOI] [PubMed] [Google Scholar]
- 14.Koschak A, Reimer D, Walter D, Hoda J-C, Heinzle T, Grabner M, Striessnig J. Cav1. 4α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McRory JE, Hamid J, Doering CJ, Garcia E, Parker R, Hamming K, Chen L, Hildebrand M, Beedle AM, Feldcamp L, Zamponi GW, Snutch TP. The CACNA1F gene encodes an L-type calcium channel with unique biophysical properties and tissue distribution. J Neurosci. 2004;24:1707–1718. doi: 10.1523/JNEUROSCI.4846-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bech-Hansen NT, Naylor MJ, Maybaum TA, Pearce WG, Koop B, Fishman GA, Mets M, Musarella MA, Boycott KM. Loss-of-function mutations in a calcium-channel α1-subunit gene in Xp11. 23 cause incomplete X-linked congenital stationary night blindness. Nat Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]