

Abstract
Multidrug and toxic compound extrusion (MATE) transporters contribute to multidrug resistance by extruding different drugs across cell membranes. The MATE transporters alternate between their extracellular and intracellular facing conformations to propel drug export, but how these structural changes occur is unclear. Here we combine site-specific cross-linking and functional studies to probe the movement of transmembrane helices in NorM from Neiserria gonorrheae (NorM-NG), a MATE transporter with known extracellular facing structure. We generated an active, cysteine-less NorM-NG and conducted pairwise cysteine mutagenesis on this variant. We found that copper phenanthroline catalyzed disulfide bond formation within five cysteine pairs and increased the electrophoretic mobility of the corresponding mutants. Furthermore, copper phenanthroline abolished the activity of the five paired cysteine mutants, suggesting that these substituted amino acids come in spatial proximity during transport, and the proximity changes are functionally indispensable. Our data also implied that the substrate-binding transmembrane helices move up to 10 Å in NorM-NG during transport and afforded distance restraints for modeling the intracellular facing transporter, thereby casting new light on the underlying mechanism.
Keywords: conformational change, drug resistance, exchanger, membrane protein, multidrug transporter, cysteine cross-linking, multidrug efflux
Introduction
The relentless rise in multidrug resistance exerts devastating human and economic tolls, raising the specter of a public health crisis (1, 2). A major mechanism underlying the rampant multidrug resistance is through integral membrane proteins named “multidrug transporters,” which can remove a plethora of therapeutic drugs from the cell (1). The spread of multidrug resistance, at a rate unprecedented in the past several decades, is outpacing drug discovery and accelerating (2). As such, we may remain on the losing side of the war against multidrug resistance until we understand how multidrug transporters export drugs and how they can be countervailed.
Multidrug and toxic compound extrusion (MATE)3 proteins constitute a ubiquitous family of multidrug transporters and couple the efflux of structurally dissimilar, typically cationic compounds to the influx of Na+ or H+ (3–5). The known MATE transporters can be separated into the NorM, DinF (DNA damage-inducible protein F) and eukaryotic subfamilies based on their amino acid sequence similarity (3). MATE transporters are appealing targets for tackling multidrug resistance because they can extrude a wide variety of antibiotic, anticancer, and diabetic drugs across the cell membrane (4, 5).
Over the past 6 years, the x-ray structures of Na+-coupled NorM from Vibrio cholera and Neiserria gonorrheae (NorM-VC and NorM-NG), and H+-coupled DinF from Pyrococcus furiosus and Bacillus halodurans (PfMATE and DinF-BH) have been reported, revealing the transporter architecture and providing important insights into the underlying transport mechanisms (6–10). Nevertheless, these findings highlight new areas of uncertainty that need to be addressed experimentally (11). Particularly, in all of the published MATE structures, the interface between the amino (N) and carboxyl (C) domains always opens into the extracellular milieu, i.e. adopting the extracellular facing conformations. Therefore, the molecular basis for the interconversion between the extracellular and intracellular facing conformations, which lies at the heart of the transport mechanism, remains poorly understood.
Previously we deduced the intracellular facing models of MATE transporters and posited how they undergo conformational changes during drug export (9). Here we utilized mutational, cross-linking, and biochemical methods to elucidate such structural changes in NorM-NG. On the basis of our new biochemical data and available crystal structures, we uncovered the movement of transmembrane helices during transport in NorM-NG, shedding new light on how MATE transporters switch between their extracellular and intracellular facing states. Our findings take our understanding of the MATE-mediated multidrug efflux to new depths and lend fresh hope for thwarting multidrug resistance.
Experimental Procedures
Generation of Mutants
Full-length NorM-NG was expressed with a decahistidine tag at the C terminus using a modified pET15b vector (7). Mutations were introduced into the norM-NG gene by using the QuikChange method (Agilent Technologies) and were confirmed by DNA sequencing. Vector-specific primers (5′-GCTAGTTATTGCTCAGCGG-3′ and 5′-TAATACGACTCACTATAGGG-3′), as well as NorM-NG-specific primers (5′-ATCGCCAAGGAAAAATTCTTCC-3′ and 5′-CCATTGTCGCCACGCCGCAACC-3′), were used. The membrane expression levels of the NorM-NG mutants discussed in this work were similar to that of the wild-type protein, based on the Western blot using an antibody against the His tag. Poorly expressed and/or unstable NorM-NG mutants had been removed from further study. For the Western blot, the antibody (Qiagen, catalog no. 34460) was diluted 2,500-fold before being mixed with the transfer membranes, and each sample examined on the SDS-PAGE gel was derived from cell membranes isolated from 80 μg of Escherichia coli BL21 (DE3) ΔacrABΔmacABΔyojHI cells expressing the NorM-NG variants.
Protein Expression and Purification
The NorM-NG variants were expressed and purified as follows. Briefly, E. coli BL21 (DE3) ΔacrABΔmacABΔyojHI cells transformed with the protein expression vectors were grown in Luria-Bertani (LB) medium to an optical density of 0.5 at 600 nm and induced with 0.5 mm isopropyl β-d-1-thiogalactopyranoside at 30 °C for 4 h. The cells were harvested by centrifugation and ruptured by multiple passages through a precooled French pressure cell. All the protein purification experiments were performed at 4 °C. Membranes were collected by ultracentrifugation and extracted with 1% (w/v) n-dodecyl-β-maltoside (Anatrace) in 20 mm HEPES-NaOH, pH7.5, 100 mm NaCl, 20% (v/v) glycerol, and 1 mm tris(2-carboxyethyl)phosphine. The soluble fraction was equilibrated with nickel-nitrilotriacetic acid resin in 20 mm HEPES-NaOH, pH 7.5, 100 mm NaCl, 25% glycerol, 0.05% n-dodecyl-β-maltoside, and 1 mm tris(2-carboxyethyl)phosphine for >3 h. Protein was eluted using the same buffer supplemented with 450 mm imidazole. The eluted protein sample was promptly buffer-exchanged into 20 mm HEPES-NaOH, pH 7.5, 0 or 100 mm NaCl, 20% glycerol, 0.05% n-dodecyl-β-maltoside by using gel filtration chromatography. The expression levels of NorM-NG variants were low, which may explain why the well characterized MATE transporters confer only modest levels of cellular resistance against drugs (4, 5).
Cysteine Cross-linking and Gel Mobility Shift Assay
Purified NorM-NG variants at 10 μm were incubated with 50 or 200 μm CuSO4 and 100 or 400 μm 1,10-phenanthroline in 20 mm HEPES, pH 7.5, 0 or 100 mm NaCl, 0 or 1 mm tetraphenylphosphonium (TPP), 20% glycerol, and 0.05% n-dodecyl-β-maltoside buffer for 10 min at 22 °C (12–17). 20 mm N-(5-fluoresceinyl)maleimide was then added to the samples and left for 10 min at 22 °C in the dark. Some samples were additionally treated with 10 mm DTT, whereas all samples were incubated in the dark for an additional 5 min at 22 °C. Prior to the nonreducing SDS-PAGE analysis, 100 mm N-ethylmaleimide was added to all the samples and incubated for 10 min at 22 °C. The gels were visualized by using both Coomassie staining and UV illumination. For studies of the membrane-embedded proteins, the crude E. coli membranes were isolated by ultracentrifugation; washed in buffers containing 20 mm HEPES, pH 7.5, 100 mm NaCl, and 20% glycerol; and cross-linked as in detergents and analyzed by using nonreducing SDS-PAGE and Western blot (16).
Drug Resistance Assay
The drug export activities of NorM-NG variants were evaluated based on their ability to confer cellular resistance against cytotoxic chemicals (7, 10). Drug susceptibility experiments were conducted in LB medium, with each assay repeated at least three times. Briefly, the exponential phase bacterial culture from freshly transformed E. coli BL21 (DE3) ΔacrABΔmacABΔyojHI cells was diluted to 1 × 104 colony-forming units/ml with LB broth containing 0.3 mm isopropyl β-d-1-thiogalactopyranoside and 200 μg/ml ampicillin, with or without 250 μm CuSO4, 500 μm 1,10-phenanthroline, and 250 μm DTT at each drug concentration. The culture was then incubated at 30 °C with shaking, and the bacterial growth was monitored after 10 h. Assays were performed in 96-well plates, and the optical density at 595 nm was measured using a microplate reader (Tecan GENios Plus). We defined the minimal inhibitory concentration as the lowest concentration of antimicrobial compounds that precludes the growth of E. coli under our experimental conditions (7).
Drug Accumulation Assay
Cultures of the E. coli BL21 (DE3) ΔacrABΔmacABΔyojHI cells expressing NorM-NG variants were grown to an A600 nm of 0.5 and induced with 1 mm isopropyl β-d-1-thiogalactopyranoside at 30 °C for 4 h. The cells were harvested; washed three times with 100 mm Tris-HCl, pH 7.0; resuspended in the same buffer to an A600 nm of 1.0/ml. Rhodamine 6G (R6G) or ethidium was added to the cells at a final concentration of 4 μg/ml, with or without the inclusion of 300 μm CuSO4, 600 μm 1,10-phenanthroline, and 750 μm DTT. After incubation on ice for 10 min, a 1-ml sample was withdrawn every 2.5 min, and cells were harvested by centrifugation and washed twice with 100 mm Tris-HCl, pH 7.0. The R6G or ethidium fluorescence was then measured with the excitation and emission wavelengths of 485 and 590 nm, respectively (10).
Results
Generation of Cys-less NorM-NG
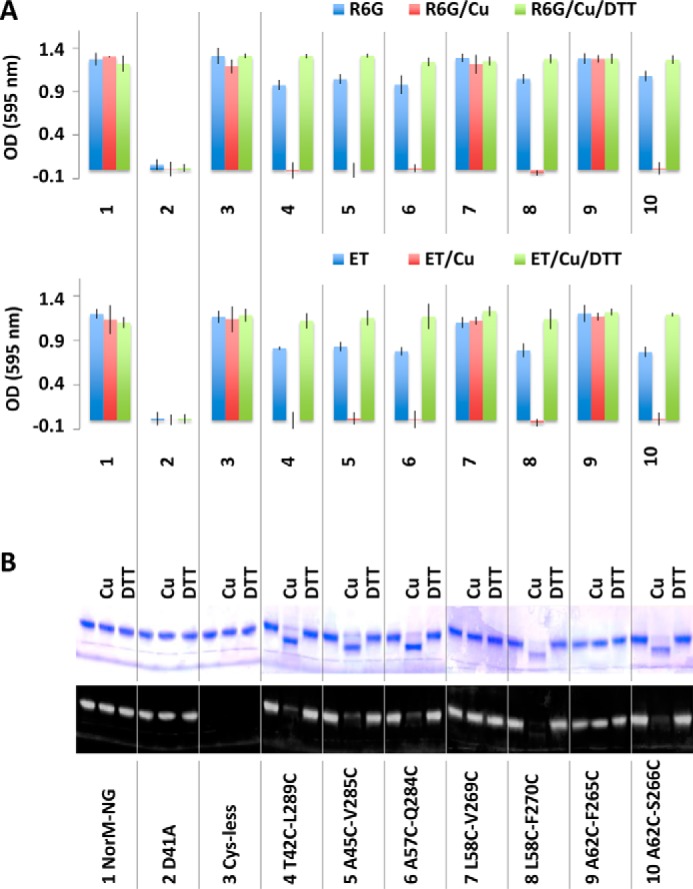
In this work, we aimed to use our intracellular facing model of NorM-NG as a guide for site-directed mutagenesis (9) and then “freeze” NorM-NG in its intracellular facing conformation by utilizing disulfide cross-linking (16). To accomplish this aim, we first attempted to remove all the endogenous cysteines in NorM-NG and then to carry out paired cysteine substitutions to generate mutants wherein disulfide bonds can be formed in the intracellular facing conformation but not the extracellular facing conformation. NorM-NG has four endogenous cysteines: Cys202 (TM6), Cys381 (TM10), Cys407 (TM11), and Cys444 (TM12), most of which are removed from the substrate- and Na+-binding sites and not conserved even in the NorM branch (7). Therefore, the replacement of these cysteines by isosteric serine or valine, we reasoned, would be unlikely to affect the protein folding or transport function. Much to our amazement, although the single NorM-NG mutants C202S and C381S were both expressed to the same level as that of the wild-type protein, the replacement of Cys407 or Cys444 in NorM-NG, by serine or valine, completely abolished the protein expression (data not shown).
To overcome this difficulty, we aligned the amino acid sequence of NorM-NG with those of >30 NorM orthologues. Based on the sequence alignment, we substituted Cys407 and Cys444 in NorM-NG individually with the equivalent residues found in other NorM orthologues and then examined the membrane expression and solution behaviors of those single mutants by using Western blot and gel filtration chromatography, respectively (18). Well behaved single mutations were then selected and combined to generate double, triple, and finally quadruple mutants (data not shown). After screening >40 different NorM-NG mutants, we identified a quadruple mutant, C202S/C381S/C407F/C444R, hereafter termed “Cys-less NorM-NG,” that was expressed to a similar level to that of NorM-NG and exhibited good solution behavior. Based on the drug resistance and accumulation assays (see below), we found that the Cys-less NorM-NG retained the multidrug efflux activity, suggesting that the quadruple mutation had not disrupted the transporter structure or function and thus was suitable for further mutagenesis and functional studies.
Selection of Positions for Paired Cysteine Mutagenesis
Previously we hypothesized that NorM-NG switches from its extracellular to intracellular facing conformation upon substrate release and Na+ binding (7). Our studies further predicted that in the intracellular facing NorM-NG, the extracellular portions of TM1 and TM2 are in close proximity to those of TM8 and TM7, respectively (9). If our prediction is correct, once we replace the amino acids envisioned to be close enough in the intracellular facing NorM-NG with cysteines, we may be able to employ disulfide cross-linking to “stitch” together the extracellular portions of TM1 and TM8, TM2 and TM7, or TM2 and TM8.
Along this line of reasoning, we identified 14 pairs of amino acids in NorM-NG whose Cα positions are far apart in the extracellular facing structure (7), but close to each other in the intracellular facing model (Fig. 1). The intracellular facing model of NorM-NG was constructed by rotating the C domain as a rigid body in the extracellular facing structure by 20° relative to the N domain (9). Among the 14 pairs of amino acids, 8 are located within TM1 and TM8, 5 are within TM2 and TM7, and 1 is within TM2 and TM8. Notably, most of these amino acids are removed from the substrate- and Na+-binding sites (7), thereby diminishing the possibility of interfering with substrate- and/or Na+-binding and hence giving rise to inactive proteins upon cysteine substitutions. We then replaced these amino acids in the Cys-less NorM-NG with cysteines singularly or in pairs, yielding a total of 28 and 14 of what we denoted “mono-” and “dicysteine” mutants, respectively. Among the 14 dicysteine mutants, only 7 were expressed to similar levels to that of NorM-NG (Fig. 2) and were well folded based on their profiles on gel filtration chromatography (data not shown). Importantly, these 7 mutants retained the ability to extrude different drugs (see below) and were therefore suitable for further investigation.
FIGURE 1.
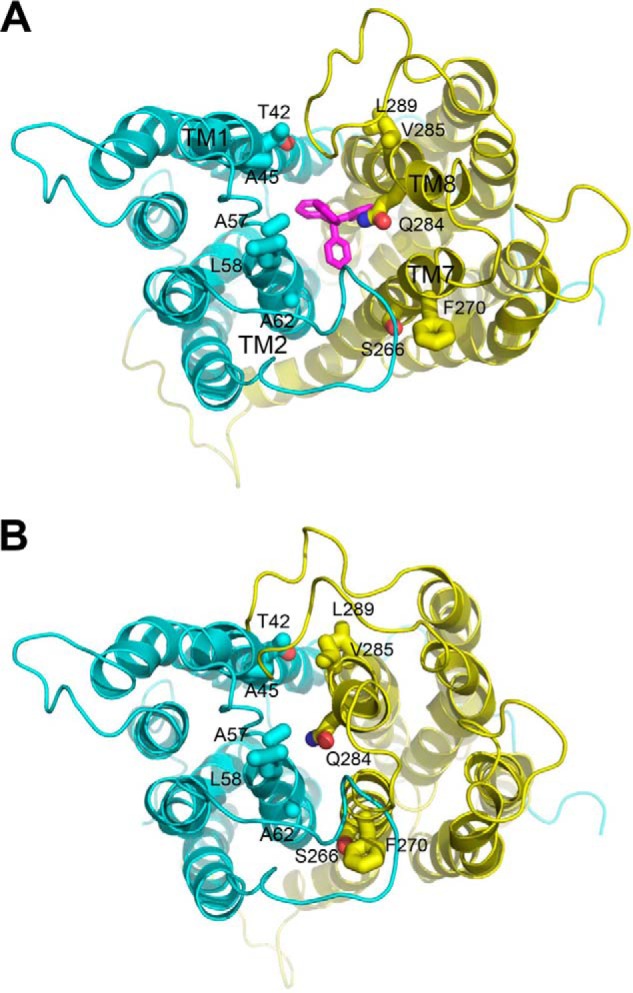
Structural basis for the design of paired cysteine mutagenesis in NorM-NG. A, periplasmic view of the extracellular facing NorM-NG structure (Protein Data Bank code 4HUK), which shows the bound substrate tetraphenylphosphonium (magenta) sandwiched between the N (cyan) and C (yellow) domains of the transporter. B, the intracellular facing model of NorM-NG highlights the relative movement between the N and C domains. Relevant amino acids (sticks) and transmembrane helices are labeled. This figure was prepared using the software PyMOL (version 1.5.0.4, Schrodinger).
FIGURE 2.
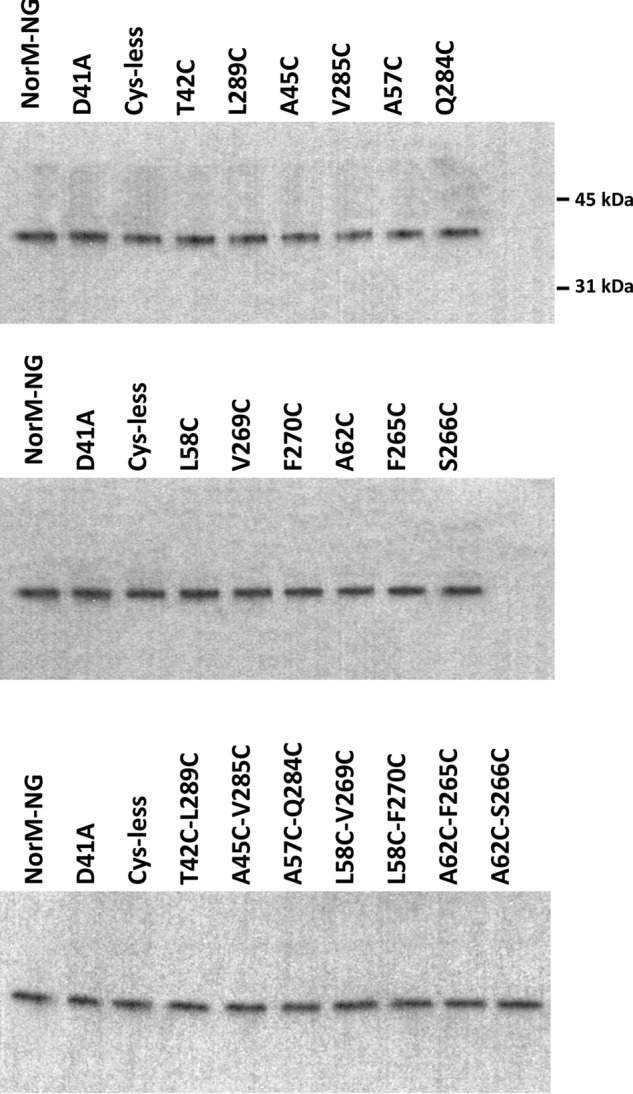
Western blot analysis of the NorM-NG variants. Western blot analysis of NorM-NG variants in membrane preparations was performed by using an antibody against the His tag. This analysis suggested that all of the NorM-NG variants investigated in this work were expressed at similar levels in the E. coli membrane. The positions of two molecular weight markers are also indicated.
Electrophoretic Mobility Shift of Purified NorM-NG Mutants
We first asked whether the seven dicysteine mutants, namely T42C/L289C, A45C/V285C (TM1 and TM8), L58C/V269C, L58C/F270C, A62C/F265C, A62C/S266C (TM2 and TM7), and A57C/Q284C (TM2 and TM8) can form intramolecular disulfide bonds in vitro. We expressed and purified these dicysteine mutants in the presence of Na+. We then incubated these proteins with an oxidizing agent (copper phenanthroline) and performed nonreducing SDS-PAGE analysis (Fig. 3A). We observed that for five mutants, T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C, copper phenanthroline treatment yielded distinct protein species with markedly higher electrophoretic mobility. Moreover, the faster migrating protein species contained no free cysteine thiols, because they could no longer react with a thiol-reactive fluorophore, N-(5-fluoresceinyl)maleimide (Fig. 3B).
FIGURE 3.
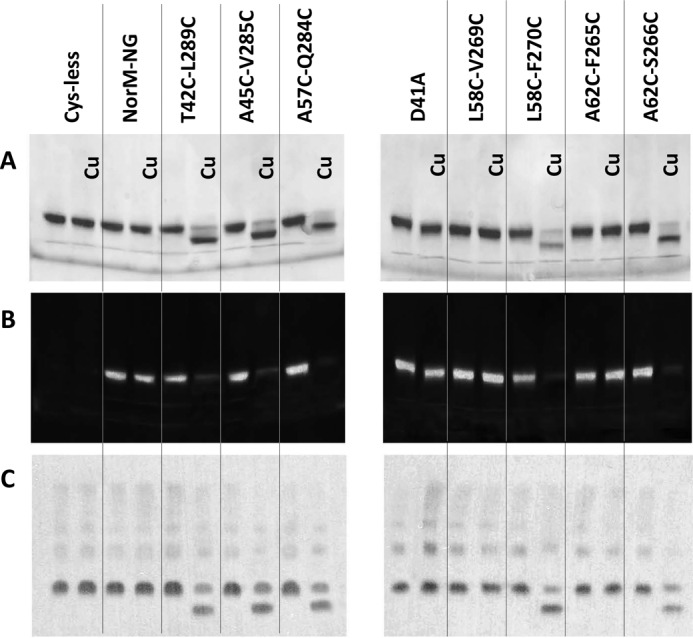
Cross-linking of five NorM-NG mutants revealed by gel mobility shift assays. A and B, nonreducing SDS-PAGE analysis of detergent-purified NorM-NG variants with and without copper phenanthroline (Cu) treatment. The proteins were purified in the presence of 100 mm NaCl and incubated with and without 200 μm CuSO4 plus 400 μm phenanthroline. The proteins were visualized by Coomassie staining (A) or UV illumination (B). Free cysteine thiol groups in the protein reacted with N-(5-fluoresceinyl)maleimide and thus gave rise to fluorescence signals (B). C, Western blot of membrane-embedded NorM-NG variants with and without copper phenanthroline treatment. Copper phenanthroline stimulated the formation of disulfide bonds and the loss of free cysteine thiol groups in five dicysteine NorM-NG mutants and markedly increased their electrophoretic mobility.
Notably, the electrophoretic mobility shift unlikely resulted from protein aggregation and/or denaturation induced by copper phenanthroline, because the five dicysteine mutants remained monomeric and well folded after the copper phenanthroline treatment, as judged by their behavior on gel filtration chromatography (data not shown). Indeed, the changes in electrophoretic mobility could be completely reversed by incubating the protein samples subsequently with the reducing agent DTT (see below), arguing that the gel mobility shift was due to oxidative cross-linking of the paired cysteines, and the disulfide bonds in a well folded transporter were readily reduced by DTT. Moreover, the cross-linking of the five NorM-NG mutants depended on the presence of copper phenanthroline, because neither the gel mobility shift nor the loss of free cysteine thiols could be detected for the untreated protein samples even after prolonged storage (data not shown).
By contrast, copper phenanthroline treatment failed to alter the electrophoretic mobility of another two dicysteine mutants L58C/V269C and A62C/F265C (Fig. 3A). Additionally, these two mutants retained the free cysteine thiols even after the exposure to copper phenanthroline (Fig. 3B). Because NorM-NG is a monomer in solution (7, 10), our data were consistent with the five cysteine pairs: T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C, each forming an intramolecular disulfide bond. The formation of such disulfide bonds likely prevented complete unfolding of the protein under nonreducing and yet denaturing conditions and thus substantially altered the electrophoretic mobility of the transporter (13, 16). Significantly, under our experimental conditions, the five detergent-purified dicysteine mutants could be converted into the faster migrating species almost completely, further attesting to the validity of our experimental design and the intracellular facing model (9).
Gel Shift Assay on Other NorM-NG Variants
In comparison with the five shifted dicysteine mutants, copper phenanthroline had no measurable effect on the gel mobility of the wild-type or Cys-less NorM-NG or the single mutant D41A (Fig. 3A). D41A exhibited severely crippled transport function (7, 10) and was used as a negative control in our functional assays. Because both NorM-NG and D41A bear the four endogenous cysteines, our data implied that under our experimental conditions, copper phenanthroline did not cause the formation of intramolecular disulfide bond(s) among the endogenous cysteines or intermolecular disulfide bond(s) between different NorM-NG molecules. Nonetheless, the Cys-less NorM-NG vastly simplified the interpretation of our experimental data (e.g. the detection of free cysteine thiols; Fig. 3B) and also ruled out the possibility of forming disulfide bond(s) between the endogenous and newly introduced cysteines. Therefore, we deemed the Cys-less NorM-NG better suited for our cross-linking studies than the wild-type transporter.
Unlike the five cross-linkable dicysteine mutants, the electrophoretic mobility of the corresponding monocysteine mutants was not affected by the copper phenanthroline and/or DTT treatment (Fig. 4). Additionally, the 10 monocysteine mutants retained their free cysteine thiols even after the copper phenanthroline treatment, in marked contrast to the dicysteine mutants (Fig. 3B), indicating that under our experimental conditions, copper phenanthroline catalyzed the oxidation of the paired cysteines rather than the single cysteines in NorM-NG.
FIGURE 4.
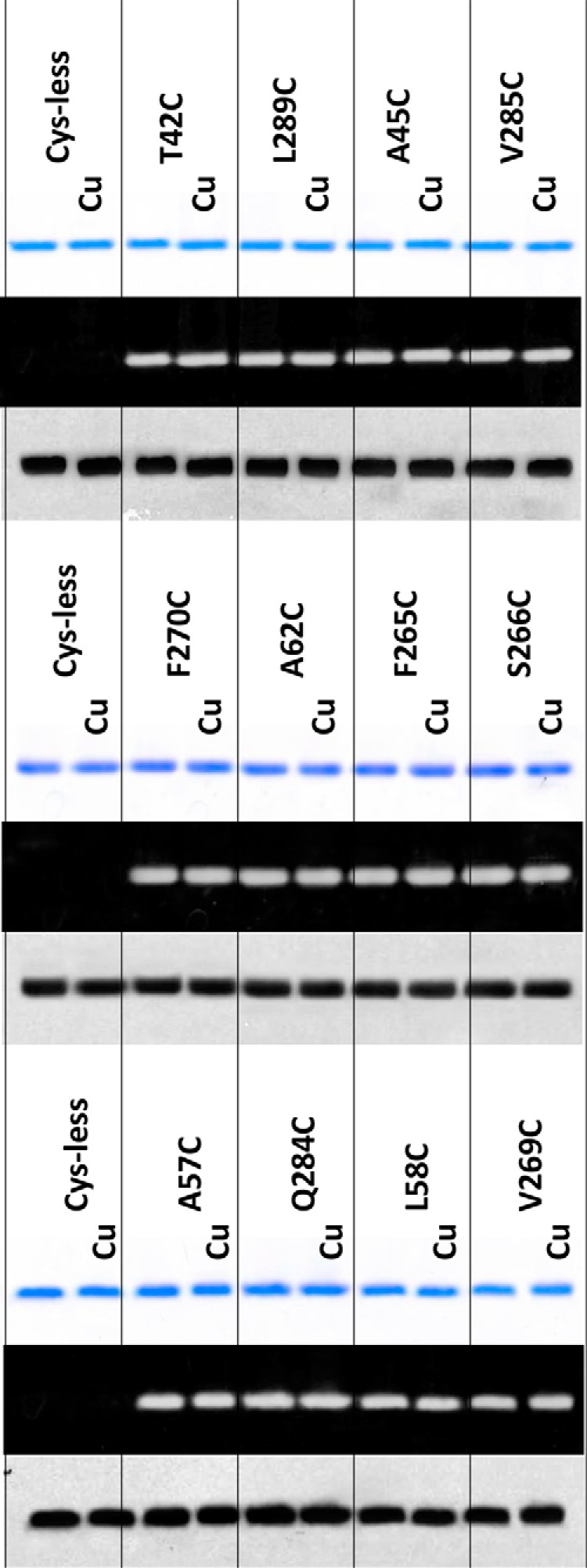
SDS-PAGE analysis revealed no cross-linking in monocysteine mutants of NorM-NG. Nonreducing SDS-PAGE analysis of detergent-purified NorM-NG variants with and without copper phenanthroline (Cu) treatment (200 μm CuSO4 plus 400 μm phenanthroline). 100 mm NaCl was included during this analysis. The proteins were visualized either by Coomassie staining (top panel) or UV illumination (middle panel). Western blot of membrane-embedded NorM-NG variants with and without copper phenanthroline treatment is shown (bottom panel). Notably, copper phenanthroline had no measurable effect on the electrophoretic mobility of the 10 monocysteine mutants of NorM-NG.
Effects of Ligand Binding on the Cross-linking of NorM-NG Mutants
We further examined the impact of substrates on the cross-linking. We found that TPP, a substrate of NorM-NG (7), stimulated the gel mobility shift of the dicysteine mutants T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C both in the absence (Fig. 5) and in the presence (data not shown) of added NaCl. In the TPP-bound structure of NorM-NG (7), these five cysteine pairs are unlikely to be cross-linked (Fig. 1A), our data thus implied that TPP promoted the cross-linking of NorM-NG by triggering the transporter to adopt a new conformation in which the five cysteine pairs are in sufficient spatial proximity to be stitched together by virtue of disulfide bond formation.
FIGURE 5.
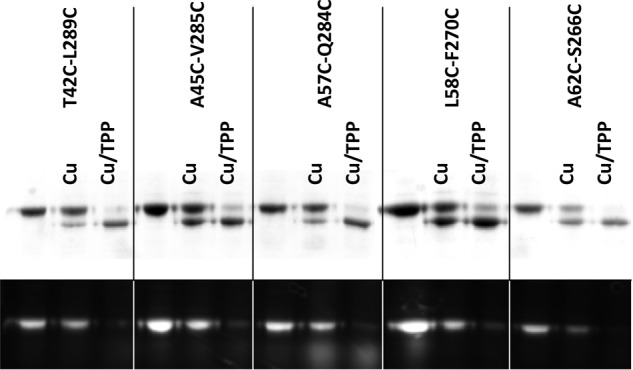
Effects of substrate-binding on the cross-linking of NorM-NG mutants. Nonreducing SDS-PAGE analysis of detergent-purified NorM-NG variants with and without copper phenanthroline (Cu) treatment (50 μm CuSO4 plus 100 μm phenanthroline), in the absence and presence of 1 mm TPP. No NaCl was added to the samples during this analysis. The proteins were visualized by Coomassie staining (top panel) and UV illumination (bottom panel). TPP stimulated the electrophoretic mobility shift in the NorM-NG mutants and enhanced their loss of free cysteine thiols.
Furthermore, we found that Na+, the countertransported cation (7), also promoted the cross-linking of those five pairs of cysteines (data not shown). Our findings were unexpected, because the prevailing view holds that substrate and countertransported cation exert different effects on an antiporter, so that the protein can assume distinct conformations upon association with the substrate versus the cation during transport (11). These results thus implied that NorM-NG extrudes drugs via an antiport mechanism that is considerably different from that of other well characterized antiporters (7, 19).
Cysteine Cross-linking of NorM-NG Mutants in Membranes
We next asked whether disulfide bonds can be formed in the five dicysteine mutants while they are still embedded in the lipid bilayer. We incubated the E. coli membranes harboring the five NorM-NG mutants with copper phenanthroline, conducted the gel mobility shift assay and visualized the proteins by using immunoblot (Fig. 3C). We found that copper phenanthroline treatment also increased the electrophoretic mobility of the five membrane-embedded, dicysteine mutants, although less completely than the detergent-purified proteins. This difference may reflect the distinct steady-state structures of NorM-NG, and/or the different accessibility of the introduced cysteines to copper phenanthroline in membrane bilayers versus solution. Nevertheless, similar to the detergent-purified proteins, the gel mobility shift of the five mutants was obliterated if the bacterial membranes were incubated with the reducing agent DTT after the copper phenanthroline treatment (data not shown).
By contrast, copper phenanthroline failed to elicit any gel mobility shift in the corresponding monocysteine NorM-NG mutants (Fig. 4), affirming that the shift was indeed due to the formation of specific intramolecular disulfide bond formation rather than nonspecific cross-linking caused by for example intermolecular disulfide bridges. Moreover, no cross-linking was observed for membrane-embedded NorM-NG, Cys-less NorM-NG, and D41A, similarly to the purified proteins (Fig. 3A). Taken together, our data suggested that the observed disulfide cross-linking or lack thereof is unlikely attributed to the isolation of the NorM-NG variants from their native environment, i.e. the lipid bilayer, and that copper phenanthroline likely catalyzes disulfide bond formation in the five dicysteine mutants in vivo.
Transport Function of NorM-NG Mutants
The foregoing results implied that copper phenanthroline catalyzes the disulfide bridge formation within T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C both in detergents and in lipid bilayers. Because these five pairs of amino acids are spatially too far apart from each other to form disulfide bonds in the extracellular facing NorM-NG (Fig. 1A), we concluded that the formation of those disulfide bridges locked the transporter into a conformational state that has yet to be captured by x-ray crystallography and is likely to be the intracellular facing state (Fig. 1B). If this assertion held up, we predicted that copper phenanthroline would inactivate the five dicysteine mutants by constraining the motions of the corresponding transmembrane helices and that the addition of reducing agent DTT would overcome the inactivation by precluding the disulfide bond formation. Additionally, we anticipated that the function of the monocysteine mutants would be unaffected by the copper phenanthroline and/or DTT treatment.
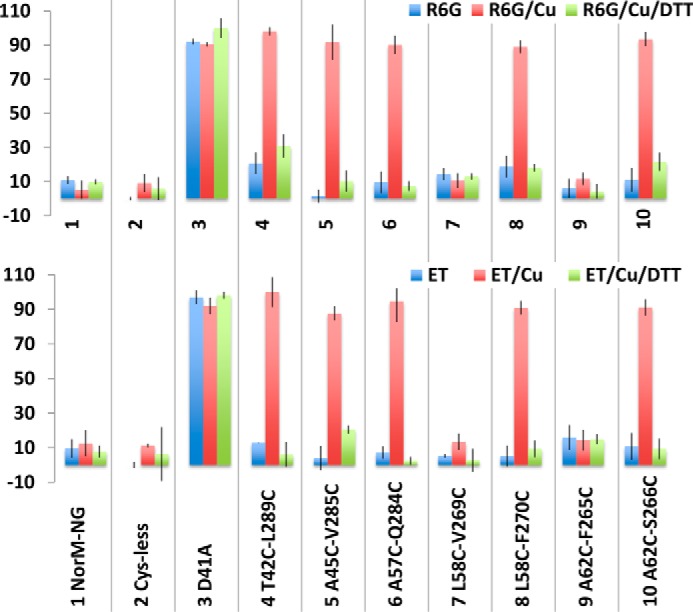
We first tested whether the five dicysteine mutants can confer cellular resistance to drugs, with and without copper phenanthroline. We observed that similar to the NorM-NG and its Cys-less variant, mutants T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C rendered bacteria resistant to cytotoxic R6G or ethidium, indicating that the cysteine substitutions did not perturb the transporter structure and function to any significant degree (Fig. 6A). Furthermore, copper phenanthroline abolished the ability of these five mutants to impart cellular resistance to drugs but exerted no measurable effect on the wild-type or Cys-less NorM-NG, D41A, dicysteine mutant L58C/V269C or A62C/F265C (Fig. 6A).
FIGURE 6.
Functional consequences of cysteine cross-linking as measured in the drug resistance assay. A, optical density measurement of bacteria expressing NorM-NG variants in the absence and presence of copper phenanthroline (Cu) and DTT. 3 μg/ml of R6G or 0.5 μg/ml of ethidium (ET) was used. The results represent means ± S.D. (error bars) of at least three independent experiments performed in duplicate. B, nonreducing SDS-PAGE analysis of the NorM-NG variants, with and without copper phenanthroline and DTT treatment. Copper phenanthroline gave rise to gel mobility shift of five dicysteine NorM-NG mutants, which could be reversed by DTT.
Because the inhibitory effects exerted by copper phenanthroline are limited to only the cross-linkable dicysteine NorM-NG mutants, the inhibition is unlikely to be caused by the obstruction of the transport path in NorM-NG by copper phenanthroline, which is a bulky and positively charged molecule that resembles the typical MATE substrates (4, 5). Indeed, in the presence of DTT, the T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C retained their ability to confer drug resistance even with copper phenanthroline being present (Fig. 6A), indicating that DTT reversed the inhibition of multidrug efflux by copper phenanthroline, most probably by keeping the five cysteine pairs reduced. By contrast, copper phenanthroline or DTT had no measurable effect on the 10 monocysteine mutants, all of which endowed bacteria with resistance against R6G or ethidium (Table 1).
TABLE 1.
Minimal inhibitory concentrations (μg/ml) of cells expressing the NorM-NG variants
R6G, rhodamine 6G; ET, ethidium; Cys-less, cysteine-less NorM-NG; Cu, copper phenanthroline.
| Variants | R6G | R6G + Cu | R6G + Cu + DTT | ET | ET + Cu | ET + Cu + DTT |
|---|---|---|---|---|---|---|
| pET15b | 3.0 | 3.0 | 3.0 | 0.5 | 0.5 | 0.5 |
| NorM-NG | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| Cys-less | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| D41A | 3.0 | 3.0 | 3.0 | 0.5 | 0.5 | 0.5 |
| T42C/L289C | 6.0 | 3.0 | 6.0 | 2.0 | 0.5 | 2.0 |
| A45C/V285C | 6.0 | 3.0 | 6.0 | 2.0 | 0.5 | 2.0 |
| A57C/Q284C | 6.0 | 3.0 | 6.0 | 2.0 | 0.5 | 2.0 |
| L58C/V269C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| L58C/F270C | 6.0 | 3.0 | 6.0 | 2.0 | 0.5 | 2.0 |
| A62C/F265C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| A62C/S266C | 6.0 | 3.0 | 6.0 | 2.0 | 0.5 | 2.0 |
| T42C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| L289C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| A45C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| V285C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| A57C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| Q284C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| L58C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| V269C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| F270C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| A62C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| F265C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
| S266C | 6.0 | 6.0 | 6.0 | 2.0 | 2.0 | 2.0 |
Altogether, our experiments demonstrated that copper phenanthroline abrogated the ability of the five cross-linkable mutants to confer drug resistance, likely by promoting the formation of the disulfide bonds (Fig. 6B), which inactivated NorM-NG by precluding any further structural changes required for multidrug efflux. Although we observed that the oxidation-elicited cross-linking was incomplete in the bacterial membranes (Fig. 3B), the inactivation of the five NorM-NG mutants by copper phenanthroline appeared complete in vivo (Fig. 6A). These seemingly conflicting observations may be understood in light of the different time frames of the two sets of experiments, i.e. the cysteine cross-linking became complete over longer lengths of time (hours versus minutes).
Impact of Cross-linking on Drug Accumulation
We next examined the multidrug efflux function of the NorM-NG mutants in the drug accumulation assay, which provides yet another means to measure the impact of cross-linking on the ability of NorM-NG variants to export drugs (9, 10). We found that NorM-NG, Cys-less NorM-NG and the seven dicysteine mutants, but not D41A, reduced the level of R6G or ethidium within the bacterial cells, presumably by extruding these cytotoxic compounds (Fig. 7). Furthermore, copper phenanthroline treatment substantially enhanced the accumulation of R6G or ethidium in bacteria expressing the five cross-linking-competent dicysteine mutants but had no such effect on those cells expressing the wild-type or Cys-less NorM-NG, D41A, or cross-linking-incompetent dicysteine mutant L58C/V269C or A62C/F265C (Fig. 7).
FIGURE 7.
Functional impact of disulfide cross-linking as measured in the drug accumulation assay. The accumulation of R6G or ethidium (ET) in cells expressing the NorM-NG variants, in the absence or presence of copper phenanthroline (Cu) and DTT. The results represent means ± S.D. (error bars) of at least three independent experiments performed in duplicate. The data are given as percentages of the values for cells expressing the inactive D41A in the presence of both copper phenanthroline and DTT.
In addition, the five cross-linking-competent NorM-NG mutants could still reduce the accumulation of R6G or ethidium within the bacterial cells, as long as the copper phenanthroline treatment was accompanied by the addition of DTT (Fig. 7). By contrast, copper phenanthroline had little, if any, effect on the cells expressing the 10 monocysteine mutants (Fig. 8). Overall, these results concurred with our drug resistance data (Fig. 6) and suggested that copper phenanthroline rendered T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C incapable of reducing drug accumulation by catalyzing the disulfide cross-linking.
FIGURE 8.
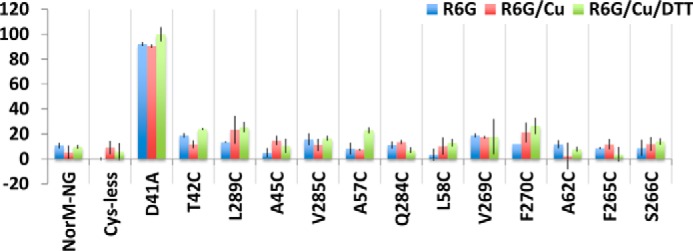
Lack of functional impact of copper phenanthroline on monocysteine NorM-NG mutants. The accumulation of R6G in cells expressing the NorM-NG variants, in the absence or presence of copper phenanthroline (Cu) and DTT. The results represent means ± S.D. (error bars) of at least three independent experiments performed in duplicate. The data are given as percentages of the values for cells expressing D41A in the presence of copper phenanthroline and DTT. In stark contrast to the five dicysteine mutants of NorM-NG, copper phenanthroline or DTT had little, if any, effect on the accumulation of R6G in bacteria expressing the 10 monocysteine mutants.
Discussion
In this study, we introduced five pairs of cysteines, T42C/L289C, A45C/V285C (TM1 and TM8), L58C/F270C, A62C/S266C (TM2 and TM7), and A57C/Q284C (TM2 and TM8) into the Cys-less NorM-NG. Although these amino acid substitutions exerted little effect on the expression or transport function of NorM-NG (Figs. 2, 6, and 7), we found copper phenanthroline substantially altered the migration of the purified dicysteine mutants in SDS-PAGE (Fig. 3A). The gel mobility shift, which is likely to be caused by the formation of disulfide bonds within the mutants, is not confined to the detergent-purified proteins, because similar observations were made for the mutants embedded in the bacterial membranes (Fig. 3C). Furthermore, copper phenanthroline inactivated the five dicysteine mutants in our drug resistance and accumulation assays, which could be nevertheless prevented by the addition of a reducing agent (Figs. 6 and 7).
Our data further argued in favor of the close proximity of T42C/L289C, A45C/V285C, L58C/F270C, A62C/S266C, and A57C/Q284C in NorM-NG during excursions to a crystallographically uncharacterized and yet functionally relevant state. In the extracellular facing structure of NorM-NG, the five pairs of substituted amino acids are not in sufficient proximity to form disulfide bonds (Fig. 1A). Therefore, it seems likely that during transport, the extracellular facing NorM-NG undergoes structural changes that bring these five pairs of amino acids closer together. In this cross-linking-competent state, copper phenanthroline triggers the formation of disulfide bonds and prohibits the transporter from undergoing any further structural changes necessary for multidrug efflux, thereby abolishing the transport function.
During transport, MATE transporters alternate between their extracellular and intracellular facing conformations (6–11). Therefore, we suggest that the cross-linking-permitting conformation of NorM-NG is the intracellular facing state (Fig. 1B). To form the five experimentally observed disulfide bridges, TM1 and TM2 in the extracellular facing NorM-NG need to move substantially toward TM8 and TM7, respectively. Upon such movement, the substrate-binding cavity is likely to be sealed off from the extracellular side. Concomitantly, the transporter likely exposes its substrate-binding site toward the cytoplasm, i.e. adopting the intracellular facing conformation, to acquire a new substrate. In prior studies, we suggested that the N and C domains of NorM-NG rotate relative toward each other during these structural changes (9).
The observation that T42C and L289C, A45C and V285C (TM1 and TM8), L58C and F270C, A62C and S266C (TM2 and TM7), and A57C and Q284C (TM2 and TM8) formed disulfide bonds places their Cα atoms within 6 Å of each other in the intracellular facing NorM-NG. Our estimation was based on the fact that the average Cα-Cα distance for 6,874 unique disulfide bonds in the protein database is 5.6 Å (20). This spatial requirement implies that when the extracellular facing NorM-NG transitions into the intracellular facing state, its N and C domains need to rotate at least 30° relative to each other (Table 2). Our data also implied that among the five pairs of amino acids, L58C/F270C would make the largest movement during transport, corresponding to an almost 10 Å shift between the substrate-binding TM2 and TM7 (7). For L58C/F270C in particular, a rotation of 30° or more between the N and C domains would be needed to bring the two cysteines into sufficient proximity for the cross-linking to take place.
TABLE 2.
Predicted Cα-Cα distances for cysteine pairs in the NorM-NG mutants
| NorM-NG Mutant | Outward facing conformation | Inward facing model with 20° rotation | Inward facing model with 30° rotation |
|---|---|---|---|
| Å | Å | Å | |
| T42C/L289C | 12.5 | 8.1 | 5.9 |
| A45C/V285C | 13.9 | 7.7 | 4.6 |
| A57C/Q284C | 14.0 | 8.2 | 5.4 |
| L58C/F270C | 15.5 | 9.5 | 6.9 |
| A62C/S266C | 10.8 | 6.4 | 4.3 |
| L58C/V269C | 12.9 | 6.8 | 4.1 |
| A62C/F265C | 10.0 | 6.0 | 4.0 |
A 30° rotational movement between the N and C domains, however, would lead to severe steric clash between the TM1/TM2 and TM7/TM8 in NorM-NG. To avoid such a clash, the degree of rotation must be limited to 20° or less (9), which in turn would render the formation of four experimentally observed disulfide bonds, T42C/S289C, A45C/V285C, L58C/F270C, and A57C/Q284C unlikely (Table 2). To reconcile this discrepancy, we invoke two possibilities. First, we might have substantially underestimated the Cα-Cα distance required for disulfide bond formation. Second, several transmembrane helices may bend or kink when the N and C domains rotate 30° relative to each other in NorM-NG (Fig. 9).
FIGURE 9.
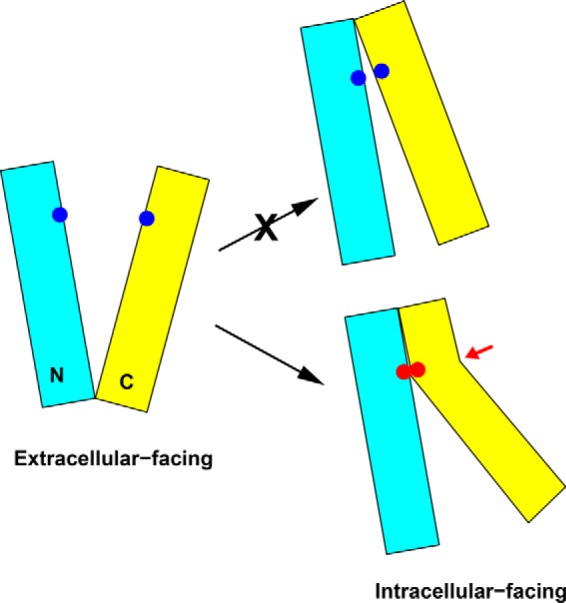
Proposed model for the interconversion between the extracellular and intracellular facing NorM-NG. Rotational movement alone between the N (cyan) and C (yellow) domains is unlikely to bring all the five cysteine pairs studied in this work (blue dots) into sufficient proximity for disulfide formation. Helix bending (highlighted by a red arrow) likely occurs as the transporter adopts the intracellular facing conformation, which enables the disulfide bond formation (red dots). Although helix bending may occur in the N and/or C domain, for simplicity the bending of helices is only shown in the C domain.
Because the first possibility is at odds with the known protein structures (20), we contend that during transport the extracellular and intracellular portions of TM1, TM2, TM7, TM8, and/or other transmembrane helices in NorM-NG undergo movement to different extents, i.e. rather than simply rotating or tilting, they bend or kink, to enable the formation of the five disulfide bonds, as well as to avoid steric clash between the N and C domains. Consistent with this mechanism, TM2, TM4, TM7, and TM8 in NorM-NG each contain a conserved proline in the mid-section of the helix, which may facilitate the bending or kinking of the helices when N and C domains rotate relative to each other. In line with our findings, studies on several other secondary membrane transporters also supported the notion that the transition from one conformational state to another entails not only the en bloc rotational movement of structural domains but also the bending of individual transmembrane helices (21, 22).
On the basis of these considerations, we conclude that the transition from the extracellular facing NorM-NG to the intracellular facing state involves “asymmetric” conformational changes, i.e. a mixture of both rotational movement and helix bending (Fig. 9), rather than merely “symmetric” rotational movement (9). Of note, such movement in principle would bring two cross-linking-incompetent cysteine pairs, L58C/V269C and A62C/F265C, into spatial proximity (Table 2). However, under our experimental conditions, no disulfide bond was detected within these two cysteine pairs (Fig. 3). Because the formation of the disulfide bonds depends on not only the spatial proximity but also the orientation of the cysteine side chains (23), it is plausible that the side chain orientation of L58C/V269C or A62C/F265C is not productive to form a disulfide bond.
Interestingly, we found that both TPP and NaCl promoted NorM-NG to assume its intracellular facing conformation in solution (Fig. 5), despite the fact that all of the ligand-bound NorM-NG structures captured the detergent-purified transporter in its extracellular facing conformation (7, 10). It thus seems likely that the extracellular facing state of NorM-NG is favored by the crystal packing forces, even though the intracellular facing conformation is a well populated state in solution. Furthermore, our results implied that the binding of substrate is compatible with the intracellular facing conformation of NorM-NG, whereas the unbinding of Na+ favors the extracellular facing state. Evidently, these findings are consistent with our previously proposed mechanism in which the intracellular facing, substrate-bound NorM-NG switches to its extracellular facing conformation upon the dissociation of Na+ (7).
Although we still lack a full portrait of the intracellular facing NorM-NG, the data presented here provide specific structural constraints for refining the intracellular facing model of NorM-NG and give fresh insights into how a MATE transporter changes its conformations during transport. As such, our findings take us one step closer toward elucidating the mechanism underlying MATE-driven multidrug extrusion and may spark new prospects for curbing the widespread multidrug resistance.
Author Contributions
M. L., M. R., and R. N. designed the study. R. N., M. R., and M. L. performed the experiments. M. L. and M. R. wrote the paper.
Acknowledgment
We thank Jindrich Symersky for helpful discussions.
This work was supported in part by National Institutes of Health Grant R01-GM094195 (to M. L.). The authors declare that they have no conflicts of interest with the contents of this article.
- MATE
- multidrug and toxic compound extrusion
- TPP
- tetraphenylphosphonium
- R6G
- rhodamine 6G.
References
- 1. Higgins C. F. (2007) Multiple molecular mechanisms for multidrug resistance transporters. Nature 446, 749–757 [DOI] [PubMed] [Google Scholar]
- 2. Fischbach M. A., and Walsh C. T. (2009) Antibiotics for emerging pathogens. Science 325, 1089–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brown M. H., Paulsen I. T., and Skurray R. A. (1999) The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol. Microbiol. 31, 394–395 [DOI] [PubMed] [Google Scholar]
- 4. Omote H., Hiasa M., Matsumoto T., Otsuka M., and Moriyama Y. (2006) The MATE proteins as fundamental transporters of metabolic and xenobiotic organic cations. Trends Pharmacol. Sci. 27, 587–593 [DOI] [PubMed] [Google Scholar]
- 5. Kuroda T., and Tsuchiya T. (2009) Multidrug efflux transporters in the MATE family. Biochim. Biophys. Acta 1794, 763–768 [DOI] [PubMed] [Google Scholar]
- 6. He X., Szewczyk P., Karyakin A., Evin M., Hong W. X., Zhang Q., and Chang G. (2010) Structure of a cation-bound multidrug and toxic compound extrusion transporter. Nature 467, 991–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lu M., Symersky J., Radchenko M., Koide A., Guo Y., Nie R., and Koide S. (2013) Structures of a Na+-coupled, substrate-bound MATE multidrug transporter. Proc. Natl. Acad. Sci. U.S.A. 110, 2099–2104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tanaka Y., Hipolito C. J., Maturana A. D., Ito K., Kuroda T., Higuchi T., Katoh T., Kato H. E., Hattori M., Kumazaki K., Tsukazaki T., Ishitani R., Suga H., and Nureki O. (2013) Structural basis for the drug extrusion mechanism by a MATE multidrug transporter. Nature 496, 247–251 [DOI] [PubMed] [Google Scholar]
- 9. Lu M., Radchenko M., Symersky J., Nie R., and Guo Y. (2013) Structural insights into H+-coupled multidrug extrusion by a MATE transporter. Nat. Struct. Mol. Biol. 20, 1310–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radchenko M., Symersky J., Nie R., and Lu M. (2015) Structural basis for the blockade of MATE multidrug efflux pumps. Nat. Commun. 6, 7995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu M. (2016) Structures of multidrug and toxic compound extrusion transporters and their mechanistic implications. Channels 10, 88–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kubo Y., Konishi S., Kawabe T., Nada S., and Yamaguchi A. (2000) Proximity of periplasmic lops in the metal-tetracycline/H+ antiporter of Escherichia coli observed on site-directed chemical cross-linking. J. Biol. Chem. 275, 5270–5274 [DOI] [PubMed] [Google Scholar]
- 13. Brocke L., Bendahan A., Grunewald M., and Kanner BI. (2002) Proximity of two oppositely oriented reentrant loops in the glutamate transporter GLT-1 identified by paired cysteine mutagenesis. J. Biol. Chem. 277, 3985–3992 [DOI] [PubMed] [Google Scholar]
- 14. Ryan R. M., Mitrovic A. D., and Vandenberg R. J. (2004) The chloride permeation pathway of a glutamate transporter and its proximity to the glutamate translocation pathway. J. Biol. Chem. 279, 20742–20751 [DOI] [PubMed] [Google Scholar]
- 15. Tao Z., Zhang Y. W., Agyiri A., and Rudnick G. (2009) Ligand effects on cross-linking support a conformational mechanism for serotonin transport. J. Biol. Chem. 284, 33807–33814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reyes N., Ginter C., and Boudker O. (2009) Transport mechanism of a bacterial homologue of glutamate transporters. Nature 462, 880–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Valdés R., Shinde U., and Landfear S. M. (2012) Cysteine cross-linking defines the extracellular gate for the Leishmania denovani nucleoside transporter 1.1 (LdNT1.1). J. Biol. Chem. 287, 44036–44045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li X., Dang S., Yan C., Gong X., Wang J., and Shi Y. (2013) Structure of a presinilin family intramembrane aspartate protease. Nature 493, 56–61 [DOI] [PubMed] [Google Scholar]
- 19. Steed P. R., Stein R. A., Mishra S., Goodman M. C., and McHaourab H. S. (2013) Na+-substrate coupling in the multidrug antiporter NorM probed with a spin-labeled substrate. Biochemistry 52, 5790–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmidt B., Ho L., and Hogg P. J. (2006) Allosteric disulfide bonds. Biochemistry 45, 7429–7433 [DOI] [PubMed] [Google Scholar]
- 21. Krishnamurthy H., and Gouaux E. (2012) X-ray structures of LeuT in substrate-free outward-open and apo inward-open states. Nature 481, 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Perez C., Koshy C., Yildiz O., and Ziegler C. (2012) Alternating-access mechanism in conformationally asymmetric trimers of the betain transporter BetP. Nature 490, 126–130 [DOI] [PubMed] [Google Scholar]
- 23. Gudlur A., Quintana A., Zhou Y., Hirve N., Mahapatra S., and Hogan P. G. (2014) STIM1 triggers a gating rearrangement at the extracellular mouth of the ORAl1 channel. Nat. Commun. 5, 5164. [DOI] [PMC free article] [PubMed] [Google Scholar]