

Abstract
Insulin resistance and β cell dysfunction contribute to the pathogenesis of type 2 diabetes. Unlike insulin resistance, β cell dysfunction remains difficult to predict and monitor, because of the inaccessibility of the endocrine pancreas, the integrated relationship with insulin sensitivity, and the paracrine effects of incretins. The goal of our study was to survey the plasma response to a metabolic challenge in order to identify factors predictive of β cell dysfunction. To this end, we combined (i) the power of unbiased iTRAQ (isobaric tag for relative and absolute quantification) mass spectrometry with (ii) direct sampling of the portal vein following an intravenous glucose/arginine challenge (IVGATT) in (iii) mice with a genetic β cell defect. By so doing, we excluded the effects of peripheral insulin sensitivity as well as those of incretins on β cells, and focused on the first phase of insulin secretion to capture the early pathophysiology of β cell dysfunction. We compared plasma protein profiles with ex vivo islet secretome and transcriptome analyses. We detected changes to 418 plasma proteins in vivo, and detected changes to 262 proteins ex vivo. The impairment of insulin secretion was associated with greater overall changes in the plasma response to IVGATT, possibly reflecting metabolic instability. Reduced levels of proteins regulating redox state and neuronal stress markers, as well as increased levels of coagulation factors, antedated the loss of insulin secretion in diabetic mice. These results suggest that a reduced complement of antioxidants in response to a mixed secretagogue challenge is an early correlate of future β cell failure.
Keywords: beta cell (B-cell), biomarker, diabetes, FOXO, neurodegeneration, oxidative stress
Introduction
The incidence of diabetes has increased considerably (1). Type 2 diabetes is characterized by insulin resistance and impaired β cell function (2). Both abnormalities contribute to the onset and progression of the disease. However, progression from pre-diabetes to diabetes is characterized by a steep decrease of insulin secretory function, whereas insulin resistance remains relatively constant (3, 4). It appears that even modest elevations of plasma glucose levels are toxic to β cells (5). In addition, outcome studies have consistently demonstrated that diabetic patients treated with sulfonylurea-type secretagogues experience faster therapeutic failure rates that require the addition of a second medication, when compared with those treated with insulin sensitizers (6). These facts point to two conclusions: (i) that there is an intrinsic impairment of β cell function in diabetes; and (ii) that promoting insulin secretion does not redress the problem.
To develop better therapies and increase the sensitivity of probing β cell function, it would be desirable to have markers predictive of β cell failure. Sensitive tests of insulin secretion are rarely applicable as routine diagnostics, and have limited predictive value of response to treatment. Given the renewed focus on durability as a key criterion to develop more efficacious diabetes treatments (7), it is essential to evaluate new methods of assessing and predicting β cell function. A recurring problem in this regard is that β cell function is linked to insulin sensitivity, and it is difficult to dissect the effects of impaired β cell function from those of impaired insulin action (8).
We recently reported that mice lacking the three isoforms of FoxO transcription factors in pancreatic β cells develop a MODY-like2 form of diabetes, characterized by impaired insulin response to metabolic challenge and resulting in mild, non-progressive hyperglycemia (9). These mice maintained normal insulin sensitivity, thus eliminating the secondary effects of peripheral insulin resistance on β cell function. Therefore, they serve as an ideal model to investigate changes associated with pure β cell dysfunction.
13–20% of the human proteome is composed of secretory proteins (10). Among them, plasma proteins represent a major class involved in cell signaling and communication, and can serve as diagnostic and therapeutic biomarkers. To identify patterns associated with the progression of β cell failure, we performed an unbiased iTRAQ (isobaric tag for relative and absolute quantification)-based survey of plasma proteins released in response to a mixed arginine/glucose tolerance test, before and after the onset of hyperglycemia, in FoxO-deficient mice and their littermate controls.
This experimental setup has several advantages: (i) it is a pure model of β cell dysfunction, bereft of the confounders of peripheral insulin resistance; (ii) each mouse serves as its own control; (iii) by collecting plasma from the portal vein, it allows enrichment of factors released from the endocrine pancreas prior to their hepatic extraction; (iv) intravenous challenge eliminates incretin effects; and (v) it can be focused on the distinctive deficit of first phase insulin release of pre- and early diabetes (11). To distinguish between proteins of pancreatic provenance and those secreted by other tissues and cell types, we compared overall plasma protein patterns with ex vivo secretome analyses of isolated islets and RNA profiling of purified islets. Surprisingly, our analysis identified abnormalities of antioxidants, neurodegeneration, and coagulation markers as early correlates of β cell dysfunction.
Experimental Procedures
Animals
We generated mice lacking the three FoxO isoforms in pancreas (TKO) using Tg(Ipf1-cre)1Tuv transgenics to excise the floxed Foxo1, Foxo3a, and Foxo4 genes. We chose this Cre line by virtue of its minimal hypothalamic expression (12). Mice were maintained on a 129J x C57BL/6 background and then genotyped as described (9). Mice were weaned at 3 weeks of age, fed normal chow diet, and maintained on a 12-h light-dark cycle (lights on at 7 a.m.). The Columbia University Institutional Animal Care and Utilization Committee approved all animal procedures.
Metabolic Analyses
We performed intraperitoneal glucose tolerance tests (ipGTT) by injecting glucose (2 g/kg) after an overnight fast, and we performed insulin tolerance tests by injecting insulin (0.75 units/kg) after a 5-h fast (13). We developed a method to sample directly pre- and post-challenge portal vein blood by placing needles in the inferior vena cava (IVC) and portal vein (PV), termed the intravenous glucose/arginine tolerance test (IVGATT) (see Fig. 2, A–D). On the day of the experiment, we fasted mice for 3 h, and then performed anesthesia with a mixture of ketamine (80 mg/kg) and xylazine (8 mg/kg) administered intraperitoneally. Throughout the procedure, mice were kept on a heating pad to prevent anesthesia-induced hypothermia and maintain a constant blood flow for optimal cardiovascular function and to facilitate blood collection. The abdominal area was opened to expose the PV and IVC (see Fig. 2A). A catheter was inserted into the PV, and the needle was removed (see Fig. 2B). A 200-μl pipette tip rinsed with EDTA was used to draw basal blood. A plastic stopper was used to prevent blood overflow. A mixture of glucose (1 g/kg) and arginine (0.25 g/kg) (Sigma A5006) was injected through the IVC (see Fig. 2C). After 2 min, the plastic stopper was removed from the catheter, residual blood was cleared, and post-challenge blood was collected (see Fig. 2D). Samples were centrifuged, and plasma insulin levels were determined by ELISA (Mercodia 10-1247-01). Samples that met quality control criteria were subjected to BSA/IgG depletion, protein reduction and digestion, iTRAQ labeling, and nanoLC-MS/MS. Raw spectra were processed using Proteome Discoverer 1.4 (Thermo) and searched against Mascot Daemon version 2.3.02 (Matrix Science). Complete experimental protocols are described in the supplemental Experimental Procedures.
FIGURE 2.

Intravenous glucose and arginine tolerance test. A, the ventral side of the abdominal area of a mouse, indicating the positions of the IVC and PV. B, following a 3-h fast, a basal plasma sample was drawn from the PV under anesthesia. C, a mixture of glucose (1 g/kg of body weight) and l-arginine (0.25 g/kg of body weight) was injected into the IVC. D, 2 min after injection, a post-challenge sample was collected from PV. E, correlation of mass spectrometry analysis with different iTRAQ-labeled single sets of WT pre- and post-challenge plasma samples.
iTRAQ Labeling
To survey protein levels in an unbiased fashion, we utilized iTRAQ followed by mass spectrometry (14). iTRAQ was developed to survey quantitative and qualitative differences in protein levels in a biological system, and it does not require prior labeling of samples. Digested and reduced peptides were labeled with iTRAQ reagent (AB SCIEX) following the manufacturer's instructions (14). An iTRAQ reagent consists of three groups: (i) a reporter group that allows relative quantification upon MS; (ii) a balance group that maintains the isobaric nature of the tags to avoid mass shift in MS (designated 114, 115, 116, and 117); and (iii) a peptide reactive group that allows attachment to proteins. For in vivo secretome, iTRAQ 4plex was used, where WT basal, WT post-challenge, TKO basal, and TKO post-challenge plasma were each labeled with an isobaric tag, pooled, and analyzed concurrently.
Protein Identification by Mass Spectrometry and Data Analysis
To reduce the probability of false peptide identification, we used a 0.5% false discovery rate and a confidence interval of 95% (p < 0.05) for peptides defined by the Mascot probability analysis. Intensities of the reporter ions from iTRAQ tags upon fragmentation were used for quantification, and the relative quantitation ratios were normalized at the median ratio for the 4plex. Between-group comparison was carried as described in the statistical section.
Data Analysis
We analyzed changes to plasma protein patterns using a 2 × 2 × 2 design, with IVGATT, age, and genotype as the variables. We compared pre- and post-IVGATT data within genotypes and at the two time points of our study, and compared genotypes at different ages. Regression analyses showed strong correlation between two ratios of post- to pre-IVGATT from different iTRAQ-labeled single sets of WT plasma samples (114- and 115-labeled basal plasma and 116- and 117-labeled post-challenge plasma), which indicates reproducible labeling efficiency (see Fig. 2E). Following mass spectrometry, we set an arbitrary threshold of 2-fold to declare differences.
Islet Isolation and Secretome Analysis
Collagenase-purified, size-matched islets (∼200 μm) were isolated and handpicked as described (15). 20 islets were allowed to recover for 1 h at 37 °C, and then incubated for the same length of time in 0.1 ml of fresh Krebs buffer supplemented with 2.8 mm glucose. This medium was collected to determine basal protein patterns. Thereafter, islets were incubated in 0.1 ml of fresh Krebs buffer supplemented with glucose (16.8 mm) and l-arginine (10 mm) for 30 min at 37 °C. This sample was collected to determine post-challenge protein patterns. Insulin content was determined by ELISA, and total protein content was determined by colorimetric assay (Pierce 23225). For secretome analysis, samples were subjected to BSA/IgG depletion, in-gel trypsin digestion and extraction, nanoLC-MS/MS, and MaxQuant-based label free quantification and protein identification. The false discovery rate was set to <1%. Up to two missing cleavage points were allowed. Protein quantitation was performed using unique peptides and razor peptides. Complete experimental protocols are described in the supplemental Experimental Procedures.
RNA Profiling
We performed GeneChip mouse exon arrays (Affymetrix) and analyzed the data with the Partek Genomics Suite (Partek, Inc.) and Ingenuity Pathway Analysis (Ingenuity System, Inc.). We used a threshold of p < 0.05 and >1.3-fold change to determine significance. Each array was performed with pooled islets from three mice per genotype.
Statistical Analysis
We analyzed the data with two-tailed Student's t test or one-way ANOVA using the Prism GraphPad software. Error bars represent S.E., and statistical significance was declared when p < 0.05.
Results
Pan-pancreatic Ablation of FoxO1, FoxO3a, and FoxO4 Impairs Insulin Secretion
We have shown that ablating the three FoxO isoforms in terminally differentiated β cells results in MODY-like diabetes (9). We observed a similar metabolic phenotype in mice carrying an embryonic deletion of FoxO1 in the pancreas (16), as these mice displayed fasting hyperglycemia, age-dependent glucose intolerance, and compromised arginine-stimulated insulin secretion. The difference between the two models is the developmental stage at which FoxO1 is inactivated (β cell versus pancreatic progenitor) (17), and the presence of FoxO3a and FoxO4. As we have shown previously, single ablation of FoxO1 results in a compensatory increase of FoxO3a and FoxO4, which may partly mask the full range of FoxO functions in β cells (18).
For this study, we generated triple FoxO knock-outs at the pancreatic progenitor stage by mating Tg(Ipf1-cre)1Tuv transgenics with mice bearing Foxo1, Foxo3a, and Foxo4 floxed alleles (henceforth, TKO). TKO mice recapitulated the metabolic features of pan-pancreatic single FoxO1 knock-outs (16), displaying normal glucose tolerance at 12 weeks (Fig. 1A) and frank glucose intolerance at 30 weeks of age (Fig. 1B). Although TKO mice showed elevated fed glucose levels at both ages, insulin tolerance was normal, indicated by similar slope of insulin tolerance test and area under the curve, and the end-point glucose levels (Fig. 1, C and D).
FIGURE 1.
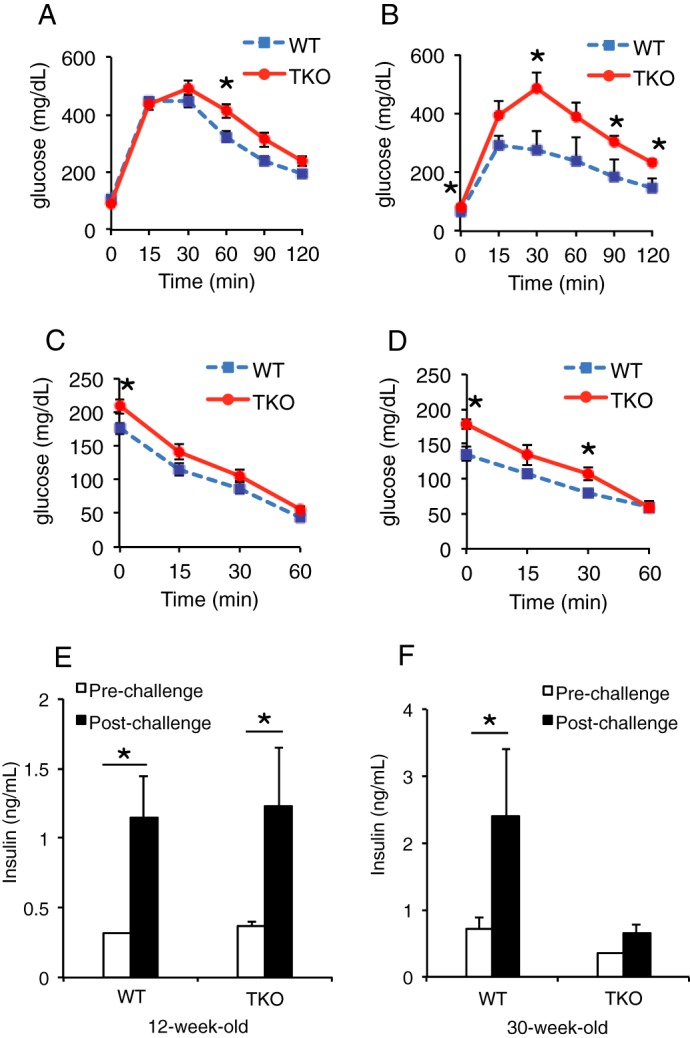
Metabolic parameters of TKO mice. A, ipGTT at 12 weeks of age. B, ipGTT at 30 weeks of age. C, insulin tolerance tests at 12 weeks of age. D, insulin tolerance tests at 30 weeks of age. E and F, insulin levels in pre- and post-challenge plasma collected from the portal vein. E, 12-week-old group. F, 30-week-old group. Error bars represent S.E., *, p < 0.05 by Student's t test and ANOVA.
Next, we evaluated insulin secretion by sampling the portal vein following an IVGATT (see “Experimental Procedures” and Fig. 2). 12-week-old WT and TKO mice showed similar basal and post-challenge insulin levels (Fig. 1E). In contrast, 30-week-old TKO mice showed decreased basal insulin, as well as severely blunted response to challenge (Fig. 1F). These results show that portal collection during IVGATT is a sensitive approach to detect pancreatic endocrine secretion and that TKO mice are a model of impaired insulin secretion.
RNA Profiling of TKO Islets
To understand the pathogenesis of the secretory abnormality in TKO mice, we surveyed islet transcriptomes in 12-week-old mice, prior to the onset of glucose intolerance (supplemental Table S1). The transcriptional profile of TKO islets closely resembled that of the recently reported β cell-specific triple FoxO knock-outs generated via insulin-Cre-mediated recombination (9). Specifically, three MODY genes, Hnf4α, Hnf1α, and Pdx1, topped the list of altered transcription networks. In addition, we detected alterations of the glucocorticoid receptor (Nr3c1) and Mist1 (Bhlha15) networks (Table 1). Interestingly, Ingenuity Pathway Analysis identified the “neurological disease” pathway as the most pronounced alteration among disease pathways. Similar to β cell-specific triple FoxO knock-outs, there were alterations in metabolic pathways related to mitochondrial dysfunction and oxidation, as well as branched-chain amino acid degradation. Unlike β cell-specific triple FoxO knock-outs, we detected a profound decrease in genes involved in glucose uptake and metabolism (Table 1). This finding suggests that the main developmental stage-specific role of FoxO in pancreatic progenitors is to regulate acquisition of glucose-sensing capabilities by the β cell. These findings help explain the secretory defect seen in IVGATT.
TABLE 1.
Summary of pathway analysis of transcriptome data
BCAA, branched-chain amino acid; TOR, target of rapamycin; PPAR, peroxisome proliferator-activated receptor; RXR, retinoid X receptor.
| p value | |
|---|---|
| Transcriptional regulator | |
| Hnf4α | 1.15 × 10−14 |
| Hnf1α | 2.11 × 10−8 |
| Nr3c1 | 5.47 × 10−6 |
| Pdx1 | 8.05 × 10−5 |
| Bhlha15 | 2.99 × 10−4 |
| Diseases and disorders | |
| Neurological disease | 3.97 × 10−6 to 2.90 × 10−2 |
| Skeletal and muscular disorders | 6.97 × 10−6 to 1.88 × 10−2 |
| Gastrointestinal disease | 2.46 × 10−5 to 2.40 × 10−2 |
| Genetic disorder | 2.46 × 10−5 to 2.32 × 10−2 |
| Inflammatory disease | 2.46 × 10−5 to 1.57 × 10−2 |
| Molecular functions | |
| Cellular assembly and organization | 4.69 × 10−6 to 2.90 × 10−2 |
| Lipid metabolism | 9.90 × 10−6 to 2.76 × 10−2 |
| Small molecule biochemistry | 9.90 × 10−6 to 2.76 × 10−2 |
| Carbohydrate metabolism | 2.00 × 10−5 to 2.71 × 10−2 |
| Free radical scavenging | 3.36 × 10−5 to 2.25 × 10−2 |
| Top canonical pathways | |
| Mitochondrial dysfunction | 9.05 × 10−7 |
| BCAA degradation | 3.44 × 10−6 |
| FFA mitochondrial oxidation | 6.64 × 10−6 |
| TOR signaling | 2.62 × 10−5 |
| Glycerolipid metabolism | 6.11 × 10−5 |
| “Toxic” list | |
| Mitochondrial dysfunction | 1.71 × 10−6 |
| PPARα regulation | 4.03 × 10−3 |
| PPARα/RXRα activation | 1.28 × 10−2 |
| Renal necrosis/cell death | 1.85 × 10−2 |
| Liver proliferation | 2.24 × 10−2 |
Plasma Protein Profiles before and after Onset of β Cell Dysfunction
Next, we carried out iTRAQ analyses of plasma samples obtained following IVGATT. First, we analyzed differences as a function of genotype at 12 weeks (Fig. 3A). We detected 85 changes in WT and 166 changes in TKO; of these, 54 were common to both genotypes (overlap). Surprisingly, all shared proteins changed in opposite directions in WT versus TKO mice: 19 were enriched in the latter, and 35 were enriched in the former (supplemental Table S2).
FIGURE 3.
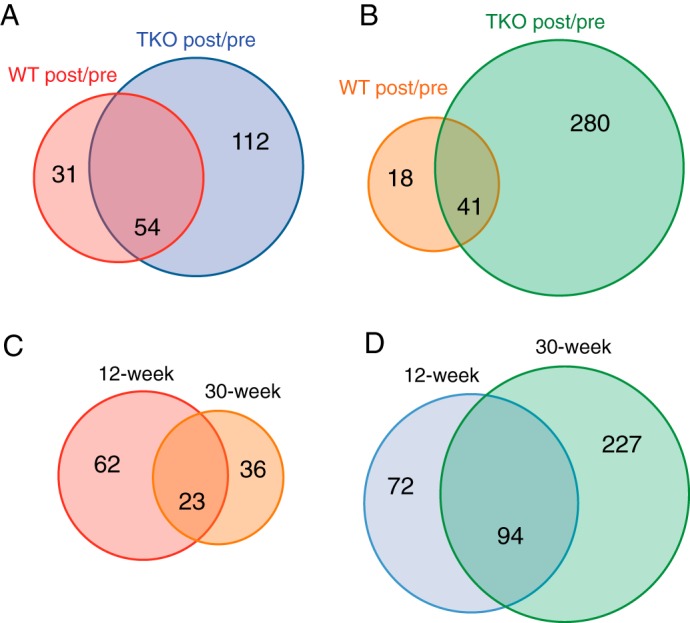
Venn diagrams summarizing the ratios of plasma proteins detected by iTRAQ. A, 12-week-old mice: proteins showing a post-IVGATT/basal ratio ≥2 in WT and TKO mice. B, 30-week-old mice: proteins showing a post-IVGATT/basal ratio ≥2 in WT and TKO mice. C, WT mice: proteins showing a post-IVGATT/basal ratio ≥2 at 12 and 30 weeks of age. D, TKO mice: proteins showing a post-IVGATT/basal ratio ≥2 at 12 and 30 weeks of age. p < 0.05 by ANOVA in all experiments.
In 30-week-old animals, the number of changes detected decreased to 59 in WT but increased to 321 in TKO (Fig. 3B). Interestingly, 14 of 18 proteins unique to WT rose post-challenge, whereas 270 of 280 decreased in TKO. There were 41 common proteins, of which 18 changed in a similar fashion, 6 had different magnitudes, and 17 changed in opposite directions (supplemental Table S2). The conclusion from these data is that the impairment of insulin secretion in TKO is associated with greater overall changes in the plasma response to IVGATT, possibly reflecting metabolic instability in TKO mice. Moreover, differences between WT and TKO mice increased with age, resulting in a lower percentage of shared changes. Finally, although WT mice tended to show fewer changes at older ages, TKO mice showed more.
Next, we compared changes to post-IVGATT plasma profiles at 12 versus 30 weeks to identify factors associated with the progression of β cell dysfunction in TKO mice (Fig. 3C). In WT controls, we found that 23 proteins changed at both ages, 62 changed only at 12 weeks, and 36 changed only at 30 weeks. Proteins with overlapping patterns changed in the same direction and with similar magnitude, providing an important quality control (supplemental Table S3). In TKO mice, 72 proteins changed only at 12 weeks, 227 changed only at 30 weeks, and 94 changed at both ages (Fig. 3D). Among the latter, 12 proteins changed in opposite directions and 3 changed in the same direction, but with different magnitude (supplemental Table S3).
Analyses of Selected Proteins
We performed a four-way comparison among animals of different genotype and age (Fig. 4A and supplemental Table S4). We found 79 proteins that changed in 12-week-old WT versus TKO mice, 221 that changed only in 30-week-old animals, and 118 that changed at both ages. We focused subsequent detailed candidate gene analyses on the latter.
FIGURE 4.
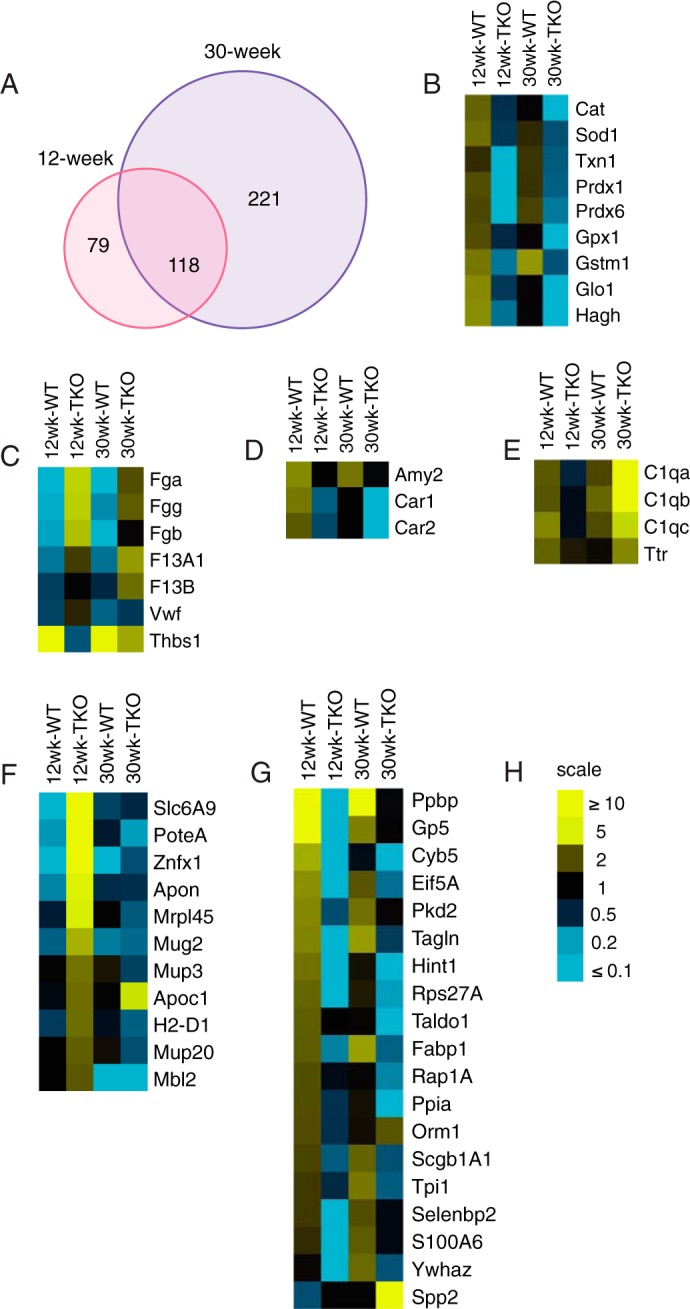
Analysis of selected plasma proteins. A, Venn diagrams summarizing the ratios of plasma proteins detected by iTRAQ in 12- and 30-week-old WT and TKO mice. B–E, heat maps of selected candidate proteins involved in redox reactions and oxidative stress (B), coagulation factors (C), exocrine pancreatic function (D), and complement component and transthyretin (E). F and G, heat maps of selected candidate proteins that increased (F) or decreased (G) in response to IVGATT in 12-week TKO mice. H, scale of -fold change represented in B–H.
Surprisingly, in WT mice, we found a significant induction of antioxidant enzymes that was greatly blunted in TKO at both 30 and 12 weeks of age. These included catalase (Cat), superoxide dismutase 1 (Sod1), thioredoxin (Txn), peroxiredoxins (Prdx) 1 and 6, glutathione peroxidase 1 (Gpx1), glutathione S-transferase Mu1 (Gstm1), as well as two additional proteins required to metabolize glutathione, lactoylglutathione lyase (Glo1) and mitochondrial hydroxyacylglutathione hydrolase (Hagh) (Fig. 4B). It is unclear how these enzymes are released into the circulation, as they are unlikely to follow the conventional secretory pathway, but they are all present in human plasma (19). The blunted induction of oxidative enzymes in TKO mice appears to precede the onset of β cell dysfunction.
Among proteins over-represented in TKO mice, we found coagulation factors including fibrinogen precursor α (Fga), β (Fgb), and γ (Fgg) chains, coagulation factor XIII A1 (F13A1) and B (F13B), as well as Von Willebrand factor (Vwf) (Fig. 4C), whereas Thbs1 was decreased when compared with WT (Fig. 4C). These factors may be released in response to the venipuncture, and their decrease may reflect the effect of lower insulin levels on the coagulation response.
Pancreatic secretory products secretogranin1/chromogranin B (Chgb) precursor (supplemental Table S4), amylase (Amy2), and carbonic anhydrase 1 and 2 (Car1–2) increased post-IVGATT in WT, but not TKO mice (Fig. 4D). Interestingly, genetic ablation of Chgb results in widespread endocrine cell abnormities (20).
Complement component C1q functions as a pattern recognition molecule and binds to proteases C1r and C1s to activate the complement cascade (21). Complement component C3a has been implicated in stimulating insulin secretion (22). Interestingly, levels of C1q subunits a, b, and c in 12-week-old TKO were lower than WT but became higher at 30 weeks of age. This was not due to changes in their WT levels, which were comparable at both ages (Fig. 4E). Plasma transthyretin (Ttr) is a transporter for thyroxine and retinol with retinol-binding protein. It has been shown to regulate circulating retinol-binding protein 4 (RBP4) levels, and through this function, it may contribute to glucose homeostasis (23). Ttr levels decreased in WT from 12 to 30 weeks, whereas they increased in TKO mice (Fig. 4E).
We next analyzed proteins that increased exclusively in 12-week-old TKO mice, and may therefore represent biomarkers of β cell strain. This list included proteins of diverse function, such as MHC components and carbohydrate-binding proteins, but none that could easily be identified as β cell-specific (Fig. 4F). Finally, we list proteins elevated in 12-week WT but reduced in 12-week TKO mice. The top protein is platelet basic protein precursor (Ppbp), also known as chemokine (CXC motif) ligand 7 and variously associated pancreatic cancer and chronic pancreatitis (Fig. 4G) (24).
In Vivo Plasma Profiles Versus ex Vivo Islet Secretome
Next, we asked which plasma proteins detected by iTRAQ derived from endocrine islets. First, we demonstrated that islets isolated from 40-week-old TKO mice had reduced insulin response to arginine (Fig. 5A), glucose (Fig. 5B), or both, following a mixed glucose and arginine challenge in vitro (Fig. 5C). Due to interference from the albumin in the culture medium, we could not use iTRAQ to determine quantitative differences between basal and post-challenge secretomes. We used a label-free approach that provided a list of islet-secreted proteins in response to metabolic challenge.
FIGURE 5.

Comparison of in vivo plasma proteins and ex vivo secretome. A–C, insulin secretion from isolated WT and TKO islets stimulated with arginine (A), glucose (B), or a combination of arginine and glucose (C). Error bars represent S.E., *, p < 0.05 by Student's t test and ANOVA. D, Venn diagrams summarizing significantly changed proteins identified in vivo and proteins detected in the supernatant of isolated islets following the experiment described in C. p < 0.05 by ANOVA in all experiments. E and F, heat maps of proteins detected in the overlap of the Venn diagrams in D showing proteins that changed in WT and TKO mice over time (E), or those that changed only at 30 weeks (F). Red arrows indicate redox proteins, and the red box indicates macrophage migration inhibitory factor (Mif).
We detected changes to 418 proteins using iTRAQ analysis in vivo, and detected changes to 262 proteins using label-free mass spectrometry ex vivo. 54 proteins were common between the two approaches, indicating potential islet-derived proteins (Fig. 5D and supplemental Fig. S5). We then queried the iTRAQ database for differences in their expression according to age and genotype. Interestingly, antioxidant enzymes topped the list of proteins altered at both time points, indicating that at least part of their variation may reflect islet cell-intrinsic changes (Fig. 5E). In addition, a separate list of proteins that changed only after the onset of islet dysfunction included macrophage migration inhibitory factor (Mif) (Fig. 5F), a protein co-secreted with insulin (25, 26).
In Vivo Secretome Versus Transcriptome
As an alternative approach to find islet-specific proteins, we queried islet RNA profiles (supplemental Table S1) from 12-week-old mice for transcripts encoding proteins that changed post-IVGATT. We found 985 genes displaying a significant >1.4-fold change that were altered in TKO mice. Of them, 29 encoded proteins that were also detected in vivo (Fig. 6A and supplemental Table S6), and 6 of 29 proteins increased at least at one age following IVGATT in WT or TKO (Fig. 6B). Interestingly, this group included α-synuclein (Snca) and Park7 (DJ-1), two markers of Parkinson disease (Fig. 6, B and C). This result echoes the transcriptome data, in which neurological disease topped the predicted disease pathways (Table 1).
FIGURE 6.

Comparison of in vivo plasma proteins and islet transcriptome. A, Venn diagrams summarizing significantly changed proteins identified in vivo and differentially expressed mRNAs detected in RNA profiling studies of purified islets. p < 0.05 by ANOVA in all experiments. B, heat map of proteins detected in the overlap of the Venn diagrams in B showing proteins that increased in WT mice, and failed to increase in TKO mice. C, heat map of mRNA expression corresponding to the proteins shown in B. Red arrows indicate proteins involved in Parkinson disease.
Discussion
By cross-comparisons of in vivo and ex vivo secretomes with transcriptomes of pan-pancreatic triple FoxO knock-out mice and littermate controls, we found three surprising abnormalities of the plasma response to an acute IVGATT that antedate the onset of insulin secretory failure: decreased oxidoreductases, increased markers of neuronal damage, and increased clotting factors. Although the first two classes of proteins have been linked to β cell activity, our study suggests that they are also plasma markers of evolving β cell dysfunction. We cannot determine the contribution of the β cell itself to the amount of protein detected in the plasma, even among proteins that we detected in isolated islets. It could be, for example, that a protein is released from multiple sources including β cells, but its main source lies elsewhere. Nonetheless, given the absence of additional defects (e.g. insulin resistance), the portal vein sampling, and the use of ex vivo and in vivo analyses, our approach tried to enrich the plasma samples for β cell-released proteins.
A first consideration emerging from our analysis is that TKO mice display a much broader pattern of changes to plasma protein profiles following IVGATT. This is especially evident after the onset of β cell dysfunction, and may reflect the metabolic instability of TKO mice in response to a metabolic challenge. This metabolic instability is associated with decreased insulin release, rather than insulin resistance, and probably reflects the tissue homeostatic effect of insulin, independent of its metabolic effects on nutrient utilization, because it occurs within minutes of glucose/arginine injection.
The first major finding of our work is that plasma levels of proteins regulating the oxidoreductive state (Cat, Sod1, Txn1, Prdx1, Prdx2, and Prdx6, Gpx1, Gstm1, Glo1, and Hagh) increase after glucose and arginine challenge in healthy animals, while failing to do so in TKO mice. These changes are seemingly independent of insulin levels, peripheral insulin sensitivity, and incretin effect. These proteins have been detected in a high-confidence human plasma proteome reference set, obtained from 91 high-quality LC-MS/MS datasets that yielded 1929 highly non-redundant protein sequences with an estimated false discovery rate of 1% (supplemental Table S7), lending support to our findings (19).
Impaired oxidoreductase activity is detrimental to β cell function, possibly because of the low basal levels of these enzymes in β cells when compared with other tissues (27). The redox state of β cells correlates with their secretory function (28), NADPH levels (29), and exocytosis of insulin granules (30). Low antioxidant activity might suppress NADPH levels and decrease insulin secretion. Whether changes in plasma antioxidant levels can be explained entirely by secretion of these proteins from β cells, and what triggers the release of these enzymes, remains to be clarified.
In humans, genetic defects of catalase are associated with diabetes (31), and low levels of catalase activity have been reported in diabetics (32). Acatalasemic mice are more sensitive to alloxan-induced diabetes and pancreatic cell apoptosis (33). Moreover, Sod1 null mice display decreased plasma insulin, increased glucose, and blunted insulin response to glucose, associated with decreased Pdx1 and FoxA2 (34). Sod1 and Gpx1 knock-out mice show decreased β cell mass with impaired glucose-stimulated insulin secretion from isolated islets (34). Conversely, overexpression of Gpx1 in β cells of db/db mice lowers glucose levels and prevents loss of β cell transcription factor MafA (35).
The second striking finding of our work is the induction of markers of neuronal stress, Snca and Park7, by IVGATT, and its failure in TKO mice. Pancreatic β cells share several features with neurons (36), including mechanisms of membrane depolarization-induced insulin secretion (37). Park7 is an antioxidant protein with neuroprotective functions that maintains mitochondrial integrity (38, 39). Its mutations have been linked to familial forms of Parkinson disease (40, 41). Park7 expression is reduced in pancreatic islets of type 2 diabetic patients, where it may be required for mitochondrial function and glucose-stimulated insulin secretion (42). Cerebrospinal, but not plasma, Park7 levels have been proposed as a biomarker of Parkinson disease (43, 44), whereas 4-hydroxy-2-nonenal-modified Park7 could be a plasma biomarker of late-stage Parkinson (45).
Snca expression has been found in transcriptomic and proteomic analyses of pancreatic islets (46). Snca gain of function in β cells of transgenic mice decreases glucose-stimulated insulin secretion and glucose tolerance, suggesting that elevated levels of Snca in β cells negatively affect insulin secretion (47). Mechanistically, Snca has been shown to interact with KATP channels and to localize to insulin secretory granules (48). Furthermore, low plasma Snca levels are associated with insulin resistance (49). Whether islet-secreted Snca is a marker of inappropriate or impaired insulin secretion requires further investigation.
The third key finding of our work is the elevation of plasma levels of several clotting factors, including Fga, Fgb, Fgg, F13A1, F13B, and Vwf in 12- and/or 30-week-old TKO mice. Notably, five of these six factors increased prior to the onset of insulin secretory abnormalities. Diabetes is a hypercoagulable state. Our data appear to indicate that an impairment of insulin secretion can also adversely affect the clotting profile, possibly paving the way for the cardiovascular complications of this disease (50).
In summary, we report a new method to probe β cell secretory function in genetically altered rodents that should prove helpful to detect and optimize biomarkers of the progression of β cell failure. The findings of our work, implicating oxidoreductive enzymes and neuronal damage markers as readouts of impaired insulin secretion, are surprisingly and not easily imputable to known mechanisms of β cell function. Validation of these proteins as biomarkers will require more extensive investigations in humans than we are able to currently perform. Moreover, it will be essential to understand the cell biology underpinning of the release of these proteins in the bloodstream. The data provide a testable hypothesis for the onset of β cell dysfunction and its mechanism that we will pursue in subsequent studies.
Author Contributions
T. K. designed and performed experiments, analyzed data and wrote the manuscript. J. Y. K. contributed reagents and RNA profiling data. T. E. M. reviewed experiments and edited the manuscript. D. A. designed experiments, reviewed data and wrote the manuscript. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Sheng Zhang (Cornell University) for iTRAQ and MS analysis, Thomas Kolar and Ana Flete-Castro (Columbia University) for excellent technical support, and members of the Accili laboratory for discussions. The Columbia University Diabetes Research Center was supported by National Institutes of Health Grant DK63608.
This work was supported by National Institutes of Health Grants T32DK07328 (to T. K.) and DK64819 (to D. A.), and the JPB Foundation. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Experimental Procedures and supplemental Tables S1–S7.
- MODY
- maturity onset diabetes of the young
- iTRAQ
- isobaric tag for relative and absolute quantification
- IVGATT
- intravenous glucose/arginine challenge
- ipGTT
- intraperitoneal glucose tolerance tests
- IVC
- inferior vena cava
- PV
- portal vein
- TKO
- triple knock-out
- ANOVA
- analysis of variance.
References
- 1. Abraham T. M., Pencina K. M., Pencina M. J., and Fox C. S. (2015) Trends in diabetes incidence: the Framingham Heart Study. Diabetes Care 38, 482–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Accili D. (2004) Lilly lecture 2003: the struggle for mastery in insulin action: from triumvirate to republic. Diabetes 53, 1633–1642 [DOI] [PubMed] [Google Scholar]
- 3. Weyer C., Bogardus C., Mott D. M., and Pratley R. E. (1999) The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J. Clin. Invest. 104, 787–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Levy J., Atkinson A. B., Bell P. M., McCance D. R., and Hadden D. R. (1998) β-Cell deterioration determines the onset and rate of progression of secondary dietary failure in type 2 diabetes mellitus: the 10-year follow-up of the Belfast Diet Study. Diabet. Med. 15, 290–296 [DOI] [PubMed] [Google Scholar]
- 5. Wajchenberg B. L. (2007) β-Cell failure in diabetes and preservation by clinical treatment. Endocr. Rev. 28, 187–218 [DOI] [PubMed] [Google Scholar]
- 6. U.K. Prospective Diabetes Study Group. (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352, 837–853; Correction (1999) 354, 602 [PubMed] [Google Scholar]
- 7. Kahn S. E., Haffner S. M., Heise M. A., Herman W. H., Holman R. R., Jones N. P., Kravitz B. G., Lachin J. M., O'Neill M. C., Zinman B., and Viberti G. (2006) Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N. Engl. J. Med. 355, 2427–2443; Correction (2007) N. Engl. J. Med.356, 1387–1388 [DOI] [PubMed] [Google Scholar]
- 8. Kahn S. E. (2003) The relative contributions of insulin resistance and β-cell dysfunction to the pathophysiology of Type 2 diabetes. Diabetologia 46, 3–19 [DOI] [PubMed] [Google Scholar]
- 9. Kim-Muller J. Y., Zhao S., Srivastava S., Mugabo Y., Noh H. L., Kim Y. R., Madiraju S. R., Ferrante A. W., Skolnik E. Y., Prentki M., and Accili D. (2014) Metabolic inflexibility impairs insulin secretion and results in MODY-like diabetes in triple FoxO-deficient mice. Cell Metab. 20, 593–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mukherjee P., and Mani S. (2013) Methodologies to decipher the cell secretome. Biochim. Biophys. Acta 1834, 2226–2232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gerich J. E. (2002) Is reduced first-phase insulin release the earliest detectable abnormality in individuals destined to develop type 2 diabetes? Diabetes 51, Suppl. 1, S117–S121 [DOI] [PubMed] [Google Scholar]
- 12. Wicksteed B., Brissova M., Yan W., Opland D. M., Plank J. L., Reinert R. B., Dickson L. M., Tamarina N. A., Philipson L. H., Shostak A., Bernal-Mizrachi E., Elghazi L., Roe M. W., Labosky P. A., Myers M. G. Jr., et al. (2010) Conditional gene targeting in mouse pancreatic β-cells: analysis of ectopic Cre transgene expression in the brain. Diabetes 59, 3090–3098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsuchiya K., Westerterp M., Murphy A. J., Subramanian V., Ferrante A. W. Jr., Tall A. R., and Accili D. (2013) Expanded granulocyte/monocyte compartment in myeloid-specific triple FoxO knockout increases oxidative stress and accelerates atherosclerosis in mice. Circ. Res. 112, 992–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., and Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 15. Kido Y., Burks D. J., Withers D., Bruning J. C., Kahn C. R., White M. F., and Accili D. (2000) Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. J. Clin. Invest. 105, 199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Talchai S. C., and Accili D. (2015) Legacy effect of Foxo1 in pancreatic endocrine progenitors on adult β-cell mass and function. Diabetes 64, 2868–2879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kitamura T., Kitamura Y. I., Kobayashi M., Kikuchi O., Sasaki T., Depinho R. A., and Accili D. (2009) Regulation of pancreatic juxtaductal endocrine cell formation by FoxO1. Mol. Cell. Biol. 29, 4417–4430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talchai C., Xuan S., Lin H. V., Sussel L., and Accili D. (2012) Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 150, 1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Farrah T., Deutsch E. W., Omenn G. S., Campbell D. S., Sun Z., Bletz J. A., Mallick P., Katz J. E., Malmström J., Ossola R., Watts J. D., Lin B., Zhang H., Moritz R. L., and Aebersold R. (2011) A high-confidence human plasma proteome reference set with estimated concentrations in PeptideAtlas. Mol. Cell. Proteomics 10, M110.006353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Obermüller S., Calegari F., King A., Lindqvist A., Lundquist I., Salehi A., Francolini M., Rosa P., Rorsman P., Huttner W. B., and Barg S. (2010) Defective secretion of islet hormones in chromogranin-B deficient mice. PLoS ONE 5, e8936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stephan A. H., Barres B. A., and Stevens B. (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389 [DOI] [PubMed] [Google Scholar]
- 22. Lo J. C., Ljubicic S., Leibiger B., Kern M., Leibiger I. B., Moede T., Kelly M. E., Chatterjee Bhowmick D., Murano I., Cohen P., Banks A. S., Khandekar M. J., Dietrich A., Flier J. S., Cinti S., et al. (2014) Adipsin is an adipokine that improves β cell function in diabetes. Cell 158, 41–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zemany L., Bhanot S., Peroni O. D., Murray S. F., Moraes-Vieira P. M., Castoldi A., Manchem P., Guo S., Monia B. P., and Kahn B. B. (2015) Transthyretin antisense oligonucleotides lower circulating RBP4 levels and improve insulin sensitivity in obese mice. Diabetes 64, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Matsubara J., Honda K., Ono M., Tanaka Y., Kobayashi M., Jung G., Yanagisawa K., Sakuma T., Nakamori S., Sata N., Nagai H., Ioka T., Okusaka T., Kosuge T., Tsuchida A., et al. (2011) Reduced plasma level of CXC chemokine ligand 7 in patients with pancreatic cancer. Cancer Epidemiol. Biomarkers. Prev. 20, 160–171 [DOI] [PubMed] [Google Scholar]
- 25. Stojanovic I., Saksida T., and Stosic-Grujicic S. (2012) β Cell function: the role of macrophage migration inhibitory factor. Immunol. Res. 52, 81–88 [DOI] [PubMed] [Google Scholar]
- 26. Vujicic M., Senerovic L., Nikolic I., Saksida T., Stosic-Grujicic S., and Stojanovic I. (2014) The critical role of macrophage migration inhibitory factor in insulin activity. Cytokine 69, 39–46 [DOI] [PubMed] [Google Scholar]
- 27. Tiedge M., Lortz S., Drinkgern J., and Lenzen S. (1997) Relation between antioxidant enzyme gene expression and antioxidative defense status of insulin-producing cells. Diabetes 46, 1733–1742 [DOI] [PubMed] [Google Scholar]
- 28. Van De Winkel M., and Pipeleers D. (1983) Autofluorescence-activated cell sorting of pancreatic islet cells: purification of insulin-containing B-cells according to glucose-induced changes in cellular redox state. Biochem. Biophys. Res. Commun. 114, 835–842 [DOI] [PubMed] [Google Scholar]
- 29. Trus M., Warner H., and Matschinsky F. (1980) Effects of glucose on insulin release and on intermediary metabolism of isolated perifused pancreatic islets from fed and fasted rats. Diabetes 29, 1–14 [DOI] [PubMed] [Google Scholar]
- 30. Ivarsson R., Quintens R., Dejonghe S., Tsukamoto K., in 't Veld P., Renström E., and Schuit F. C. (2005) Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54, 2132–2142 [DOI] [PubMed] [Google Scholar]
- 31. Góth L., Lenkey A., and Bigler W. N. (2001) Blood catalase deficiency and diabetes in Hungary. Diabetes care 24, 1839–1840 [DOI] [PubMed] [Google Scholar]
- 32. Atalay M., Laaksonen D. E., Niskanen L., Uusitupa M., Hänninen O., and Sen C. K. (1997) Altered antioxidant enzyme defences in insulin-dependent diabetic men with increased resting and exercise-induced oxidative stress. Acta Physiol. Scand 161, 195–201 [DOI] [PubMed] [Google Scholar]
- 33. Kikumoto Y., Sugiyama H., Inoue T., Morinaga H., Takiue K., Kitagawa M., Fukuoka N., Saeki M., Maeshima Y., Wang D. H., Ogino K., Masuoka N., and Makino H. (2010) Sensitization to alloxan-induced diabetes and pancreatic cell apoptosis in acatalasemic mice. Biochim. Biophys. Acta 1802, 240–246 [DOI] [PubMed] [Google Scholar]
- 34. Wang X., Vatamaniuk M. Z., Roneker C. A., Pepper M. P., Hu L. G., Simmons R. A., and Lei X. G. (2011) Knockouts of SOD1 and GPX1 exert different impacts on murine islet function and pancreatic integrity. Antioxid. Redox Signal. 14, 391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harmon J. S., Bogdani M., Parazzoli S. D., Mak S. S., Oseid E. A., Berghmans M., Leboeuf R. C., and Robertson R. P. (2009) β-Cell-specific overexpression of glutathione peroxidase preserves intranuclear MafA and reverses diabetes in db/db mice. Endocrinology 150, 4855–4862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arntfield M. E., and van der Kooy D. (2011) β-Cell evolution: How the pancreas borrowed from the brain: the shared toolbox of genes expressed by neural and pancreatic endocrine cells may reflect their evolutionary relationship. BioEssays 33, 582–587 [DOI] [PubMed] [Google Scholar]
- 37. Eberhard D. (2013) Neuron and β-cell evolution: learning about neurons is learning about β-cells. BioEssays 35, 584. [DOI] [PubMed] [Google Scholar]
- 38. Kim R. H., Smith P. D., Aleyasin H., Hayley S., Mount M. P., Pownall S., Wakeham A., You-Ten A. J., Kalia S. K., Horne P., Westaway D., Lozano A. M., Anisman H., Park D. S., and Mak T. W. (2005) Hypersensitivity of DJ-1-deficient mice to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrindine (MPTP) and oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 102, 5215–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Farrer M. J. (2006) Genetics of Parkinson disease: paradigm shifts and future prospects. Nat. Rev. Genet. 7, 306–318 [DOI] [PubMed] [Google Scholar]
- 40. Tsuboi Y., Munemoto H., Ishikawa S., Matsumoto K., Iguchi-Ariga S. M., and Ariga H. (2008) DJ-1, a causative gene product of a familial form of Parkinson's disease, is secreted through microdomains. FEBS Lett. 582, 2643–2649 [DOI] [PubMed] [Google Scholar]
- 41. Bonifati V., Rizzu P., van Baren M. J., Schaap O., Breedveld G. J., Krieger E., Dekker M. C., Squitieri F., Ibanez P., Joosse M., van Dongen J. W., Vanacore N., van Swieten J. C., Brice A., Meco G., et al. (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299, 256–259 [DOI] [PubMed] [Google Scholar]
- 42. Jain D., Jain R., Eberhard D., Eglinger J., Bugliani M., Piemonti L., Marchetti P., and Lammert E. (2012) Age- and diet-dependent requirement of DJ-1 for glucose homeostasis in mice with implications for human type 2 diabetes. J. Mol. Cell Biol. 4, 221–230 [DOI] [PubMed] [Google Scholar]
- 43. Hong Z., Shi M., Chung K. A., Quinn J. F., Peskind E. R., Galasko D., Jankovic J., Zabetian C. P., Leverenz J. B., Baird G., Montine T. J., Hancock A. M., Hwang H., Pan C., Bradner J., et al. (2010) DJ-1 and α-synuclein in human cerebrospinal fluid as biomarkers of Parkinson's disease. Brain 133, 713–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shi M., Zabetian C. P., Hancock A. M., Ginghina C., Hong Z., Yearout D., Chung K. A., Quinn J. F., Peskind E. R., Galasko D., Jankovic J., Leverenz J. B., and Zhang J. (2010) Significance and confounders of peripheral DJ-1 and α-synuclein in Parkinson's disease. Neurosci. Lett. 480, 78–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lin X., Cook T. J., Zabetian C. P., Leverenz J. B., Peskind E. R., Hu S. C., Cain K. C., Pan C., Edgar J. S., Goodlett D. R., Racette B. A., Checkoway H., Montine T. J., Shi M., and Zhang J. (2012) DJ-1 isoforms in whole blood as potential biomarkers of Parkinson disease. Sci. Rep. 2, 954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Waanders L. F., Chwalek K., Monetti M., Kumar C., Lammert E., and Mann M. (2009) Quantitative proteomic analysis of single pancreatic islets. Proc. Natl. Acad. Sci. U.S.A. 106, 18902–18907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Steneberg P., Bernardo L., Edfalk S., Lundberg L., Backlund F., Ostenson C. G., and Edlund H. (2013) The type 2 diabetes-associated gene Ide is required for insulin secretion and suppression of α-synuclein levels in β-cells. Diabetes 62, 2004–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Geng X., Lou H., Wang J., Li L., Swanson A. L., Sun M., Beers-Stolz D., Watkins S., Perez R. G., and Drain P. (2011) α-Synuclein binds the KATP channel at insulin-secretory granules and inhibits insulin secretion. Am. J. Physiol. Endocrinol. Metab. 300, E276–E286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Rodriguez-Araujo G., Nakagami H., Takami Y., Katsuya T., Akasaka H., Saitoh S., Shimamoto K., Morishita R., Rakugi H., and Kaneda Y. (2015) Low α-synuclein levels in the blood are associated with insulin resistance. Sci. Rep. 5, 12081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pomero F., Di Minno M. N., Fenoglio L., Gianni M., Ageno W., and Dentali F. (2015) Is diabetes a hypercoagulable state? A critical appraisal. Acta Diabetol. 52, 1007–1016 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.