

Abstract
A major focus for our laboratory is identifying the molecules and mechanisms that regulate polarized apical protein sorting in hepatocytes, the major epithelial cells of the liver. These trafficking pathways are regulated, in part, by small molecular weight rab GTPases. We chose to investigate rab17, whose expression is restricted to polarized epithelial cells, is enriched in liver, and has been implicated in regulating basolateral to apical transcytosis. To initiate our studies, we generated three recombinant adenoviruses expressing wild type, constitutively active (GTP bound), or dominant-negative (GDP bound) rab17. Immunoblotting revealed rab17 immunoreactive species at 25 kDa (the predicted rab17 molecular mass) and 40 kDa. We determined that mono-sumoylation of the 25-kDa rab17 is responsible for the shift in molecular mass, and that rab17 prenylation is required for sumoylation. We further determined that sumoylation selectively promotes interactions with syntaxin 2 (but not syntaxins 3 or 4) and that these interactions are nucleotide dependent. Furthermore, a K68R-mutated rab17 led to the redistribution of syntaxin 2 and 5′ nucleotidase from the apical membrane to subapical puncta, whereas multidrug resistance protein 2 distributions were not changed. Together these data are consistent with the proposed role of rab17 in vesicle fusion with the apical plasma membrane and further implicate sumoylation as an important mediator of protein-protein interactions. The selectivity in syntaxin binding and apical protein redistribution further suggests that rab17 and syntaxin 2 mediate fusion of transcytotic vesicles at the apical surface.
Keywords: hepatocyte, Rab, small GTPase, SNARE proteins, sumoylation
Introduction
Our research is focused on identifying and defining the molecules and mechanisms that regulate polarized protein sorting in hepatic cells. In particular, we have been interested in the molecular machinery that participates in the delivery of newly synthesized apical residents to the apical surface along the transcytotic pathway. Recently, we turned our attention to the small molecular weight GTPase, rab17, as a candidate regulator of the final steps of vesicle delivery and fusion with the apical surface. Over two decades ago, rab17 was identified as a polarized epithelia-specific rab isoform with enriched expression in liver, kidney, and intestine (1, 2). It has been localized to subapical structures in a variety of cell types and tissues (2–5), and in intact hepatocytes, it was found associated with purified transcytotic vesicles (6). This expression pattern and distribution led many to hypothesize that rab17 functioned in transcytosis, which was confirmed in studies from polarized mammary gland-derived Eph4 cells and in Madin-Darby canine kidney cells (3, 5).
Interest has been rekindled in rab17 over the last few years as it was discovered that this GTPase is also expressed in hippocampus and in melanocytic cells (7, 8). As observed for polarized epithelial cells of the kidney, rab17 expression in hippocampus and melanocytes is developmentally regulated with increased expression upon maturation and differentiation (1, 2, 7, 8). Functional studies have implicated rab17 as an important regulator of dendrite formation in hippocampal neurons by mediating cargo delivery to developing dendrites and dendritic spines (8). In melanocytes, rab17 knockdown led to increased cellular melanin content without corresponding changes in melanosome maturation or movement. Based on alterations in filipodia formation (the putative sites of melanin release) in knockdown cells, the authors proposed that increased melanin content could be explained by impaired melanosome fusion (7). These newer findings therefore implicate rab17 as an important mediator of membrane fusion with the cell surface. Thus, combined with results in polarized epithelial cells and its apical/subapical distributions, we suggest that rab17 functions in vesicle docking and fusion with the apical membrane in polarized hepatocytes by mediating interactions with the SNARE-mediated fusion machinery.
Over 15 years ago, we determined that syntaxins 2, 3, and 4 exhibit polarized surface expression in hepatocytes with syntaxins 2 and 3 being the apical isoforms and syntaxin 4 the sole, basolateral Q-SNARE (9). Thus, to test our hypothesis that rab17 mediates apical vesicle fusion, we took a biochemical approach and assayed rab17 interactions with the plasma membrane-associated SNAREs. For these studies, we generated three recombinant adenoviruses expressing wild type, GTP-bound/Q77L, or GDP-bound/N132I rab17. Immunoblotting revealed immunoreactive species at 25 kDa (the predicted rab17 molecular mass) and 40 kDa for all three rab17 constructs. Using pharmacological agents that impair sumoylation, lysine mutational analysis, and immunoblotting for SUMO conjugates, we determined that the rab17 molecular mass shift is due to sumoylation of the 25-kDa species, and that rab17 prenylation is required for sumoylation. We further determined that sumoylation selectively promotes interactions with syntaxin 2 and that these interactions are nucleotide dependent. Furthermore, a K68R-mutated rab17 led to the redistribution of syntaxin 2 and 5′ nucleotidase (5′NT)2 from the apical membrane to subapical puncta, whereas multidrug resistance protein 2 (MRP2) distributions were not changed. Together these data are consistent with the proposed role of rab17 in vesicle fusion with the apical plasma membrane and further implicate sumoylation as an important mediator of protein-protein interactions. The selectivity in syntaxin binding and apical protein redistribution further suggests that rab17 and syntaxin 2 mediate fusion of apically directed vesicles delivered via transcytosis, but not via the direct pathway.
Experimental Procedures
Reagents and Antibodies
F-12 and F-12 (Coon's modification) medium, nocodazole, latrunculin B, methyl-β-cyclodextrin (mβCD) HRP-conjugated secondary antibodies, and monoclonal antibodies against the FLAG epitope and α-tubulin were purchased from Sigma. Hepes and Alexa 466- and 568-conjugated secondary antibodies were purchased from Invitrogen Life Technologies. GST-tagged SENP1 and SENP2 catalytic domains (human/recombinant) and anacardic acid was from Enzo Life Sciences (Farmingdale, NY). Monoclonal antibodies against the V5 epitope tag or full-length rab17 were from AbD Serotec (Raleigh, NC) and Proteintech (Chicago, IL), respectively. Polyclonal antibodies against histone deacetylase 6 (HDAC6) were from Santa Cruz Biotechnologies (Dallas, TX). Monoclonal antibodies against SUMO1 (21C7) or SUMO 2/3 (8A2) epitopes were from Thermo Fisher Scientific (Waltham, MA) and AbCam, respectively (Cambridge, UK). Monoclonals against RanGAP (1B4) were from AbCam, whereas monoclonals against MRP2 (M2III-6) were from Alexis Biochemicals (San Diego, CA). Fetal bovine serum (FBS) and newborn calf serum were from Gemini Bio-Products (Woodland, CA). Antibodies against the myc epitope, aminopeptidase N (APN), 5′NT, albumin, syntaxins 2, 3, and 4, and the GST-syntaxin constructs (9) were kindly provided by Dr. Ann Hubbard (Johns Hopkins University School of Medicine, Baltimore, MD).
Cell Culture
WIF-B cells were grown in a humidified 7% CO2 incubator at 37 °C as described (10). Briefly, cells were grown in F-12 medium (Coon's modification), pH 7.0, supplemented with 5% FBS, 10 μm hypoxanthine, 40 nm aminoterpin, and 1.6 μm thymidine. Clone 9 cells were grown at 37 °C in a 5% CO2 incubator in F-12 medium supplemented with 10% newborn calf serum. WIF-B cells were seeded onto glass coverslips at 1.3 × 104 cells/cm2, whereas Clone 9 cells were seeded onto coverslips in 6-well dishes at 0.5–1.0 × 106 cells/well. Clone 9 cells were cultured for 1–2 days and WIF-B cells for 8–12 days until they reached maximal density and polarity.
Virus Production and Infection
Full-length mouse rab17 (MGC clone ID 2648145) in a pCMV-SPORT6 vector was purchased from Invitrogen Life Technologies. Recombinant adenoviruses encoding myc or FLAG epitope-tagged full-length wild type rab17, and the previously characterized full-length GTP-bound/Q77L and GDP-bound/N132I rab17 mutants (2) were generated using the ViraPower Adenoviral Expression System (Life Technologies) according to the manufacturer's instructions. The myc and FLAG epitopes were added to the N terminus of all rab17 constructs except for one full-length wild type construct with a V5 tag at the C terminus. The Q77L and N132I mutations were made using the QuikChange Lightening Site-directed Mutagenesis kit from Agilent Technologies (Wilmington, DE). Sumoylation sites Lys-41 and Lys-68 were determined by the Sumoplot analysis program (Abgent) and the lysines substituted with arginines using the Q5 Site-directed Mutagenesis kit (New England Biolabs, Ipswich, MA) according to the manufacturer's instructions. All mutations were verified by plasmid sequencing (Retrogen, San Diego, CA). WIF-B or Clone 9 cells were infected with recombinant adenovirus particles for 60 min at 37 °C as described (11). Complete medium was added to the cells and they were incubated an additional 16–24 h to allow for protein expression.
Immunoblotting
Samples were mixed with Laemmli sample buffer (12) and boiled for 3 min. Proteins were electrophoretically separated using SDS-PAGE, transferred to nitrocellulose using a Transblot Turbo (Bio-Rad), and immunoblotted with the indicated antibodies. HRP-conjugated secondary antibodies were used at 5 ng/ml, and immunoreactivity was detected with enhanced chemiluminescence (PerkinElmer Life Sciences, Waltham, MA) using the ChemiDoc Touch Imager (Bio-Rad). Relative protein levels were determined by densitometric analysis of immunoreactive bands using ImageJ software (National Institutes of Health).
MALDI-MS Analysis
Mass spectrometry was performed by the Protein Chemistry Core Facility at Columbia University (New York, NY). Gel bands were prepared for digestion by washing twice with 50 mm Tris, pH 8.5, 30% acetonitrile. Gel pieces were subsequently dried in a SpeedVac concentrator and digested with trypsin (Roche Molecular Biochemicals, Indianapolis, IN) in 25 mm Tris, pH 8.5. Tubes were placed in a heating block at 32 °C and incubated overnight. Peptides were extracted with 50% acetonitrile, 2% trifluoroacetic acid (TFA) and suspended in a matrix solution containing 10 mg/ml of 4-hydroxy-α-cyanocinnamic acid and 50% acetonitrile, 0.1% TFA. The dried sample was analyzed by MALDI-MS analysis (Applied Biosystems Voyager DE Pro Mass spectrometer in linear mode). The MALDI spectra were manually searched against the NCBI database for protein matches. Parameters used in the search were Database: NBCI: taxonomy, musculus; enzyme, trypsin. MOWSE scores were generated with the MS-Fit program of Protein Prospector, version 5.3.0 (University of California San Francisco Mass Spectrometry Facility). Mascot (Ions) scores were generated from the Mascot search program. Up to one missed tryptic cleavage was allowed and cysteine propionamidation and methionine oxidation were considered. The peptide mass tolerance was 0.5 Da.
Cell Treatments
To disrupt SDS-resistant complexes, WIF-B cells were either directly lysed in Laemmli sample buffer containing 8 m urea or in Hepes lysis buffer (150 mm NaCl, 25 mm Hepes, 1% Triton X-100 (v/v), 5 mm CaCl2, and 2 mm MgSO4, pH 7.4) containing 6 n guanidine hydrochloride for 30 min followed by centrifugation for 5 min at 1,000 × g to remove nuclei. The supernatant was precipitated with 9 volumes of absolute ethanol at −20 °C for 60 min. The sample was centrifuged for 15 min at 4 °C at 15,000 × g, the supernatant was removed and the pellet air-dried. The pellet was washed twice with 90% ethanol at −20 °C and air dried. The final pellet was resuspended in Laemmli sample buffer. Alternatively, cells were extracted in chloroform/methanol. Briefly, WIF-B cells grown on coverslips were lysed in 100 μl of Hepes lysis buffer to which 400 μl of methanol was added. After vortexing, 100 μl of chloroform and 300 μl H2O were added and the sample was centrifuged for 1 min at 14,000 × g. The aqueous layer was removed and mixed with 400 μl of methanol and recentrifuged at 2 min at 14,000 × g. The methanol was removed and the pellet dried with a SpeedVac. The final pellet was resuspended in Laemmli sample buffer.
To inhibit sumoylation, cells were treated with 5 μm anacardic acid or 1 mm H2O2 in serum-free medium at 37 °C for up to 60 min. Proteosomal degradation was inhibited by addition of 5 μm lactacystin for 4 h at 37 °C in complete medium. Protein synthesis was inhibited by addition of 50 μg/ml of cycloheximide in complete medium for 2–4 h at 37 °C. Cells were also treated with 33 μm nocodazole, 10 μm latrunculin B, or 5 mm mβCD for 60 min at 37 °C to disrupt microtubules, actin, or deplete cholesterol, respectively. In all cases, cells were lysed directly in Laemmli sample buffer after treatments and immunoblotted.
Immunofluorescence Microscopy and Imaging
Control or infected cells were fixed on ice with chilled PBS containing 4% paraformaldehyde for 1 min and permeabilized with ice-cold methanol for 10 min or were pre-permeabilized with 0.1% Triton X-100, PBS for 2 min at 37 °C and then fixed with ice-cold methanol for 5 min at −20 °C. Cells were processed for indirect immunofluorescence as previously described (13). Alexa 488- or 568-conjugated secondary antibodies were used at 3–5 μg/ml. Labeled cells were visualized at room temperature by epifluorescence with an Olympus BX60 Fluorescence Microscope (OPELCO, Dulles, VA) using an UPlanFl ×60/NA 1.3, phase 3, oil immersion objective. Images were taken with an HQ2 CoolSnap digital camera (Roper Scientific, Germany) and IP Labs software (BD Biosciences, Rockville, MD). Adobe Photoshop (Adobe Systems Inc., Mountain View, CA) was used to process images and compile figures.
Immunoprecipitations
WIF-B cells grown on 6 coverslips were pooled and lysed in 0.5 ml of Hepes lysis buffer with added protease inhibitors (1 μg/ml each of leupeptin, antipain, PMSF, and benzamidine) and incubated on ice for 30 min. Lysates were cleared by centrifugation at 120,000 × g for 30 min at 4 °C. Supernatants were incubated with anti-FLAG antibodies (1 μg/ml) overnight at 4 °C on a fixed-speed tube rotator. Protein G-Sepharose (50 μl of a 50% (v/v) slurry) was added for 2 h at 4 °C. The beads were recovered by centrifugation (1,000 × g for 2 min at 4 °C). Beads were washed once with Hepes lysis buffer containing 5% BSA, twice with Hepes lysis buffer, and once with PBS (14).
SENP1/2 Proteolysis Assays
WIF-B cells grown on 5 coverslips were pooled and lysed in 0.5 ml of Hepes lysis buffer containing 3 mm MgCl2 and 1 mm dithiothreitol, pH 7.5, with protease inhibitors (1 μg/ml each of leupeptin, antipain, PMSF, and benzamidine) and incubated on ice for 30 min. Lysates were cleared by centrifugation at 120,000 × g for 30 min at 4 °C. The supernatant was divided into 100-μl aliquots to which GST-SENP1 or −2 was added (1 or 2 μg of each) and the reaction mixtures were incubated at 37 °C for 1 h. The reactions were stopped by the addition of Laemmli sample buffer.
Cell Extraction and Fractionation
For extractions, WIF-B cells grown on coverslips were placed in 1 ml of Hepes lysis buffer (with only 0.1% Triton X-100) containing protease inhibitors (2 μg/ml each of leupeptin, antipain, PMSF, and benzamidine) at 37 °C for up to 150 s. The buffer with extracted cellular contents was collected and immunoblotted for rab17, α-tubulin, or HDAC6. For fractionation, WIF-B cells grown on 6 coverslips were scraped into 1 ml of 0.25 m sucrose, 3 mm imidazole, pH 7.4, with added protease inhibitors (2 μg/ml each of leupeptin, antipain, PMSF, and benzamidine). The cells were homogenized with a BeadBug Homogenizer (Benchmark, South Plainfield, NJ) in microcentrifuge tubes with 0.5-mm glass beads for 30 s at 2,800 rpm. The homogenate was centrifuged for 5 min at 1,000 × g at 4 °C to prepare a postnuclear supernatant. The postnuclear supernatant was centrifuged at 60,000 × g for 60 min at 4 °C to prepare a membrane pellet (excluding nuclei) and a cytosolic fraction.
GST-syntaxin Expression and Pulldown Assays
Syntaxins 2, 3, and 4 lacking their transmembrane domains and fused in frame to GST were expressed in Escherichia coli using standard methods of growth and isopropyl 1-thio-β-d-galactopyranoside induction (9). Cells were harvested by centrifugation (12,000 × g for 20 min at 4 °C) and resuspended in PBS containing 1% (v/v) Triton X-100, 5 mm benzamidine, 2 mm EDTA, 0.2 mm PMSF, and 0.1% (v/v) 2-mercaptoethanol. After sonication and centrifugation (12,000 × g for 10 min at 4 °C), the supernatant was mixed with an equal volume of a 50% (v/v) slurry of glutathione-agarose equilibrated in PBS containing 1% (v/v) Triton X-100. The mixture was incubated for 2 h to overnight at 4 °C with gentle rotation. The agarose with bound fusion proteins was washed 4–6 times by resuspension in PBS containing 1% (v/v) Triton X-100 followed by sedimentation at 1,000 × g for 5 min at 4 °C.
For pulldowns, WIF-B cells grown on 6 coverslips were scraped and pooled in 1 ml of Hepes lysis buffer as described above and extracted on ice for 30 min. To pre-clear the extracts of endogenous GST, each lysate was incubated with 60 μl of a 50% (v/v) glutathione-agarose slurry for 2 h at 4 °C on a fixed-speed tube rotator. The extracts were recovered by centrifugation (1,000 × g for 2 min at 4 °C) and mixed with 60 μl of a 50% (v/v) slurry of GST-syntaxin 2-, 3-, or 4-agarose. The extracts were incubated overnight at 4 °C while gently mixing on a fixed-speed tube rotator. The agarose was recovered by centrifugation (1,000 × g for 2 min at 4 °C). The unbound sample was collected and the agarose was washed once with Hepes lysis buffer containing 5% BSA, twice with Hepes lysis buffer, and once with PBS (14). The agarose pellet was eluted with Laemmli sample buffer.
Statistical Analysis
Results were expressed as the mean ± S.E. from at least three independent experiments. Comparisons between experimental groups were made using the Student's t test for paired data. p values ≤ 0.05 were considered significant.
Results
Rab17 Is Expressed as a Low and High Molecular Weight Protein
For our analysis, we generated recombinant adenoviruses expressing myc-tagged wild type rab17. When immunoblotted with anti-myc antibodies, a 25-kDa species was detected corresponding to the predicted rab17 molecular mass (Fig. 1A). Of particular interest was an additional species at 40 kDa detected in overexpressing cells (Fig. 1A, upper arrow). Another 37-kDa protein was detected in both control and expressing cells (marked with an asterisk) that was not observed when lysates were immunoblotted with rab17 antibodies (Fig. 1B) indicating it is an anti-myc cross-reactive band. To rule out that the 40-kDa immunoreactive species was an artifact of the addition of a myc tag, we also immunoblotted lysates expressing FLAG-tagged versions of wild type rab17. As shown in Fig. 1C, both the 25- and 40-kDa species of the FLAG-tagged rab17 were detected. Both molecular mass species of the wild type rab17 were also detected in lysates from Clone 9 cells (non-polarized hepatic cells that lack endogenous rab17 expression) (Fig. 1D) indicating that expression of the 40-kDa protein is independent of cell polarity and not specific to WIF-B cells. To rule out that the higher molecular weight band was an artifact of overexpression, we infected cells with decreasing concentrations of virus. As shown in Fig. 1E (and see Fig. 4), both bands were visible even at the lowest detectable levels of expression. The percentage of the 40-kDa species relative to total rab17 was remarkably consistent among the different virus dilutions (34.4, 35.7, and 31.7%, respectively) and among differently tagged constructs and cell types, ranging from ∼30 to 40% (Fig. 1F).
FIGURE 1.
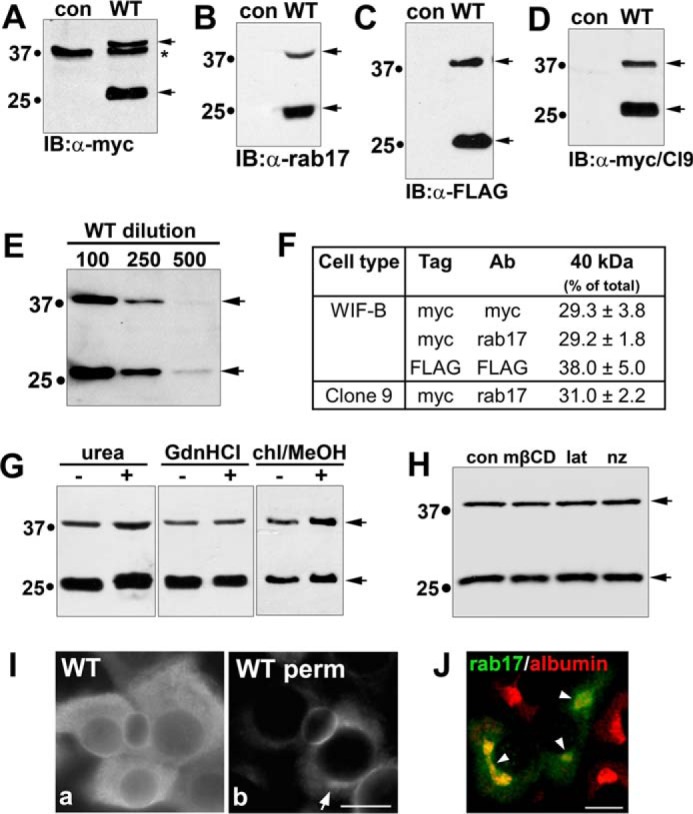
Rab17 is expressed as a low and high molecular weight protein. WIF-B lysates expressing an N terminally myc-tagged rab17 were immunoblotted with antibodies against myc (A) or rab17 (B). In C, WIF-B lysates from cells expressing an N terminally FLAG-tagged rab17 were immunoblotted with antibodies against FLAG, whereas in D, Clone 9 cells expressing the N terminally myc-tagged rab17 were blotted with anti-myc antibodies. Arrows mark the 40- and 25-kDa immunoreactive species. Molecular mass markers are indicated on the left of each immunoblot in kDa. In A, the asterisk is marking a myc cross-reactive band. E, WIF-B cells were infected with decreasing concentrations of virus (1:100, 1:250, and 1:500) for 24 h. Lysates were immunoblotted for rab17. Even at the lowest infection levels, the 40 kDa was observed. In F, the relative levels of the 40-kDa immunoreactive band were determined by densitometry and calculated as the percent of total rab17. Values represent the average of at least 3 independent experiments ± S.E. In G, to disrupt possible SDS-resistant complexes, WIF-B lysates from cells expressing wild type myc-tagged rab17 were mixed with 8 m urea or 6 n guanidine hydrochloride (GdnHCl) or cells were pre-extracted with chloroform/methanol (chl/MeOH) and immunoblotted (IB) for rab17 using anti-rab17 antibodies. H, WIF-B cells expressing myc-tagged wild type rab17 were treated with 5 mm methyl-β-cyclodextrin (mβCD; to deplete cholesterol), 10 μm latrunculin B (lat; to depolymerize actin), or 33 μm nocodazole (nz; to depolymerize microtubules) for 60 min at 37 °C. Lysates were immunoblotted for rab17 with anti-rab17 antibodies. In I, WIF-B cells expressing FLAG-tagged rab17 were stained with anti-FLAG antibodies. In a, cells were permeabilized after fixation, whereas in b, cells were pre-permeabilized with Triton X-100 (see ”Experimental Procedures“). The arrow in b is pointing to intracellular distributions of rab17. Bar = 10 μm. In H, WIF-B cells overexpressing FLAG-tagged rab17 were double labeled with the hepatic Golgi marker, albumin. The merged image reveals partial distribution of rab17 to the Golgi. Arrows are pointing to colocalized rab17 and albumin. Bar = 10 μm.
FIGURE 4.

Rab17 sumoylation is a reversible modification and its levels are specifically maintained. A, WIF-B cells were infected for the indicated times with recombinant adenovirus expressing FLAG-tagged rab17. Lysates were collected and immunoblotted for rab17 using anti-FLAG antibodies. The relative levels of the 25- and 40-kDa bands were determined by densitometry and the arbitrary units (a.u.) plotted (B). In C, the percent of total rab17 detected as the 40-kDa species for each time was determined and plotted. B and C are representative of at least three independent experiments. D, WIF-B cells expressing myc-tagged wild type rab17 were treated for the indicated times with 50 μg/ml of cycloheximide (CHX). Lysates were prepared and immunoblotted for rab17 using anti-rab17 antibodies. The relative levels of the 25- and 40-kDa bands were determined by densitometry, the values plotted as the percent present at 0 min (E). In F, the percent of total rab17 detected as the 40-kDa species for each time was determined. The values in E and F represent the average from at least 3 independent experiments ± S.E.
To confirm that both the 25- and 40-kDa immunoreactive species were indeed rab17, MALDI-MS was performed on gel-purified proteins. As shown in Table 1, multiple rab17 tryptic fragments were recovered for each band representing 46% coverage for the 25-kDa band and 29% for the 40-kDa band. The relatively high ions scores (457 and 136) calculated for each sample were well above the threshold of 35 (with 95% confidence interval) thereby confirming the presence of rab17 at both molecular weights.
TABLE 1.
Mass spectrometry confirms that both immunoreactive species are rab17
The 25- and 40-kDa bands of all three rab17 proteins were excised from Coomassie Blue-stained gels, eluted, and processed for mass spectrometry. From the different tryptic fragments (Frags.) analyzed for each sample, the percent coverage of the rab17 sequence was calculated. The high ions scores confirm that each sample contains rab17.
| Species | Construct | Frags. | % Coverage | Ions score |
|---|---|---|---|---|
| 25 kDa | Wild type | 10 | 46.0 | 457 |
| Q77L | 12 | 67.0 | 503 | |
| N132I | 7 | 38.0 | 123 | |
| 40 kDa | Wild type | 6 | 29.0 | 136 |
| Q77L | 5 | 22.0 | 155 | |
| N132I | 5 | 24.0 | 107 |
To rule out the possibility that the 40-kDa rab17 was present in an SDS-resistant complex with itself or other proteins as observed for members of the SNARE family of proteins (15, 16), we used three treatments known to disrupt highly resistant complexes: solubilization in 8 m urea, treatment with 6 n guanidine chloride, or extraction in chloroform/methanol (17). As shown in Fig. 1G, the 40-kDa protein persisted in all three conditions suggesting that the 40-kDa species represents a monomeric, post-translationally modified rab17. Furthermore, no changes in the relative levels of the 40-kDa species were observed in cells treated with mβCD (to deplete cholesterol), latrunculin B (to disrupt actin), or nocodazole (to disrupt microtubules) indicating the modification is not dependent on acute changes in cellular distribution (Fig. 1H).
Finally as part of our initial characterization, we examined wild type rab17 distributions by indirect immunofluorescence. In methanol-permeabilized cells, a considerable diffuse cytosolic rab17 staining pattern was observed (Fig. 1I, panel a). This diffuse labeling was lost in detergent pre-permeabilized cells, indicating it represents a soluble population (Fig. 1I, panel b). In pre-permeabilized cells, discrete rab17 labeling mainly at or near the apical plasma membrane was observed with a smaller intracellular subpopulation (Fig. 1I, panel b). Co-labeling with albumin, a hepatic Golgi marker (18), confirmed that this intracellular rab17 was at the Golgi (Fig. 1J). Similar predominantly apical rab17 distributions have been reported for other polarized epithelial cells (2–5) and is consistent with syntaxin binding at the canalicular surface.
Rab17 Is Reversibly Sumoylated
Mono/di-ubiquitination and sumoylation are two common post-translational modifications of high enough molecular mass (8.5 and 11–12 kDa each subunit, respectively) to account for the apparent 12-kDa shift in molecular mass in rab17. To discriminate between the two possibilities, we first treated overexpressing cells with two pharmacological agents: the proteosomal inhibitor, lactacystin (to accumulate ubiquitinated species) or H2O2 (a cellular stress that impairs sumoylation (19–21)). As shown in Fig. 2A, no change in the levels of either the 25- or 40-kDa species was observed in lactacystin-treated cells indicating the molecular weight shift is not due to ubiquitination. Although only a loose ubiquitination consensus sequence has been identified (a lysine near multiple polar, but not hydrophobic residues) (22), no such sequence patterns are present in rab17 consistent with this result. In contrast, treatment with 1 mm H2O2 for 60 min led to a 50% (p ≤ 0.002) reduction in the 40-kDa species relative to total rab17 (Fig. 2, B and C) suggesting the shift in molecular mass could be accounted for by sumoylation.
FIGURE 2.

Rab17 is sumoylated. A, Clone 9 cells expressing myc-tagged wild type rab17 were treated with 5 μm lactacystin for 4 h at 37 °C. Lysates were collected and immunoblotted (IB) for rab17 with anti-rab17 antibodies or α-tubulin as a control. B, Clone 9 cells expressing FLAG-tagged wild type rab17 were treated with 1 mm H2O2 for 1 h at 37 °C. Lysates were collected and immunoblotted for rab17 using FLAG antibodies or for α-tubulin as a control. C, the percent of rab17 relative to total was determined by densitometry and plotted as the percent of the control ratio. The values represent the average from at least three independent experiments ± S.E. *, p ≤ 0.002. D, Clone 9 cells expressing myc-tagged wild type rab17 were treated for the indicated times with 5 μm anacardic acid at 37 °C. Lysates were collected and immunoblotted for rab17 with anti-rab17 antibodies or α-tubulin as a control. In E, the percent of 40- or 25-kDa rab17 relative to total was determined by densitometry for each sample and plotted as the percent of the control ratio. The values represent the average from at least three independent experiments ± S.E. *, p ≤ 0.003. F, wild type rab17 was immunoprecipitated from cell lysates with anti-FLAG antibodies and immunolabeled for rab17 (with anti-FLAG antibodies) or antibodies against SUMO1 and SUMO2/3. Lysates without added antibodies (−Ab) served as negative controls for nonspecific protein binding to the Sepharose (middle panels). A lighter exposure of the SUMO immunoblots are shown (right panel). Arrows are marking the cross-reactive IgG heavy (HC) and light chains (LC), the SUMO-positive 40-kDa rab17, and unmodified 25-kDa rab17. The asterisk is marking a cross-reactive species in both bound and unbound samples. Molecular mass markers are indicated on the left in kDa. G, cleared lysates were treated with the indicated concentrations of either SENP1 or SENP2 proteases for 1 h at 37 °C. Reactions were stopped by addition of Laemmli sample buffer and immunoblotted for RanGAP (positive control; upper panel) or rab17 (lower panel). Arrows indicate the sumoylated (top-most arrow for each panel) and desumoylated (bottom-most arrow) forms of the proteins. A representative immunoblot from at least three independent experiments is shown. In H, the levels of the 40-kDa band relative to total rab17 from the immunoblot in G were determined by densitometry and plotted.
To directly determine whether the 40-kDa band represents a sumoylated form of rab17, we first treated cells with anacardic acid. This compound directly binds and deactivates the SUMO activating enzyme (E1) thereby inhibiting the first step of the three-step sumoylation reaction. Also importantly, it is specific for sumoylation with no effects on ubiquitination (23). As shown in Fig. 2D, anacardic acid led to a marked decrease in the levels of the 40-kDa rab17 with a reciprocal increase in the levels of the 25-kDa species. When quantitated, we determined that the 40-kDa levels relative to total rab17 were decreased significantly (p ≤ 0.003) by nearly 60% (59.3 ± 8.8%) (Fig. 2E).
To confirm rab17 sumoylation, we probed rab17 immunoprecipitates with antibodies against SUMO1 and SUMO2/3. As shown in Fig. 2F (left panel), both the rab17 species were quantitatively immunoprecipitated with anti-FLAG antibodies. Furthermore, no nonspecific bands were detected in samples without added antibodies (Fig. 2F, left panel). When a corresponding precipitate was immunoblotted for SUMOs 1 and 2/3, only the 40-kDa rab17 was detected in the samples containing antibodies (Fig. 2F, middle panel). Lighter exposures where the 40-kDa band is dimly detected and the IgG light chain (LC) cross-reactivity is decreased, confirm that the 25 kDa is not detected (Fig. 2F, right panel). Furthermore, no immunoreactive bands were detected in rab17 precipitates immunoblotted with antibodies specific to ubiquitin conjugates (data not shown) confirming specificity of sumoylation.
In an effort to discriminate whether SUMO1 or SUMO2/3 was the preferred modification, we treated cleared lysates with the sumoylation-specific proteases, SENP1 (for SUMO1) or SENP2 (for SUMO2/3, but has some cross-specificity for SUMO1). Incubation with SENP1 efficiently cleaved SUMO1 from RanGAP (our positive control) and led to the dose-dependent decrease in the 40-kDa rab17 with a reciprocal increase in the 25-kDa rab17 (Fig. 2G). Although much less efficient cleavage, incubation with 2 μg of SENP1 led to the reproducible and significant loss of the 40-kDa rab17 relative to the total 70% of control ratios (Fig. 2H). When calculated across at least three independent experiments, these findings were found to be significant (p ≤ 0.002). SENP2 led to a more modest SUMO cleavage for both RanGap and rab17 (Fig. 2, G and H) suggesting that both proteins are preferentially modified by SUMO1 consistent with reports that SUMO1 is the main conjugate in mammalian cells and that RanGAP is a known SUMO1-modified protein (24).
Sequence analysis revealed two highly predicted sumoylation consensus sites at lysines 68 (score = 0.93) and 41 (score = 0.8) (Fig. 3A). To determine whether these represented bona fide sumoylation sites, we mutated each lysine to an arginine alone or together. Although the levels of the 40-kDa rab17 were not changed for the K41R mutant, a highly reproducible 28.4 ± 7.0% decrease (p ≤ 0.03) in the levels of the 40-kDa relative to total rab17 were observed for the K68R mutant (Fig. 3B). This decrease is highly consistent with the 30% decrease in rab17 sumoylation observed after SENP1 cleavage (Fig. 2, G and H). Similar decreases have been reported for single site lysine mutants in other known non-nuclear sumoylated proteins, and in some cases, only when 2 or more lysines are mutated is a complete loss of sumoylation observed (25–28). However, the K41R/K68R double mutant formed large aggregates that were unable to enter the resolving gel (Fig. 3C, upper most arrow) and that were readily visible by indirect immunofluorescence (Fig. 3D, panel c). These complexes were partially disrupted by addition of 8 m urea (Fig. 3C) suggesting they are present in aggresomes and represent unfolded, non-functional proteins. Nonetheless, like wild type rab17, the K41R mutant distributed mainly to the apical surface. However, the K68R mutant was additionally observed in small subapical puncta (Fig. 3D) indicating that even a partial loss of sumoylation may impact rab17 function.
FIGURE 3.

Lysine 68 in rab17 is sumoylated. In A, the two highly predicted sumoylation sites in rab17 at lysines 68 and 41 using the Sumoplot Analysis program are shown with their respective scores. For comparison, the consensus sumoylation site is indicated. B, WIF-B cells expressing FLAG-tagged wild type, K68R, or K41R rab17 were lysed and immunoblotted for rab17 using anti-FLAG antibodies. In C, WIF-B cells expressing the FLAG-tagged wild type or K68R/K41R double mutant (dbl) were lysed in the absence (con) or presence of urea (+ urea) to disrupt protein aggregates. Lysates were probed for rab17 using anti-FLAG antibodies. The top-most arrow points to a large aggregate in both control and treated cells. The additional arrows point to species disrupted by urea treatment. D, cells expressing the K41R or K68R point mutants or the double mutant were labeled with anti-FLAG antibodies after pre-permeabilization in Triton X-100 and fixation (see ”Experimental Procedures“). Arrows in panel c are marking large K41R/K68R rab17-positive intracellular aggregates. Bar = 10 μm.
To determine whether the sumoylated rab17 reflected a mature form of the GTPase, we monitored the kinetics of the modifications appearance using two methods. The first was to immunoblot lysates from cells at early time points after infection with recombinant adenoviruses. If the higher molecular weight rab17 represents a mature form, the prediction is that its appearance should lag behind that of the lower molecular weight rab17. As shown in Fig. 4A, both the 25- and 40-kDa species were first detected 14 h post-infection. However, even at this early time point, the 40-kDa protein represented ∼30% of total rab17, similar to that observed at steady state (see Fig. 1F). The kinetics of appearance of the two species were nearly identical up to 24 h (Fig. 4, A and B) and the proportion of the 40-kDa species to total rab17 remained constant at ∼35–40% at each time point (Fig. 4C). Because no lag in the appearance of the 40-kDa species was observed, we conclude that it does not represent a mature form of rab17.
To confirm this result, we monitored possible maturation of the rab17 modification by immunoblotting lysates from cells after incubation with cycloheximide to prevent protein synthesis. The prediction is that the upper band should increase in relative intensity as the newly synthesized 25-kDa band is modified. However, as shown in Fig. 4, D and E, no increase in the upper band was detected. Although degradation of both bands was detected at later time points, both rab17 species were degraded with the same kinetics and importantly, the proportion of the 40 kDa to total rab17 remained constant (Fig. 4F). Together, these data indicate the upper band is not a mature form of the protein, but rather suggest that rab17 sumoylation is a highly regulated, reversible modification that is maintained at specific steady state levels.
Rab17 Sumoylation Is Nucleotide-independent, but Prenylation-dependent
To examine the effects of the rab17 nucleotide-bound state on sumoylation, we immunoblotted lysates from cells overexpressing the previously characterized GTP-bound/Q77L or GDP-bound/N132I rab17 mutants (2). As for wild type, the 40-kDa rab17 species was observed in WIF-B cells expressing the myc-tagged (Fig. 5A) or FLAG-tagged (Fig. 5B) mutants. Also as for wild type, the 40-kDa species for both mutants was also observed in Clone 9 cells (Fig. 5C). The percentage of the 40-kDa species relative to total rab17 was also remarkably consistent among the differently tagged constructs, mutants, and between cell types (Fig. 5D) irrespective of expression levels (N132I was consistently expressed at lower levels than wild type or Q77L rab17).
FIGURE 5.

Rab17 sumoylation is nucleotide independent, but prenylation dependent. A, WIF-B lysates expressing the N terminally myc-tagged GTP-bound/Q77L or GDP-bound/N132I rab17 mutants were immunoblotted with antibodies against myc. In B, lysates from cells expressing the N terminally FLAG-tagged GTP-bound/Q77L or GDP-bound/N132I rab17 mutants were immunoblotted (IB) with antibodies against FLAG, whereas in C, Clone 9 cells expressing the myc-tagged mutants were blotted with anti-myc antibodies. Arrows are marking the 40- and 25-kDa immunoreactive species. In A, the asterisk is marking a myc cross-reactive band. D, the relative levels of the 40-kDa immunoreactive species was determined by densitometry and calculated as the percent of total rab17. Values represent the average of at least 3 independent experiments ± S.E. E, lysates from cells expressing a C terminally V5-tagged rab17 were immunoblotted with antibodies against V5. Only the 25-kDa band was detected. F, WIF-B cells expressing rab17-V5 were fractionated by differential centrifugation and fractions were immunoblotted with anti-V5 antibodies (top panel) or antibodies specific for APN (middle) or HDAC6 (bottom). PNS, post-nuclear supernatant; nuc, nuclei; cyt, cytosol; mem, total membranes excluding nuclei.
MALDI-MS confirmed that both the 25- and 40-kDa immunoreactive species were rab17 for both mutants (Table 1). Multiple fragments for each sample were recovered with ions score well above the 35 threshold value thereby confirming the presence of rab17 at both molecular weights. Taken together, these results indicate that sumoylation is independent of the GTP/GDP-bound state of rab17.
Because rab17 is known to be prenylated at two C-terminal cysteines, and that its acylation is required for its role in transcytosis (3), we examined whether prenylation was required for sumoylation. For these experiments, we expressed wild type rab17 with a C-terminal V5 tag (rab17-V5) that disrupts proper CAAX box processing and prenylation (29, 30). As shown in Fig. 5E, only the low molecular mass 25-kDa form of rab17 was detected in cells overexpressing rab17-V5. To confirm that the expressed C terminally tagged protein was soluble (not prenylated), we assayed the distributions of rab17-V5 in nuclear, cytosolic, and membrane fractions (excluding nuclei) recovered by differential centrifugation. Immunoblotting for APN (a plasma membrane protein) and HDAC6 (a cytosolic protein) confirmed the fraction identities (Fig. 5F, bottom panels). Although a small amount of each marker was detected in the nuclear fraction, this likely reflects sedimentation of intact cells at low speed. Nonetheless, as predicted, the rab17-V5 exclusively distributed to the cytosolic fraction confirming its solubility and its unprenylated state (Fig. 5F). Thus, rab17 sumoylation is dependent on prenylation.
Sumoylated Wild Type and GTP-bound rab17, but Not GDP-bound rab17, Are Membrane Associated
Because protein sumoylation is often correlated with enhanced intermolecular binding interactions (reviewed in Ref. 24), we examined whether the sumoylated rab17 was exclusively membrane bound. Immunoblotting medium aliquots from permeabilized cells not only confirmed the wild type distributions (see Fig. 1I), but also revealed that the detergent-resistant/membrane-associated population is enriched for the sumoylated rab17 species. Within 30 s of permeabilization, the 25-kDa rab17 was detected in the medium, and by 90 s, robust levels were observed (Fig. 6A). This pattern bears a striking resemblance to that of the release of HDAC6, a cytosolic marker (Fig. 6A). In contrast, low levels of the 40-kDa rab17 species were not detected until after permeabilization for 90 s, and more robust levels were not released until after 150 s (Fig. 6A) correlating with the release of tubulin (from the polymer) in permeabilized cells (Fig. 6A).
FIGURE 6.

Sumoylated wild type and GTP-bound, but not GDP-bound rab17, are membrane associated. A, live WIF-B cells expressing myc-tagged wild type rab17 were extracted with 0.1% Triton X-100 in Hepes buffer at 37 °C for the indicated times. The buffer with extracted cellular contents was collected and immunoblotted for rab17, α-tubulin, or HDAC6 as indicated. Arrows mark the two rab17 species. B, WIF-B cells expressing wild type myc-tagged rab17 were fractionated as described in the legend to Fig. 5 and fractions were immunoblotted for rab17, APN, and HDAC6 as indicated. Numbers on the right indicate the percent total of either species detected in the final membrane pellet. C, WIF-B cells expressing FLAG-tagged Q77L or N132I rab17 were detergent pre-permeabilized, fixed, and stained with anti-FLAG antibodies. Arrows mark the subapically located Q77L rab17. Asterisks mark bile canaliculi in panels c and d. Bar = 10 μm.
To test membrane association directly, we assayed the distributions of rab17 in nuclear, cytosolic, and membrane fractions recovered by differential centrifugation. Blotting for APN (a plasma membrane protein) and HDAC6 (a cytosolic protein) confirmed the fraction identities (Fig. 6B, lower panels). As shown in Fig. 6B, nearly all of the sumoylated rab17 was detected in the membrane fraction (96.6 ± 1.7%). In contrast, only about one-third of the 25-kDa species was pelleted (33.9 ± 5.1%), which likely reflects a prenylated (but not sumoylated) rab17.
Like wild type, robust levels of both the GTP-bound/Q77L and GDP-bound/N132I mutants were detected in the cytosol in methanol-permeabilized cells (Fig. 6C). However, only the GTP-bound Q77L mutant was observed associated with membranes in pre-permeabilized cells (Fig. 6C); the GDP-bound N132I mutant staining was mostly lost (Fig. 6C). Notably, Q77L staining appeared less restricted to the apical surface than wild type and was detected in subapical puncta (arrows). Because both rab17 mutants are sumoylated, these results indicate that membrane association is additionally nucleotide-dependent.
Sumoylated Wild Type and GTP-bound rab17, but Not GDP-bound rab17, Selectively Bind Syntaxin 2
To determine whether rab17 interacts with apically located syntaxins 2 and/or 3, we performed GST pulldown assays, as previously described (9). Syntaxin 4 (basolaterally located) served as our negative control. As shown in Fig. 7A, only syntaxin 2 effectively pulled down wild type rab17. Interestingly, only the 40-kDa sumoylated rab17 was recovered in the bound samples. Although only 0.28 ± 0.09% of total rab17 added was recovered in the bound fractions, this was a highly reproducible result and likely reflects transient and/or indirect interactions between rab17 and syntaxin 2. Colocalization of rab17 with syntaxin 2 at the apical surface confirms these results (Fig. 7B).
FIGURE 7.
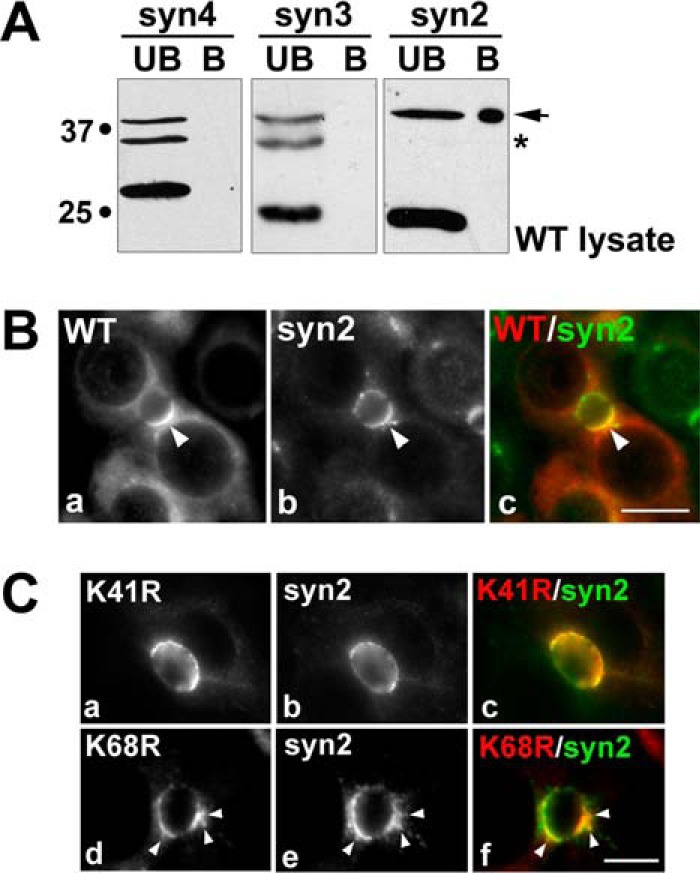
Sumoylated wild type rab17 selectively binds syntaxin 2 at the apical surface. A, detergent lysates from cells expressing wild type myc-tagged rab17 were applied to syntaxin-GST immobilized on glutathione-agarose and allowed to bind. The beads were pelleted and the unbound (UB) fraction collected. After extensive washing, the beads were mixed in sample buffer and represent the bound (B) fraction. The unbound and bound fractions were immunoblotted for rab17 with anti-myc antibodies. The arrow marks the sumoylated 40-kDa rab17 bound to syntaxin 2-GST. The asterisk marks the myc cross-reactive band. B, WIF-B cells expressing wild type FLAG-tagged rab17 were pre-permeabilized, fixed, and stained for rab17 (a) (with anti-FLAG antibodies) and for endogenous syntaxin 2 (b). Merged images are shown in c. Bar = 10 μm. C, WIF-B cells expressing FLAG-tagged K68R or K41R rab17 were pre-permeabilized, fixed, and stained for rab17 (a and d) (with anti-FLAG antibodies) and endogenous syntaxin 2 (b and e). Merged images are shown in c and f. Arrows are marking K68R rab17 present in subapical puncta that colocalizes with syntaxin 2. Bar = 10 μm.
Localization of the lysine mutants further implicates sumoylation in the regulation of rab17 and syntaxin 2 interactions. Like wild type, K41R rab17 colocalized with syntaxin 2 at the apical surface (Fig. 7C). However, syntaxin 2 colocalized with the K68R rab17 at both the apical surface and in subapical puncta (arrows in Fig. 7C).
To determine whether sumoylated rab17 binding to syntaxin 2 was nucleotide dependent, lysates expressing either the GTP-bound/Q77L rab17 or the GDP-bound/N132I or mutants were added to the syntaxin-GST batch columns. Like for wild type, only the 40-kDa GTP-bound rab17 selectively bound syntaxin 2 (Fig. 8A) at similar levels (0.21 ± 0.10% of total added). Consistent with its lack of membrane association, the GDP-bound rab17 did not bind the syntaxins (Fig. 8B) despite its sumoylated status. Similarly, the soluble rab17 acylation deficient mutant was recovered only in the unbound fractions (Fig. 8C). The soluble N132I distributions were confirmed in immunolabeled cells with no changes in syntaxin 2 apical distributions (Fig. 8D). Although the GTP-bound rab17 and syntaxin 2 colocalized in WIF-B cells, the labeling was less confined to the apical membrane than wild type rab17, and was also detected in subapical puncta (Fig. 8D).
FIGURE 8.
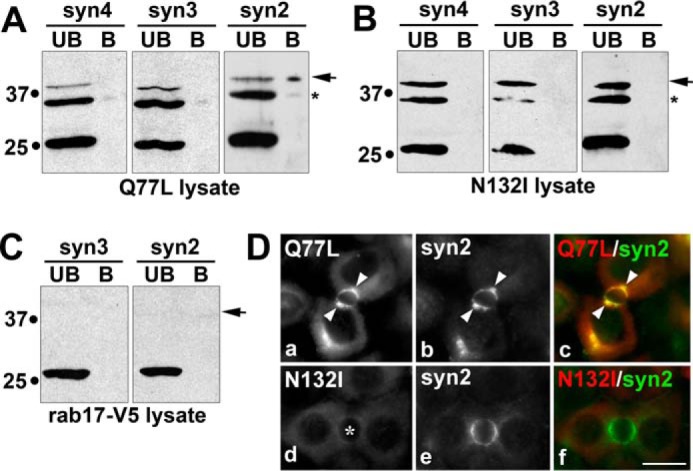
Sumoylated rab17 binding to syntaxin 2 is nucleotide dependent. Detergent lysates from cells expressing myc-tagged GTP-bound/Q77L rab17 (A), GDP-bound/N132I (B), or C terminally tagged rab17 (C) were applied to syntaxin-GST immobilized on glutathione-agarose and allowed to bind. The beads were pelleted and the unbound (UB) fraction collected. After extensive washing, the beads were mixed in sample buffer and represents the bound (B) fraction. The unbound and bound fractions were immunoblotted for rab17 with anti-FLAG (A and B) or anti-V5 antibodies (C). D, WIF-B cells expressing FLAG-tagged Q77L or N132I rab17 were pre-permeabilized, fixed, and stained for rab17 (a and d) (with anti-FLAG antibodies) and endogenous syntaxin 2 (b and e). Merged images are shown in c and f. Arrows mark subapically located Q77L that colocalizes with syntaxin 2. The asterisk in panel d marks the bile canaliculus. Bar = 10 μm.
K68R rab17 Expression Alters the Distributions of a Transcytosing Apical Protein
Because rab17 has been implicated as an important regulator of basolateral-to-apical transcytosis in a variety of polarized cell types (2, 5, 6), we examined the distributions of two apical resident proteins in cells expressing the K68R mutant: 5′NT (a resident delivered via transcytosis) (31) and MRP2 (a resident directly delivered) (32) as our negative control. The simple prediction is that if rab17 sumoylation is required for syntaxin 2-mediated vesicle fusion at the apical membrane, increased labeling in subapical puncta (a compartment immediately upstream of transcytotic apical delivery) will be observed for 5′NT (but not MRP2) in cells expressing K68R rab17. As predicted, MRP2 distributions did not change in cells expressing K68R rab17 (Fig. 9A). As in uninfected cells and in cells expressing wild type or K41R rab17, discrete MRP2 apical labeling was observed in more than 90% of K68R-expressing cells (Fig. 9B). As for MRP2, discrete 5′NT apical labeling was also observed in uninfected cells and in cells expressing wild type or K41R rab17 (Fig. 9C). In contrast, and as predicted, a subpopulation of 5′NT was observed in subapical puncta in cells expressing K68R rab17 (Fig. 9C, panels g and h). In almost 75% of cells expressing K68R rab17, 5′NT was detected in subapical puncta (Fig. 9D, p ≤ 0.001) consistent with impaired apical delivery. This result is also consistent with syntaxin 2 redistribution to and colocalization with K68R rab17 (but not K41R) in subapical puncta (Fig. 7).
FIGURE 9.

The distributions of 5′NT, but not MRP2, are altered in cells overexpressing K68R rab17. Uninfected cells (con) and WIF-B cells expressing FLAG-tagged wild type (WT), K68R, or K41R rab17 were fixed and stained for rab17 (not shown) or endogenous MRP2 (A) or 5′NT (C). Arrows mark the intracellular, subapical 5′NT labeling in cells overexpressing K68R rab17. The images were intentionally overexposed to highlight the subapical labeling. Bar = 10 μm. In B and D, the percent of infected cells were scored for intracellular subapical staining and the percent of total infected cells with such staining was calculated. Values are expressed as the average from at least 3 independent experiments ± S.E. *, p ≤ 0.001.
Discussion
We have determined that wild type, GTP-bound and GDP-bound rab17 are all sumoylated. Only the wild type and GTP-bound sumoylated proteins exhibit syntaxin 2 associations indicating the interactions are additionally GTP-dependent. Rab17 prenylation is required for sumoylation shortly after protein synthesis, but the modification is reversible and maintained at specific levels. The selectivity in syntaxin binding and apical protein redistribution further suggests that rab17 and syntaxin 2 mediate fusion of apical proteins delivered via transcytosis, but not via the direct pathway and raises the interesting possibility that syntaxin 3 and an unknown rab isoform regulate vesicle fusion from the direct pathway.
Rab17 Is Reversibly Sumoylated
During the initial characterization of the recombinant adenoviruses expressing the wild type, GTP-bound/Q77L and GDP-bound/N132I rab17s, we noticed that all three enzymes were expressed as 25- and 40-kDa species. Mass spectrometry confirmed that both bands are rab17 for all three constructs. By monitoring the appearance of the modifications in cells early after infection, we noticed that the 40-kDa band appeared with the same kinetics as the 25-kDa one, and that the relative amounts of the 40-kDa species to total rab17 remained constant suggesting sumoylation is reversible. Similarly, the proportions remained constant in cells treated with cycloheximide and both species were degraded with identical kinetics. Thus, not only is the modification reversible, it is maintained at very specific steady state levels. How this is regulated and what it signifies is not clear, but will be an interesting area of further investigation. Also interesting is the finding that rab17 prenylation is required for sumoylation. Again, the significance of this finding is not clear, but may represent sumoylation ligase(s) specificity in rab17 binding interactions.
Sequence analysis using the Sumoplot Analysis program revealed two near-perfect sumoylation consensus sites at lysines 41 and 68. Only the K68R point mutant decreased sumoylation suggesting it may be the important lysine. Although sumoylation was not completely abolished, a reproducible and statistically significant 30% decrease was observed (28.4 ± 7.0% decrease, p ≤ 0.03) consistent with the 30% decrease in rab17 sumoylation observed after SENP1 cleavage (Fig. 2, G and H). Because treatment with anacardic acid led to a ∼60% decrease of rab17 sumoylation, Lys-68 is likely not the sole sumoylated residue. Similar decreases have been reported for single site lysine mutants in other known non-nuclear sumoylated proteins (25–28) and some have argued that these incomplete losses can be explained by the presence of sumoylation at non-conventional sites. In some cases, only when two or more lysines are mutated, is sumoylation fully lost. However, the K41R/K68R double mutant is not folded properly and forms large aggregates such that the experiments were not interpretable, but indicate that these are critical structural residues. Nonetheless, the K68R mutant distribution is different from wild type and is additionally observed in small subapical puncta. Furthermore, its expression promotes partial redistribution of syntaxin 2 to the same subapical puncta and leads to altered distributions of 5′NT (but not MRP2) into subapical puncta indicating that even a partial loss of sumoylation may impact rab17 function.
Sumoylated and GTP-bound rab17 Selectively Interact with Syntaxin 2
In 1998 we determined that the hepatic apical domain expresses both syntaxins 2 and 3 (9). Although they displayed overlapping distributions on gradients of purified plasma membrane sheets, the patterns were distinct. Not only were their peak apical distributions at different densities, each syntaxin was also detected on other smaller vesicles (9). These results prompted us at the time to speculate that these differential distributions likely reflected differences in their functions in vesicle docking and fusion, i.e. each syntaxin participated in the docking and fusion of distinct vesicle populations. This hypothesis is supported by our finding that rab17 selectively interacts with syntaxin 2. Thus, a compelling extension of this hypothesis is that each syntaxin receives apical cargo delivered by distinct pathways. In hepatocytes, newly synthesized apical proteins take two pathways to the apical surface. So far, all ABC transporters and other polytrophic membrane proteins studied take the direct route, where they are delivered directly to the apical membrane from the trans-Golgi network (32, 33). In contrast, all newly synthesized single spanning residents (including the “professional” transcytosing proteins like polymeric IgA receptor) and glycosylphosphatidylinositol-anchored proteins examined take the indirect route where they are delivered from the trans-Golgi network to the basolateral surface, selectively retrieved by endocytosis and transcytosed to the apical surface by first traversing basolateral early endosomes and then the subapical compartment (14, 31, 34–36). Because rab17 has been implicated in mediating transcytosis in other epithelial cells (3, 5) and immunoisolated on transcytotic vesicles in hepatocytes (6), it is compelling to speculate that syntaxin 2 accepts the transcytosing, rab17 positive vesicles, whereas syntaxin 3 (and an unidentified rab) accept the directly targeted proteins. We are currently performing the functional studies to test this hypothesis.
The Expanding List of Non-nuclear Sumoylated Proteins
Sumoylation was first identified on nuclear proteins and identified as an important modification mediating multiple nuclear-based functions such as translocation through nuclear pores, chromatin organization, and DNA transcription and repair. To date, the list of known nuclear sumoylated proteins is long, but in recent years, non-nuclear proteins have been added to the list and that list is rapidly expanding (reviewed in Refs. 24, 37, and 38). Thus, sumoylation is likely a common post-translation modification regulating a host of cellular functions.
A notable non-nuclear protein recently identified as a sumoylated protein is the actin-associated, small GTPase, rac1. As for rab17, sumoylation enhanced membrane associations of the GTP-bound form of the enzyme (39). This in turn led to optimal hepatocyte growth factor-induced lamellipodia formation and cell migration. Similarly, sumoylation of the dynamin-related protein, DRP1, is thought to recruit the GTPase to mitochondrial membranes possibly allowing for enhanced mitochondrial fission (40). Additionally, sumoylation of the ciliary small GTPase, ARL-13/ARL13B, has been implicated in regulating ciliary targeting of sensory receptors in Caenorhabditis elegans and mammalian cells (41). Thus, one emerging general function of sumoylation is promoting and stabilizing intermolecular interactions that lead to formation of larger protein machineries that mediate complex cellular functions. Based on the result presented here, we propose that vesicle docking is one such function that is regulated by protein sumoylation. This hypothesis is made even more provocative given that all three syntaxin isoforms we examined encode putative sumo-interacting domains, yet sumoylated rab17 only bound syntaxin 2. Thus, the simple prediction is that other yet-to-be-identified sumoylated proteins selectively enhance interactions with syntaxins 3 and 4 thereby mediating plasma membrane docking and fusion.
Author Contributions
A. C. S. and P. L. T. are responsible for the experimental design. A. C. S. performed all experiments. Both authors analyzed and interpreted data and compiled figures. P. L. T. wrote and edited the manuscript with assistance from A. C. S.
Acknowledgments
We thank Dr. Ann Hubbard (Johns Hopkins University School of Medicine, Baltimore, MD) for providing the many antibodies and reagents used in this study. We are also grateful to Alfonso López-Coral (The Catholic University of America, Washington, DC) for expression and purification of the syntaxin-GST fusion proteins.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 DK082890 (to P. L. T.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The authors declare that they have no have no conflicts of interest with the contents of this article.
- 5′NT
- 5′ nucleotidase
- APN
- aminopeptidase N
- HDAC6
- histone deacetylase 6
- mβCD
- methyl-β-cyclodextrin
- MRP2
- multidrug associated protein-2
- SUMO
- small ubiquitin related modifier.
References
- 1. Lehtonen S., Lehtonen E., and Olkkonen V. M. (1999) Vesicular transport and kidney development. Int. J. Dev. Biol. 43, 425–433 [PubMed] [Google Scholar]
- 2. Lütcke A., Jansson S., Parton R. G., Chavrier P., Valencia A., Huber L. A., Lehtonen E., and Zerial M. (1993) Rab17, a novel small GTPase, is specific for epithelial cells and is induced during cell polarization. J. Cell Biol. 121, 553–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hunziker W., and Peters P. J. (1998) Rab17 localizes to recycling endosomes and regulates receptor-mediated transcytosis in epithelial cells. J. Biol. Chem. 273, 15734–15741 [DOI] [PubMed] [Google Scholar]
- 4. Peters P. J., and Hunziker W. (2001) Subcellular localization of Rab17 by cryo-immunogold electron microscopy in epithelial cells grown on polycarbonate filters. Methods Enzymol. 329, 210–225 [DOI] [PubMed] [Google Scholar]
- 5. Zacchi P., Stenmark H., Parton R. G., Orioli D., Lim F., Giner A., Mellman I., Zerial M., and Murphy C. (1998) Rab17 regulates membrane trafficking through apical recycling endosomes in polarized epithelial cells. J. Cell Biol. 140, 1039–1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jin M., Saucan L., Farquhar M. G., and Palade G. E. (1996) Rab1a and multiple other Rab proteins are associated with the transcytotic pathway in rat liver. J. Biol. Chem. 271, 30105–30113 [DOI] [PubMed] [Google Scholar]
- 7. Beaumont K. A., Hamilton N. A., Moores M. T., Brown D. L., Ohbayashi N., Cairncross O., Cook A. L., Smith A. G., Misaki R., Fukuda M., Taguchi T., Sturm R. A., and Stow J. L. (2011) The recycling endosome protein Rab17 regulates melanocytic filopodia formation and melanosome trafficking. Traffic 12, 627–643 [DOI] [PubMed] [Google Scholar]
- 8. Mori Y., Matsui T., Furutani Y., Yoshihara Y., and Fukuda M. (2012) Small GTPase Rab17 regulates dendritic morphogenesis and postsynaptic development of hippocampal neurons. J. Biol. Chem. 287, 8963–8973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fujita H., Tuma P. L., Finnegan C. M., Locco L., and Hubbard A. L. (1998) Endogenous syntaxins 2, 3 and 4 exhibit distinct but overlapping patterns of expression at the hepatocyte plasma membrane. Biochem. J. 329, 527–538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shanks M. R., Cassio D., Lecoq O., Hubbard A. L. (1994) An improved rat hepatoma hybrid cell line: generation and comparison with its hepatoma relatives and hepatocytes in vivo. J. Cell Sci. 107, 813–825 [DOI] [PubMed] [Google Scholar]
- 11. Bastaki M., Braiterman L. T., Johns D. C., Chen Y. H., and Hubbard A. L. (2002) Absence of direct delivery for single transmembrane apical proteins or their “secretory” forms in polarized hepatic cells. Mol. Biol. Cell 13, 225–237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Laemmli U. K. (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685 [DOI] [PubMed] [Google Scholar]
- 13. Ihrke G., Neufeld E. B., Meads T., Shanks M. R., Cassio D., Laurent M., Schroer T. A., Pagano R. E., and Hubbard A. L. (1993) WIF-B cells: an in vitro model for studies of hepatocyte polarity. J. Cell Biol. 123, 1761–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bartles J. R., Feracci H. M., Stieger B., and Hubbard A. L. (1987) Biogenesis of the rat hepatocyte plasma membrane in vivo: comparison of the pathways taken by apical and basolateral proteins using subcellular fractionation. J. Cell Biol. 105, 1241–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kubista H., Edelbauer H., and Boehm S. (2004) Evidence for structural and functional diversity among SDS-resistant SNARE complexes in neuroendocrine cells. J. Cell Sci. 117, 955–966 [DOI] [PubMed] [Google Scholar]
- 16. Otto H., Hanson P. I., and Jahn R. (1997) Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc. Natl. Acad. Sci. U.S.A. 94, 6197–6201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gentile F., Amodeo P., Febbraio F., Picaro F., Motta A., Formisano S., and Nucci R. (2002) SDS-resistant active and thermostable dimers are obtained from the dissociation of homotetrameric β-glycosidase from hyperthermophilic Sulfolobus solfataricus in SDS: stabilizing role of the A-C intermonomeric interface. J. Biol. Chem. 277, 44050–44060 [DOI] [PubMed] [Google Scholar]
- 18. In J. G., Striz A. C., Bernad A., and Tuma P. L. (2014) Serine/threonine kinase 16 and MAL2 regulate constitutive secretion of soluble cargo in hepatic cells. Biochem. J. 463, 201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bossis G., and Melchior F. (2006) Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell 21, 349–357 [DOI] [PubMed] [Google Scholar]
- 20. Datta S., Snow C. J., and Paschal B. M. (2014) A pathway linking oxidative stress and the Ran GTPase system in progeria. Mol. Biol. Cell 25, 1202–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xu Z., Lam L. S., Lam L. H., Chau S. F., Ng T. B., and Au S. W. (2008) Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 22, 127–137 [DOI] [PubMed] [Google Scholar]
- 22. Tokmakov A. A., Kurotani A., Takagi T., Toyama M., Shirouzu M., Fukami Y., and Yokoyama S. (2012) Multiple post-translational modifications affect heterologous protein synthesis. J. Biol. Chem. 287, 27106–27116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fukuda I., Ito A., Hirai G., Nishimura S., Kawasaki H., Saitoh H., Kimura K., Sodeoka M., and Yoshida M. (2009) Ginkgolic acid inhibits protein SUMOylation by blocking formation of the E1-SUMO intermediate. Chem. Biol. 16, 133–140 [DOI] [PubMed] [Google Scholar]
- 24. Flotho A., and Melchior F. (2013) Sumoylation: a regulatory protein modification in health and disease. Annu. Rev. Biochem. 82, 357–385 [DOI] [PubMed] [Google Scholar]
- 25. Caron D., Boutchueng-Djidjou M., Tanguay R. M., and Faure R. L. (2015) Annexin A2 is SUMOylated on its N-terminal domain: regulation by insulin. FEBS Lett. 589, 985–991 [DOI] [PubMed] [Google Scholar]
- 26. Fuhs S. R., and Insel P. A. (2011) Caveolin-3 undergoes SUMOylation by the SUMO E3 ligase PIASy: sumoylation affects G-protein-coupled receptor desensitization. J. Biol. Chem. 286, 14830–14841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. González-Santamaría J., Campagna M., Ortega-Molina A., Marcos-Villar L., de la Cruz-Herrera C. F., González D., Gallego P., Lopitz-Otsoa F., Esteban M., Rodríguez M. S., Serrano M., and Rivas C. (2012) Regulation of the tumor suppressor PTEN by SUMO. Cell Death Dis. 3, e393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Qu Y., Chen Q., Lai X., Zhu C., Chen C., Zhao X., Deng R., Xu M., Yuan H., Wang Y., Yu J., and Huang J. (2014) SUMOylation of Grb2 enhances the ERK activity by increasing its binding with Sos1. Mol. Cancer 13, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robibaro B., Stedman T. T., Coppens I., Ngô H. M., Pypaert M., Bivona T., Nam H. W., and Joiner K. A. (2002) Toxoplasma gondii Rab5 enhances cholesterol acquisition from host cells. Cell Microbiol. 4, 139–152 [DOI] [PubMed] [Google Scholar]
- 30. Fiordalisi J. J., Johnson R. L. 2nd, Ulkü A. S., Der C. J., and Cox A. D. (2001) Mammalian expression vectors for Ras family proteins: generation and use of expression constructs to analyze Ras family function. Methods Enzymol. 332, 3–36 [DOI] [PubMed] [Google Scholar]
- 31. Schell M. J., Maurice M., Stieger B., and Hubbard A. L. (1992) 5′ Nucleotidase is sorted to the apical domain of hepatocytes via an indirect route. J. Cell Biol. 119, 1173–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kipp H., and Arias I. M. (2000) Newly synthesized canalicular ABC transporters are directly targeted from the Golgi to the hepatocyte apical domain in rat liver. J. Biol. Chem. 275, 15917–15925 [DOI] [PubMed] [Google Scholar]
- 33. Kipp H., Pichetshote N., and Arias I. M. (2001) Transporters on demand: intrahepatic pools of canalicular ATP binding cassette transporters in rat liver. J. Biol. Chem. 276, 7218–7224 [DOI] [PubMed] [Google Scholar]
- 34. Bartles J. R., and Hubbard A. L. (1988) Plasma membrane protein sorting in epithelial cells: do secretory pathways hold the key? Trends Biochem. Sci. 13, 181–184 [DOI] [PubMed] [Google Scholar]
- 35. Ihrke G., Martin G. V., Shanks M. R., Schrader M., Schroer T. A., and Hubbard A. L. (1998) Apical plasma membrane proteins and endolyn-78 travel through a subapical compartment in polarized WIF-B hepatocytes. J. Cell Biol. 141, 115–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tuma P., and Hubbard A. L. (2003) Transcytosis: crossing cellular barriers. Physiol. Rev. 83, 871–932 [DOI] [PubMed] [Google Scholar]
- 37. Martin S., Wilkinson K. A., Nishimune A., and Henley J. M. (2007) Emerging extranuclear roles of protein SUMOylation in neuronal function and dysfunction. Nat. Rev. Neurosci. 8, 948–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wasik U., and Filipek A. (2014) Non-nuclear function of sumoylated proteins. Biochim. Biophys. Acta 1843, 2878–2885 [DOI] [PubMed] [Google Scholar]
- 39. Castillo-Lluva S., Tatham M. H., Jones R. C., Jaffray E. G., Edmondson R. D., Hay R. T., and Malliri A. (2010) SUMOylation of the GTPase Rac1 is required for optimal cell migration. Nat. Cell. Biol. 12, 1078–1085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chang C. R., and Blackstone C. (2010) Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N.Y. Acad. Sci. 1201, 34–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Zhang Q., Wei Q., Zhang Y., Ling K., and Hu J. (2012) SUMOylation of the small GTPase ARL-13 promotes ciliary targeting of sensory receptors. J. Cell Biol. 199, 589–598 [DOI] [PMC free article] [PubMed] [Google Scholar]