

ABSTRACT
Conjugate vaccines are known to be one of the most effective and safest types of vaccines against bacterial pathogens. Previously, vaccine biosynthesis has been performed by using N-linked glycosylation systems. However, the structural specificity of these systems for sugar substrates has hindered their application. Here, we report a novel protein glycosylation system (O-linked glycosylation via Neisseria meningitidis) that can transfer virtually any glycan to produce a conjugate vaccine. We successfully established this system in Shigella spp., avoiding the construction of an expression vector for polysaccharide synthesis. We further found that different protein substrates can be glycosylated using this system and that the O-linked glycosylation system can also effectively function in other Gram-negative bacteria, including some strains whose polysaccharide structure was not suitable for conjugation using the N-linked glycosylation system. The results from a series of animal experiments show that the conjugate vaccine produced by this O-linked glycosylation system offered a potentially protective antibody response. Furthermore, we elucidated and optimized the recognition motif, named MOOR, for the O-glycosyltransferase PglL. Finally, we demonstrated that the fusion of other peptides recognized by major histocompatibility complex class II around MOOR had no adverse effects on substrate glycosylation, suggesting that this optimized system will be useful for future vaccine development. Our results expand the glycoengineering toolbox and provide a simpler and more robust strategy for producing bioconjugate vaccines against a variety of pathogens.
IMPORTANCE
Recently, the rapid development of synthetic biology has allowed bioconjugate vaccines with N-linked protein glycosylation to become a reality. However, the difficulty of reestablishing the exogenous polysaccharide synthetic pathway in Escherichia coli hinders their application. Here, we show that an O-linked protein glycosylation system from Neisseria meningitidis, which has a lower structure specificity for sugar substrates, could be engineered directly in attenuated pathogens to produce effective conjugate vaccines. To facilitate the further design of next-generation bioconjugate vaccines, we optimized a novel short motif consisting of 8 amino acids that is sufficient for glycosylation. Our results expand the application potential of O-linked protein glycosylation and demonstrate a simpler and more robust strategy for producing bioconjugate vaccines against different pathogens. In the future, bacterial antigenic polysaccharides could be attached to major histocompatibility complex binding peptides to improve immunological memory or attached to protein subunit vaccine candidates to provide double immune stimulation.
INTRODUCTION
Conjugate vaccines, created by covalently attaching a bacterial antigenic polysaccharide to a carrier protein, can activate a T-cell-dependent immune response (1–3) and are one of the most successful weapons for the prevention of infectious disease, especially in infants and older people (4). To date, many types of conjugate vaccines against Haemophilus influenzae type b, Streptococcus pneumoniae, Neisseria meningitidis, and Salmonella enterica serovar Typhi, among others, have been licensed and have superb safety and efficacy, especially the seven-valent pneumococcal conjugate vaccine Prevnar (PCV7) for infant immunization, which was licensed in the United States in 2000. By 2004, the rates of all-cause pneumonia admission and of hospitalizations for pneumococcal meningitis decreased by 39% and 66%, respectively, in children younger than 2 years (5, 6). To our knowledge, all of the licensed conjugate vaccines, such as Hiberix, Menveo, Prevnar, and Synflorix, are created by chemical methods. However, such methods involve a multistep strategy that includes several purification processes, which greatly increases their cost and thereby limits the market for these vaccines in developing countries.
Biosynthesis of polysaccharide conjugate vaccine production is evolving. In the last two decades, glycosylation pathways have been discovered in bacteria. The two best understood of these are the N-linked glycosylation system discovered in Campylobacter jejuni (7–9) and the O-linked glycosylation system found in Neisseria species (10, 11). In these two systems, the bacterial polysaccharide is transferred from an undecaprenyl pyrophosphate (UndPP) carrier onto the protein acceptor. This process is similar to the Wzy-dependent O-antigen biosynthetic system that transfers glycans onto the lipid A core (12, 13). Further, the two glycosyltransferases, PglB from C. jejuni (N linked) and PglL from Neisseria (O linked), also can be functionally transferred into Escherichia coli alone and are capable of mediating long glycan transfer (8, 11). PglB, which is homologous to the Stt3 component of the oligosaccharyltransferase (OTase) complex in eukaryotic cells (14), was the first to be used to produce conjugate vaccines because its glycosylation sequon was clear, a conserved pentapeptide motif, D/E-X1-N-X2-S/T (where X1 and X2 are any residues except proline), unlike the tripeptide motif NXS/T (where X can be any amino acid except proline) that is present in eukaryotic cells (15). This motif can be fused to a carrier protein to generate a glycoprotein (16). A promising bioconjugate against Shigella dysenteriae produced by genetically modified E. coli using this N-linked glycosylation system has been developed by the company GlycoVaxyn AG and was recently tested in a phase 1 clinical trial (16, 17). However, PglB works only if the sugar substrate contains an acetamido group at position C-2 of the reducing end and does not possess a β1-4 linkage between the first two sugars (18, 19). Polysaccharides in some bacteria, such as S. pneumoniae, Streptococcus suis, and Salmonella, lack appropriate reducing ends (20–23) and cannot be used in this system. Additionally, in many other bacteria, the gene cluster associated with the assembly of antigenic polysaccharides is still unknown, making the expression of exogenous polysaccharides difficult.
In Neisseria meningitidis, the glycosyltransferase PglL has been shown to be responsible for the attachment of the UndPP-glycan to pilin (PilE), which generates a glycoprotein, and one of the glycosylation sites on PilE is Ser63 (24, 25). Previous work has demonstrated that PglL can transfer virtually any glycan to pilin in vivo (26), and the only requirement for this process is that the glycan must be carried by a lipid carrier, UndPP. PglL, therefore, has more potential applications than PglB. However, unlike PglB, the structural determinants intrinsic to PglL have been difficult to characterize (27), and this has prevented the O-linked system from being used to produce conjugate vaccines. In N. meningitidis, at least seven proteins can be glycosylated by PglL (28), but as in eukaryotes, the sequons around glycosylation sites are of low complexity and do not follow a single pattern (29). The peptide-glycan combination is important for antigen recognition and presentation (3), and the next generation of conjugate vaccines might be designed based on the location of this sugar-peptide glycosidic bond (30). In cases where the recognition sequence for O-link glycosylation is known, the bacterial antigenic polysaccharide can be attached to major histocompatibility complex (MHC) binding peptides to further enhance vaccine efficacy.
The goal of this study was to develop a novel synthetic biology strategy to produce bioconjugate vaccines, providing an alternative to conventional N-glycosylation biological methods. Here, we report a novel protein glycosylation system that uses O-linked glycosylation from N. meningitidis to produce a vaccine. We show that glycoproteins with different carriers can be achieved by engineering directly in attenuated pathogens, and this type of bioconjugate can evoke a protective and specific immune response. Further, we elucidated and optimized the recognition motif, named MOOR, for the O-glycosyltransferase PglL, and then we generated a novel bioconjugate by fusing it to peptides that are recognized by MHC class II (MHC-II) to improve its presentation ability. Our results demonstrate a simpler and more robust strategy for producing bioconjugate vaccines against a variety of pathogens, expanding the application potential of O-linked protein glycosylation and, by site optimization, enabling the use of O-linked glycosylation to produce the next generation of conjugate vaccines.
RESULTS
Establishment of an O-linked glycosylation system in the attenuated Shigella flexneri strain 301DWP.
Escherichia coli strains, such as CLM24 (16), are the bacteria that have been most commonly employed as host strains in biomethods to produce conjugate vaccines, and multiple plasmids are required to express carrier proteins, PglB, and the glycan gene cluster together in these strains. To make the production of conjugate vaccines simpler, we adopted a new strategy that involves using the attenuated pathogen as the host strain to produce glycoproteins. Here, we used S. flexneri 2a strain 301 as the host, and because the O-antigen ligase gene waaL from this strain is responsible for the transformation of O-polysaccharide (OPS) from UndPP to the lipid A core, we used the λ-red recombination system (31) to delete this gene (see Text S1 in the supplemental material), resulting in a lipopolysaccharide (LPS)-deficient attenuated strain. For biosafety, this strain was further attenuated by curing the virulence plasmid using plasmid incompatibility, and the resulting strain was named 301DWP (see Fig. S1A in the supplemental material). To confirm the phenotype of this mutant, the LPS of each strain was analyzed by silver staining, and no LPS ladder was detected in strain 301DWP (Fig. 1A). To evaluate virulence, Sereny test and plaque formation experiments were performed (see also Text S1 in the supplemental material). The results show that compared with the wild-type strain, strain 301DWP did not cause inflammation of the cornea (see Fig. S1B in the supplemental material), and plaques were hardly detectable (see Fig. S1C in the supplemental material). These results indicate that the virulence of S. flexneri 2a strain 301 was mostly determined by the virulence plasmid and the surface glycan and that the 301DWP strain, which lacks these, is not virulent.
FIG 1 .

Establishment of an O-linked glycosylation system in the attenuated S. flexneri strain 301DWP. (A) Silver staining of LPS from S. flexneri 2a strains 301 and 301DWP. (B and C) Coomassie blue staining and Western blot assays to analyze the glycosylation of the natural substrate protein, PilE (B), or of recombinant substrates, CTB protein fused with the PilE S45-K73 fragment (4573) at the C terminus or recombinant rEPA or TTc proteins fused with 4573 at the N terminus (C), when each was coexpressed with the corresponding O-linked glycosyltransferase PglL.
PilE, a structural component of type IV pilin, is the natural substrate of PglL that catalyzes the O-linked glycosylation reaction in Neisseria. To verify that this system could work effectively in other pathogens, such as S. flexneri 2a strain 301DWP, plasmids expressing an inducible PglL (pET-pglL) and an inducible substrate protein PilE (pMM-pilE) were introduced into strain 301DWP. The Western blot results show the typical ladders caused by different sugar repeat units for the isopropyl-β-d-thiogalactopyranoside (IPTG)-induced 301DWP strain coexpressing PilE and PglL (Fig. 1B). The molecular mass was about 30 kDa for OPS chains with a modal length of 11 to 17 repeated units controlled by the chromosomally encoded Wzz protein (32, 33).
PilE can be glycosylated at Ser63 (24, 25). To determine whether or not this O-linked glycosyltransferase could attach an antigenic polysaccharide to widely used commercial carrier proteins, such as recombinant Pseudomonas aeruginosa exotoxin A (rEPA), tetanus toxin C fragment (TTc), and cholera toxin b subunit (CTB), we added a peptide fragment (45SAVTEYYLNHGEWPGNNTSAGVATSSEIK73) surrounding S63 (underlined) onto the N termini of rEPA and TTc and onto the C terminus of CTB (pMM-CTB4573C, pMM-rEPA4573N, and pMM-TTc4573N) (Table 1). rEPA and TTc have been widely used for vaccine research, and some conjugate vaccines using these carriers, such as ActHib, Hiberix, and StaphVAX, have been licensed or are under investigation in a phase 3 clinical trial. CTB also has been widely used as carrier because of its adjuvant activity (34). The Western blot results show that, when the corresponding plasmids were transformed into the 301DWP strain harboring the pET-pglL plasmid, our recombinant carrier proteins were efficiently glycosylated following induction with IPTG (Fig. 1C). Notably, the glycosylation efficacy was highest when CTB4573C was used as a carrier, in which case almost all of the substrate proteins were glycosylated (Fig. 1C).
TABLE 1 .
Main plasmids used in this study
| Plasmid | Characteristica | Source |
|---|---|---|
| pKD46 | Used for λ-red recombination, araC-ParaB, Apr | Laboratory stock |
| pET-kan | Carries a kanamycin resistance gene flanked by FRT sites | Laboratory stock |
| pCP20 | Used for the removal of kanamycin resistance gene | Laboratory stock |
| pMM-pilE | Encodes 6×His-tagged PilE under control of Tac promoter, Ampr | This work |
| pET-pglL | Encodes PglL under control of Tac promoter, Kanr | This work |
| pMM-CTB4573C | Encodes 6×His-tagged CTB and fuse DsbA signal peptide at N terminus and PilE4573 fragment at C terminus under control of Tac promoter, Ampr | This work |
| pMM-rEPA4573N | Encodes 6×His-tagged rEPA and fuse DsbA signal peptide at N terminus and PilE4573 fragment at N terminus under control of Tac promoter, Ampr | This work |
| pMM-TTc4573N | Encodes 6×His-tagged TTc and fuse DsbA signal peptide at N terminus and PilE4573 fragment at N terminus under control of Tac promoter, Ampr | This work |
| pET-pglL-CTB4573C | Encodes PglL and 6×His-tagged CTB from pMM-CTB4573C, both of them under control of Tac promoter, Kanr | This work |
| pET-pglL-rEPA4573N | Encodes PglL and 6×His-tagged rEPA from pMM-rEPA4573N, both of them under control of Tac promoter, Kanr | This work |
Abbreviations: araC, 1-β-d-arabinofuranosylcytosine; FRT, FLP recombination target.
Application potential of the O-linked glycosylation system.
To determine if this system could be applied to other pathogens, we used E. coli O157:H7, a serious gut pathogen, and Salmonella enterica serovar Paratyphi A strain CMCC 50973, whose OPS cannot be recognized by PglB because the sugar substrate does not contain an acetamido group at position C-2 of the reducing end. The structures of their repeating OPS units are shown in Fig. 2A (35, 36). The waaL gene was deleted in each of these strains using the method described above, and the plasmids pET-pglL-rEPA4573N and pET-pglL-CTB4573C (Table 1) were then introduced into these mutants. The Western blot results show that the carrier proteins rEPA4573N and CTB4573C were efficiently glycosylated in both strains (Fig. 2B), indicating that this modified O-linked glycosylation system might be adopted by many other Gram-negative pathogens. Furthermore, the E. coli strain CLM24, which is commonly used in glycosylation research, was also compatible with our O-linked glycosylation system (see Fig. S2 in the supplemental material). That is, all reported applications of N-linked glycosylation could be theoretically imitated in our O-linked glycosylation system. Furthermore, some cases that could not be completed in the PglB system might be achieved using the PglL system.
FIG 2 .
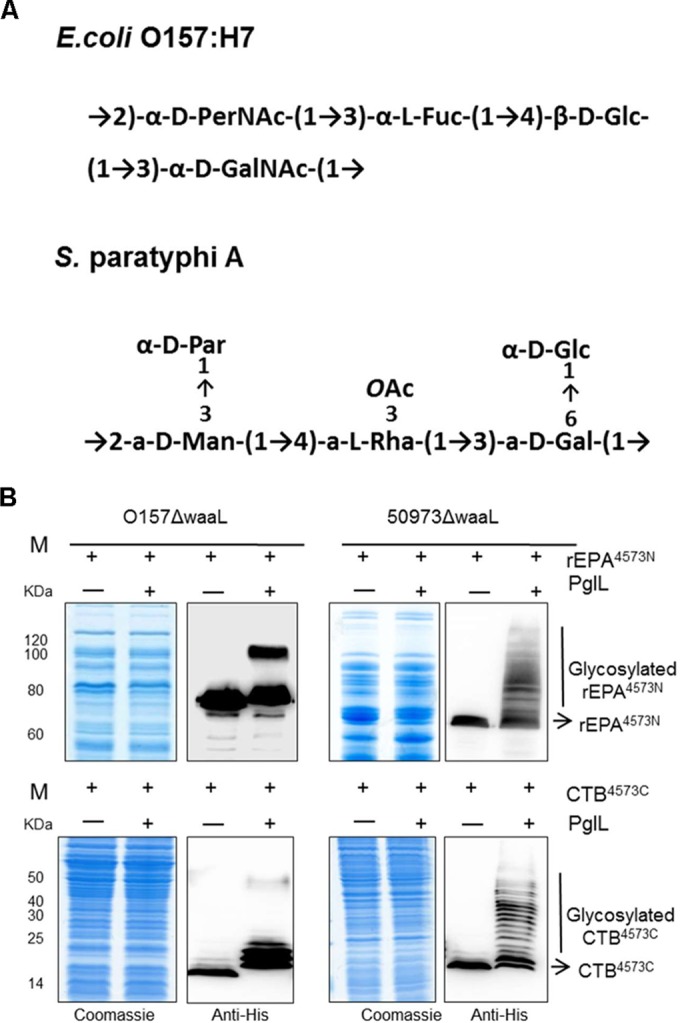
O-linked glycosylation in different pathogens. (A) The repeating unit structures of the OPS of E. coli O157:H7 (top) (35) and Salmonella serovar Paratyphi A (bottom) (36). (B) The O-linked glycosylation system was introduced into E. coli O157:H7 or Salmonella serovar Paratyphi A strain CMCC 50973, each of which lacked waaL, to test the universality of this system. Coomassie blue staining and Western blot assays were used to analyze the glycosylation of the substrate proteins rEPA4573N (top) and CTB4573C (bottom) expressed alone or coexpressed with PglL.
Purification of the glycoproteins.
To detect the immunogenicity of our bioconjugates, purification was performed to obtain glycosylated rEPA4573N and CTB4573C, and the levels of their production were detected by Coomassie blue and periodic acid-Schiff (PAS) staining (Pierce glycoprotein staining kit; Thermo Scientific) (Fig. 3A). Their purities were further detected as 99.7% and 93.2%, respectively, by size exclusion high-performance liquid chromatography (SEC-HPLC) (Fig. 3B), and these purities were sufficient for our subsequent experiments. Next, protein and glycan concentrations were measured via the micro-bicinchoninic acid (micro-BCA) method (MicroBCA protein assay kit; Thermo Scientific) and anthrone-sulfuric acid method (see also Text S1 in the supplemental material), and the results show that the concentrations of rEPA4573-OPS and CTB4573-OPS were 673 µg/ml and 965 µg/ml, respectively. Additionally, approximately 9 mg and 6 mg of high-purity rEPA4573N-OPS and CTB4573C-OPS, respectively, could be achieved per liter of culture broth at the experimental stage. Furthermore, it is likely that the glycoconjugate yields could be increased by optimizing the medium and culture conditions in medium-scale experiments.
FIG 3 .

Evaluation of the glycoproteins isolated from animal experiments. (A) Purified CTB4573C-OPS and rEPA4573N-OPS were separated by 12% and 10% SDS-PAGE, respectively. The proteins were then stained by Coomassie blue, and the OPSs were visualized by PAS staining. (B) For analysis with SEC-HPLC (TSK G4000SWXL, diameter [φ], 7.8 × 300 mm), 25 µl of purified glycoproteins was added, and the mobile phase consisted of 50 mM phosphate buffer and 0.9% NaCl. (C) The IgG subtype titers (IgG1, IgG2a, IgG2b, and IgG3) against the LPS from S. flexneri 2a strain 301 were measured in the serum of BALB/c mice immunized with CTB4573C-OPS, rEPA4573N-OPS, OPS, or adjuvant alone. Data are shown as points, and the lines indicate the means. Compared with the OPS group by t test, t was 15.4 for CTB4573C-OPS and t was 2.76 for rEPA4573N-OPS (*, P < 0.05; ***, P < 0.0001). (D) Complement bactericidal activity in different dilutions of serum from the mice vaccinated with CTB4573C-OPS. The error bars indicate the range for the percentage of deaths. (E) Mice that had been vaccinated as described for panel C were infected intraperitoneally with approximately 3 × 107 CFU/mouse of S. flexneri 2a strain 301, and their survival was monitored. (F) BN-PAGE analysis of CTB4573C-OPS and rEPA4573N-OPS.
Immune evaluation of the glycoproteins.
The glycoproteins (rEPA4573N-OPS and CTB4573C-OPS) and OPS were used, along with 10% Al(OH)3 adjuvant (General Chemical), to immunize BALB/c mice (5 weeks old, 10 per group; 2.5 µg polysaccharide/mouse) subcutaneously on days 1, 15, and 29, with the adjuvant-only group as a control. Serum was harvested on day 39 by tail snip, and the subtype concentrations of antibodies to the LPS of S. flexneri 2a strain 301 were evaluated by enzyme-linked immunosorbent assays (ELISAs) using peroxidase-conjugated goat anti-mouse IgG1, IgG2a, IgG2b, and IgG3 as the secondary antibodies. Among these subtypes, the titer of IgG1 induced by the CTB4573C-OPS group was dramatically higher than that of the rEPA4573N-OPS group, although the titers of both groups were higher than those of the OPS and control groups (Fig. 3C).
Additionally, because IgG1 can activate the classical complement pathway, complement bactericidal activity assays were performed to further investigate the immune effects induced by CTB4573C-OPS. When the antigens and antibodies were combined, the classical complement pathway was activated, and CTB4573C-OPS serum induced a high level of complement bactericidal activity. The sterilization rate reached 90% at serum dilutions of less than 640 (Fig. 3D). Further, to confirm the protective effects of this vaccine, a survival experiment was performed. By intraperitoneally infecting mice with approximately 3 × 107 CFU/mouse of S. flexneri 2a strain 301 on day 43 (14 days after the last vaccine or control injection), we observed that deaths mainly occurred in the first few days, and the protection rate for the CTB4573C-OPS group was 70%, which is higher than those of the other vaccine candidates or control substances (Fig. 3E). Taken together, these experiments show that vaccination with CTB4573C-OPS is potentially protective.
CTB exists in a pentameric form (37), whereas rEPA is monomeric in nature. We hypothesized that CTB4573C-OPS performed better as a vaccine than rEPA4573N-OPS because purified CTB4573C-OPS also formed a pentamer. To test this idea, blue native-polyacrylamide gel electrophoresis (BN-PAGE) was performed using a Ready-Gel with a linear 4 to 15% gradient (catalog no. 1611104; Bio-Rad) according to the manufacturer’s instructions, and the results show that the molecular mass of the polymer was more than 1,000 kDa, while the molecular mass of the CTB4573C-OPS monomer was only about 40 kDa (Fig. 3F). Interestingly, the measured molecular masses of both CTB4573C-OPS and rEPA4573N-OPS were higher than predicted, possibly because of the poor electrophoretic mobility of the glycan or the branching structure of the glycoproteins (Fig. 3F). In N-glycosylation studies, rEPA has been successfully used as a carrier protein to produce conjugate vaccines (16). However, our results show that CTB is a more effective carrier protein than rEPA.
The original glycosylation sequence could be truncated to 10 amino acids.
All of our above data support the use of the modified O-linked glycosylation system for the development of bacterial bioconjugate vaccines. However, two significant problems still hindered its application: the O-glycosylation site was not clear, and the amino acids conserved within the glycosylation motif remained to be determined. In our previous work, the peptide recognized by PglL was 29 amino acids in length, which is longer than the peptides that can be presented by MHC-II molecules (generally 15 to 24 amino acids long). Because the efficacy of these types of bioconjugates might be largely determined by the immunogenicity of the 29 amino acids, irrespective of which region of the carrier protein is attached to the polysaccharide antigen (30), we determined the minimum and optimal motif recognized by PglL.
First, we truncated the sequence step by step according to its secondary structure (38) using the N-terminus-fused recombinant protein rEPA4573N as a model (Fig. 4A). By using nested PCR (see Fig. S3 in the supplemental material), G55 to K73 (5573), G55 to S69 (5569), G55 to V66 (5566), and P58 to V66 (5866) were amplified, and new plasmids containing a single one of each of these sequences were created by replacing the fragment S45 to K73 in pET-PglL-rEPA4573N with these sequences. These various expression vectors were then introduced into strain 301DWP. Results from Western blot analyses of these strains reveal that the amount of glycosylated protein decreased as the length of the peptide decreased. Hardly any glycosylated peptides were detected when the peptide was only 12 amino acids long (rEPA5566) (Fig. 4B). One possible reason for this might be that the loss of flanking regions inhibited the correct folding, and hence presentation, of the core recognition motif. Thus, we chose two hydrophilic fragments from the rEPA sequence (592DPRNVGGDLD601 and 621QPGKPPR627) to add before and after the 12-amino-acid sequence described above. As expected, glycosylation of this recombinant protein (named rEPA5566AA) was significantly improved compared with that of rEPA5566, and its level of glycosylation was comparable to that of rEPA4573N (Fig. 4C). Based on this finding, we further shortened the recognition motif to ensure that the minimum recognition motif was obtained. By further deleting the amino acids one by one from both sides based on the rEPA5566AA sequence (Fig. 4D; see also Fig. S4 in the supplemental material), we confirmed that a minimum recognition motif of 10 amino acids (57WPGNNTSAGV66) is required for efficient glycosylation.
FIG 4 .

Determination of the minimum recognition motif for the O-linked glycosylation system. (A) A schematic of how the sequence of PilE 45S-73K was shortened step by step from 29 amino acids (aa) down to 9 amino acids, according to its secondary structure. (B) Coomassie blue staining and Western blotting assays were used to analyze the glycosylation of the sequences depicted in panel A. 4573 indicates a fusion of the S45-K73 fragment at the N terminus of rEPA (pET-pglL-rEPA4573N), and other fusion patterns are indicated similarly. The control was strain 301DWP lacking plasmid, and 4573PglLmut indicates the expression of PglLmut (pET-pglLmut-rEPA4573N). (C) Two hydrophilic fragments were added before and after the 12-amino-acid sequence (pET-pglL-rEPA5566AA), and glycosylation was assessed as in panel B. (D) The recognition motif was shortened to 10 amino acids (57WPGNNTSAGV66), based on 5566AA, and glycosylation was assessed as in panel B.
Optimization of the recognition motif of the O-linked glycosylation system.
Next, we aimed to optimize the shortened sequence by shuffling some amino acids within the minimum core motif. First, in the NCBI protein database, a total of 63 nonredundant homolog sequences of the native carrier protein PilE of N. meningitidis were aligned for a conservation analysis of the shortened motif. According to the results, the seventh amino acid, S, was the site of glycosylation, and the second, fifth, eighth, and ninth amino acids supported the uniqueness of this motif (Fig. 5A). Therefore, we performed alanine scanning to rapidly identify the importance of particular residues for glycosylation. All amino acids from W57 to V66, except S63 and A64, were mutated to alanine one by one, and Western blot analyses were used to detect glycosylated proteins.
FIG 5 .
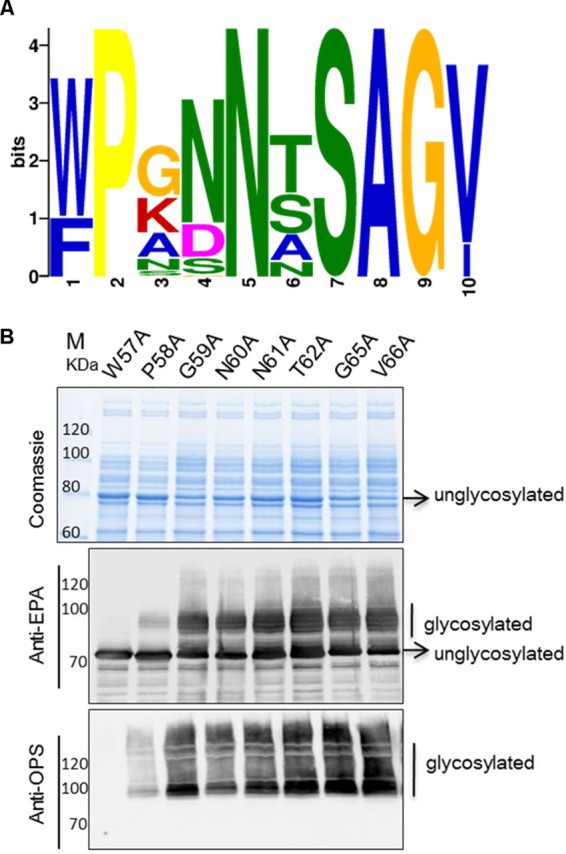
Analysis of the minimum recognition motif. (A) Conservation analysis of the 63 nonredundant PilE homolog sequences around S63 in Neisseria meningitidis (http://meme-suite.org/). (B) Alanine scanning of the glycosylated sequence. Plasmids containing these sequences were transformed into strain 301DWP, and glycosylation was detected by Western blotting.
To our surprise, the glycosylation of rEPA was abolished only when W57 was mutated to alanine, even though decreased glycosylation levels were evident when P58 was mutated to alanine instead of when the amino acids nearer S63 were mutated (Fig. 5B). That is, several of the other amino acid residues had no significant influence on glycosylation. In accordance with the results of the previous conservation analysis, W57 was not unique, and in some cases, it was replaced with phenylalanine. To determine if the sequence containing phenylalanine could also be glycosylated, we generated a W57F variant. Although the difference between tryptophan and phenylalanine is only an aromatic ring, decreased levels of glycosylation were observed in the W57F variant (see Fig. S5 in the supplemental material).
We had previously shown that each residue between 57WP58 and S63 (GNNT) had no obvious influence on glycosylation, implying that these residues may be interchangeable or dispensable. However, when we created a series of variants in which G, GN, N, or NN at 57 to 66 were deleted or the NN was mutated into an A, glycoproteins were hardly detectable (see Fig. S6A and B in the supplemental material), except in the cases with the amino acid sequence GAT (see also Fig. S6B in the supplemental material). Therefore, we performed further optimization, and the results from a Western blot analysis show that the peptide containing AAA between 57WP58 and S63 displayed a glycosylated serine (see Fig. S6C in the supplemental material). Finally, based on the WPAAAS sequence, we generated sequences with all combinations of A residues (no A, a single A, a double A [AA], or a triple A [AAA]) between 57WP58 and S63. As the number of alanine residues decreased, so did the level of protein glycosylation (Fig. 6A). From this set of experiments, we concluded that the optimal amino acid sequence between 57WP58 and S63 was AAA.
FIG 6 .

Optimization of the recognition motif. (A) The middle 4 amino acids (GNNT) of the recognition motif were replaced with single, double, or triple alanines or were removed completely. Plasmids containing these sequences were transformed into strain 301DWP, and glycosylation was detected by Western blotting. (B) A G65P mutant was constructed based on the sequence 57WPAAASAG65. This sequence was used and assessed as described for panel A. (C) The A after S63 was mutated into one of each of the other 19 amino acids. After plasmids with each of these sequences were transformed into strain 301DWP and protein expression was induced, periplasmic fractions were extracted and a Western blot analysis was performed to detect which sequences resulted in glycosylation. Control indicates the control group that expressed the carrier protein only (pET-pglLmut-rEPAMOOR-AA). (D) Western blotting assays were performed as in panel C. The glycosylated and unglycosylated bands of each lane were framed and used in a volume analysis performed by using the software Quantity One. The bar graph shows the percentage of glycosylated proteins for each transformant of 301DWP. “Co” indicates the control group, and the error bars indicate the standard deviations.
Our previous work confirmed that the number of amino acids after S63 influences glycosylation (see also Fig. S4 in the supplemental material). We achieved the same result here when the amino acids between 57WP58 and S63 were mutated to AAA (see Fig. S7A in the supplemental material). Therefore, we suspected that the amino acids after S form another flexible structure to separate the recognition motif from the downstream domains. In some studies in eukaryotes, proline residues have had a positive effect on O-linked glycosylation. Thus, we hypothesized that if we added an inflexible proline, it would inhibit this type of unexpected secondary structure. To test this idea, we created a variant based on the WPAAASAG sequence by replacing G65 with a proline. The results of a Western blot analysis indicate that this change induced increased levels of glycoproteins (Fig. 6B). In contrast, further deleting A64 abolished all glycosylation (see Fig. S7B in the supplemental material).
Based on these findings, we hypothesized that a simple and efficient sequence for O-glycosylation would be W-P-Xn-S-Xm-P, in which Xn is a flexible amino acid and AAA is optimal. To test if the alanine between S63 and the last proline is optimal for glycosylation, we evaluated the glycosylation efficiencies following the mutation of A64 to each of the other 19 amino acids in turn. After induction, periplasmic fractions were extracted and Western blot analyses were performed to detect glycoprotein. The results show that a higher glycosylation efficiency could be achieved only when glycine, alanine, or methionine was inserted between S63 and the last proline (Fig. 6C). Additionally, almost no glycoprotein was detectable when proline, cysteine, aspartic acid, or lysine was inserted (Fig. 6C). To evaluate the glycosylation efficiency, the bands of glycosylated and unglycosylated proteins were quantized with gray-scale values by using the software Quantity One. These results show that higher glycosylation efficiency (>40%) could be achieved only when glycine, alanine, or methionine was inserted between S63 and the last proline (Fig. 6D). Furthermore, an ELISA (see also Text S1 in the supplemental material), which used the extracted periplasmic fractions to coat 96-well plates and detected the optical density at 490 nm (OD490) of EPA and OPS using anti-EPA and anti-OPS antibodies, was also performed, and its results were used to calculate the glycosylation effect relative to the control group; these findings were similar to the Western blot assay results (see Fig. S7C in the supplemental material). Taking into account the stability of the structure, the final recognition motif was optimized to be 8 amino acids long (WPAAASAP) and was named the MOOR (minimum optimal O-linked recognition) motif.
When conjugate vaccines are produced by chemical methods, the substrate proteins are usually toxin proteins and the glycans can combine on many protein surface regions with strong immunogenicity. The acquisition of a minimum motif makes the concatenation of MHC binding peptides possible, and we believe that this may be an important step forward in the design of bioconjugates. We attempted to replace the hydrophilic fragments around the MOOR with different peptides that are recognized by MHC-II, such as gp10044–59 (WNRQLYPEWTEAQRLD) (39), HA307–319 (PKYVKQNTLKLAT) (40), P30 (TT947–967; FNNFTVSFWLRVPKVSASHLE) (41, 42), and P2 (TT830–843; QYIKANSKFIGITE) (41, 42), to design the glycan peptide. For example, the hydrophilic fragment at the C terminus of the core glycosylation sequence was replaced with gp10044–59, HA307–319, or P30, and the hydrophilic fragments at the N and C termini of the core glycosylation sequence were replaced with P30 and P2, respectively. We then tested the glycosylation of these designed carrier proteins, and the results of Western blot analyses show that all combinations were efficiently glycosylated (see Fig. S8 in the supplemental material). Evaluation of the immune response induced by these recombinant bioconjugates is under way in our laboratory.
DISCUSSION
We demonstrated that O-linked glycosylation system were a better way to create conjugate vaccines, and we further optimized the glycosylation recognition motif for this system. We found that CTB is a better carrier protein than rEPA and that glycosylated CTB, similar to natural CTB, is a polymer. Because polymeric particles may lead to a preferred interaction with antigen-presenting cells (43), glycosylated CTB has the potential to be an ideal carrier protein. An example of using polymeric particles to create vaccine was hepatitis B virus core particle, which, acting as a carrier, was fused with a conserved region of human influenza A virus to create a vaccine; it provided complete protection against lethal challenge in animal experiments (44–46). Potentially, particle carriers could also be applied to conjugate vaccines in the future.
Glycoproteins are thymus-dependent antigens which can induce T-cell, Ig isotype switch, and memory responses against the polysaccharide moiety. In this process, complement-activating, bactericidal IgG1 can be produced. In our animal experiments, most of the IgG antibody produced was subtype IgG1. Similarly, a previous clinical study of a pneumococcal conjugate vaccine also showed that IgG1 was the main antibody subtype elicited in infants following vaccination (47). So, conjugate vaccines show a better immune reaction than previous polysaccharide vaccines which are thymus-independent antigens and the main inducers of IgM production.
Although the production of bioconjugate vaccines is still in its initial stages, it offers huge advantages over chemical methods, as previously discussed (16, 48, 49). Currently, there are two known types of glycosyltransferases (PglL and PglB), both with the potential to produce conjugate vaccines. PglB has been more widely employed to date, mainly owing to the fact that its glycosylation recognition sequence, a 5-amino-acid motif, has been clearly defined. However, some polysaccharides are not suitable for PglB. Group I and IV capsules and OPS can synthesize UndPP (50), but some, including 50 of 54 known structural capsules in S. pneumoniae (20), 24 of 35 structural capsules in S. suis (21, 22), and 9 of 47 OPSs in Salmonella (23, 36), also lack appropriate reducing ends and cannot be transferred in N-linked glycosylation systems. Recently, a study showed that by the mutation of some amino acids in PglB, a Salmonella Gal-initiated O antigen could be transferred to proteins (51). Compared with PglB, PglL has more potential applications because of its lower structural specificity for sugar substrates and because it can transfer virtually any glycan in vivo (26). For example, a heptavalent conjugate vaccine, Prevnar, which is produced by chemical methods and consists of conjugates of diphtheria toxin mutant (CRM197) and 4, 6B, 9V, 14, 18C, 19F, and 23F capsular polysaccharides, is a successful vaccine for pneumococcal disease and has been licensed in more than 100 countries around the world. The results of clinical trials prove that this heptavalent pneumococcal conjugate vaccine successfully prevents pneumococcal infections in children (52). Theoretically, our O-glycosylation system also could be used to create S. pneumoniae conjugate vaccines because the capsular polysaccharides are similarly synthesized through the Wzy-dependent pathway (20). Of course, many difficulties, such as protein expression, remain to be addressed.
Here, we also elucidated that an 8-amino-acid motif named MOOR (sequence WPAAASAP) is sufficient for glycosylation. Statistical analyses of the different peptides containing O-glycans show that proline residues in positions −3 and +1 of the glycosylation sites have a positive effect on eukaryotic glycosylation (53). However, we found that proline at positions −4 and +2 increased glycosylation in prokaryotes. This motif is similar to a sequence in protein AniA (28), whose O-glycosylation sites are the S residues in the sequence 367GAAPAASAPAASAP380. Given that this sequence differs significantly from 45WPGNNTSAGV73, we propose that one reason for the diversity in natural O-glycosylation is that the amino acids surrounding serine may have other biological functions, such as delivering an external signal. Our motif provides new insight into the use of O-linked glycosylation for the biological production of conjugate vaccines.
The biological strategy for producing conjugate vaccines directly in attenuated strains described here is different from previously reported biological methods (16), in which three vectors need to be constructed and introduced into E. coli. In contrast, only one vector is needed in our strategy, and so the process is easier and more convenient. Not only could the production of conjugate vaccines directly in attenuated strains avoid the difficulty of cloning a long polysaccharide gene cluster, but this strategy could also be applied in strains whose polysaccharide gene cluster remains unknown. In summary, using O-linked glycosylation to produce polysaccharide conjugate vaccines in attenuated strains will be a breakthrough in bacterial vaccine production.
MATERIALS AND METHODS
Animals.
All animals were purchased from the Laboratory Animal Center of the Academy of Military Medical Sciences and were also housed in the Center, which had a constant ambient temperature (23 ± 3°C) and humidity (55% ± 5%). Food, bedding, and water were changed every 4 days. All animal experiments were approved by and performed in accordance with the recommendations of the Academy of Military Medical Sciences Institutional Animal Care and Use Committee.
Strains, growth conditions, and plasmids.
All bacterial strains were grown in lysogeny broth medium or on solid medium containing 1.5% agar. Escherichia coli DH5α was used in cloning experiments. Cells were cultured at 37°C to an OD600 of 0.4 to 0.5, and 1 mM IPTG was used at 30°C for 10 h to induce protein expression. The main plasmids used in this study are listed in Table 1. PglLmut was constructed using a Fast Mutagenesis System kit (Transgen Biotech) according to the manufacturer’s instructions, with primers pglL-C-A-F/pglL-C-A-R (see Table S1 in the supplemental material) to mutate the 57th base, C, to an A. This resulted in a TAA sequence, which could stop protein translation. The vectors used in the glycosylation site optimization experiments were created based on pET-pglL-rEPA4573N using nested PCR (see also Fig. S3 in the supplemental material).
LPS silver staining.
Shigella flexneri 2a strains, cultured at 37°C in 5 ml of lysogeny broth medium for 12 h, were washed with phosphate-buffered saline (PBS) three times and then resuspended in 100 µl of buffer (2% [wt/vol] sodium dodecyl sulfate [SDS], 4% [vol/vol] 2-mercaptoethanol, 1 M Tris-HCl, pH 6.8, and 0.02% [wt/vol] bromophenol blue). After boiling in a water bath for 10 min, 4 mg of proteinase K was added and incubated at 60°C for 2 h. The whole-cell lysates were then separated by SDS-PAGE. LPS was fixed in the gel with 40% (vol/vol) ethanol and 10% (vol/vol) acetic acid for 30 min and then sensitized for 30 min in a solution containing 7% (wt/vol) sodium acetate, 0.2% (wt/vol) sodium thiosulfate, 30% (vol/vol) ethanol, and 0.25% (vol/vol) glutaraldehyde. The gels were subsequently washed with double-distilled water (ddH2O) three times for 10 min and were then stained with 100 ml of staining solution (0.25 g of silver nitrate and 40 µl of formaldehyde) for 20 min. The gels were washed twice for 1 min each and then transferred into 100 ml of solution (2.5 g of sodium carbonate, 3 µl of 5% [wt/vol] sodium thiosulfate, and 40 µl of formaldehyde). The reactions were stopped with 1.5% (wt/vol) EDTA-Na2 for 10 min followed by repeated washings with ddH2O. All solutions were prepared with ddH2O.
Western blot analyses.
After protein expression was induced with IPTG, whole-cell extracts were heated to 100°C with 1× SDS-PAGE loading buffer containing 50 mM Tris-HCl (pH 6.8), 1.6% (wt/vol) SDS, 0.02% (wt/vol) bromophenol blue, 8% (vol/vol) glycerol, and 20 mM dl-dithiothreitol. Proteins were separated by SDS-PAGE, and Western blot analysis was performed using polyvinylidene fluoride membranes (GE Healthcare) as previously described (54). Anti-6×His antibodies conjugated to horseradish peroxidase (HRP) (Abmart) were used to detect the proteins that contained a 6×His tag. Anti-EPA antibody (P2318; Sigma) (1:7,500) was used to detect rEPA and rEPA mutants. Antiserum (catalog no. 210227; Denka Seiken) (1:50) specific for the S. flexneri 2a OPS was used to detect the glycans of glycoproteins. Both the anti-EPA antibody and the antiserum were produced in rabbits, and HRP-conjugated anti-rabbit IgG (Transgen Biotech) was used as the secondary antibody.
Production and purification of glycosylated proteins.
The IPTG-induced bacteria were harvested by centrifugation (at 7,000 × g for 10 min), and the resulting cell pellets (10 g) were resuspended in buffer A1 (20 mM Tris-HCl, pH 7.5, 10 mM imidazole, and 500 mM NaCl) to a volume of 100 ml, after which the cells were broken by sonication. Following sonication, the product was centrifuged at 12,000 × g for 10 min, and the resulting supernatant, which contained CTB-OPS, was applied on a chelating column (φ, 1.6 by 15 cm; GE Healthcare) that had been equilibrated with buffer A1. After the column was washed with buffer A1, the CTB-OPS sample was eluted with 100% buffer B1 (20 mM Tris-HCl, pH 7.5, 500 mM imidazole, and 500 mM NaCl). The eluent was desalted through a G25 column (φ, 2.5 by 30 cm; GE Healthcare) with buffer A3 (20 mM HAc-NaAc, pH 5.4) or, for rEPA-OPS, with buffer A2 (20 mM Tris-HCl, pH 7.5) and then applied to a ProteinPak SP8HR column (φ, 1 by 10 cm; Waters) or, for rEPA-OPS, to a ProteinPak DEAE8HR column (φ, 1 by 10 cm; Waters). After the ProteinPak SP8HR (DEAE8HR for rEPA-OPS) column was washed with buffer A3, or with buffer A2 for rEPA-OPS, the glycoprotein sample was eluted with a gradient of 0 to 100% buffer B3 (20 mM HAc-NaAc, pH 5.4, and 1 M NaCl) or of buffer B2 (20 mM Tris-HCl, pH 7.5, and 1 M NaCl) for rEPA-OPS. The elutriated samples were analyzed by Western blotting assays, and the sample containing protein-OPS was separated by Superdex 75 fast-performance liquid chromatography (FPLC) (φ, 1 by 30 cm; GE Healthcare) with buffer A4 (20 mM phosphate buffer, pH 7.4, and 150 mM NaCl).
Immunization experiments.
Groups of 10 female BALB/c mice, 5 weeks old, were used in immunization experiments. The purified bioconjugates were diluted with PBS and mixed with alum (10%; General Chemical). The dose of polysaccharide was 2.5 µg per injection. Immunizations were performed on days 1, 15, and 29. Blood samples were taken by tail snip on day 39, and the serum was stored at 4°C.
ELISA.
Ninety-six-well immunoplates were coated overnight with 100 µl of 100 µg/ml LPS from S. flexneri 2a strain 301 diluted in carbonate coating buffer (50 mM Na2CO3-NaHCO3, pH 9.6) at 4°C. The wells were then washed with 200 µl of wash buffer (1× PBS with 0.05% Tween 20) three times, and the plates were patted dry. Then, 200 µl of ELISA blocking buffer (1× PBS with 5% milk) was added to each well, and the plates were incubated at 37°C for 2 h. After washing and drying, 100 µl of immune serum, diluted to different concentrations in dilution buffer (1× PBS with 0.5% milk), was added to each well and incubated for 1 h at 37°C. After another washing and drying step, 100 µl of HRP-conjugated goat anti-mouse IgG antibodies (Abcam) diluted 1:50,000 in dilution buffer was added to each well, and the plates were incubated for 1 h at 37°C. After each well was washed five times and dried, 100 µl of color solution (o-phenylenediamine [OPD]–H2O2 solution) was added, and the samples were left to react in the dark for 15 min at room temperature. The reaction was stopped with 50 µl of ELISA stop solution (2 mol/liter H2SO4). A microplate reader was used to measure the OD490.
Complement sterilization experiments.
S. flexneri 2a strain 301 was cultured at 37°C to an OD600 of approximately 2.0 and then diluted 40,000-fold with normal saline. Immune sera for each group were mixed and incubated at 56°C for 30 min to inactivate complement and then diluted to various concentrations. After the addition of 10 µl immune serum (using normal saline as a negative control) to 10 µl diluted cells, the mixture was incubated for 1 h at room temperature, and then 20 µl of complement was added to each group. The samples were incubated at 37°C for 1 h, plated on LB solid medium, and cultured overnight at 37°C. The number of single colonies was counted to calculate the percentage of survivors.
Survival experiment.
A survival experiment was performed on day 43 (14 days after the last injection). S. flexneri 2a strain 301 was cultured at 37°C at an OD600 of 2.0 and then diluted with normal saline to approximately 3 × 107 CFU/200 µl. The immunized mice (described above) were infected intraperitoneally with 200 µl/mouse of the diluted strain and then observed for death every 8 h.
Preparation of periplasmic extracts.
Periplasmic extracts were prepared by osmotic shock lysis as described previously (9). Briefly, 5 ml of IPTG-induced cells was harvested by centrifugation and washed with PBS twice. The cells were then incubated in 100 µl of buffer (30 mM Tris-HCl, pH 8.5, 20% [wt/vol] sucrose, 1 mM EDTA, and 1 mg/ml lysozyme) on ice for 30 min and centrifuged for 20 min to yield periplasmic proteins in the supernatants.
SUPPLEMENTAL MATERIAL
Supplemental methods. The supplemental methods used in our experiments included the construction of ligase-defective strains, plaque assays, Sereny tests, determination of sugar content, and calculation of the relative glycosylation. Download
Toxicity test for strain 301DWP. (A) PCR was performed to amplify the virulence factors (IpaA, IpgB, MxiD, and VirG), located in the virulence plasmid from S. flexneri 2a strains 301 and 301DWP. Bands in the marker lane from top to bottom show fragments of 7,000, 5,000, 3,000, 2,000, 1,000, and 500 bp. (B) Sereny test in guinea pigs at 36 and 48 h postinfection. Representative images of the cornea after infection with S. flexneri 2a strain 301, 301ΔwaaL, or 301DWP. Control animals were treated with normal saline. (C) Plaque assays were performed to detect the virulence of S. flexneri 2a strains 301, 301ΔwaaL, and 301DWP in HeLa cells. The diameters of the plaques were measured using a microscope. Bar, 1,000 µm. Download
O-linked glycosylation in E. coli strain CLM24. Western blot analysis with anti-His antibodies to detect glycosylation in E. coli strain CLM24 coexpressing WbbL (pACU184-wbbL) and the recombinant substrate protein rEPA4573N (pMM-rEPA4573N), with or without glycosyltransferase PglL (pET-PglL). Download
Using nested PCR to mutate the amino acids in the glycosylation sequence. The upper line is a schematic of pET-pglL-rEPA4573N, which contains a SacI site at the C terminus of Ptac in rEPA and a SpeI site between 4573 and rEPA. We designed an upstream primer containing a SacI site and downstream primers 1, 2, and 3 containing a SpeI site, as shown in the bottom line. pET-pglL-rEPA4573N was amplified using nested PCR to mutate position 4573. Download
Glycosylation of the sequences truncated after S63. After shortening of the original glycosylation sequence to 10 amino acids, the amino acids from the C terminus were deleted one by one, which left three (AGV), two (AG), or one (A) amino acid after S63. Plasmids containing the mutant sequences were transformed into strain 301DWP, and the amounts of glycosylated protein in each group were assessed by Western blotting. Download
Glycosylation status when W57 was replaced with F. W57 in the 57WPGNNTSAGV66 sequence (pET-pglL-rEPA5766AA) was mutated to F, and either the original sequence or the W57F mutant sequence was transformed into strain 301DWP. Western blot analyses with anti-EPA (middle) and anti-OPS (right) antibodies were performed to compare the glycosylation statuses between transformed 301DWP strains. The corresponding gel stained with Coomassie blue is shown on the left. Download
Optimization of the amino acids between 57WP58 and S63. New plasmids were generated by deleting specific residues, and then these plasmids were transformed into strain 301DWP, and glycosylation was detected by Western blot analysis. (A) The G or GN of the original sequence GNNT (control) between 57WP58 and S63 was deleted, resulting in NNT or NT. (B) One or two of the N residues between 57WP58 and S63 were deleted, or the remaining N was mutated into an A, resulting in the amino acid sequence GNT, GT, or GAT. (C) Based on the results from GAT in panel B, the G alone or both the G and the T were mutated into A’s, and so the amino acids between 57WP58 and S63 became GAA or AAA. Download
Glycosylation status of sequences that were truncated or mutated after S63. (A) Based on the sequence WPAAASAGV, amino acids from the C terminus were deleted one by one, leaving three (AGV), two (AG), or one (A) amino acid after S63. Plasmids containing the mutant sequences were transformed into strain 301DWP, and the amounts of glycosylated proteins in each group were assessed by Western blotting. (B) Based on the sequence containing amino acids AAA between 57WP58 and S63, two new plasmids were created by mutating the original AG after S63 into AP or P. Plasmids containing the mutant sequences were transformed into strain 301DWP, and the amounts of glycosylated proteins in these two groups were assessed by Western blotting. (C) The A after S63 was mutated into one of each of the 19 other amino acids. After plasmids with each of these sequences were transformed into strain 301DWP and protein expression was induced, the extracted periplasmic fractions of the stains were used to coat 96-well plates. The OD490s of EPA and OPS were measured by ELISAs using anti-EPA and anti-OPS antibodies. “Co” indicates the control group. The glycosylation level in each group was compared with that of the control, and the histogram indicates the relative glycosylation levels. The dotted line indicates the corresponding value of the control group. Download
Glycosylation status of sequences with different peptides in the flanking scaffold sequence. (A) The hydrophilic fragment at the C terminus of the MOOR was replaced with HA307–319 (PKYVKQNTLKLAT). The mutant plasmid was transformed into strain 301DWP, and a Western blot analysis was used to detect glycosylation. The control expressed the carrier protein alone. (B) The hydrophilic fragment at the C terminus of the core glycosylation sequence was replaced with gp10044–59 (WNRQLYPEWTEAQRLD). This sequence was used and assessed as described in panel A. (C) The hydrophilic fragments at the N and C termini of the core glycosylation sequence were replaced with P30 (TT947–967; FNNFTVSFWLRVPKVSASHLE) and P2 (TT830–843; QYIKANSKFIGITE), respectively, or the C terminus only was replaced with P30. Glycosylation was detected as described in panels A and B. Download
Primers used in our experiments. Sequences and the introductions about the primers used in our experiments. The underlined bases indicate restriction sites.
ACKNOWLEDGMENTS
This work was supported by the National Key Basic Research Program of China (973 Program, numbers 2011CB504901 and 2013CB910804) and the National Natural Science Foundation of China (numbers 81125012, 81373316, and 81471912).
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Footnotes
Citation Pan C, Sun P, Liu B, Liang H, Peng Z, Dong Y, Wang D, Liu X, Wang B, Zeng M, Wu J, Zhu L, Wang H. 2016. Biosynthesis of conjugate vaccines using an O-linked glycosylation system. mBio 7(2):e00443-16. doi:10.1128/mBio.00443-16.
REFERENCES
- 1.Avery OT, Goebel WF. 1931. Chemo-immunological studies on conjugated carbohydrate-proteins: V. The immunological specificity of an antigen prepared by combining the capsular polysaccharide of type III pneumococcus with foreign protein. J Exp Med 54:437–447. doi: 10.1084/jem.54.3.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mond JJ, Vos Q, Lees A, Snapper CM. 1995. T cell independent antigens. Curr Opin Immunol 7:349–354. doi: 10.1016/0952-7915(95)80109-X. [DOI] [PubMed] [Google Scholar]
- 3.Avci FY, Li X, Tsuji M, Kasper DL. 2011. A mechanism for glycoconjugate vaccine activation of the adaptive immune system and its implications for vaccine design. Nat Med 17:1602–1609. doi: 10.1038/nm.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Finn A. 2004. Bacterial polysaccharide-protein conjugate vaccines. Br Med Bull 70:1–14. doi: 10.1093/bmb/ldh021. [DOI] [PubMed] [Google Scholar]
- 5.Grijalva CG, Nuorti JP, Arbogast PG, Martin SW, Edwards KM, Griffin MR. 2007. Decline in pneumonia admissions after routine childhood immunisation with pneumococcal conjugate vaccine in the USA: a time-series analysis. Lancet 369:1179–1186. doi: 10.1016/S0140-6736(07)60564-9. [DOI] [PubMed] [Google Scholar]
- 6.Tsai CJ, Griffin MR, Nuorti JP, Grijalva CG. 2008. Changing epidemiology of pneumococcal meningitis after the introduction of pneumococcal conjugate vaccine in the United States. Clin Infect Dis 46:1664–1672. doi: 10.1086/587897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szymanski CM, Yao R, Ewing CP, Trust TJ, Guerry P. 1999. Evidence for a system of general protein glycosylation in Campylobacter jejuni. Mol Microbiol 32:1022–1030. doi: 10.1046/j.1365-2958.1999.01415.x. [DOI] [PubMed] [Google Scholar]
- 8.Wacker M, Linton D, Hitchen PG, Nita-Lazar M, Haslam SM, North SJ, Panico M, Morris HR, Dell A, Wren BW, Aebi M. 2002. N-linked glycosylation in Campylobacter jejuni and its functional transfer into E. coli. Science 298:1790–1793. doi: 10.1126/science.298.5599.1790. [DOI] [PubMed] [Google Scholar]
- 9.Feldman MF, Wacker M, Hernandez M, Hitchen PG, Marolda CL, Kowarik M, Morris HR, Dell A, Valvano MA, Aebi M. 2005. Engineering N-linked protein glycosylation with diverse O antigen lipopolysaccharide structures in Escherichia coli. Proc Natl Acad Sci U S A 102:3016–3021. doi: 10.1073/pnas.0500044102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stimson E, Virji M, Makepeace K, Dell A, Morris HR, Payne G, Saunders JR, Jennings MP, Barker S, Panico M, Blench I, Moxon ER. 1995. Meningococcal pilin: a glycoprotein substituted with digalactosyl 2,4-diacetamido-2,4,6-trideoxyhexose. Mol Microbiol 17:1201–1214. doi: 10.1111/j.1365-2958.1995.mmi_17061201.x. [DOI] [PubMed] [Google Scholar]
- 11.Faridmoayer A, Fentabil MA, Mills DC, Klassen JS, Feldman MF. 2007. Functional characterization of bacterial oligosaccharyltransferases involved in O-linked protein glycosylation. J Bacteriol 189:8088–8098. doi: 10.1128/JB.01318-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hug I, Feldman MF. 2011. Analogies and homologies in lipopolysaccharide and glycoprotein biosynthesis in bacteria. Glycobiology 21:138–151. doi: 10.1093/glycob/cwq148. [DOI] [PubMed] [Google Scholar]
- 13.Power PM, Seib KL, Jennings MP. 2006. Pilin glycosylation in Neisseria meningitidis occurs by a similar pathway to wzy-dependent O-antigen biosynthesis in Escherichia coli. Biochem Biophys Res Commun 347:904–908. doi: 10.1016/j.bbrc.2006.06.182. [DOI] [PubMed] [Google Scholar]
- 14.Young NM, Brisson JR, Kelly J, Watson DC, Tessier L, Lanthier PH, Jarrell HC, Cadotte N, St Michael F, Aberg E, Szymanski CM. 2002. Structure of the N-linked glycan present on multiple glycoproteins in the gram-negative bacterium, Campylobacter jejuni. J Biol Chem 277:42530–42539. doi: 10.1074/jbc.M206114200. [DOI] [PubMed] [Google Scholar]
- 15.Kowarik M, Young NM, Numao S, Schulz BL, Hug I, Callewaert N, Mills DC, Watson DC, Hernandez M, Kelly JF, Wacker M, Aebi M. 2006. Definition of the bacterial N-glycosylation site consensus sequence. EMBO J 25:1957–1966. doi: 10.1038/sj.emboj.7601087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ihssen J, Kowarik M, Dilettoso S, Tanner C, Wacker M, Thöny-Meyer L. 2010. Production of glycoprotein vaccines in Escherichia coli. Microb Cell Fact 9:61. doi: 10.1186/1475-2859-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatz CF, Bally B, Rohrer S, Steffen R, Kramme S, Siegrist CA, Wacker M, Alaimo C, Fonck VG. 2015. Safety and immunogenicity of a candidate bioconjugate vaccine against Shigella dysenteriae type 1 administered to healthy adults: a single blind, partially randomized phase I study. Vaccine 33:4594–4601. doi: 10.1016/j.vaccine.2015.06.102. [DOI] [PubMed] [Google Scholar]
- 18.Wacker M, Feldman MF, Callewaert N, Kowarik M, Clarke BR, Pohl NL, Hernandez M, Vines ED, Valvano MA, Whitfield C, Aebi M. 2006. Substrate specificity of bacterial oligosaccharyltransferase suggests a common transfer mechanism for the bacterial and eukaryotic systems. Proc Natl Acad Sci U S A 103:7088–7093. doi: 10.1073/pnas.0509207103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen MM, Glover KJ, Imperiali B. 2007. From peptide to protein: comparative analysis of the substrate specificity of N-linked glycosylation in C. jejuni. Biochemistry 46:5579–5585. doi: 10.1021/bi602633n. [DOI] [PubMed] [Google Scholar]
- 20.Bentley SD, Aanensen DM, Mavroidi A, Saunders D, Rabbinowitsch E, Collins M, Donohoe K, Harris D, Murphy L, Quail MA, Samuel G, Skovsted IC, Kaltoft MS, Barrell B, Reeves PR, Parkhill J, Spratt BG. 2006. Genetic analysis of the capsular biosynthetic locus from all 90 pneumococcal serotypes. PLoS Genet 2:e31. doi: 10.1371/journal.pgen.0020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang K, Fan W, Cai L, Huang B, Lu C. 2011. Genetic analysis of the capsular polysaccharide synthesis locus in 15 Streptococcus suis serotypes. FEMS Microbiol Lett 324:117–124. doi: 10.1111/j.1574-6968.2011.02394.x. [DOI] [PubMed] [Google Scholar]
- 22.Okura M, Takamatsu D, Maruyama F, Nozawa T, Nakagawa I, Osaki M, Sekizaki T, Gottschalk M, Kumagai Y, Hamada S. 2013. Genetic analysis of capsular polysaccharide synthesis gene clusters from all serotypes of Streptococcus suis: potential mechanisms for generation of capsular variation. Appl Environ Microbiol 79:2796–2806. doi: 10.1128/AEM.03742-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu B, Knirel YA, Feng L, Perepelov AV, Senchenkova SN, Reeves PR, Wang L. 2014. Structural diversity in salmonella O antigens and its genetic basis. FEMS Microbiol Rev 38:56–89. doi: 10.1111/1574-6976.12034. [DOI] [PubMed] [Google Scholar]
- 24.Parge HE, Forest KT, Hickey MJ, Christensen DA, Getzoff ED, Tainer JA. 1995. Structure of the fibre-forming protein pilin at 2.6-A resolution. Nature 378:32–38. doi: 10.1038/378032a0. [DOI] [PubMed] [Google Scholar]
- 25.Musumeci MA, Faridmoayer A, Watanabe Y, Feldman MF. 2014. Evaluating the role of conserved amino acids in bacterial O-oligosaccharyltransferases by in vivo, in vitro and limited proteolysis assays. Glycobiology 24:39–50. doi: 10.1093/glycob/cwt087. [DOI] [PubMed] [Google Scholar]
- 26.Faridmoayer A, Fentabil MA, Haurat MF, Yi W, Woodward R, Wang PG, Feldman MF. 2008. Extreme substrate promiscuity of the Neisseria oligosaccharyl transferase involved in protein O-glycosylation. J Biol Chem 283:34596–34604. doi: 10.1074/jbc.M807113200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vik A, Aas FE, Anonsen JH, Bilsborough S, Schneider A, Egge-Jacobsen W, Koomey M. 2009. Broad spectrum O-linked protein glycosylation in the human pathogen Neisseria gonorrhoeae. Proc Natl Acad Sci U S A 106:4447–4452. doi: 10.1073/pnas.0809504106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schulz BL, Jen FE, Power PM, Jones CE, Fox KL, Ku SC, Blanchfield JT, Jennings MP. 2013. Identification of bacterial protein O-oligosaccharyltransferases and their glycoprotein substrates. PLoS One 8:e62768. doi: 10.1371/journal.pone.0062768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Julenius K, Mølgaard A, Gupta R, Brunak S. 2005. Prediction, conservation analysis, and structural characterization of mammalian mucin-type O-glycosylation sites. Glycobiology 15:153–164. doi: 10.1093/glycob/cwh151. [DOI] [PubMed] [Google Scholar]
- 30.Cuccui J, Wren B. 2015. Hijacking bacterial glycosylation for the production of glycoconjugates, from vaccines to humanised glycoproteins. J Pharm Pharmacol 67:338–350. doi: 10.1111/jphp.12321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morona R, van den Bosch L, Manning PA. 1995. Molecular, genetic, and topological characterization of O-antigen chain length regulation in Shigella flexneri. J Bacteriol 177:1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morona R, Van Den Bosch L, Daniels C. 2000. Evaluation of Wzz/MPA1/MPA2 proteins based on the presence of coiled-coil regions. Microbiology 146:1–4. doi: 10.1099/00221287-146-1-1. [DOI] [PubMed] [Google Scholar]
- 34.Lebens M, Holmgren J. 1994. Mucosal vaccines based on the use of cholera toxin B subunit as immunogen and antigen carrier. Dev Biol Stand 82:215–227. [PubMed] [Google Scholar]
- 35.Perry MB, MacLean L, Griffith DW. 1986. Structure of the O-chain polysaccharide of the phenol-phase soluble lipopolysaccharide of Escherichia coli 0:157:H7. Biochem Cell Biol 64:21–28. [DOI] [PubMed] [Google Scholar]
- 36.Konadu E, Shiloach J, Bryla DA, Robbins JB, Szu SC. 1996. Synthesis, characterization, and immunological properties in mice of conjugates composed of detoxified lipopolysaccharide of Salmonella paratyphi A bound to tetanus toxoid with emphasis on the role of O acetyls. Infect Immun 64:2709–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang RG, Westbrook ML, Westbrook EM, Scott DL, Otwinowski Z, Maulik PR, Reed RA, Shipley GG. 1995. The 2.4-A crystal structure of cholera toxin B subunit pentamer: choleragenoid. J Mol Biol 251:550–562. doi: 10.1006/jmbi.1995.0455. [DOI] [PubMed] [Google Scholar]
- 38.Craig L, Volkmann N, Arvai AS, Pique ME, Yeager M, Egelman EH, Tainer JA. 2006. Type IV pilus structure by cryo-electron microscopy and crystallography: implications for pilus assembly and functions. Mol Cell 23:651–662. doi: 10.1016/j.molcel.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 39.Chen S, Li Y, Depontieu FR, McMiller TL, English AM, Shabanowitz J, Kos F, Sidney J, Sette A, Rosenberg SA, Hunt DF, Mariuzza RA, Topalian SL. 2013. Structure-based design of altered MHC class II-restricted peptide ligands with heterogeneous immunogenicity. J Immunol 191:5097–5106. doi: 10.4049/jimmunol.1300467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lamb JR, Eckels DD, Lake P, Woody JN, Green N. 1982. Human T-cell clones recognize chemically synthesized peptides of influenza haemagglutinin. Nature 300:66–69. doi: 10.1038/300066a0. [DOI] [PubMed] [Google Scholar]
- 41.Valmori D, Pessi A, Bianchi E, Corradin G. 1992. Use of human universally antigenic tetanus toxin T cell epitopes as carriers for human vaccination. J Immunol 149:717–721. [PubMed] [Google Scholar]
- 42.Cai H, Chen MS, Sun ZY, Zhao YF, Kunz H, Li YM. 2013. Self-adjuvanting synthetic antitumor vaccines from MUC1 glycopeptides conjugated to T-cell epitopes from tetanus toxoid. Angew Chem Int Ed Engl 52:6106–6110. doi: 10.1002/anie.201300390. [DOI] [PubMed] [Google Scholar]
- 43.Yue H, Ma G. 2015. Polymeric micro/nanoparticles: particle design and potential vaccine delivery applications. Vaccine 33:5927–5936. doi: 10.1016/j.vaccine.2015.07.100. [DOI] [PubMed] [Google Scholar]
- 44.De Filette M, Min Jou W, Birkett A, Lyons K, Schultz B, Tonkyro A, Resch S, Fiers W. 2005. Universal influenza A vaccine: optimization of M2-based constructs. Virology 337:149–161. doi: 10.1016/j.virol.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 45.Fiers W, De Filette M, Birkett A, Neirynck S, Min Jou W. 2004. A “universal” human influenza A vaccine. Virus Res 103:173–176. doi: 10.1016/j.virusres.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 46.De Filette M, Martens W, Smet A, Schotsaert M, Birkett A, Londoño-Arcila P, Fiers W, Saelens X. 2008. Universal influenza A M2e-HBc vaccine protects against disease even in the presence of pre-existing anti-HBc antibodies. Vaccine 26:6503–6507. doi: 10.1016/j.vaccine.2008.09.038. [DOI] [PubMed] [Google Scholar]
- 47.Wuorimaa T, Dagan R, Väkeväinen M, Bailleux F, Haikala R, Yaich M, Eskola J, Käyhty H. 2001. Avidity and subclasses of IgG after immunization of infants with an 11-valent pneumococcal conjugate vaccine with or without aluminum adjuvant. J Infect Dis 184:1211–1215. doi: 10.1086/323648. [DOI] [PubMed] [Google Scholar]
- 48.Terra VS, Mills DC, Yates LE, Abouelhadid S, Cuccui J, Wren BW. 2012. Recent developments in bacterial protein glycan coupling technology and glycoconjugate vaccine design. J Med Microbiol 61:919–926. doi: 10.1099/jmm.0.039438-0. [DOI] [PubMed] [Google Scholar]
- 49.Frasch CE. 2009. Preparation of bacterial polysaccharide-protein conjugates: analytical and manufacturing challenges. Vaccine 27:6468–6470. doi: 10.1016/j.vaccine.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 50.Whitfield C. 2006. Biosynthesis and assembly of capsular polysaccharides in Escherichia coli. Annu Rev Biochem 75:39–68. doi: 10.1146/annurev.biochem.75.103004.142545. [DOI] [PubMed] [Google Scholar]
- 51.Ihssen J, Haas J, Kowarik M, Wiesli L, Wacker M, Schwede T, Thöny-Meyer L. 2015. Increased efficiency of Campylobacter jejuni N-oligosaccharyltransferase PglB by structure-guided engineering. Open Biol 5:140227. doi: 10.1098/rsob.140227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oosterhuis-Kafeja F, Beutels P, Van Damme P. 2007. Immunogenicity, efficacy, safety and effectiveness of pneumococcal conjugate vaccines (1998–2006). Vaccine 25:2194–2212. doi: 10.1016/j.vaccine.2006.11.032. [DOI] [PubMed] [Google Scholar]
- 53.Nehrke K, Ten Hagen KG, Hagen FK, Tabak LA. 1997. Charge distribution of flanking amino acids inhibits O-glycosylation of several single-site acceptors in vivo. Glycobiology 7:1053–1060. doi: 10.1093/glycob/7.8.1053-c. [DOI] [PubMed] [Google Scholar]
- 54.Aebi M, Gassenhuber J, Domdey H, te Heesen S. 1996. Cloning and characterization of the ALG3 gene of Saccharomyces cerevisiae. Glycobiology 6:439–444. doi: 10.1093/glycob/6.4.439. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. The supplemental methods used in our experiments included the construction of ligase-defective strains, plaque assays, Sereny tests, determination of sugar content, and calculation of the relative glycosylation. Download
Toxicity test for strain 301DWP. (A) PCR was performed to amplify the virulence factors (IpaA, IpgB, MxiD, and VirG), located in the virulence plasmid from S. flexneri 2a strains 301 and 301DWP. Bands in the marker lane from top to bottom show fragments of 7,000, 5,000, 3,000, 2,000, 1,000, and 500 bp. (B) Sereny test in guinea pigs at 36 and 48 h postinfection. Representative images of the cornea after infection with S. flexneri 2a strain 301, 301ΔwaaL, or 301DWP. Control animals were treated with normal saline. (C) Plaque assays were performed to detect the virulence of S. flexneri 2a strains 301, 301ΔwaaL, and 301DWP in HeLa cells. The diameters of the plaques were measured using a microscope. Bar, 1,000 µm. Download
O-linked glycosylation in E. coli strain CLM24. Western blot analysis with anti-His antibodies to detect glycosylation in E. coli strain CLM24 coexpressing WbbL (pACU184-wbbL) and the recombinant substrate protein rEPA4573N (pMM-rEPA4573N), with or without glycosyltransferase PglL (pET-PglL). Download
Using nested PCR to mutate the amino acids in the glycosylation sequence. The upper line is a schematic of pET-pglL-rEPA4573N, which contains a SacI site at the C terminus of Ptac in rEPA and a SpeI site between 4573 and rEPA. We designed an upstream primer containing a SacI site and downstream primers 1, 2, and 3 containing a SpeI site, as shown in the bottom line. pET-pglL-rEPA4573N was amplified using nested PCR to mutate position 4573. Download
Glycosylation of the sequences truncated after S63. After shortening of the original glycosylation sequence to 10 amino acids, the amino acids from the C terminus were deleted one by one, which left three (AGV), two (AG), or one (A) amino acid after S63. Plasmids containing the mutant sequences were transformed into strain 301DWP, and the amounts of glycosylated protein in each group were assessed by Western blotting. Download
Glycosylation status when W57 was replaced with F. W57 in the 57WPGNNTSAGV66 sequence (pET-pglL-rEPA5766AA) was mutated to F, and either the original sequence or the W57F mutant sequence was transformed into strain 301DWP. Western blot analyses with anti-EPA (middle) and anti-OPS (right) antibodies were performed to compare the glycosylation statuses between transformed 301DWP strains. The corresponding gel stained with Coomassie blue is shown on the left. Download
Optimization of the amino acids between 57WP58 and S63. New plasmids were generated by deleting specific residues, and then these plasmids were transformed into strain 301DWP, and glycosylation was detected by Western blot analysis. (A) The G or GN of the original sequence GNNT (control) between 57WP58 and S63 was deleted, resulting in NNT or NT. (B) One or two of the N residues between 57WP58 and S63 were deleted, or the remaining N was mutated into an A, resulting in the amino acid sequence GNT, GT, or GAT. (C) Based on the results from GAT in panel B, the G alone or both the G and the T were mutated into A’s, and so the amino acids between 57WP58 and S63 became GAA or AAA. Download
Glycosylation status of sequences that were truncated or mutated after S63. (A) Based on the sequence WPAAASAGV, amino acids from the C terminus were deleted one by one, leaving three (AGV), two (AG), or one (A) amino acid after S63. Plasmids containing the mutant sequences were transformed into strain 301DWP, and the amounts of glycosylated proteins in each group were assessed by Western blotting. (B) Based on the sequence containing amino acids AAA between 57WP58 and S63, two new plasmids were created by mutating the original AG after S63 into AP or P. Plasmids containing the mutant sequences were transformed into strain 301DWP, and the amounts of glycosylated proteins in these two groups were assessed by Western blotting. (C) The A after S63 was mutated into one of each of the 19 other amino acids. After plasmids with each of these sequences were transformed into strain 301DWP and protein expression was induced, the extracted periplasmic fractions of the stains were used to coat 96-well plates. The OD490s of EPA and OPS were measured by ELISAs using anti-EPA and anti-OPS antibodies. “Co” indicates the control group. The glycosylation level in each group was compared with that of the control, and the histogram indicates the relative glycosylation levels. The dotted line indicates the corresponding value of the control group. Download
Glycosylation status of sequences with different peptides in the flanking scaffold sequence. (A) The hydrophilic fragment at the C terminus of the MOOR was replaced with HA307–319 (PKYVKQNTLKLAT). The mutant plasmid was transformed into strain 301DWP, and a Western blot analysis was used to detect glycosylation. The control expressed the carrier protein alone. (B) The hydrophilic fragment at the C terminus of the core glycosylation sequence was replaced with gp10044–59 (WNRQLYPEWTEAQRLD). This sequence was used and assessed as described in panel A. (C) The hydrophilic fragments at the N and C termini of the core glycosylation sequence were replaced with P30 (TT947–967; FNNFTVSFWLRVPKVSASHLE) and P2 (TT830–843; QYIKANSKFIGITE), respectively, or the C terminus only was replaced with P30. Glycosylation was detected as described in panels A and B. Download
Primers used in our experiments. Sequences and the introductions about the primers used in our experiments. The underlined bases indicate restriction sites.