

Abstract
Proteostasis maintenance of γ-aminobutyric acid type A (GABAA) receptors dictates their function in controlling neuronal inhibition in mammalian central nervous systems. However, as a multisubunit, multispan, integral membrane protein, even wild type subunits of GABAA receptors fold and assemble inefficiently in the endoplasmic reticulum (ER). Unassembled and misfolded subunits undergo ER-associated degradation (ERAD), but this degradation process remains poorly understood for GABAA receptors. Here, using the α1 subunits of GABAA receptors as a model substrate, we demonstrated that Grp94, a metazoan-specific Hsp90 in the ER lumen, uses its middle domain to interact with the α1 subunits and positively regulates their ERAD. OS-9, an ER-resident lectin, acts downstream of Grp94 to further recognize misfolded α1 subunits in a glycan-dependent manner. This delivers misfolded α1 subunits to the Hrd1-mediated ubiquitination and the valosin-containing protein-mediated extraction pathway. Repressing the initial ERAD recognition step by inhibiting Grp94 enhances the functional surface expression of misfolding-prone α1(A322D) subunits, which causes autosomal dominant juvenile myoclonic epilepsy. This study clarifies a Grp94-mediated ERAD pathway for GABAA receptors, which provides a novel way to finely tune their function in physiological and pathophysiological conditions.
Keywords: Cys-loop receptor, endoplasmic reticulum-associated protein degradation (ERAD), epilepsy, γ-aminobutyric acid (GABA), protein misfolding, proteostasis, Grp94, Hrd1, OS-9, VCP
Introduction
Proteostasis maintenance of membrane proteins is essential for their function in normal physiology (1–4). Because of the complex structure of membrane proteins, their folding and assembly in the endoplasmic reticulum (ER)2 are often inefficient. Many pathogenic mutations aggravate such inefficient processes. Misfolded and unassembled membrane proteins are rapidly disposed via the ER-associated degradation (ERAD) pathway (5–8). Excessive ERAD is linked to an increasing number of human diseases (9). The ER proteostasis network regulates the protein folding, assembly, and degradation processes. However, such processes and ER proteostasis network components for integral membrane proteins remain poorly understood.
We use γ-aminobutyric acid type A (GABAA) receptors as a model to elucidate the ERAD pathway. GABAA receptors are the primary inhibitory neurotransmitter-gated ion channels in the mammalian central nervous system (10), belonging to the Cys loop superfamily, which also includes nicotinic acetylcholine receptors (11, 12). Remarkably, under physiological conditions only 20–30% of newly synthesized wild type (WT) subunits of nicotinic acetylcholine receptors reach their intended destination at the plasma membrane (13). This is likely the case for GABAA receptors as well (14). Moreover, proteasome inhibition substantially accumulates WT α1 subunits of GABAA receptors in cells, supporting that they are an ERAD substrate (15). Such an unproductive surface trafficking and extensive ERAD is also observed for many other WT membrane proteins, including cystic fibrosis transmembrane conductance regulator (16, 17) and peripheral myelin protein 22 (18). In humans, GABAA receptors have at least 19 subunits (α1–6, β1–3, γ1–3, δ, ϵ, θ, π, ρ1–3). The most common type in the human brain contains two α1 subunits, two β2 subunits, and one γ2 subunit. Individual subunits need to fold into their native structures in the ER (19, 20) and assemble with other subunits correctly on the ER membrane to form a heteropentamer (14, 21, 22) for subsequent trafficking to the plasma membrane. The recent crystal structure of human β3 subunits confirms the long-predicted GABAA receptor subunit topology (23). Each subunit has the following: a large extracellular (or ER luminal) N terminus; four transmembrane (TM) helices (TM1–TM4, with the TM2 domain lining the interior of the pore); a large intracellular loop connecting TM3 and TM4; and a short extracellular (or ER luminal) C terminus (Fig. 1A). Loss of function of GABAA receptors causes idiopathic epilepsies (24–26) and many other psychiatric and neurological disorders (27). Numerous mutations in GABAA receptors predispose them to misfolding or insufficient assembly and thus excessive ERAD (28, 29). For example, the missense A322D mutation in the TM3 domain of the α1 subunit leads to its misfolding and thus loss of functional channels on the plasma membrane (30), causing autosomal dominant juvenile myoclonic epilepsy (31).
FIGURE 1.

Grp94 positively regulates the ERAD of the α1 subunit of GABAA receptors. A, subunit topology of GABAA receptors. The schematic is built from the crystal structure of the β3 subunit (Protein Data Bank code 4COF). It has a large extracellular (or ER luminal) N terminus, four transmembrane (TM) helices (TM1–TM4), a large intracellular loop connecting TM3 and TM4, and a short extracellular (or ER luminal) C terminus. The two cysteines that form the signature disulfide bond are shown as sphere models. B, knocking down Grp94 increases the total and cell surface protein levels of WT α1 subunits. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting (NT) control siRNA or siRNA against Grp94. Forty eight hours post-siRNA transfection, cells were lysed, and the total cell lysates were subjected to SDS-PAGE and immunoblotting (IB) with mouse anti-α1 or rat anti-Grp94 antibodies (n = 3). β-Actin serves as a total protein loading control. Alternatively, the surface α1 subunits were measured using a cell surface protein biotinylation assay (n = 3). The Na+/K+-ATPase α chain serves as a loading control for biotinylated membrane proteins. Protein band quantifications using ImageJ are shown on the bottom panel. C, stably depleting Grp94 increases the total and cell surface protein levels of WT α1 subunits. HEK293T cells stably expressing control (Ctr) or Grp94 shRNA were transiently transfected with WT α1 plasmids together with β2 and γ2 subunit plasmids. Forty eight hours post-transfection, cells were lysed and treated as in B (n = 3). D, Grp94 knockdown does not significantly influence the mRNA level of CHOP by using quantitative RT-PCR analysis. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting control siRNA or siRNA against Grp94 for 48 h, or cells were treated with thapsigargin (1 μm, 6 h) as a positive control, which induces CHOP mRNA expression. Then total RNA was extracted from the cells and reverse-transcribed to cDNA before being subjected to quantitative PCR analysis. The experiments were done using two biological replicates in triplicate each time. NS, not significantly. E, knocking down Grp94 increases the half-life of the α1 subunit determined by CHX chase analysis. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting control siRNA or siRNA against Grp94 for 48 h and then chased for the indicated time with CHX (100 μg/ml), which inhibits protein synthesis. Cells were lysed and subjected to SDS-PAGE and immunoblotting analysis (n = 3). Degradation kinetics was plotted by quantifying α1 intensity against time after CHX addition on the bottom panel. F, stably depleting Grp94 increases the half-life of the α1 subunit determined by CHX chase analysis. HEK293T cells stably expressing control (Ctr) or Grp94 shRNA were transiently transfected with WT α1 plasmids together with β2 and γ2 subunit plasmids for 48 h and then chased for the indicated time with CHX (100 μg/ml). Cells were then treated as in E (n = 3). G, overexpression of HA-tagged Grp94 decreases the half-life of the α1 subunit determined by CHX chase analysis. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with empty vector (EV) controls or HA-tagged Grp94 plasmids for 48 h and then chased with CHX (100 μg/ml) for the indicated time (n = 3). H, Grp94 knockdown decreases the relative ubiquitination level of α1 subunits. HEK293 cells stably expressing (FLAG-α1)β2γ2 GABAA receptors were transfected with an HA-tagged ubiquitin plasmid together with non-targeting control siRNA or siRNA against Grp94 for 48 h. Total cell lysates were then immunoprecipitated with anti-FLAG M2 magnetic beads before being subjected to SDS-PAGE and blotted for ubiquitin. Quantification of the ratio of ubiquitin/α1 band intensity post-immunoprecipitation, representing the relative ubiquitin level of the α1 subunits, is shown on the bottom (n = 3). IB, immunoblotting. IP, immunoprecipitation. Each data point in B–H is reported as mean ± S.E. *, p < 0.05.
Cells use the ERAD pathway to recognize misfolded proteins in the ER, dislocate them from the ER membrane, ubiquitinate them using various ubiquitin E3 ligases, and target them for degradation by cytosolic 26S proteasome (5, 6, 32–34). This process is accomplished by the synchronized action of a series of chaperones and ERAD factors in the ER and cytosol, which are collectively called the ERAD machinery. Much of our understanding about the basic principles of ERAD comes from genetic and biochemical studies in yeast (8, 35), and recently, more knowledge has been gained in mammalian cells (36). The core ERAD machinery is well conserved from yeast to human, although the ERAD system in mammalian cells is much more complex. Here, we focused on elucidating the ERAD pathway of GABAA receptors in HEK293 cells, which is largely unexplored. We demonstrated that glucose-regulated protein 94 (Grp94) and osteosarcoma amplified 9 (OS-9) recognize misfolded WT α1 subunits in the ER lumen and deliver them for ubiquitination by Hrd1 (gene name SYVN1).
Experimental Procedures
Chemicals and Plasmids
MG-132 was obtained from Sigma. Eeyarestatin I came from Tocris Bioscience. BNIM, a specific Grp94 inhibitor, was synthesized according to the published procedure (37). Dithiobis(succinimidyl propionate) (DSP) was obtained from Thermo Scientific. The pCMV6 plasmids containing the human GABAA receptor α1 (Uniprot no. P14867-1), β2 (isoform 2, Uniprot no. P47870-1), and γ2 (isoform 2, Uniprot no. P18507-2) subunits and pCMV6 Entry Vector plasmid (pCMV6-EV) were obtained from Origene. The human GABAA receptor α1 subunit missense mutation A322D or N38Q/N138Q was constructed using a QuickChange II site-directed mutagenesis kit (Agilent Genomics). A FLAG tag was inserted between Leu-31 and Gln-32 in the α1 subunit, which did not interfere with its trafficking (38). The pCMV6-XL4 plasmid containing human Grp94 was obtained from Origene. An HA tag was inserted between Ser-796 and Thr-797 in Grp94, keeping the KDEL ER retention signal intact using the QuickChange II site-directed mutagenesis kit (Agilent Genomics). The forward and reverse primers for HA tag inserted are asfollows: forward (5′ to 3′), agatgaagaagaagaaacagcaaaggaatcttacccatacgatgttccagattacgctacagctgaaaaagatg, and reverse, catctttttcagctgtagcgtaatctggaacatcgtatgggtaagattcctttgctgtttcttcttcttcatct. Grp94 variants were constructed by deletions of the C-terminal domain (602–785 amino acids), N-terminal domain (22–341 amino acids), MC domain (342–785 amino acids), N and C domains (22–341 and 602–785 amino acids), and NM domain (22–601 amino acids) from the full-length HA-tagged Grp94 plasmid using QuickChange II site-directed mutagenesis kit (Agilent Genomics). Primers for Grp94 mutants are listed as follows: Grp94-N domain forward (5′ to 3′), gggaacttatgaatgatatcaaaccaacagatgaagaagaagaaacagc, and reverse, gctgtttcttcttcttcatctgttggtttgatatcattcataagttccc; Grp94-NM domain forward (5′ to 3′), ccaaggaaggagtgaagttcgatgaaagtacagatgaagaagaagaaacagc, and reverse, gctgtttcttcttcttcatctgtactttcatcgaacttcactccttccttgg; Grp94-MC domain forward (5′ to 3′), cgggtcggtcagagctatatggcagagaccat, and reverse, atggtctctgccatatagctctgaccgacccg; Grp94-C domain forward (5′ to 3′), cgggtcggtcagagctgagaaaactaaggagag, and reverse, ctctccttagttttctcagctctgaccgacccg. The Grp94-M domain plasmid was constructed from the Grp94-MC domain plasmid using primers for Grp94-NM domain. All cDNA sequences were confirmed by DNA sequencing. The pcDNA3.1− plasmids containing human WT Hrd1 and C291A Hrd1 are a gift from Professor Ron Kopito (Stanford University). The GFP and pCS2-mCherry and the pRK5-HA-ubiquitin-WT plasmids were obtained from Addgene.
Antibodies
The mouse monoclonal anti-α1 subunit antibody (clone BD24) was obtained from Millipore (catalogue no. MAB339); the rabbit polyclonal anti-α1 antibody was from R&D Systems (catalogue no. PPS022), and the goat polyclonal anti-α1 subunit antibody (A-20) was from Santa Cruz Biotechnology (catalogue no. SC-31405). The mouse monoclonal anti-β-actin antibody came from Sigma (catalogue no. A1978). The rabbit polyclonal anti-calnexin (catalogue no. ADI-SPA-860-F) and rat polyclonal anti-Grp94 (catalogue no. ADI-SPA-850-F) antibodies were from Enzo Life Sciences. The rabbit polyclonal anti-Hrd1 (catalogue no. Ap2184e) and anti-HA tag (catalogue no. Ap1012a) antibodies were obtained from Abgent. The rabbit monoclonal anti-OS-9 (catalogue no. 3705-1) and anti-VCP (catalogue no. 3339-1) antibodies were obtained from Epitomics. The mouse monoclonal anti-His tag (catalogue no. 2366S)and anti-ubiquitin (catalogue no. 3936) antibodies were obtained from Cell Signaling. The rabbit monoclonal anti-sodium potassium ATPase (catalogue no. ab76020) antibody came from Abcam. The mouse anti-gp78 (catalogue no. SC-166358) antibody was obtained from Santa Cruz Biotechnology. The mouse anti-GST (catalogue no. NB600-446) antibody was obtained from Novus Biologicals.
Cell Culture and Transfection
HEK293 cells and SH-SY5Y cells came from the ATCC and were maintained in Dulbecco's modified Eagle's medium (DMEM) (Hyclone) with 10% heat-inactivated fetal bovine serum (Sigma) and 1% penicillin/streptomycin (Hyclone) at 37 °C in 5% CO2. Monolayers were passaged upon reaching confluency with TrypLE Express (Invitrogen). Cells were grown in 6-well plates or 10-cm dishes and allowed to reach ∼70% confluency before transient transfection using FuGENE 6 (Roche Applied Science) or Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. Stable cell lines for α1β2γ2 and α1(A322D)β2γ2 receptors were generated using the G-418 selection method. Briefly, cells were transfected with α1:β2:γ2 (1:1:1) or α1(A322D):β2:γ2 (1:1:1) plasmids and then maintained in DMEM supplemented with 0.6 mg/ml G418 (Enzo Life Sciences) for 15 days. G-418-resistant cells were selected for follow-up experiments.
To generate monoclonal HEK293 cells stably expressing α1(A322D)β2γ2 receptors, the α1(A322D) sequence was subcloned into a pIRES2-EGFP bicistronic vector (Clontech) using EcoRI and SacII restriction sites, which would allow the simultaneous expression of α1 subunits and enhanced GFP separately but from the same RNA transcript. After transfection and G-418 treatment, cells that are GFP-positive were considered as those successfully transfected with the α1(A322D) subunit. GFP-positive cells were further diluted into 96-well plates, allowing a single cell distribution in each well. Cells with robust GFP signals were further selected to grow to population.
Stable shGrp94 and shControl HEK293T cells were a kind gift of Professor Yair Argon (University of Pennsylvania) (39). They were maintained in DMEM with 10% heat-inactivated fetal bovine serum (Sigma) and 1% penicillin/streptomycin (Hyclone) at 37 °C in 5% CO2. Two μg/ml puromycin (Dot Scientific) was added to maintain the colony.
siRNA Transfection
HEK293 cells were seeded at ∼2.5 × 105 cells per well in 6-well plates for the siRNA treatment. Cells were allowed to reach ∼70% confluency before transfection. The following small interfering RNA (siRNA) duplexes were obtained from Dharmacon: Grp94 (J-006417-08); Hrd1 (J-007090-08); gp78 (J-006522-08); OS-9 (J-010811-09); and non-targeting siRNA D-001810-01-20), which was used as a negative control. Cells were transfected with corresponding 50 nm siRNA using the HiPerfect transfection reagent (Qiagen) according to the manufacturer's transfection protocol. Forty eight hours post-transfection, cells were harvested for further analysis.
Western Blot Analysis
Cells were harvested with TrypLE Express and then lysed with lysis buffer (50 mm Tris, pH 7.5, 150 mm NaCl, and 1% Triton X-100) supplemented with the Roche Applied Science complete protease inhibitor mixture. Lysates were cleared by centrifugation (15,000 × g, 10 min, 4 °C). Protein concentration was determined by MicroBCA assay (Pierce). Aliquots of cell lysates were separated in an SDS-8% polyacrylamide gel, and Western blot analysis was performed using the appropriate antibodies. Band intensity was quantified using ImageJ software from the National Institutes of Health.
Quantitative RT-PCR
The relative expression levels of target genes were analyzed using quantitative RT-PCR. Total RNA was extracted from cells using the RNeasy mini kit (Qiagen catalogue no. 74104). cDNA was synthesized from 500 ng of total RNA using the QuantiTect reverse transcription kit (Qiagen catalogue no. 205311). Quantitative PCRs (40 cycles of 15 s at 94 °C, 30 s at 57 °C, and 30 s at 72 °C) were performed using cDNA, the QuantiTect SYBR Green PCR kit (Qiagen catalogue no. 204143), and corresponding primers in the StepOnePlus system (Applied Biosystems). The results were analyzed using StepOne version 2.2 software (Applied Biosystems). The forward and reverse primers for CHOP are 5′-GGAAACAGAGTGGTCATTCCC-3′ and 5′-CTGCTTGAGCCGTTCATTCTC-3′; the forward and reverse primers for Grp94 are 5′-GGCCAGTTTGGTGTCGGTTT-3′ and 5′-CGTTCCCCGTCCTAGAGTGTT-3′; the forward and reverse primers for GAPDH (housekeeping gene control) are 5′-GTCGGAGTCAACGGATT-3′ and 5′-AAGCTTCCCGTTCTCAG-3′. The threshold cycle (CT) was extracted from the PCR amplification plot, and the ΔCT value was defined as follows: ΔCT = CT (target gene) − CT (housekeeping gene). The relative mRNA expression level of target genes of treated cells was normalized to that of control cells as follows: relative mRNA expression level = 2 exp(−(ΔCT (treated cells) − ΔCT (control cells))). Each data point was evaluated in triplicate and measured using two biological replicates.
Cycloheximide (CHX) Chase Assay
HEK293 cells were seeded at 2.5 × 105 cells per well in 6-well plates and incubated at 37 °C overnight. Cells were then transfected with the indicated siRNAs or plasmids for 48 h prior to CHX chase. To stop protein translation, cells were treated with 100 μg/ml CHX (Amresco). Cells were then chased for the indicated time, harvested, and lysed for SDS-PAGE and Western blot analysis.
Biotinylation of Cell Surface Proteins
HEK293 cells and SH-SY5Y cells stably overexpressing α1β2γ2 or α1(A322D)β2γ2 receptors were plated in 10-cm dishes for surface biotinylation experiments according to our published procedure (40). Briefly, intact cells were washed twice with ice-cold PBS and incubated with the membrane-impermeable biotinylation reagent Sulfo-NHS SS-Biotin (0.5 mg/ml; Pierce) in PBS containing 0.1 mm CaCl2 and 1 mm MgCl2 (PBS + CM) for 30 min at 4 °C to label surface membrane proteins. To quench the reaction, cells were incubated with 10 mm glycine in ice-cold PBS + CM twice for 5 min at 4 °C. Sulfhydryl groups were blocked by incubating the cells with 5 nm N-ethylmaleimide in PBS for 15 min at room temperature. Cells were solubilized for 1 h at 4 °C in lysis buffer (1% Triton X-100, 50 mm Tris-HCl, 150 mm NaCl, and 5 mm EDTA, pH 7.5) supplemented with the complete protease inhibitor mixture (Roche Applied Science) and 5 mm N-ethylmaleimide. The lysates were cleared by centrifugation (16,000 × g, 10 min at 4 °C) to pellet cellular debris. The supernatant contained the biotinylated surface proteins. The concentration of the supernatant was measured using MicroBCA assay (Pierce). Biotinylated surface proteins were affinity-purified from the above supernatant by incubating for 1 h at 4 °C with 100 μl of immobilized neutravidin-conjugated agarose bead slurry (Pierce). The samples were then subjected to centrifugation (16,000 × g, 10 min, at 4 °C). The beads were washed six times with buffer (0.5% Triton X-100, 50 mm Tris-HCl, 150 mm NaCl, and 5 mm EDTA, pH 7.5). Surface proteins were eluted from beads by boiling for 5 min with 200 μl of LSB/urea buffer (2× Laemmli sample buffer (LSB) with 100 mm DTT and 6 m urea, pH 6.8) for SDS-PAGE and Western blotting analysis. The Na+/K+-ATPase α chain serves as a loading control for biotinylated membrane proteins.
Immunoprecipitation
For immunoprecipitation using cell lysates (500 μg), they were pre-cleared with 30 μl of protein A/G plus-agarose beads (Santa Cruz Biotechnology) and 1.0 μg of normal mouse IgG for 1 h at 4 °C to remove nonspecific binding proteins. The pre-cleared cell lysates were incubated with 2.0 μg of mouse anti-α1 antibody (clone BD24) for 1 h at 4 °C and then with 30 μl of protein A/G plus agarose beads overnight at 4 °C. For immunoprecipitation using the mouse brain homogenates (1 mg), they were pre-cleared with 30 μl of protein A/G plus-agarose beads (Santa Cruz Biotechnology) and 1.0 μg of normal rat IgG for 1 h at 4 °C to remove nonspecific binding proteins. The pre-cleared complex was incubated with 2.0 μg of rat anti-Grp94 antibody for 1 h at 4 °C and then with 30 μl of protein A/G plus agarose beads overnight at 4 °C. Afterward, the beads were collected by centrifugation at 8000 × g for 30 s and washed three times with lysis buffer. The complex was eluted by incubation with 30 μl of SDS loading buffer in the presence of DTT. The immunopurified eluents were separated in SDS-8% polyacrylamide gel, and Western blot analysis was performed using appropriate antibodies.
HEK293 cells stably expressing (FLAG-α1)β2γ2 GABAA receptors were transfected with the indicated siRNA or plasmids for 48 h. Then Triton X-100 cell extracts (500 μg) were pre-cleared with 30 μl of protein A/G plus-agarose beads (Santa Cruz Biotechnology) and 1.0 μg of normal mouse IgG for 1 h at 4 °C to remove nonspecific binding proteins. The pre-cleared cell lysates were incubated with anti-FLAG M2 magnetic beads (Sigma) for 1 h at room temperature. Afterward, the beads were collected by a magnetic separator (Promega) and washed three times with lysis buffer. The complex was eluted by incubation with 30 μl of FLAG peptides (1 mg/ml) or SDS loading buffer in the presence of DTT. The immunopurified eluents were separated in SDS-8% polyacrylamide gel, and Western blot analysis was performed using appropriate antibodies.
The cross-linking reaction was carried out as before with modification (41). Cells in 10-cm dishes were treated with 10 μm proteasome inhibitor MG-132 for 2 h before harvesting. Cells were then washed with DPBS and cross-linked by incubation with 1.5 mm dithiobis(succinimidyl propionate) (DSP) in DPBS for 15 min at room temperature. The DSP reaction was quenched by the addition of 10 mm Tris buffer, pH 7.5. Cells were then scraped into microtubes, pelleted, and washed with DPBS. Then cells were lysed in the Triton X-100 lysis buffer, and the total cell lysates were immunoprecipitated with a mouse anti-α1 antibody or anti-FLAG M2 magnetic beads. The immunoisolated eluents were blotted with rabbit anti-OS-9 and rabbit anti-α1 subunit antibodies.
In Vivo Ubiquitination Assay
HEK293 cells stably expressing (FLAG-α1)β2γ2 GABAA receptors were transfected with HA-tagged ubiquitin constructs together with the indicated siRNAs or plasmids. Two hours before harvesting cells, 10 μm proteasome inhibitor MG-132 was added. Forty eight hours post-transfection, Triton X-100 cell extracts (500 μg) were immunoprecipitated with anti-FLAG M2 magnetic beads, and the immunoisolated eluents were subjected to SDS-PAGE and blotted with a mouse anti-ubiquitin antibody.
In Vitro Binding Assay
One μg of GST epitope tag protein (GST) (Novus Biologicals, catalogue no. NBC1-18537) or GST-tagged GABAA receptor α1 subunit protein (GST-α1) (Abnova, catalogue no. H00002554-P01) was mixed with 4 μg of recombinant His-tagged Grp94 (ProSpec, catalogue no. hsp-091) protein in 500 μl of buffer (50 mm Tris, pH 7.5, 150 mm NaCl, and 1% Triton X-100). The protein complex was isolated by immunoprecipitation using anti-His antibody followed by SDS-PAGE and Western blot analysis with a rabbit anti-GABAAR α1 subunit antibody (R&D Systems PPS022) or a mouse anti-GST (Novus Biologicals NB600-446) antibody.
Mouse Brain Homogenization
C57BL/6J mice (The Jackson Laboratory) at 6–10 weeks were sacrificed, and the cortex was isolated and homogenized in lysis buffer (25 mm Tris, pH 7.6, 150 mm NaCl, 1 mm EDTA, and 2% Triton X-100) supplemented with the complete protease inhibitor mixture from Roche Applied Science. The sample was centrifuged at 800 × g for 10 min at 4 °C. The pellet was re-homogenized in the same lysis buffer and centrifuged at 800 × g for 10 min at 4 °C. The combined supernatants were placed on a rotating device for 2 h at 4 °C and then centrifuged at 15,000 × g for 30 min at 4 °C. The resulting supernatant was collected as mouse brain homogenate, and its protein concentration was determined by a MicroBCA assay (Pierce). This animal study followed the guidelines of the Institutional Animal Care and Use Committees (IACUC) at Case Western Reserve University and was carried out in agreement with the recommendation of the American Veterinary Medical Association Panel on Euthanasia.
Whole-cell Patch Clamp Electrophysiology Recording
Whole-cell currents were recorded using monoclonal HEK293 cells stably expressing α1(A322D)β2γ2 receptors. The glass electrodes were pulled from thin-walled borosilicate capillary glass (Kimble-Chase) and fire-polished on a DMZ Universal puller (Zeitz Instruments), having a tip resistance of 3–5 megohms. The internal solution contained 153 mm KCl, 1 mm MgCl2, 5 mm EGTA, 10 mm HEPES, and 2 mm MgATP, pH 7.3. The external solution contained 142 mm NaCl, 8 mm KCl, 6 mm MgCl2, 1 mm CaCl2, 10 mm glucose, 10 mm HEPES, and 120 nm fenvalerate, pH 7.4. Coverslips containing HEK293 cells were placed in an RC-25 recording chamber (Warner Instruments) on the stage of an Olympus IX-71 inverted fluorescence microscope and superfused with external solution. Fast GABA application was accomplished with a pressure-controlled perfusion system (Warner Instruments) positioned within 50 μm of the cell utilizing a Quartz MicroManifold with 100-μm inner diameter inlet tubes (ALA Scientific). The whole-cell GABA-induced currents were recorded at a holding potential of −60 mV in voltage clamp mode using an Axopatch 200B amplifier (Molecular Devices). The signals were filtered at 2 kHz and detected at 10 kHz using pClamp10 acquisition software.
Statistical Analysis
All data are presented as mean ± S.E., and any statistical significance was calculated using two-tailed Student's t test.
Results
Grp94 Positively Regulates the ERAD of the α1 Subunit of GABAA Receptors
Grp94, a metazoan-specific member of the Hsp90 family in the ER lumen, plays an important role in the recognition of the following two ERAD substrates: mutant α1-antitrypsin (42, 43) and mutant myocilin (44). Probably a more prominent role of Grp94 is to assist the folding of a very limited subset of client proteins, including immunoglobulins (45), multiple Toll-like receptors and integrins (46, 47), insulin-like growth factors (48), and low density lipoprotein receptor-related protein 6 (LRP6) (49). Therefore, we began to clarify whether Grp94 promotes the folding or degradation of the WT α1 subunit of GABAA receptors. Transient knockdown of Grp94 using small interfering RNA (siRNA) (49% knockdown efficiency) significantly increased the total α1 subunit protein level in HEK293 cells stably expressing WT α1β2γ2 GABAA receptors (Fig. 1B). This suggested a possible role of Grp94 in positively regulating the ERAD of α1 subunits. Furthermore, stable depletion of Grp94 using shRNA (>95% knockdown efficiency) in HEK293T cells resulted in a more dramatic increase of the steady state level of the α1 subunit (Fig. 1C), indicating that the effect of Grp94 on the α1 subunit does not depend on whether the depletion of Grp94 is short term or long term (also see under “Discussion”).
Quantitative RT-PCR analysis showed that reducing the Grp94 level genetically did not significantly increase the mRNA level of CHOP, a pro-apoptotic marker, indicating that this operation did not cause cell toxicity (Fig. 1D). Also, it has been reported that silencing Grp94 only induces the expression of a limited set of proteostasis network components, which is distinct from activating the unfolded protein response (39). In response to misfolded proteins in the ER, the unfolded protein response adjusts the cellular proteostasis by translational attenuation and transcriptional remodeling of the ER proteostasis network, including some ERAD components (50, 51). Therefore, it is unlikely that Grp94 acts as a general ERAD substrate-recruiting chaperone.
We next manipulated the Grp94 level genetically and evaluated the influence on the degradation rate of α1 subunits using the CHX chase assay. The α1 subunit has a half-life of 1.6 h when fitted to a single exponential function (Fig. 1E). Remarkably, transiently depleting Grp94 using siRNA substantially increased its half-life to 3.5 h (Fig. 1E), indicating that Grp94 positively regulates the α1 subunit degradation. Consistently, permanent inactivation of Grp94 reduced the degradation rate of the α1 subunit (Fig. 1F), and overexpression of HA-tagged Grp94 significantly decreased the half-life of the α1 subunit to 0.8 h (Fig. 1G).
Moreover, we determined the effect of Grp94 in regulating the ubiquitination of the α1 subunit. HEK293 cells expressing FLAG-α1β2γ2 GABAA receptors were transfected with HA-tagged ubiquitin, immunoprecipitated with anti-FLAG magnetic beads, and blotted for ubiquitin. The ratio of ubiquitin to α1 subunit post-immunoprecipitation represents the relative ubiquitination level of the α1 subunit. This in vivo ubiquitination assay clearly demonstrated that knockdown of Grp94 (55% knockdown efficiency) decreased the relative ubiquitination level of the α1 subunit (Fig. 1H), indicating that Grp94 positively regulates the ubiquitination and ERAD of the α1 subunit. As a result, transiently or stably depleting Grp94 significantly increased the surface α1 subunit protein level using a cell surface biotinylation assay (Fig. 1, B and C).
Grp94 Uses Its Middle Domain to Bind the α1 Subunit of GABAA Receptors
Because operating Grp94 genetically could influence the ER proteostasis network, we asked whether Grp94 directly binds the α1 subunit. We carried out an in vitro binding assay using recombinant GST-tagged α1 subunits and recombinant His-tagged Grp94 proteins. The His tag antibody pulldown led to the detection of the α1 subunit in the GST-α1 complex using an anti-α1 antibody or an anti-GST antibody, although we did not detect any α1 band in the GST control complex (Fig. 2A), indicating that Grp94 directly binds the α1 subunit. However, because the folding degree of the recombinant α1 subunit was not characterized, whether such an interaction is functionally relevant to its ERAD remains to be established. To show an endogenous interaction between Grp94 and the α1 subunit in the mammalian central nervous systems, we performed a co-immunoprecipitation assay using the mouse brain cortex homogenates. Pulling down Grp94 led to the detection of the α1 subunit (Fig. 2B), confirming an endogenous interaction between Grp94 and the α1 subunit.
FIGURE 2.

Grp94 uses its middle domain to bind the α1 subunit of GABAA receptors. A, recombinant His-tagged Grp94 binds the recombinant GST-tagged α1 subunit of GABAA receptors in vitro. One μg of GST or GST-α1 was mixed with 4 μg of His-Grp94 in buffers containing 1% Triton X-100. The protein complex was isolated by immunoprecipitation using an anti-His antibody, and the immunopurified eluents were separated by SDS-PAGE and blotted with a rabbit anti-GABAAR α1 subunit antibody and a mouse anti-GST antibody (n = 4). B, endogenous Grp94 binds the endogenous α1 subunit of GABAA receptors in the mouse brain. Total lysates from the cortex of mouse brains were used for immunoprecipitation with a rat anti-Grp94 antibody or control rat IgG, and the immunoisolated eluents were subjected to SDS-PAGE and Western blot analysis with rat anti-Grp94 and goat anti-α1 antibodies (n = 2). C, schematic representation of HA-tagged Grp94 constructs. Full-length Grp94 contains a signal peptide (SP, residues 1–21), an N-terminal domain (N, residues 22–341), a middle domain (M, residues 342–601), and a C-terminal domain (C, 602–785). All constructs contain a signal peptide (residues 1–21), an HA tag that was inserted between residues 796 and 797, and a sequence containing KDEL for ER localization (786–803). FL, full-length, 1–803. NM, N and M domains, 1–601 and 786–803. MC, M and C domains, 1–21 and 342–803. N, N domain, 1–341 and 786–803. M, M domain, 1–21, 342–601, and 786–803. C, C domain, 1–21 and 602–803. D, interaction between different Grp94 domains and the α1 subunit. HEK293 cells stably expressing (FLAG-α1)β2γ2 GABAA receptors and empty vector-transfected HEK293 cells were transiently transfected with different HA-tagged variant Grp94 plasmids for 48 h. Then Triton X-100 cell extracts were immunoprecipitated with anti-FLAG M2 magnetic beads, and the immunoisolated eluents were subjected to SDS-PAGE and blotted with rabbit anti-HA and rabbit anti-α1 subunit antibodies (n = 2). EV, empty vector. IB, immunoblotting. IP, immunoprecipitation.
We next characterized the cellular interaction between Grp94 and the α1 subunit. FLAG-tagged α1 subunits strongly interacted with endogenous Grp94 in HEK293 cells (Fig. 1H). Grp94 is divided into three domains as follows: an N-terminal domain containing an ATP-binding site (N domain), a middle domain (M domain), and a C-terminal domain containing dimerization sites (C domain) (52). The specific domain that Grp94 uses to interact with its substrates may vary for different substrates. For example, it was reported that Grp94 uses its C-terminal domain to bind Toll-like receptors (53) but uses its M domain to interact with OS-9 (54). To determine which domain of Grp94 interacts with the α1 subunit in the ER lumen, we constructed plasmids containing an HA-tagged full-length N domain, M domain, C domain, NM domain, or MC domain of Grp94 (Fig. 2C). HEK293 cells expressing (FLAG-α1)β2γ2 GABAA receptors were transiently transfected with these HA-tagged Grp94 variant plasmids for 48 h before being immunoprecipitated using an anti-FLAG antibody and blotted for HA. Clearly, the M domain, but not the N domain or the C domain, was pulled down with the α1 subunit (Fig. 2D, cf. lanes 13 to 12 and 14), indicating that the M domain alone is sufficient to bind the α1 subunit. The M construct migrated as two bands in the SDS-polyacrylamide gel; the lower band corresponds to the molecular weight of a monomer, and the upper band corresponds to that of a dimer, which was resistant to the SDS denaturation (Fig. 2D, lane 6). Only the fast migrating M species interacted with the α1 subunit (Fig. 2D, lane 13), possibly because the putative dimerization of the M domain prevented its association with the α1 subunit. Intriguingly, the α1 subunit interacted with the M domain and the MC domain much stronger than the full-length Grp94 (Fig. 2D, cf. lanes 11 and 13 to 9). However, the NM domain was not pulled down with the α1 subunit (Fig. 2D, lane 10), which could be due to the possibilities that the addition of the N domain to the M domain blocked the interaction between the M domain and the α1 subunit or altered the conformation of the M domain.
OS-9 Acts Downstream of Grp94 to Recognize Misfolded α1 Subunits of GABAA Receptors through a Glycan-dependent Manner
The α1 subunit has two N-linked glycosylation sites, Asn-38 and Asn-138. The oligosaccharyltransferase complex catalyzes this glycosylation and transfers 14-monosaccharide residues Glc3Man9GlcNAc2 to an Asn residue contained in an Asn-Xaa-(Ser/Thr) sequence motif (Xaa can be any residue except Pro). N-Glycans serve as sensors of glycoprotein folding in the ER (55, 56). The recognition of terminally misfolded N-linked glycoproteins is mediated by mannose trimming events, which are facilitated by ER-mannosidase I and EDEM proteins after the release of glycoproteins from the calnexin/calreticulin cycles (57, 58). Afterward, two ER-resident lectins, OS-9 and XTP3-B, recognize the trimmed oligosaccharides (42). OS-9 and XTP3-B seem to have distinct sets of substrates. OS-9, but not XTP3-B, robustly binds Grp94 (42). Therefore, it is possible the Grp94-mediated ERAD substrates depend on OS-9. Because Grp94 positively regulates the ERAD of the α1 subunit, we next determined the role of OS-9 in this degradation process and the potential sequential effect of OS-9 with Grp94. Knockdown of OS-9 (71% knockdown efficiency) significantly increased the total α1 subunit protein level 3.0-fold (Fig. 3A), supporting that OS-9 recognizes the misfolded α1 subunit for ERAD. To access the role of N-glycans in the function of OS-9, we introduced the N38Q/N138Q double mutation at both glycosylation sites (Asn-38 and Asn-138) in the α1 subunit. This QQ mutation reduced the mass and protein level of the α1 subunit (Fig. 3B, cf. 2nd to 1st lane and 6th to 5th lane), indicating that glycosylation is required for the maturation of this subunit. A co-immunoprecipitation assay revealed that the QQ mutation significantly decreased the interaction between OS-9 and the α1 subunit according to the quantification of the ratio of OS-9 to α1 after immunoprecipitation using anti-α1 antibody in HEK293 cells (Fig. 3B, cf. 6th to 5th lane). This indicates that OS-9 binds the N-glycans in the α1 subunit. The above data (Fig. 3, A and B) supported the assertion that OS-9 recognizes misfolded α1 subunit in a glycan-dependent manner.
FIGURE 3.

OS-9 acts downstream of Grp94 to promote ERAD of the α1 subunit of GABAA receptors. A, OS-9 knockdown increases the total protein level of WT α1 subunits. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting (NT) control siRNA or siRNA against OS-9. Forty eight hours post-siRNA transfection, cells were lysed, and the total cell lysates were subjected to SDS-PAGE and immunoblotting with mouse anti-α1 or rabbit anti-OS-9 antibodies (n = 3). β-Actin serves as a protein loading control. Quantification of the total protein level of α1 is shown on the bottom. B, OS-9 interacts with the α1 subunit in a glycan-dependent way. The α1 subunit has two N-linked glycosylation sites at Asn-38 and Asn-138. The N38Q/N138Q double mutations in the α1 subunit disrupt both glycosylation sites. HEK293 cells were transiently transfected with WT α1 or N38Q/N138Q α1 plasmids (together with β2 and γ2 subunit plasmids). Cells were treated with 10 μm proteasome inhibitor MG-132 for 2 h before harvesting. Forty eight hours post-transfection, cells were washed with DPBS and cross-linked by incubation with 1.5 mm DSP for 15 min at room temperature. DSP was quenched by the addition of 10 mm Tris buffer, pH 7.5. Then cells were lysed, and the total cell lysates were immunoprecipitated with a mouse anti-α1 antibody, and the immunoisolated eluents were blotted with rabbit anti-OS-9 and rabbit anti-α1 subunit antibodies (n = 4). IgG was included as a negative control for immunoprecipitation. Quantification of the intensity of OS-9/α1 subunit post-IP represents the relative interaction between OS-9 and the α1 subunit and is shown on the bottom. The QQ mutation reduces the mass of the α1 subunit (cf. 4th to 3rd lane). Consequently, it leads to significantly weaker interactions between the α1 subunit and OS-9. C, Grp94 knockdown decreases the interaction between OS-9 and the α1 subunit. HEK293 cells stably expressing (FLAG-α1)β2γ2 GABAA receptors were transfected with non-targeting control siRNA or siRNA against Grp94. Cells were treated with MG-132 (10 μm, 2 h) before harvesting. Forty eight hours post-transfection, cells were cross-linked with DSP and lysed. Then Triton X-100 cell extracts were immunoprecipitated with anti-FLAG M2 magnetic beads, and the immunoisolated eluents were blotted with rabbit anti-OS-9 and rabbit anti-α1 subunit antibodies (n = 4). Quantification of the relative intensity of OS-9/α1 post-IP is shown on the bottom. D, OS-9 knockdown, MG-132 treatment, or EER1 treatment enhances the interaction between the α1 subunit and Grp94. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting siRNA and siRNA against Grp94 for 48 h or treated with MG-132 (2 μm, 6 h), a potent proteasome inhibitor, and eeyarestatin I (EER1) (5 μm, 24 h), a potent VCP inhibitor. The cell lysates were then subjected to immunoprecipitation before SDS-PAGE and Western blot analysis with rat anti-Grp94 and rabbit anti-α1 antibodies (n = 3). Quantification of the intensity of Grp94/α1 subunit post-IP represents their relative interaction and is shown on the bottom. E, Grp94 knockdown enhances the interaction between the α1 subunit and calnexin. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting control siRNA or siRNA against Grp94 for 48 h. Then Triton X-100 cell extracts were immunoprecipitated with a mouse anti-α1 antibody, and the immunoisolated eluents were blotted with rabbit anti-calnexin (CNX) and rabbit anti-α1 subunit antibodies (n = 3). Quantification of the relative intensity of calnexin/α1 post-IP is shown on the bottom. Each data point is reported as mean ± S.E. *, p < 0.05. IB, immunoblot.
We next determined the sequential interactions of calnexin, Grp94, and OS-9 with the α1 subunit during its biogenesis pathway. We began to evaluate the binding sequence of Grp94 and OS-9 with the α1 subunit. Knocking down Grp94 decreased the interaction between the α1 subunit and OS-9 significantly (Fig. 3C, cf. 6th to 5th lane), and consistently depleting OS-9 increased the interaction between the α1 subunit and Grp94 significantly in HEK293 cells (Fig. 3D), indicating that Grp94 acts upstream of OS-9 during the recognition of misfolded α1 subunits. Previously, we reported that calnexin binds the α1 subunits through the glycans and promotes their maturation (40). If the α1 subunits that exit the calnexin cycle fail to fold into their native structures, they are likely the substrate of Grp94. To determine whether Grp94 functions downstream of calnexin, we carried out a co-immunoprecipitation assay. Knockdown of Grp94 significantly increased the interaction between the α1 subunit and calnexin in HEK293 cells expressing α1β2γ2 receptors (Fig. 3E), indicating that Grp94 acts downstream of calnexin and accepts misfolded α1 subunits after they depart from calnexin. Afterward, Grp94 delivers the substrate to OS-9 for processing, and then the substrate will be further tagged by ubiquitin for degradation.
Hrd1 Ubiquitinates the α1 Subunit of GABAA Receptors
The mammalian ERAD systems are organized primarily by two E3 ubiquitin ligases, Hrd1 and gp78 (36), and additional ubiquitin ligases contribute to the degradation of a limited amount of substrates. Hrd1 and gp78 are both homologous to Hrd1p, an essential E3 ligase in yeast (59), but Hrd1 and gp78 have largely distinct interaction networks (36). Moreover, gp78 acts downstream of Hrd1 to enhance the ERAD of a model luminal substrate, a truncated major histocompatibility complex class I heavy chain molecule (MHC-1–147) (60). We evaluated the role of Hrd1 and gp78 in degrading the α1 subunit. Knockdown of Hrd1 using siRNA increased the total protein level of the α1 subunit in HEK293 cells stably expressing α1β2γ2 GABAA receptors (Fig. 4, A and B), whereas knockdown of gp78 did not (Fig. 4C), indicating that Hrd1, but not gp78, plays an essential role for this ERAD substrate. Consistently, overexpression of Hrd1 substantially decreased the total protein level of the α1 subunit (Fig. 4, D and E). Moreover, we utilized a C291A mutation, which resides in the critical RING domain in Hrd1, exhibiting a dominant negative effect on its ubiquitin ligase activity (61). As a result, overexpression of C291A Hrd1 significantly increased the total α1 subunit level (Fig. 4, D and E). We next employed the cycloheximide chase assay to evaluate the degradation rate of the α1 subunit. As expected, overexpression of Hrd1 accelerates the degradation of the α1 subunit, but repressing Hrd1 activity by overexpressing C291A Hrd1 slows its degradation (Fig. 4F). We then determined the effect of Hrd1 in ubiquitinating the α1 subunit. Cell ubiquitination assays showed that knockdown of Hrd1 (Fig. 4G) or overexpression of C291A Hrd1 (Fig. 4H) decreased the ubiquitinated subunit as well as the ratio of ubiquitinated to total α1 subunit. Consistently, overexpression of Hrd1 enhanced the ubiquitination of the α1 subunit (Fig. 4H). The above data (Fig. 4, A–H) unambiguously demonstrated that Hrd1 plays an essential role in ubiquitinating the α1 subunit during its ERAD pathway. As a result, inhibiting Hrd1 by using siRNA or overexpressing C291A Hrd1 increased the surface protein level of the α1 subunit, and overexpressing Hrd1 decreased its surface level significantly (Fig. 4, A and D).
FIGURE 4.

Hrd1 ubiquitinates the α1 subunit of GABAA receptors. A and B, Hrd1 knockdown increases the total and cell surface protein level of α1 subunit. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with non-targeting (NT) control siRNA or siRNA against Hrd1. Forty eight hours post-siRNA transfection, cells were lysed, and the total cell lysates were subjected to SDS-PAGE and immunoblotting (IB) with mouse anti-α1 or rabbit anti-Hrd1 antibodies (n = 3). β-Actin serves as a total protein loading control. Alternatively, the surface α1 subunits were measured using cell surface protein biotinylation assay (n = 3). The Na+/K+-ATPase α chain serves as a loading control for biotinylated membrane proteins. Protein band quantifications using ImageJ are shown in B. C, knocking down gp78 does not change the total protein level of the α1 subunit. HEK293 cells stably expressing α1β2γ2 receptors were transfected with non-targeting control siRNA or siRNA against gp78 for 48 h. Then the cell lysates were subjected to SDS-PAGE and Western blotting assay (n = 2). D and E, effect of overexpressing Hrd1 (WT and C291A) on the total and cell surface protein levels of α1 subunits. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with a GFP control plasmid, a WT Hrd1 plasmid, or a C291A Hrd1 plasmid. Forty eight hours post-transfection, cells were treated as in A to detect the total and cell surface α1 protein levels (n = 3). GFP serves as a negative control. The C291A mutation in Hrd1 has a dominant negative effect on its E3 ligase activity. Protein band quantifications are shown in E. F, influence of overexpressing Hrd1 (WT and C291A) on the degradation rate of the α1 subunit determined by CHX chase analysis. HEK293 cells stably expressing α1β2γ2 GABAA receptors were transfected with a GFP control plasmid, a WT Hrd1 plasmid, or a C291A Hrd1 plasmid for 48 h and then chased for the indicated time with CHX (100 μg/ml). Cells were lysed and subjected to SDS-PAGE and immunoblotting analysis (n = 3). The degradation kinetics were plotted by quantifying the α1 protein intensity against time after CHX addition on the bottom panel. G and H, influence of knocking down or overexpressing Hrd1 on the relative ubiquitination level of α1 subunits. HEK293 cells stably expressing (FLAG-α1)β2γ2 GABAA receptors were transfected with an HA-tagged ubiquitin plasmid together with non-targeting control siRNA or siRNA against Hrd1 for 48 h (G) or transfected with a HA-tagged ubiquitin plasmid together with a GFP control plasmid, a WT Hrd1 plasmid, or a C291A Hrd1 plasmid for 48 h (H). Then the total cell lysates were immunoprecipitated with anti-FLAG M2 magnetic beads. The immunoisolated complexes were subjected to SDS-PAGE and Western blot analysis. Quantification of the ratio of ubiquitin/α1 band intensity post-IP, representing relative ubiquitin level of the α1 subunit, is shown on the bottom (n = 3). I, Grp94 knockdown decreases the interaction between VCP or HRD1 and the α1 subunit. HEK293 cells expressing (FLAG-α1)β2γ2 GABAA receptors were transfected with non-targeting control siRNA or siRNA against Grp94 for 48 h. Then the total cell lysates were immunoprecipitated for SDS-PAGE and Western blot analysis. Quantification of the ratio of VCP or HRD1/α1 band intensity post-IP is shown on the bottom (n = 3). Each data point in B and E–I is reported as mean ± S.E. *, p < 0.05; **, p < 0.01.
VCP, an AAA+ ATPase, plays an essential role in extracting polyubiquitinated substrates from the ER to the cytosol and driving this retrotranslocation process (62, 63). Recently, we showed that VCP binds the WT α1 subunit and that inhibiting VCP increased its protein level, supporting VCP's critical role in degrading the α1 subunit (15). Because Grp94 acts early in the ERAD recognition step for the α1 subunit, we expected that Grp94 functions upstream of Hrd1 and VCP. Indeed, a co-immunoprecipitation assay demonstrated that knockdown of Grp94 significantly decreased the interaction between the α1 subunit and VCP/Hrd1 (Fig. 4I), confirming such a sequential binding. Moreover, application of eeyarestatin I, a potent VCP inhibitor (64), increased the interaction between the α1 subunit and Grp94 (Fig. 3D, 5th lane), further supporting the idea that Grp94 operates upstream of VCP. In addition, as expected, treatment with MG-132, a potent proteasome inhibitor, accumulated more α1 subunits in the ER and increased their interaction with Grp94 (Fig. 3D, 4th lane).
Inhibiting Grp94 Enhances the Functional Surface Expression of the Pathogenic α1(A322D) Subunit of GABAA Receptors
Numerous mutations in GABAA receptors predispose them to misfolding and excessive ERAD (29). A prototype of such mutations is the A322D mutation in the α1 subunit, causing autosomal dominant juvenile myoclonic epilepsy (31). Recently, we demonstrated that reducing ERAD by inhibiting VCP slows the degradation of the α1(A322D) subunit and promotes its forward trafficking (15). Because Grp94 acts early in the ERAD recognition step, which is upstream of VCP (Fig. 4I), we next evaluated whether inhibiting Grp94 enhances the functional surface expression of the α1(A322D) subunit. Strikingly, knockdown of Grp94 (42% knockdown efficiency) significantly increased the total α1(A322D) protein level 2.4-fold and the cell surface α1(A322D) protein level 2.8-fold in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors (Fig. 5A). Notably, the effect of Grp94 knockdown on the α1 protein level increase is more dramatic in cells expressing the α1(A322D) subunit than those expressing the WT α1 subunit (Figs. 1B and 5A), presumably because the α1(A322D) subunit degrades much faster than the WT α1 subunit. Moreover, modest knockdown of Grp94 (36% knockdown efficiency) significantly increased the total α1(A322D) protein level 2.0-fold and the cell surface α1(A322D) protein level 1.5-fold in human neuronal SH-SY5Y cells stably overexpressing α1(A322D)β2γ2 GABAA receptors (Fig. 5B). Consistently, application of BNIM (5 μm), a specific, potent Grp94 inhibitor (37), increased the surface expression of the α1(A322D) subunit 2.0-fold (Fig. 5C).
FIGURE 5.

Grp94 inhibition enhances the functional surface expression of the α1(A322D) subunit of GABAA receptors. A and B, Grp94 knockdown increases the total and cell surface protein levels of α1(A322D) subunits in HEK293 cells (A) and neuronal SH-SY5Y cells (B) (n = 3). Cells stably expressing α1(A322D)β2γ2 receptors were transfected with non-targeting (NT) control siRNA or siRNA against Grp94 for 48 h before total protein analysis by Western blot assay and surface protein analysis by a surface biotinylation assay. Protein band quantifications are shown on the bottom. The Na+/K+-ATPase α chain serves as a loading control for biotinylated membrane proteins. C, treatment with a Grp94-specific inhibitor BNIM (5 μm, 24 h) increases the surface protein level of the α1(A322D) subunit in HEK293 cells stably expressing α1(A322D)β2γ2 receptors according to surface biotinylation analysis (n = 3). The chemical structure of BNIM is shown on top. D, Grp94 knockdown significantly increases the peak amplitude of GABA (3 mm)-induced chloride currents in HEK293 cells expressing α1(A322D)β2γ2 GABAA receptors using a whole-cell patch clamp recording. The holding potential was set at −60 mV. Representative traces are shown on the top panel, and quantification of the peak currents (Imax) is shown on the bottom panel. The number of patches cells in each group is shown at the top of the bar. Each data point is reported as mean ± S.E. *, p < 0.05; **, p < 0.01.
To determine whether the increased surface α1(A322D) subunits form functional receptors on the plasma membrane, we performed a whole-cell patch clamping experiment to record GABA-induced chloride current. To ensure that recording was done for successfully transfected cells, Grp94 siRNA was co-transfected with an mCherry plasmid in monoclonal HEK293 cells expressing the α1(A322D) β2γ2 GABAA receptors, and only mCherry-positive cells were selected for recording. Grp94 knockdown significantly increased the peak GABA-induced chloride current to 46 pA (Fig. 5D), demonstrating a partial restoration of function for epilepsy-associated α1(A322D) GABAA receptors.
Discussion
Fig. 6 illustrates the first ERAD pathway model of the α1 subunit of GABAA receptors. Upon entering the ER, newly synthesized α1 subunits tend to fold with the assistance of BiP (also termed Grp78) in a glycan-independent manner and with the support from calnexin in a glycan-dependent way. BiP is known to play a role very early in protein biogenesis by binding to the hydrophobic patches of unfolded proteins in the ER, facilitating their folding while preventing aggregation (45, 65). Calnexin, a membrane-bound lectin chaperone, binds monoglucosylated N-glycans installed on a glycoprotein and facilitates its folding with the assistance of protein-disulfide isomerases, such as ERp57 (66). Recently, we demonstrated that both BiP and calnexin facilitate the maturation of WT α1 subunits (40). After the collaborative operations of BiP and calnexin, if native structures are obtained, α1 subunits assemble with other subunits, such as β2 subunits and γ2 subunits, to form a pentamer on the ER membrane. This pentamer traffics efficiently to the plasma membrane in a fully functional state. However, if α1 subunits misfold, Grp94 recognizes their non-native states in the ER lumen. OS-9 acts downstream of Grp94 to further recognize the misfolded subunits in a glycan-dependent manner. Grp94 and OS-9 deliver misfolded α1 subunits to Hrd1, which ubiquitinates them. VCP facilities the dislocation of the α1 subunits from the ER member to the cytosol, where they will be targeted to the 26S proteasome for degradation.
FIGURE 6.
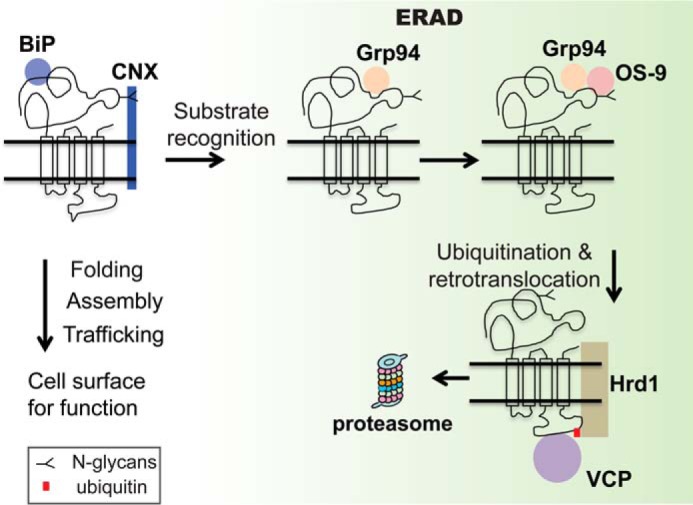
Proposed ERAD model of the α1 subunit of GABAA receptors. The α1 subunits are co-translationally translocated to the ER membrane. They tend to fold with the assistance of BiP in a glycan-independent manner and calnexin (CNX) in a glycan-dependent manner. Properly folded α1 subunits assemble with other subunits to form a heteropentamer on the ER membrane for trafficking to the plasma membrane. However, if α1 subunits misfold, Grp94 recognizes the non-native states in the ER lumen. In the ER lumen, OS-9 further recognizes the misfolded subunits in a glycan-dependent manner and delivers them to Hrd1. Hrd1 ubiquitinates α1 subunits and translocates them from the ER member to the cytosol with the assistance of VCP. Then misfolded α1 subunits are targeted to the 26S proteasome for degradation.
Many important questions remain to be answered for the ERAD pathway of the α1 subunit. E3 ligases play a central role in organizing the ERAD machinery. There are over 500 ubiquitin E3 ligases in mammals. It is believed that one protein uses only a subset of those E3 ligases. What other E3 ligases play an important role in the ubiquitination of the α1 subunits? Recently, it was reported that ring finger protein 34 (RNF34) promotes the degradation of the γ2 subunit by ubiquitinating them (67). Furthermore, our recent proteomics study identified several E3 ligases that interact with the ρ1 subunit, such as HUWE1 and UBR5 (68). It will be of great interest to determine whether these E3 ligases complement the function of Hrd1 in ubiquitinating the α1 subunits. In addition, presumably Grp94 and OS-9 act on the misfolded domains of the α1 subunit in the ER lumen. Whether and how the transmembrane domains of the α1 subunit contribute to the ERAD recognition steps remain to be determined. Beyond the ERAD factors that were identified here, we still need to decipher the role of many known ERAD factors in the context of degrading the α1 subunit, such as EDEM and Derlin family proteins. Therefore, substantial further studies need to be done to fully understand ERAD of the α1 subunit. Nonetheless, our report represents a significant progress toward that end.
We clarified that Grp94 promotes the ERAD of the α1 subunits of GABAA receptors. Unlike its cytosolic Hsp90 family members, Grp94 only has a few known client proteins or substrates in the ER, and the exact function and mechanism of Grp94 on the folding or ERAD of its substrates remains to be established (52). The effect of Grp94 on the GABAA receptor ERAD does not depend on whether the depletion of Grp94 is short term or long term, which is different from that on the null Hong Kong variant of α1-antitrypsin (NHK). Previously, it was reported that transient knockdown of Grp94 delayed the degradation and increased the steady state level of NHK (42, 43), whereas stably depleting Grp94 had no apparent effect (54). Up-regulation of BiP by transient knockdown of Grp94 might contribute to such a discrepancy on NHK; alternatively, long term depletion of Grp94 might result in cell adaption for the altered proteostasis network. A similar discrepancy was reported between short term or long term depletion of gp78; transient knockdown of gp78 using siRNA attenuated the degradation of a model ERAD substrate, MHC(1–147), whereas permanent depletion of gp78 using CRISPR cells had no effect (60). It was proposed that cells can adapt to compensate for the function of gp78 during the permanent gp78 inactivation process (60). Therefore, one possibility is that if an ERAD factor is essential for the disposal of one substrate, cells cannot adapt to the loss of that ERAD factor; if not essential, cells can adjust by adapting the proteostasis network. Our results supported that the ERAD of GABAA receptors critically depends on Grp94, adding GABAA receptor subunits to the very limited pool of Grp94 substrates. Because the β-sheet-rich α1 subunit has distinct structures compared with other known Grp94 substrates, how this subunit is recognized by Grp94 remains to be investigated. Grp94 inhibition stabilizes α1 subunits and enhances their functional surface expression. Our results strongly support that Grp94 regulates GABAA receptor function directly because Grp94 binds the α1 subunit in vitro and in vivo. Grp94 inhibition has a much greater effect on α1(A322D) subunits than WT α1 subunits. Therefore, specific Grp94 inhibition provides a promising way to enhance the function of misfolding-prone α1 subunits, as a therapeutic strategy to ameliorate idiopathic epilepsy. To achieve further specificity, it is also of interest to disrupt the direct interaction between Grp94 and α1 subunits.
For fast-degraded ERAD substrates, it is desirable to enhance folding as well as reducing ERAD to achieve substantial functional rescue. Recently, we identified different classes of small molecule proteostasis regulators that enhance the folding and trafficking and thus the function of epilepsy-associated mutant GABAA receptors (40, 69). Suberanilohydroxamic acid increases BiP protein level and the interaction between calnexin and the α1(A322D) subunit, which promotes the functional cell surface expression of this mutant subunit (40). Verapamil, an L-type Ca2+ channel blocker, enhances the plasma membrane trafficking of the α1(D219N) subunit by promoting calnexin-assisted folding and inter-subunit assembly between α1(D219N) subunit and β2 subunit (69). In addition, pharmacological chaperones, which are small molecules that directly bind their target protein, stabilize it and enhance its forward trafficking (70). Both proteostasis regulators and pharmacological chaperones are expected to yield an additive/synergistic effect with ERAD inhibitors to enhance the function of GABAA receptors. Indeed, recently, we revealed that combining suberanilohydroxamic acid treatment with eeyarestatin I, a VCP inhibitor, additively restored the functional cell surface expression of the α1(A322D) subunit, providing a proof-of-principle case (15). The elucidation of the ERAD pathway of GABAA receptors allows the development of a more effective combination therapy to restore the function of disease-causing GABAA receptors.
GABAA receptors are within the superfamily of Cys-loop receptors, which contain important neuroreceptors, including nicotinic acetylcholine receptors, serotonin type 3 receptors, and glycine receptors (2, 11). They are responsible for fast excitatory and inhibitory transmission in nervous systems. Because they share common structural characteristics, much of our knowledge gained on the folding, assembly, and degradation of GABAA receptors could be extrapolated to the whole class of Cys loop receptors.
Author Contributions
X. J. D. conducted most of the experiments, analyzed the results, and wrote most of the paper. Y. J. W. conducted some immunoprecipitation experiments and wrote part of the paper. D. Y. H. conducted experiments on the electrophysiology recording. Y. L. F. carried out some molecular biology and immunoprecipitation experiments. A. S. D. synthesized BNIM. B. S. J. B. wrote part of the paper. T. W. M. conceived the idea for the project, analyzed the results, and wrote the paper with X. J. D. and Y. J. W.
Acknowledgments
We thank Professor Ron Kopito (Stanford University) for providing the human Hrd1 (WT and C291A) plasmids. We thank Professor Yair Argon (University of Pennsylvania) for stable shGrp94 HEK293 cells. We thank Professor John Christianson (Ludwig Institute for Cancer Research, Oxford, UK) for helpful discussions.
This work was supported by the Research Startup Fund from Case Western Reserve University School of Medicine (to T. W. M.), Epilepsy Foundation of America Grant 225243 (to T. W. M.), Clinical Translational Science Collaborative of Cleveland (CTSA) National Institutes of Health Grant UL1RR024989 from the NCRR and National Center for Advancing Translational Sciences (to T. W. M.), and National Institutes of Health Grants T32 HL007567 (to Y. J. W.) and CA109265 from NCI (to B. S. J. B.).
- ER
- endoplasmic reticulum
- ERAD
- ER-associated degradation
- IP
- immunoprecipitation
- TM
- transmembrane
- CHX
- cycloheximide
- VCP
- valosin-containing protein
- DPBS
- Dulbecco's PBS
- DSP
- dithiobis(succinimidyl propionate)
- NHK
- null Hong Kong.
References
- 1. Balch W. E., Morimoto R. I., Dillin A., and Kelly J. W. (2008) Adapting proteostasis for disease intervention. Science 319, 916–919 [DOI] [PubMed] [Google Scholar]
- 2. Fu Y.-L., Wang Y.-J., and Mu T.-W. (2016) Proteostasis maintenance of Cys-loop receptors. Adv. Protein Chem. Struct. Biol. 103, 1–23 [DOI] [PubMed] [Google Scholar]
- 3. Hipp M. S., Park S. H., and Hartl F. U. (2014) Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 24, 506–514 [DOI] [PubMed] [Google Scholar]
- 4. Powers E. T., and Balch W. E. (2013) Diversity in the origins of proteostasis networks–a driver for protein function in evolution. Nat. Rev. Mol. Cell Biol. 14, 237–248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Olzmann J. A., Kopito R. R., and Christianson J. C. (2013) The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 5, a013185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ruggiano A., Foresti O., and Carvalho P. (2014) Quality control: ER-associated degradation: protein quality control and beyond. J. Cell Biol. 204, 869–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Smith M. H., Ploegh H. L., and Weissman J. S. (2011) Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vembar S. S., and Brodsky J. L. (2008) One step at a time: endoplasmic reticulum-associated degradation. Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Guerriero C. J., and Brodsky J. L. (2012) The delicate balance between secreted protein folding and endoplasmic reticulum-associated degradation in human physiology. Physiol. Rev. 92, 537–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Macdonald R. L., and Olsen R. W. (1994) GABA(A) receptor channels. Annu. Rev. Neurosci. 17, 569–602 [DOI] [PubMed] [Google Scholar]
- 11. Lester H. A., Dibas M. I., Dahan D. S., Leite J. F., and Dougherty D. A. (2004) Cys-loop receptors: new twists and turns. Trends Neurosci. 27, 329–336 [DOI] [PubMed] [Google Scholar]
- 12. Mowrey D. D., Kinde M. N., Xu Y., and Tang P. (2015) Atomistic insights into human Cys-loop receptors by solution NMR. Biochim Biophys Acta 1848, 307–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Green W. N., and Millar N. S. (1995) Ion-channel assembly. Trends Neurosci. 18, 280–287 [PubMed] [Google Scholar]
- 14. Gorrie G. H., Vallis Y., Stephenson A., Whitfield J., Browning B., Smart T. G., and Moss S. J. (1997) Assembly of GABAA receptors composed of α1 and β2 subunits in both cultured neurons and fibroblasts. J. Neurosci. 17, 6587–6596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Han D. Y., Di X. J., Fu Y. L., and Mu T. W. (2015) Combining valosin-containing protein (VCP) inhibition and suberanilohydroxamic acid (SAHA) treatment additively enhances the folding, trafficking, and function of epilepsy-associated γ-aminobutyric acid, type A (GABAA) receptors. J. Biol. Chem. 290, 325–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., and Riordan J. R. (1995) Multiple proteolytic systems, including the proteasome, contribute to CFTR processing. Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 17. Ward C. L., Omura S., and Kopito R. R. (1995) Degradation of CFTR by the ubiquitin-proteasome pathway. Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 18. Pareek S., Notterpek L., Snipes G. J., Naef R., Sossin W., Laliberté J., Iacampo S., Suter U., Shooter E. M., and Murphy R. A. (1997) Neurons promote the translocation of peripheral myelin protein 22 into myelin. J. Neurosci. 17, 7754–7762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Alder N. N., and Johnson A. E. (2004) Cotranslational membrane protein biogenesis at the endoplasmic reticulum. J. Biol. Chem. 279, 22787–22790 [DOI] [PubMed] [Google Scholar]
- 20. Skach W. R. (2009) Cellular mechanisms of membrane protein folding. Nat. Struct. Mol. Biol. 16, 606–612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Barnes E. M. (2001) Assembly and intracellular trafficking of GABA(A) receptors. Int. Rev. Neurobiol. 48, 1–29 [DOI] [PubMed] [Google Scholar]
- 22. Connolly C. N., Krishek B. J., McDonald B. J., Smart T. G., and Moss S. J. (1996) Assembly and cell surface expression of heteromeric and homomeric γ-aminobutyric acid type A receptors. J. Biol. Chem. 271, 89–96 [DOI] [PubMed] [Google Scholar]
- 23. Miller P. S., and Aricescu A. R. (2014) Crystal structure of a human GABAA receptor. Nature 512, 270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Epi4K Consortium, Epilepsy Phenome/Genome Project, Allen A. S., Berkovic S. F., Cossette P., Delanty N., Dlugos D., Eichler E. E., Epstein M. P., Glauser T., Goldstein D. B., Han Y., Heinzen E. L., Hitomi Y., and Howell K. B., et al. (2013) De novo mutations in epileptic encephalopathies. Nature 501, 217–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Noebels J. L. (2003) The biology of epilepsy genes. Annu. Rev. Neurosci. 26, 599–625 [DOI] [PubMed] [Google Scholar]
- 26. Steinlein O. K. (2012) Ion channel mutations in neuronal diseases: a genetics perspective. Chem. Rev. 112, 6334–6352 [DOI] [PubMed] [Google Scholar]
- 27. Hines R. M., Davies P. A., Moss S. J., and Maguire J. (2012) Functional regulation of GABAA receptors in nervous system pathologies. Curr. Opin. Neurobiol. 22, 552–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hirose S. (2014) Mutant GABA(A) receptor subunits in genetic (idiopathic) epilepsy. Prog. Brain Res. 213, 55–85 [DOI] [PubMed] [Google Scholar]
- 29. Macdonald R. L., Kang J.-Q., and Gallagher M. J. (2010) Mutations in GABA(A) receptor subunits associated with genetic epilepsies. J. Physiol. 588, 1861–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gallagher M. J., Ding L., Maheshwari A., and Macdonald R. L. (2007) The GABA(A) receptor α1 subunit epilepsy mutation A322D inhibits transmembrane helix formation and causes proteasomal degradation. Proc. Natl. Acad. Sci. U.S.A. 104, 12999–13004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cossette P., Liu L., Brisebois K., Dong H., Lortie A., Vanasse M., Saint-Hilaire J. M., Carmant L., Verner A., Lu W. Y., Wang Y. T., and Rouleau G. A. (2002) Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat. Genet. 31, 184–189 [DOI] [PubMed] [Google Scholar]
- 32. Merulla J., Fasana E., Soldà T., and Molinari M. (2013) Specificity and regulation of the endoplasmic reticulum-associated degradation machinery. Traffic 14, 767–777 [DOI] [PubMed] [Google Scholar]
- 33. Nakatsukasa K., Kamura T., and Brodsky J. L. (2014) Recent technical developments in the study of ER-associated degradation. Curr. Opin. Cell Biol. 29, 82–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Christianson J. C., and Ye Y. (2014) Cleaning up in the endoplasmic reticulum: ubiquitin in charge. Nat. Struct. Mol. Biol. 21, 325–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xie W., and Ng D. T. (2010) ERAD substrate recognition in budding yeast. Semin. Cell Dev. Biol. 21, 533–539 [DOI] [PubMed] [Google Scholar]
- 36. Christianson J. C., Olzmann J. A., Shaler T. A., Sowa M. E., Bennett E. J., Richter C. M., Tyler R. E., Greenblatt E. J., Harper J. W., and Kopito R. R. (2012) Defining human ERAD networks through an integrative mapping strategy. Nat. Cell Biol. 14, 93–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Duerfeldt A. S., Peterson L. B., Maynard J. C., Ng C. L., Eletto D., Ostrovsky O., Shinogle H. E., Moore D. S., Argon Y., Nicchitta C. V., and Blagg B. S. (2012) Development of a Grp94 inhibitor. J. Am. Chem. Soc. 134, 9796–9804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ding L., Feng H.-J., Macdonald R. L., Botzolakis E. J., Hu N., and Gallagher M. J. (2010) GABA(A) Receptor α1 subunit mutation A322D associated with autosomal dominant juvenile myoclonic epilepsy reduces the expression and alters the composition of wild type GABA(A) receptors. J. Biol. Chem. 285, 26390–26405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Eletto D., Maganty A., Eletto D., Dersh D., Makarewich C., Biswas C., Paton J. C., Paton A. W., Doroudgar S., Glembotski C. C., and Argon Y. (2012) Limitation of individual folding resources in the ER leads to outcomes distinct from the unfolded protein response. J. Cell Sci. 125, 4865–4875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Di X. J., Han D. Y., Wang Y. J., Chance M. R., and Mu T. W. (2013) SAHA enhances Proteostasis of epilepsy-associated α1(A322D)β2γ2 GABA(A) receptors. Chem. Biol. 20, 1456–1468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ong D. S., Wang Y. J., Tan Y. L., Yates J. R. 3rd, Mu T. W., and Kelly J. W. (2013) FKBP10 depletion enhances glucocerebrosidase proteostasis in Gaucher disease fibroblasts. Chem. Biol. 20, 403–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Christianson J. C., Shaler T. A., Tyler R. E., and Kopito R. R. (2008) OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 10, 272–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhong Y., Shen H., Wang Y., Yang Y., Yang P., and Fang S. (2015) Identification of ERAD components essential for dislocation of the null Hong Kong variant of α1-antitrypsin (NHK). Biochem. Biophys. Res. Commun. 458, 424–428 [DOI] [PubMed] [Google Scholar]
- 44. Suntharalingam A., Abisambra J. F., O'Leary J. C. 3rd, Koren J. 3rd, Zhang B., Joe M. K., Blair L. J., Hill S. E., Jinwal U. K., Cockman M., Duerfeldt A. S., Tomarev S., Blagg B. S., Lieberman R. L., and Dickey C. A. (2012) Glucose-regulated protein 94 triage of mutant myocilin through endoplasmic reticulum-associated degradation subverts a more efficient autophagic clearance mechanism. J. Biol. Chem. 287, 40661–40669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Melnick J., Dul J. L., and Argon Y. (1994) Sequential interaction of the chaperones BiP and GRP94 with immunoglobulin chains in the endoplasmic reticulum. Nature 370, 373–375 [DOI] [PubMed] [Google Scholar]
- 46. Liu B., and Li Z. (2008) Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood 112, 1223–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang Y., Liu B., Dai J., Srivastava P. K., Zammit D. J., Lefrançois L., and Li Z. (2007) Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity 26, 215–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wanderling S., Simen B. B., Ostrovsky O., Ahmed N. T., Vogen S. M., Gidalevitz T., and Argon Y. (2007) GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol. Biol. Cell 18, 3764–3775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Liu B., Staron M., Hong F., Wu B. X., Sun S., Morales C., Crosson C. E., Tomlinson S., Kim I., Wu D., and Li Z. (2013) Essential roles of grp94 in gut homeostasis via chaperoning canonical Wnt pathway. Proc. Natl. Acad. Sci. U.S.A. 110, 6877–6882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schröder M., and Kaufman R. J. (2005) The mammalian unfolded protein response. Annu. Rev. Biochem. 74, 739–789 [DOI] [PubMed] [Google Scholar]
- 51. Walter P., and Ron D. (2011) The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086 [DOI] [PubMed] [Google Scholar]
- 52. Marzec M., Eletto D., and Argon Y. (2012) GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim. Biophys. Acta 1823, 774–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu S., Hong F., Gewirth D., Guo B., Liu B., and Li Z. (2012) The molecular chaperone gp96/GRP94 interacts with Toll-like receptors and integrins via its C-terminal hydrophobic domain. J. Biol. Chem. 287, 6735–6742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dersh D., Jones S. M., Eletto D., Christianson J. C., and Argon Y. (2014) OS-9 facilitates turnover of nonnative GRP94 marked by hyperglycosylation. Mol. Biol. Cell 25, 2220–2234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hebert D. N., Lamriben L., Powers E. T., and Kelly J. W. (2014) The intrinsic and extrinsic effects of N-linked glycans on glycoproteostasis. Nat. Chem. Biol. 10, 902–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Helenius A., and Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 57. Molinari M., Calanca V., Galli C., Lucca P., and Paganetti P. (2003) Role of EDEM in the release of misfolded glycoproteins from the calnexin cycle. Science 299, 1397–1400 [DOI] [PubMed] [Google Scholar]
- 58. Oda Y., Hosokawa N., Wada I., and Nagata K. (2003) EDEM as an acceptor of terminally misfolded glycoproteins released from calnexin. Science 299, 1394–1397 [DOI] [PubMed] [Google Scholar]
- 59. Bays N. W., Gardner R. G., Seelig L. P., Joazeiro C. A., and Hampton R. Y. (2001) Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat. Cell Biol. 3, 24–29 [DOI] [PubMed] [Google Scholar]
- 60. Zhang T., Xu Y., Liu Y., and Ye Y. (2015) gp78 functions downstream of Hrd1 to promote degradation of misfolded proteins of the endoplasmic reticulum. Mol. Biol. Cell 26, 4438–4450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kikkert M., Doolman R., Dai M., Avner R., Hassink G., van Voorden S., Thanedar S., Roitelman J., Chau V., and Wiertz E. (2004) Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 279, 3525–3534 [DOI] [PubMed] [Google Scholar]
- 62. Wolf D. H., and Stolz A. (2012) The Cdc48 machine in endoplasmic reticulum associated protein degradation. Biochim. Biophys. Acta 1823, 117–124 [DOI] [PubMed] [Google Scholar]
- 63. Ye Y., Meyer H. H., and Rapoport T. A. (2001) The AAA ATPase Cdc48/p97 and its partners transport proteins from the ER into the cytosol. Nature 414, 652–656 [DOI] [PubMed] [Google Scholar]
- 64. Wang Q., Li L., and Ye Y. (2008) Inhibition of p97-dependent protein degradation by Eeyarestatin I. J. Biol. Chem. 283, 7445–7454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Flynn G. C., Pohl J., Flocco M. T., and Rothman J. E. (1991) Peptide-binding specificity of the molecular chaperone BiP. Nature 353, 726–730 [DOI] [PubMed] [Google Scholar]
- 66. Hebert D. N., and Molinari M. (2012) Flagging and docking: dual roles for N-glycans in protein quality control and cellular proteostasis. Trends Biochem. Sci. 37, 404–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jin H., Chiou T. T., Serwanski D. R., Miralles C. P., Pinal N., and De Blas A. L. (2014) Ring finger protein 34 (RNF34) interacts with and promotes γ-aminobutyric acid type-A receptor degradation via ubiquitination of the γ2 subunit. J. Biol. Chem. 289, 29420–29436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wang Y. J., Han D. Y., Tabib T., Yates J. R. 3rd, and Mu T. W. (2013) Identification of GABA(C) receptor protein homeostasis network components from three tandem mass spectrometry proteomics approaches. J. Proteome Res. 12, 5570–5586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han D. Y., Guan B. J., Wang Y. J., Hatzoglou M., and Mu T. W. (2015) L-type calcium channel blockers enhance trafficking and function of epilepsy-associated α1(D219N) subunits of GABAA receptors. ACS Chem. Biol. 10, 2135–2148 [DOI] [PubMed] [Google Scholar]
- 70. Wang Y. J., Di X. J., and Mu T. W. (2014) Using pharmacological chaperones to restore proteostasis. Pharmacol. Res. 83, 3–9 [DOI] [PMC free article] [PubMed] [Google Scholar]