

Abstract
Eight pairs of enantiomeric neolignans, norlignans, and sesquineolignans (1a/1b–8a/8b), together with five known neolignans (9a/9b and 10–12), have been isolated from 70% acetone extract of the whole plants of Phyllanthus glaucus Wall. (Euphorbiaceae). The racemic or partial racemic mixtures were successfully separated by chiral HPLC using different types of chiral columns with various mobile phases. Their structures were elucidated on the basis of extensive spectroscopic data. The absolute configurations of 2a/2b were determined by computational analysis of their electronic circular dichroism (ECD) spectrum, and the absolute configurations of other isolates were ascertained by comparing their experimental ECD spectra and optical rotation values with those of structure-relevant compounds reported in literatures. Compounds 4a/4b featured unique sesquineolignan skeletons with a novel 7-4′-epoxy-8′-8′′/7′-2′′ scaffold, consisting of an aryltetrahydronaphthalene and a dihydrobenzofuran moiety. The planar structures of compounds 2, 3, 7, and 8 were documented previously; however, their absolute configurations were established for the first time in this study. The antioxidant activities of 1a/1b–8a/8b were evaluated using DPPH free radical scavenging assay, and the results demonstrated that compounds 1b and 3b showed potent DPPH radical scavenging activities with IC50 values of 5.987 ± 1.212 and 9.641 ± 0.865 μg/mL, respectively.
Lignans and neolignans are biosynthetically generated from phenylpropanoids, depending on the ways they are linked1, which present a conspicuous chemical diversity and have multifarious clinical pharmacology activities2. In nature, lignans with chiral carbon atoms are usually composed of one enantiomer or of several stereoisomers with different amount, so they present optical activities in general3,4. In the past decades, the enantioseparation and absolute configurations of natural enantiomeric lignans and neolignans have attracted the attention of researchers around the world, with the aid of application of chromatographic enantioseparation, single-crystal X-ray diffraction, electronic circular dichroism (ECD) techniques5,6,7,8.
Plants of the genus Phyllanthus (Euphorbiaceae) are widely distributed in most tropical and subtropical areas, which have been used in folk medicine for a long time to treat kidney and urinary bladder disturbances, intestinal infections, diabetes, and hepatitis B9. Previous phytochemical investigations on plants of this genus led to the isolation of terpenoids10,11,12,13, lignans14, flavonoids15, alkaloids16, and tannins17. In the course of searching for bioactive natural products from the traditional Chinese medicine plants18,19,20, our attention was drawn to P. glaucus, a kind of deciduous shrub widely distributed in south China. The roots of the plant are commonly used to treat rheumatic arthritis and infantile malnutrition by the local people21. To the best of our knowledge, phytochemical study of P. glaucus had resulted in the isolation of 33 compounds including two new lignan glucosides by Yu and co-workers21. In our present study, eight pairs of enantiomers (1a/1b–8a/8b, Fig. 1), including a pair of enantiomeric neolignans (1a/1b), two pairs of norlignans (2a/2b and 3a/3b), and five pairs of sesquilignans (4a/4b–8a/8b), along with five known compounds (9a/9b, 10, 11, and 12) were isolated from the acetone extract of the whole plants of P. glaucus. The enantioseparations of these compounds were achieved using chiral HPLC approaches, and their structures including absolute configurations were elucidated by extensive spectroscopic analyses and ECD calculations. It is notable that compounds 4a/4b possess unique sesquineolignan architecture with 7-4′-epoxy-8′-8′′/7′-2′′ scaffolds, consisting of an aryltetrahydronaphthalene and a dihydrobenzofuran moiety. In addition, the antioxidant activities of compounds 1a/1b–8a/8b were evaluated by DPPH assay.
Figure 1. Structures of the co-isolated compounds.

Results and Discussion
Structural Elucidation of Compounds 1a/1b–8a/8b
Compound 1 was isolated as a colorless gum, and its molecular formula was determined to be C20H24O7, as elucidated by HRESIMS (m/z 399.1407, calcd for [M + Na]+ 399.1420) and 13C NMR data (Table 1), requiring nine degrees of unsaturation. The IR spectrum of 1 showed the presence of hydroxyl (3418 cm−1) and aromatic functionalities (1614, 1518, and 1461 cm−1). The 1H NMR spectrum revealed the co-existence of a symmetrical 1,3,4,5-tetrasubstituted aromatic ring at δH 6.71 (2H, s), an asymmetrical 1,3,4,5-tetrasubstituted aromatic ring at δH 6.60 (1H, s) and 6.57 (1H, s), and two methoxyl groups at δH 3.81 (6H, s, each). The 13C NMR spectra (Table 1) of 1 displayed 20 carbons including two methoxyl groups, four sp3 methylenes, two sp3 methines, and two aromatic rings. The above mentioned data, together with the 1H–1H COSY correlations of H-8 [δH 3.45 (1H, ddd, J = 7.7, 6.0, 5.3 Hz)] with H-9 [δH 3.85 (1H, dd, J = 10.9, 5.3 Hz, H-9a), and 3.76 (1H, dd, J = 10.9, 7.7 Hz, H-9b)] and H-7 [δH 5.51 (1H, d, J = 6.0 Hz)], and of H-8′ [δH 1.79 (2H, m)] with H-9′ [δH 3.56 (2H, t, J = 6.5 Hz)] and H-7′ [δH 2.56 (2H, t, J = 7.6 Hz)], suggested that compound 1 was a lignan resembled cedrusin22. This prediction was further confirmed by the HMBC correlations from H-7 to C-9, C-4′, and C-5′, from H-8 to C-1, C-4′, and C-5′, and from H-7′ to C-1′, C-2′, C-6′, and C-9′. In addition, the two methoxyl groups were located at C-3 and C-5 by the HMBC interactions from protons at δH 3.81 (6H, s) to carbons at δC 149.3 (C-3 and C-5). Thus, the planar structure of 1 was established and named phyllanglaucin A.
Table 1. 1H NMR [400 MHz, δ in ppm, Mult. (J in Hz)] and 13C NMR (100 MHz, δ in ppm) Data for 1–3.
| no. | 1a | 2b | 3b | |||
|---|---|---|---|---|---|---|
| δH (J, Hz) | δC | δH (J, Hz) | δC | δH (J, Hz) | δC | |
| 1 | 129.7 | 131.9 | 131.4 | |||
| 2 | 6.71 s | 104.0 | 6.53 s | 109.5 | 6.58 d (1.8) | 109.6 |
| 3 | 149.3 | 146.4 | 146.6 | |||
| 4 | 136.3 | 144.5 | 144.7 | |||
| 5 | 149.3 | 6.72 d (8.5) | 113.9 | 6.85 d (8.1) | 113.9 | |
| 6 | 6.71 s | 104.0 | 6.52 overlap | 120.7 | 6.69 dd (8.1, 1.8) | 120.9 |
| 7 | 5.51 d (6.0) | 88.8 | 4.31 d (9.3) | 89.7 | 4.39 d (6.6) | 85.4 |
| 8 | 3.45 ddd (7.7, 6.0, 5.3) | 55.9 | 3.01 m | 55.0 | 3.07 m | 54.6 |
| 9a | 3.85 dd (10.9, 5.3) | 65.2 | 4.16 dd (11.1, 8.0) | 66.9 | 3.76 overlap | |
| 64.4 | ||||||
| 9b | 3.76 dd (10.9, 7.7) | 65.2 | 3.85 dd (11.1, 4.3) | 66.9 | 3.68 m | 64.4 |
| 1′ | 136.8 | 131.3 | 131.1 | |||
| 2′ | 6.57 s | 117.0 | 6.31 d (1.9) | 111.7 | 6.57 d (1.8) | 112.0 |
| 3′ | 141.9 | 146.3 | 146.4 | |||
| 4′ | 146.5 | 145.2 | 145.4 | |||
| 5′ | 134.4 | 6.73 d (8.2) | 114.3 | 6.85 d (8.1) | 114.3 | |
| 6′ | 6.60 s | 116.6 | 6.50 dd (8.2, 1.9) | 120.8 | 6.67 dd (8.1, 1.8) | 121.6 |
| 7′ | 2.56 t (7.6) | 32.7 | ||||
| 8′ | 1.79 m | 35.8 | ||||
| 9′ | 3.56 t (6.5) | 62.3 | ||||
| CH3O-3 | 3.81 s | 56.7 | 3.75 s | 56.1 | 3.77 s | 56.0 |
| CH3O-5 | 3.81 s | 56.7 | ||||
| CH3O-7 | 3.24 s | 56.8 | 3.16 s | 57.1 | ||
| CH3O-3′ | 3.69 s | 56.0 | 3.81 s | 56.0 | ||
aIn CD3OD; bIn CDCl3.
The coupling constant between H-7 and H-8 (J7,8 = 6.0 Hz) of 1 suggested that the preferred conformation of the two protons was trans23, which was further supported by the NOESY correlations of H-7 with H-9b (Fig. 2a). Interestingly, we found that 1 was a racemic mixture for the presence of two peaks on the chiral HPLC. In order to define their absolute configurations, ECD, a powerful and effective method in determining the absolute configuration of natural products, was applied. Performing the enantioseparation of 1 by HPLC using a chiral-pak IA column provided the enantiomers 1a and 1b with a ratio about 1:3. Given the reversed helicity rule of the 1Lb band ECD for the 7-hydroxy-2,3-dihydrobenzo[b]furan chromophore24, the negative 1Lb Cotton effect (CE) of 1a around 293 nm (Δε − 0.45) (Fig. 3) suggested a 7R configuration, and accordingly, the positive CE of 1b at 292 nm (Δε + 0.57) (Fig. 3) indicated a 7S configuration. Thus, compounds 1a and 1b were determined as (+)-(7R,8S)-phyllanglaucin A and (−)-(7S,8R)-phyllanglaucin A (Fig. 1), respectively.
Figure 2.

(a) Key NOESY correlations of compound 1; (b) 1H–1H COSY and key HMBC correlations of compound 4; (c) Key NOESY correlations of compound 4.
Figure 3. Experimental ECD spectra of 1a/1b–8a/8b (5b and 6a/6b recorded in MeCN; the others in MeOH).

Compound 2 was purified as an amorphous white powder, which showed IR absorptions for hydroxyl (3366 cm−1) and aromatic rings (1606 and 1518 cm−1). The molecular formula, C18H22O6, was determined by HRESIMS (m/z 357.1321, calcd for [M + Na]+ 357.1314) and the 13C NMR data. The 1H NMR spectrum of 2 displayed typical protons including six aromatic protons, two methines, one oxygenated methylene, and three methoxyls, and the 13C NMR and DEPT spectra resolved 18 carbon signals. The 1D NMR data (Table 1) suggested that 2 had the same planar structure as threo-2,3-bis-(4-hydroxy-3-methoxyphenyl)-3-methoxypropanol, a norlignan isolated from Aralia bipinnata25, which was further supported by detailed analyses of the 2D NMR spectra. In consequence, the structure of 2 was assigned as 4,4′-dihydroxy-3,7,3′-trimethoxy-7′,8′,9′-trinor-8,1′-neolignan-9-ol. The relative configuration of 2 was subsequently determined to be 7,8-threo due to the large coupling constant between H-7 and H-8 (J7,8 = 9.3 Hz). However, due to the lack of optical rotation and CE, compound 2 was proposed to be a racemic mixture. With the help chiral HPLC, the enantiomers of 2a (5.3 mg) and 2b (5.3 mg) were obtained with a ratio of 1:1, which showed the mirror image-like ECD curves (Fig. 3) and owned the opposite specific rotations (2a: 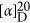 + 30.0; 2b:
+ 30.0; 2b: 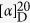 −30.8).
−30.8).
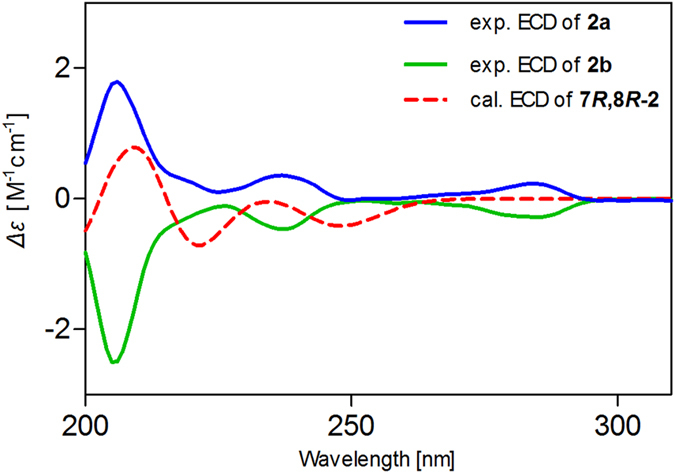
Although the planar structures of 2a/2b were reported in several literatures25,26,27,28,29, their absolute configurations remain to be determined. Our efforts to obtain fine crystals to determine the absolute configuration directly by X-ray crystallography failed. Therefore, quantum chemical ECD calculations were applied5,6, which were carried out for compound 7R,8R-2 at the level of LC-wPBE/6-311++G(2d,p)//B3LYP/6-311++G(2d,p) in MeOH with IEFPCM solvation model. The theoretical ECD curve of 7R,8R-2 matched well with the experimental ECD curve of compound 2a (Fig. 4). The result was further validated by our additional ECD calculations employing two other different functionals, i.e., CAM-B3LYP and WB97XD, in which consistent results of ECD were obtained (given in supplementary information). Thus, the absolute configuration of 2a was unambiguously assigned as 7R,8R. Finally, compound 2a was determined as (7R,8R)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol, and 2b was determined as (7S,8S)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol.
Figure 4. Assignment of the absolute configurations of 2 by comparison of the calculated ECD spectrum for (7R,8R)-2 with the experimental CD spectra for 2a/2b using TDDFT methods.
Compound 3, a white amorphous powder, had the same molecular formula (C18H22O6) as that of 2, deducing from 13C NMR and DEPT and the positive HRESIMS experiment (m/z 357.1323 [M + Na]+, calcd 357.1314). A comparison of NMR data of 3 with that of 2 indicated that the significant difference was the coupling constant between H-7 and H-8 (J7,8 = 9.3 Hz of 2 and J7,8 = 6.6 Hz of 3) (Table 1). These findings suggested that 3 was a stereoisomer of 2, and it was determined to be erythro form in comparison with structurally related compounds reported in literatures27,30. Interestingly, compound 3 was also found to be a racemic mixture, and was further resolved to yield 3a (4.5 mg) and 3b (4.1 mg) over HPLC using a chiral column. 3a and 3b exhibited mirror image-like ECD curves (Fig. 3) and opposite specific rotations (3a: 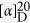 + 79.5; 3b:
+ 79.5; 3b: 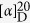 −76.9).
−76.9).
The absolute configurations of 3a and 3b were determined by comparing their optical rotations (3a: 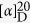 + 79.5; 3b:
+ 79.5; 3b: 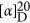 −76.9) with those of two similar compounds (1S,2R)-1,2-bis-(4-hydroxy-3-methoxyphenyl)-1,3-propanediol (3a′) and (1R,2S)-1,2-bis-(4-hydroxy-3-methoxyphenyl)-1,3-propanediol (3b′) (Fig. 1), whose absolute configurations were established by the glycosylation shift rule31 (3a′:
−76.9) with those of two similar compounds (1S,2R)-1,2-bis-(4-hydroxy-3-methoxyphenyl)-1,3-propanediol (3a′) and (1R,2S)-1,2-bis-(4-hydroxy-3-methoxyphenyl)-1,3-propanediol (3b′) (Fig. 1), whose absolute configurations were established by the glycosylation shift rule31 (3a′: 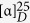 + 41.0 (c 0.7, MeOH); 3b′:
+ 41.0 (c 0.7, MeOH); 3b′: 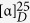 − 40.5 (c 1.2, MeOH). This conclusion was also supported by comparing the optical rotations with other analogues, such as carayensins B and C reported from Carya cathayensis32. Finally, 3a was determined as (7S,8R)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol, and 3b was determined as (7R,8S)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol.
− 40.5 (c 1.2, MeOH). This conclusion was also supported by comparing the optical rotations with other analogues, such as carayensins B and C reported from Carya cathayensis32. Finally, 3a was determined as (7S,8R)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol, and 3b was determined as (7R,8S)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol.
Compound 4 was obtained as a colorless gum. The IR spectrum displayed absorption bands for hydroxyls (3339 cm−1), and aromatic rings (1654, 1603, 1515, 1496, and 1464 cm−1), and the molecular formula C30H34O9 was deduced by the HRESIMS ion peak at m/z 561.2089 [M + Na+] (calcd 561.2101), corresponding to fourteen degrees of unsaturation. The 1H NMR spectrum revealed the presence of an ABX system [δH 6.59 (1H, d, J = 8.2 Hz, H-5), 6.57 (1H, d, J = 1.8 Hz, H-2), and 6.42 (1H, dd, J = 8.2, 1.8 Hz, H-6)], four aromatic protons [δH 6.68 (1H, s, H-6′′), 6.62 (1H, s, H-3′′), 6.62 (1H, br s, overlapped, H-6′), and 6.38 (1H, br s, H-2′)], three methoxyl groups [δH 3.85 (3H, s), 3.72 (3H, s), and 3.46 (3H, s)], one oxymethine [δH 5.46 (1H, d, J = 2.1 Hz, H-7)], three oxygenated methylenes [δH 3.83 (1H, m, H-9a), 3.46 (1H, overlapped, H-9b), 3.67 (1H, m, H-9′a)], 3.51 (1H, dd, J = 11.2, 3.3 Hz, H-9′b), 3.70 (1H, m, H-9′′a), and 3.67 (1H, m, H-9′′b)] (Table 2). The 13C NMR and DEPT spectra displayed 30 carbon resonances attributable to three benzene rings, four methylenes (three oxygenated), five methines (one oxygenated), and three methoxyl groups. Three benzene rings accounted for twelve degrees of unsaturation, and the remaining two degrees of unsaturation revealed the presence of two additional rings.
Table 2. 1H NMR Data for 4–8 and acernikol.
| no. | 4 | 5 | 6 | 7 | 8 | acernikol | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| CD3OD | CDCl3 | Me2CO-d6 | CDCl3 | Me2CO-d6 | CD3OD | Me2CO-d6 | CD3OD | Me2CO-d6 | CD3OD | |
| 2 | 6.57 d (1.8) | 6.88 d (1.5) | 6.97 d (1.2) | 6.88 d (1.4) | 6.97 d (1.2) | 6.97 d (1.7) | 7.03 d (1.5) | 6.96 d (1.7) | 7.03 d (1.6) | 6.96 d (2.0) |
| 5 | 6.59 d (8.2) | 6.86 d (8.1) | 6.86 d (8.1) | 6.86 d (8.2) | 66.86 d (8.1) | 6.74 d (8.2) | 6.77 d (8.1) | 6.74 d (8.2) | 6.76 d (8.1) | 6.74 d (8.0) |
| 6 | 6.42 dd (8.2,1.8) | 6.84 dd (8.1,1.5) | 6.81 dd (8.2,1.2) | 6.84 dd (8.2,1.4) | 6.81 dd (8.2,1.2) | 6.78 dd (8.2,1.7) | 6.83 dd (8.1,1.5) | 6.78 dd (8.2,1.7) | 6.83 dd (8.1,1.6) | 6.77 dd (8.0,2.0) |
| 7 | 5.46 d (2.1) | 4.60 d (6.1) | 4.57 d (6.3) | 4.61 d (6.1) | 4.57 d (6.3) | 4.90 d (5.1) | 4.43 d (4.2) | 4.90 overlap | 4.38 d (4.2) | 4.89 d (4.9) |
| 8 | 2.66 m | 4.05 m | 4.14 m | 4.05 m | 4.14 m | 4.24 m | 4.17 m | 4.24 m | 4.17 m | 4.23 m |
| 9a | 3.83 m | 3.97 m | 3.89 m | 3.97 m | 3.89 m | 3.90 overlap | 3.91 m | 3.90 dd (6.9,5.4) | 3.91 m | 3.90 m |
| 9b | 3.46 overlap | 3.62 m | 3.46 m | 3.62 m | 3.46 m | 3.59 overlap | 3.55 m | 3.60 overlap | 3.54 m | 3.58 m |
| 2′ | 6.38 brs | 6.61 s | 6.73 s | 6.61 s | 6.73 s | 6.71 s | 6.74 s | 6.71 s | 6.74 s | 6.70 d (2.0) |
| 6′ | 6.62 brs | 6.61 s | 6.73 s | 6.61 s | 6.73 s | 6.71 s | 6.74 s | 6.71 s | 6.74 s | 6.70 d (2.0) |
| 7′ | 4.05 d (8.8) | 5.52 d (7.4) | 5.55 d (6.6) | 5.53 d (7.3) | 5.55 d (6.6) | 5.55 d (6.0) | 5.59 d (6.5) | 5.55 d (5.8) | 5.58 d (6.5) | 5.54 d (6.1) |
| 8′ | 1.72 m | 3.57 m | 3.46 m | 3.57 m | 3.46 m | 3.46 m | 3.47 m | 3.46 m | 3.43 m | 3.45 m |
| 9′a | 3.67 m | 3.94 m | 3.81 m | 3.95 m | 3.81 m | 3.90 overlap | 3.86 overlap | 3.86 overlap | 3.86 overlap | 3.81 m |
| 9′b | 3.51 dd (11.2,3.3) | 3.91 m | 3.57 m | 3.92 m | 3.57 m | 3.76 m | 3.79 m | 3.75 m | 3.79 m | 3.75 m |
| 2′′ | 6.67 d (2.3) | 6.77 s | 6.67 brd (2.8) | 6.77 s | 6.75 brs | 6.84 s | 6.75 brs | 6.83 s | 6.75 d (2.0) | |
| 3′′ | 6.62 s | |||||||||
| 6′′ | 6.68 s | 6.67 d (2.3) | 6.77 s | 6.67 brd (2.8) | 6.77 s | 6.72 brs | 6.84 s | 6.72 brs | 6.83 s | 6.73 d (2.0) |
| 7′′ | 2.77 m | 2.67 t (7.6) | 2.61 t (7.5) | 2.67 t (7.6) | 2.61 t (7.5) | 2.63 t (7.5) | 2.61 m | 2.63 t (7.6) | 2.62 m | 2.62 t (7.3) |
| 8′′ | 1.88 m | 1.88 m | 1.78 m | 1.88 m | 1.78 m | 1.82 m | 1.79 m | 1.82 m | 1.78 m | 1.81 m |
| 9′′ | 3.67 m | 3.69 t (6.3) | 3.54 m | 3.69 t (6.3) | 3.54 m | 3.57 t (6.5) | 3.55 m | 3.57 t (6.5) | 3.54 m | 3.56 m |
| CH3O-3 | 3.72 s | 3.88 s | 3.84 s | 3.88 s | 3.84 s | 3.82s | 3.82 s | 3.80 s | 3.82 s | 3.80 s |
| CH3O-7 | 3.32 s | 3.23 s | 3.32 s | 3.24 s | ||||||
| CH3O-3′ | 3.46 s | 3.73 s | 3.77 s | 3.73 s | 3.77 s | 3.79 s | 3.84 s | 3.79 s | 3.84 s | 3.77 s |
| CH3O-5′ | 3.73 s | 3.77 s | 3.73 s | 3.77 s | 3.79 s | 3.84 s | 3.79 s | 3.84 s | 3.77 s | |
| CH3O-3′′ | 3.88 s | 3.84 s | 3.88 s | 3.84 s | 3.87 s | 3.86 s | 3.88 s | 3.86 s | 3.87 s | |
| CH3O-5′′ | 3.85 s | |||||||||
[400 MHz, δ in ppm, Mult. (J in Hz)].
Expansion of the formula to C27H25O6·(OMe)3 suggested that compound 4 was a sesquilignan. Moreover, three typical C6–C3 substructures (I: C1–C9, II: C1′–C9′, and III: C1′′–C9′′) were elucidated directly on the basis of COSY, HSQC, and HMBC spectra (Fig. 2). The connectivity of substructures I and II were established by the linkages of C7-O-C4′ and C8-C5′ to form a dihydrobenzofuran neolignan segment by the HMBC correlations from H-7 to C-4′, and C-5′, from H-8 to C-1, C-4′, and C-5′, and from H-9 to C-5′. The connectivity of substructures II and III were furnished by the direct linkages of C7′-C2′′ and C8′-C8′′ to construct an aryltetrahydronaphthalene neolignan moiety based on HMBC correlations from H-7′ to C-1′′, C-2′′, and C-8′′, and COSY correlations between H-8′ and H-8′′. In addition, HMBC correlations for CH3O-3/C-3, CH3O-3′/C-3′, and CH3O-5′′/C-5′′ revealed that these methoxyl groups were located at C-3, C-3′ and C-5′′, respectively. Thus, the planar structure of compound 4 was established as 3,3′,5′′-trimethoxy-4,4′′-dihydroxy-7,4′-epoxy-8,5′-7′,2′′-8′,8′′-sesquineolignan-9,9′,9′′-triol with an unprecedented sesquineolignan architecture, and was named phyllanglaucin B.
The relative configuration of 4 was determined by analyses of 1H–1H coupling constants and NOESY experiment. The small coupling constant of H-7/H-8 (J7,8 = 2.1 Hz) and the large coupling constant of H-7′/H-8′ (J7′,8′ = 8.8 Hz) suggested a cis configuration of H-7 and H-8 and a trans configuration of H-7′ and H-8′, respectively. The strong NOESY interactions from H-7′ to H-9′a and H-9′b confirmed the trans configuration of H-7′ and H-8′. Additional, NOESY correlation from H-7′ to H-8′′ revealed that H-7′ and H-8′′ was cis-oriented. Thus, the relative configuration of 4 was established as 7R*,8R* of the dihydrobenzofuran segment and 7′R*,8′S*,8′′S* of the aryltetrahydronaphthalene moiety.
Subsequent chiral HPLC resolution of 4 afforded the anticipated enantiomers 4a (1.5 mg) and 4b (7.5 mg), which showed mirror image-like ECD curves (Fig. 3) and specific rotations (4a: 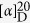 + 62.6; 4b:
+ 62.6; 4b: 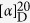 − 64.8). The absolute configurations of 4a and 4b were deduced by inspection of their ECD spectra. Generally, aryltetrahydronaphthalene lignans give two CEs in the ECD spectra at 280–290 nm and 230–245 nm, and the symbols of the two CEs are opposite33. On the basis of the published data33, CEs around 240 (+), 270 (+), and 290 (−) nm indicated the 7′S, 8′R, 8′′R configuration, in the case of 4a, it exhibited CEs around 242 (Δε + 6.72), 275 (Δε + 3.07), and 299 (Δε + 1.29) nm, the positive CE at 299 nm in the ECD spectrum of 4a was caused by S configuration at C-7, which produced a positive CE between 260–300 nm on the basis of the reversed helicity rule of the 1Lb band ECD for the 7-methoxy-2,3-dihydrobenzo[b]furan chromophore24. Namely, the positive rather than negative CE near 299 nm of 4a was ascribed to the superposition of the Cotton effects produced by 7S and 7′S. Therefore, the absolute configuration of 4a was established as 7S, 8S, 7′S, 8′R, 8′′R, and that of 4b was 7R, 8R, 7′R, 8′S, 8′′S, which were given the trival names (+)-phyllanglaucin B and (−)-phyllanglaucin B, respectively.
− 64.8). The absolute configurations of 4a and 4b were deduced by inspection of their ECD spectra. Generally, aryltetrahydronaphthalene lignans give two CEs in the ECD spectra at 280–290 nm and 230–245 nm, and the symbols of the two CEs are opposite33. On the basis of the published data33, CEs around 240 (+), 270 (+), and 290 (−) nm indicated the 7′S, 8′R, 8′′R configuration, in the case of 4a, it exhibited CEs around 242 (Δε + 6.72), 275 (Δε + 3.07), and 299 (Δε + 1.29) nm, the positive CE at 299 nm in the ECD spectrum of 4a was caused by S configuration at C-7, which produced a positive CE between 260–300 nm on the basis of the reversed helicity rule of the 1Lb band ECD for the 7-methoxy-2,3-dihydrobenzo[b]furan chromophore24. Namely, the positive rather than negative CE near 299 nm of 4a was ascribed to the superposition of the Cotton effects produced by 7S and 7′S. Therefore, the absolute configuration of 4a was established as 7S, 8S, 7′S, 8′R, 8′′R, and that of 4b was 7R, 8R, 7′R, 8′S, 8′′S, which were given the trival names (+)-phyllanglaucin B and (−)-phyllanglaucin B, respectively.
Compounds 5 and 6 were obtained as a mixture of amorphous powder and displayed only one peak on the reversed-phase HPLC. However, the carbon signals at δC 145.2, 135.7, 131.0, 127.7, 103.4, 86.1 and 54.0 were all splited in the 13C NMR spectrum. In particular, there should have been a doublet signal at δH 4.60 (H-7) in the 1H NMR spectrum of the proposed structure (Fig. 1), but it actually exhibited a de-doublet signals with the coupling constants J = 2.8, 6.0 Hz. To understand this distinctive discord, we elaborately conducted a series of chiral HPLC separations. To our delight, the results revealed that the isolated substance was a mixture of stereoisomers, which yeilded two pairs of enantiomers 5a/5b and 6a/6b, corresponding to the four peaks observed from the chiral HPLC.
The elemental composition of 5 (5a/5b) was determined as C32H40O11 by HRESIMS [m/z 623.2456 [M + Na+] (calcd 623.2468)]. The IR spectrum exhibited absorption bands of hydroxyl (3367 cm−1) and aromatic functionalities (1677, 1598, and 1460 cm−1). The 1H NMR spectrum showed the presence of a 1,3,4-trisubstituented benzene ring, a symmetrical 1,3,4,5-tetrasubstituted aromatic ring, an asymmetrical 1,3,4,5-tetrasubstituted aromatic ring, three oxymethines, two methylenes, and five methoxyl groups (Table 2). The 13C NMR of 5 showed 32 carbon resonances attributable to three benzene rings, five methylenes (three oxygenated), four methines (three oxygenated), and five methoxyl groups (Table 3). The NMR data implied that 5 (5a/5b) was a sesquineolignan, as a methoxylated derivative of dihydrobuddlenol B34. This conclusion was further confirmed by 2D NMR spectra, especially the key HMBC correlations from H-7 to C-1, C-2, and C-6, from H-8 to C-4′, from H-7′ to C-1′, C-2′, C-6′, C-4′′, and C-5′′, and from H-7′′ to C-1′′, C-2′′, and C-6′′. Thus, compound 5 (5a/5b) was assigned as 3,7,3′,5′,3′′-pentamethoxy-4-hydroxy-8,4′-oxy-7′,4′′-epoxy-8′,5′′-sesquineolignan-9,9′,9′′-triol and was named phyllanglaucin C.
Table 3. 13C NMR Data for compounds 4–8 and acernikol.
| no. | 4a | 5b | 6b | 7a | 8a | acernikola |
|---|---|---|---|---|---|---|
| 1 | 134.9 | 131.1 | 131.1 | 133.8 | 133.8 | 133.8 |
| 2 | 110.2 | 109.9 | 109.9 | 111.4 | 111.4 | 111.5 |
| 3 | 148.8 | 146.6 | 146.6 | 148.7 | 148.6 | 148.7 |
| 4 | 146.9 | 145.2 | 145.2 | 146.8 | 146.7 | 146.7 |
| 5 | 115.7 | 114.0 | 114.0 | 115.7 | 115.7 | 115.7 |
| 6 | 118.9 | 120.9 | 120.9 | 120.7 | 120.7 | 120.7 |
| 7 | 87.5 | 82.6 | 82.6 | 74.1 | 74.0 | 74.1 |
| 8 | 54.2 | 86.2 | 86.2 | 87.4 | 87.2 | 87.4 |
| 9 | 63.3 | 60.3 | 60.3 | 61.6 | 61.6 | 61.7 |
| 1′ | 138.2 | 137.4 | 137.4 | 139.7 | 139.6 | 139.8 |
| 2′ | 112.8 | 103.4 | 103.4 | 104.0 | 103.8 | 104.0 |
| 3′ | 149.0 | 153.4 | 153.4 | 154.6 | 154.5 | 154.7 |
| 4′ | 147.9 | 135.5 | 135.5 | 136.3 | 136.2 | 136.3 |
| 5′ | 128.3 | 153.4 | 153.4 | 154.6 | 154.5 | 154.7 |
| 6′ | 122.9 | 103.4 | 103.4 | 104.0 | 103.8 | 104.0 |
| 7′ | 45.5 | 87.8 | 87.8 | 88.6 | 88.6 | 88.6 |
| 8′ | 49.3 | 54.1 | 54.1 | 55.7 | 55.7 | 55.8 |
| 9′ | 61.4 | 64.0 | 64.0 | 65.1 | 65.0 | 65.1 |
| 1′′ | 131.9 | 135.8 | 135.8 | 137.2 | 137.2 | 137.2 |
| 2′′ | 130.4 | 112.6 | 112.6 | 114.2 | 114.1 | 114.3 |
| 3′′ | 115.6 | 144.4 | 144.4 | 145.3 | 145.2 | 145.3 |
| 4′′ | 145.8 | 146.5 | 146.5 | 147.5 | 147.3 | 147.5 |
| 5′′ | 144.2 | 127.6 | 127.6 | 129.5 | 129.4 | 129.6 |
| 6′′ | 113.6 | 116.1 | 116.1 | 118.0 | 118.0 | 118.0 |
| 7′′ | 34.5 | 32.2 | 32.2 | 32.9 | 32.8 | 32.9 |
| 8′′ | 39.6 | 34.7 | 34.7 | 35.8 | 35.7 | 35.8 |
| 9′′ | 65.9 | 62.4 | 62.4 | 62.2 | 62.2 | 62.3 |
| CH3O-3 | 56.4 | 56.1 | 56.1 | 56.4 | 56.4 | 56.4 |
| CH3O-7 | 57.4 | 57.4 | ||||
| CH3O-3′ | 56.0 | 56.2 | 56.2 | 56.6 | 56.6 | 56.7 |
| CH3O-5′ | 56.2 | 56.2 | 56.6 | 56.6 | 56.7 | |
| CH3O-3′′ | 56.1 | 56.1 | 56.8 | 56.8 | 56.9 | |
| CH3O-5′′ | 56.6 |
(100 MHz, δ in ppm). aIn CD3OD; bIn CDCl3.
The relative configuration of 5 (5a/5b) was deduced from analyses of the coupling constants and NOESY experiment, as well as by comparison of its chemical shifts with that of dihydrobuddlenol B34. The threo form geometry of H-7/H-8 was elucidated by the large coupling constant between H-7 and H-8 (J7,8 = 6.1 Hz in CDCl3, and J7,8 = 6.3 Hz in acetone-d6) (Table 2)35. The NOESY correlation of H-7′/H-9′, along with the large coupling constant of H-7′/H-8′ (J7′,8′ = 7.4 Hz in CDCl3, and J7′,8′ = 6.6 Hz in acetone-d6) indicated a trans configuration of H-7′/H-8′ (Table 2). The absolute configurations of 5a and 5b were defined by their ECD spectra (Fig. 3), a negative CE at 238 nm (Δε − 2.06) of 5a suggested the 8R configuration35, and the negative 1Lb band CE at 294 nm (Δε − 0.65) indicated the 7′R configuration24. Therefore, the absolute configuration of 5a was assigned as 7R, 8R, 7′R, 8′S. Since compounds 5a and 5b showed opposite ECD curves (Fig. 3) and optical rotations (5a: 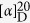 + 7.5; 5b:
+ 7.5; 5b: 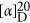 − 7.4), the absolute configuration of 5b was determined to be 7S, 8S, 7′S, 8′R. Finally, compound 5a was named (+)-phyllanglaucin C and 5b was named (−)-phyllanglaucin C, respectively.
− 7.4), the absolute configuration of 5b was determined to be 7S, 8S, 7′S, 8′R. Finally, compound 5a was named (+)-phyllanglaucin C and 5b was named (−)-phyllanglaucin C, respectively.
Compound 6 (6a/6b) had the same molecular formula as 5. The 1H and 13C NMR data of 6 were very close to those of 5 (Tables 2 and 3), assisted by the 2D NMR spectra of 6, compounds 5 and 6 elucidated to have the same planar structure and relative configurations. The 8R and 7′S configurations of 6a was established on the basis of a negative CE at 230 nm (Δε − 1.48) and a positive 1Lb CE at 282 nm (Δε + 1.41) (Fig. 3)35. Therefore, 6a (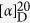 + 14.4) was elucidated as (7R,8R,7′S,8′R)-3,7,3′,5′,3′′-pentamethoxy-4-hydroxy-8,4′-oxy-7′,4′′-epoxy-8′,5′′-sesquineolignan-9,9′,9′′-triol and named (+)-phyllanglaucin D. Accordingly, 6b (
+ 14.4) was elucidated as (7R,8R,7′S,8′R)-3,7,3′,5′,3′′-pentamethoxy-4-hydroxy-8,4′-oxy-7′,4′′-epoxy-8′,5′′-sesquineolignan-9,9′,9′′-triol and named (+)-phyllanglaucin D. Accordingly, 6b (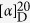 − 13.0) was elucidated to be (7S,8S,7′R,8′S)-3,7,3′,5′,3′′-pentamethoxy-4-hydroxy-8,4′-oxy-7′,4′′-epoxy-8′,5′′-sesquineolignan-9,9′,9′′-triol and named (−)-phyllanglaucin D.
− 13.0) was elucidated to be (7S,8S,7′R,8′S)-3,7,3′,5′,3′′-pentamethoxy-4-hydroxy-8,4′-oxy-7′,4′′-epoxy-8′,5′′-sesquineolignan-9,9′,9′′-triol and named (−)-phyllanglaucin D.
Given the same state as 5 and 6, chiral HPLC resolution of the mixture of 7 and 8 were performed, and afforded two pairs of sesquilignan enantiomers 7a/7b and 8a/8b.
Compound 7 (7a/7b) was isolated as a white powder, the IR spectrum of 7 showed absorption bands at 3366, 1599 and 1460 cm−1 assignable to hydroxyl and aromatic functionalities. The molecular formula was concluded to be C31H38O11 by HRESIMS [m/z 609.2294 [M + Na+] (calcd 609.2312)]. The NMR data of 7 was superimposable on those of acernikol (Tables 2 and 3), whose absolute configuration was ambiguous36. The 7,8-erythro configuration of 7 was determined by the small coupling constant of H-7 and H-8 (J7,8 = 5.1 Hz in CD3OD, and J7,8 = 4.8 Hz in acetone-d6) (Table 2)37. The strong NOESY correlation of H-7′/H-8′, and the absence of correlation between H-7′ and H-9′ indicated a cis configuration for H-7′ and H-8′.
The ECD spectrum of 7a showed a positive CE at 244 nm (Δε + 2.32) indicating an 8S configuration38, and a negative 1Lb CE at 294 nm (Δε − 0.79) suggesting a 7′R configuration (Fig. 3)24, 7b owned the opposite ECD curves (Fig. 3) and optical rotations with 7a (7a: 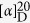 + 22.4; 7b:
+ 22.4; 7b: 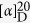 − 20.0). Thus, 7a was elucidated to be (7R,8S,7′R,8′R)-acernikol, and 7b was elucidated to be (7S,8R,7′S,8′S)-acernikol.
− 20.0). Thus, 7a was elucidated to be (7R,8S,7′R,8′R)-acernikol, and 7b was elucidated to be (7S,8R,7′S,8′S)-acernikol.
Compound 8 (8a/8b) had the same planar structure and relative configurations as 7 deducing from their closely related 1D and 2D NMR (Tables 2 and 3). 8a exhibited a positive CE at 243 nm (Δε + 6.51) and a positive CE at 281 nm (Δε + 1.76), suggestive an 8S configuration38 and a 7′S configuration24. Therefore, 8a was elucidated as (7R,8S,7′S,8′S)-acernikol, and 8b was determined to be (7S,8R,7′R,8′R)-acernikol.
The structures of known compounds 9a/9b and 10−12 were characterized as (7R,8S)-dihydrodehydroconiferyl alcohol (9a)39, (7S,8R)-dihydrodehydrodiconiferyl alcohol (9b)40, (7S,8R)-cedrusin (10)21, (7S,8R)-dihydrodehydrodiconifenyl alcohol 9-O-β-D-xylopyranoside (11)41, and (7S,8R)-4,7,9,9′-tetrahydroxy-3,3′-dimethoxy-8-O-4′-neolignan (12)42, respectively, by comparing their 1H and 13C NMR data and optical rotations with the reported literature values.
Reactive oxygen species (ROS) plays an important role in the normal physiological processes43. Oxidative stress, referring to the imbalance of pro-oxidants and antioxidant defenses, involves a wide variety of pathological processes, including cardiovascular disease44, rheumatic disease45,46, neurodegeneration47, tumor48, and so on. In addition, it was reported that the same type of lignans showed strong antioxidant activities49. In this case, compounds 1a/1b−8a/8b were evaluated for their antioxidant activities by the DPPH free radical scavenging assay50. As showed in Table 4, compound 1b showed strong activity against DPPH radical with the IC50 value of 5.987 ± 1.212 μg/mL, which was comparable with the IC50 value of the positive control (IC50 = 4.485 ± 0.157 μg/mL) (Table 4). Compounds 2a/2b, 3a/3b, and 4b also showed moderate antioxidant activity. A comparison of the structures of 7 and 8 with those of 5 and 6 showed that the antioxidant potency of the co-isolated was declined when a hydroxyl group at C-7 was replaced by a methoxy group. This fact implied that the presence of hydroxyl groups were crucial for the antioxidant activity. Comparison of the biological activities of the stereoisomers indicated that the variety of the stereochemistry had little effect on the antioxidant activity.
Table 4. Antioxidant Activities of compounds 1−8 (DPPH Test).
| Compounds | Removaleffect (%)a | IC50(μg/mL) | Compounds | Removaleffect (%)a | IC50(μg/mL) |
|---|---|---|---|---|---|
| 1a | 81.149 | (−)b | 5a | 34.069 | (−)b |
| 1b | 86.989 | 5.987 ± 1.212 | 5b | 34.299 | (−)b |
| 2a | 81.747 | 16.622 ± 0.797 | 6a | 33.103 | (−)b |
| 2b | 78.069 | 17.941 ± 1.994 | 6b | 37.471 | (−)b |
| 3a | 77.287 | 13.306 ± 0.907 | 7a | 49.563 | (−)b |
| 3b | 81.609 | 9.641 ± 0.865 | 7b | 56.736 | (−)b |
| 4a | 57.609 | (−)b | 8a | 51.540 | (−)b |
| 4b | 66.943 | 26.784 ± 0.812 | 8b | 48.598 | (−)b |
| Trolox | 95.494c | 4.485 ± 0.157 |
aConcentration of removal effect: 50 μg/mL; bIC50 value not determined; cConcentration of removal effect: 25 μg/mL.
Methods
General experimental procedures
Optical rotations were measured on a Perkin-Elmer PE-341 polarimeter (Perkin-Elmer, Waltham, MA, USA). ECD spectra were measured on a Jasco J-810 spectrometer (Jasco, Easton, MD, USA). UV spectra were taken in a Varian Cary 50 UV/VIS spectrophotometer (Varian, Salt Lake City, UT, USA). IR spectra were recorded with a Bruker Vertex 70 FT-IR spectrophotometer (Bruker, Karlsruhe, Germany). HRESIMS data were obtained on a Thermo Fisher LTQ XL LC/MS (Thermo Fisher, Palo Alto, CA, USA). NMR spectra were recorded on Bruker AM-400 spectrometers and Bruker AM-600 spectrometers (Bruker, Karlsruhe, Germany). Chemical shifts were given in ppm with reference to the residual CD3OD (δH 3.31/δC 49.0), CDCl3 (δH 7.26/δC 77.16), and CD3COCD3 (δH 2.05) signals. Silica gel (100–200 mesh and 200–300 mesh was used for column chromatography, Qingdao Marine Chemical Inc., Qingdao, China), ODS (50 μm, YMC, Japan), and Sephadex LH-20 (Pharmacia Biotech AB, Uppsala, Sweden) were used for column chromatography. Semipreparative HPLC was performed on a Dionex Ultimate 3000 HPLC (Dionex, Sunnyvale, CA, USA) with UV detector and an Ultimate XB-C18 (10 × 250 mm, 5 μm) column. The chiral HPLC isolation was accomplished on Daicel Chiralpak IA, IC, and ASH columns (4.6 × 250 mm, 5 μm; Daicel Chemical Ltd, Tokyo, Japan). TLC was carried out with glass precoated with silica gel GF254 (Qingdao Marine Chemical Inc. China). Solvents were distilled prior to use, and spectroscopic grade solvents were used.
Plant materials
The whole plants of P. glaucus were collected at Lin’an, Zhejiang Province, People’s Republic of China, in August 2012, and identified by Prof. Yunhe He of Zhejiang A&F University. A voucher specimen (No. 20120805A) was deposited in the herbarium of Hubei Key Laboratory of Natural Medicinal Chemistry and Resource Evaluation, Tongji Medical College, Huazhong University of Technology and Science.
Extraction and isolation
Part I: Air dried whole plant (25.0 kg) of Phyllanthus glaucus was chopped and soaked in 70% aqueous acetone at room temperature (3 × 7 d). The acetone extract was evaporated under reduced pressure, the residue was suspended in H2O and extracted successively with petroleum ether (60–90 °C), EtOAc, and n-BuOH. The EtOAc fraction (250.0 g) was subjected to silica gel CC using a stepwise gradient elution of CH2Cl2–MeOH (1:0–0:1) to afford four subfractions (A–D). Fraction B (35.0 g) was subjected to MCI gel CC (eluted with MeOH–H2O) to remove chlorophyll, and the residue (19.0 g) was partitioned by a RP-C18 CC (gradient elution of MeOH–H2O, 10:90–100:0) to eight subfractions (B1–B8). Fr. B4 (eluted with MeOH–H2O, 40:60) was chromatographed on a Sephadex LH-20 column (MeOH) to afford six subfractions B4-1–B4-6, B4-4 (421 mg) was subjected to silica gel CC (CH2Cl2–MeOH, from 30:1 to 5:1) to give B4-4-1 to B4-4-3. B4-4-2 was purified by semipreparative HPLC thrice (MeOH–H2O, 40:60, MeCN–H2O, 32:68, MeCN–H2O, 26:74, respectively) to afford 1 (6.7 mg). Fr. B5 (eluted with MeOH–H2O, 50:50) was separated by Sephadex LH-20 column (MeOH) to give six subfractions B5-1–B5-6. B5-3 was further separated by silica gel CC (CH2Cl2–MeOH, from 30:1 to 5:1) to give five subfractions (B5-3-1 to B5-3-5), B5-3-1 (100.8 mg) was subjected to silica gel CC (CH2Cl2–MeOH, from 40:1 to 5:1) to give B5-3-1-1 to B5-3-1-4, B5-3-1-1 (35 mg) was purified by YMC ODS and normal phase semipreparative column to afford 2 (12.0 mg) and 3 (12.0 mg). Fr. B6 (eluted with MeOH–H2O, 60:40) was chromatographed on a Sephadex LH-20 column (MeOH) to give three subfractions B6-1–B6-3. B6-2 was separated by silica gel CC (CH2Cl2–MeOH, from 40:1 to 5:1) to give six portion (B6-2-1–B6-2-6), B6-2-4 (485 mg) was further purified by silica gel CC (CH2Cl2–MeOH, 30:1) to afford 7/8 (26 mg). Fr. B7 (a mixture eluted with MeOH–H2O 70:30 and 80:20) was chromatographed on a Sephadex LH-20 column (MeOH) to give four subfractions (B7-1–B7-4). B7-2 was chromatographed on silica gel column eluting with CH2Cl2–MeOH (from 40:1 to 3:1) to afford six portion (B7-2-1–B7-2-6). B7-2-3 (217 mg) was purified by semipreparative HPLC twice (MeOH–H2O, 56:44, and MeCN–H2O, 40:60) to furnish 5/6 (25 mg). Fraction C was subjected to MCI gel CC (eluted with MeOH–H2O) to remove chlorophyll, and the residue (19.0 g) was partitioned by RP-C18 CC (gradient elution of MeOH–H2O, 15:85–80:20) to 9 subfractions (C1–C9). Fr. C5 was chromatographed on a Sephadex LH-20 column (MeOH) to give four subfractions (C5-1–C5-4). C5-2 was separated on silica gel CC (CH2Cl2–MeOH, from 25:1 to 3:1) to give C5-2-1 to C5-2-4. C5-2-4 (530 mg) was further eluted by silica gel CC (CH2Cl2-MeOH, from 20:1 to 2:1) to give seven subfractions (C5-2-4-1–C5-2-4-7). C5-2-4-2 (88 mg) was purified by semipreparative HPLC (MeCN–H2O, 30:70) to yield 4 (9.5 mg).
Part II: Chiral resolution of 1 (6.7 mg) was performed on Daicel chiral-pak IA column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 70:30:0.1; flow rate 1.0 mL/min, column temperature 27 °C) to give 1a (1.7 mg, tR 8.3 min) and 1b (4.8 mg, tR 11.4 min). 2 (12 mg) was separated by Daicel chiral-pak ASH column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 80:20:0.05; flow rate 1.0 mL/min, column temperature 25 °C) to give 2a (5.3 mg, tR 12.2 min) and 2b (5.3 mg, tR 22.3 min). 3 (12 mg) was also purified by Daicel chiral-pak ASH column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 110:10:0.05; flow rate 1.0 mL/min, column temperature 25 °C) to give 3a (4.5 mg, tR 17.8 min) and 3b (4.1 mg, tR 19.4 min). 4 (9.5 mg) was eluted by Daicel chiral-pak ASH column (n-hexane–EtOH–HCOOH, v/v/v, 100:20:0.05; flow rate 1.0 mL/min, column temperature 26 °C) to give 4a (1.5 mg, tR 8.5 min) and 4b (7.5 mg, tR 15.1 min). 5 and 6 were gained together from one peak on the semipreparative HPLC, and had been divided into four peaks on Daicel chiral-pak ASH column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 80:20:0.05, flow rate 1.0 mL/min, column temperature 26 °C). As a result, we got 5a (4.0 mg, tR 35.9 min), 5b (6.6 mg, tR 13.3 min), 6a (8.5 mg, tR 18.4 min), 6b (4.6 mg, tR 31.5 min). 7 and 8 were obtained together from one peak on the semipreparative HPLC as a mixture named A, A was partitioned to A1 (5.0 mg, tR 17 min) and A2 (6.5 mg, tR 20 min) by Daicel chiral-pak IC column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 60:40:0.1; flow rate 1.0 mL/min, column temperature 27 °C), A1 then purified by Daicel chiral-pak ASH column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 60:40:0.05; flow rate 1.0 mL/min, column temperature 26 °C) and gave 7a (2.5 mg, tR 7.7 min) and 8a (2.0 mg, tR 12.8 min), A2 was further purified on Daicel chiral-pak IA column (eluted with n-hexane–EtOH–HCOOH, v/v/v, 70:30:0.05; flow rate 1.0 mL/min, column temperature 27 °C) to furnish 7b (2.7 mg, tR 12.6 min) and 8b (2.4 mg, tR 15.6 min).
Phyllanglaucin A (1)
colorless gum, 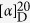 − 6.9 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 211.0 (4.65), 230.5 (4.24), 282.5 (3.62) nm; IR vmax 3418, 2938, 1614, 1518, 1461, 1380, 1338, 1218, 1115, 1037, 958 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 399.1407 [M + Na]+ (calcd for C20H24O7Na, 399.1414).
− 6.9 (c 0.28, MeOH); UV (MeOH) λmax (log ε) 211.0 (4.65), 230.5 (4.24), 282.5 (3.62) nm; IR vmax 3418, 2938, 1614, 1518, 1461, 1380, 1338, 1218, 1115, 1037, 958 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 399.1407 [M + Na]+ (calcd for C20H24O7Na, 399.1414).
(+)-Phyllanglaucin A (1a)
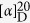 + 12.8 (c 0.065, MeOH); ECD (MeOH) λmax (Δε) 205 (+2.57), 293 (−0.45) nm.
+ 12.8 (c 0.065, MeOH); ECD (MeOH) λmax (Δε) 205 (+2.57), 293 (−0.45) nm.
(−)-Phyllanglaucin A (1b)
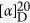 − 14.1 (c 0.19, MeOH); ECD (MeOH) λmax (Δε) 207 (−3.88), 292 (+0.57) nm.
− 14.1 (c 0.19, MeOH); ECD (MeOH) λmax (Δε) 207 (−3.88), 292 (+0.57) nm.
4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol (2)
colorless gum, 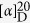 0 (c 0.40, MeOH); UV (MeOH) λmax (log ε) 206.0 (4.47), 228.5 (4.02), 280.5 (3.69) nm; IR vmax 3366, 2937, 2840, 1606, 1518, 1458, 1432, 1372, 1274, 1220, 1154, 1125, 1096, 1073, 1031 956, 856, 819 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 357.1321 [M + Na]+ (calcd for C21H26O9Na, 357.1309).
0 (c 0.40, MeOH); UV (MeOH) λmax (log ε) 206.0 (4.47), 228.5 (4.02), 280.5 (3.69) nm; IR vmax 3366, 2937, 2840, 1606, 1518, 1458, 1432, 1372, 1274, 1220, 1154, 1125, 1096, 1073, 1031 956, 856, 819 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 357.1321 [M + Na]+ (calcd for C21H26O9Na, 357.1309).
(7R,8R)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol (2a)
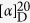 + 30.0 (c 0.18, MeOH); ECD (MeOH) λmax (Δε) 206 (+1.77), 237 (+0.35), 285 (+0.23) nm.
+ 30.0 (c 0.18, MeOH); ECD (MeOH) λmax (Δε) 206 (+1.77), 237 (+0.35), 285 (+0.23) nm.
(7S,8S)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol (2b)
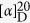 − 30.8 (c 0.22, MeOH); ECD (MeOH) λmax (Δε) 206 (−2.57), 237 (−0.48), 287 (−0.28) nm.
− 30.8 (c 0.22, MeOH); ECD (MeOH) λmax (Δε) 206 (−2.57), 237 (−0.48), 287 (−0.28) nm.
4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol (3)
colorless gum, 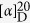 0 (c 0.40, MeOH); UV (MeOH) λmax (log ε) 207.5 (4.47), 230.0 (4.09), 280.5 (3.72) nm; IR vmax 3353, 2934, 2848, 1605, 1518, 1458, 1432, 1371, 1274, 1218, 1154, 1126, 1099, 1073, 1031 953, 860, 820 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 357.1323 [M + Na]+ (calcd for C21H26O9Na, 357.1309).
0 (c 0.40, MeOH); UV (MeOH) λmax (log ε) 207.5 (4.47), 230.0 (4.09), 280.5 (3.72) nm; IR vmax 3353, 2934, 2848, 1605, 1518, 1458, 1432, 1371, 1274, 1218, 1154, 1126, 1099, 1073, 1031 953, 860, 820 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 357.1323 [M + Na]+ (calcd for C21H26O9Na, 357.1309).
(7S,8R)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol (3a)
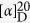 + 79.5 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 208 (+12.52), 235 (+4.80), 282 (+0.84) nm.
+ 79.5 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 208 (+12.52), 235 (+4.80), 282 (+0.84) nm.
(7R,8S)-4,4′-dihydroxy-3,7,3′-trimethoxy-8,1′-7′,8′,9′-trinor-neolignan-9-ol (3b)
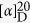 − 76.9 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 209 (−12.97), 235 (−5.81), 283 (−0.92) nm.
− 76.9 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 209 (−12.97), 235 (−5.81), 283 (−0.92) nm.
Phyllanglaucin B (4)
colorless gum, 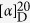 − 42.1 (c 0.34, MeOH); UV (MeOH) λmax (log ε) 206.5 (4.76), 231.5 (4.30), 283.0 (3.92) nm; IR vmax 3339, 2932, 1654, 1603, 1515, 1496, 1464, 1374, 1271, 1207, 1123, 1066, 1033, 951, 853 cm−1; 1H and 13C NMR data see Table 2 and 3. HRESIMS: m/z 561.2089 [M + Na]+ (calcd for C21H26O9Na, 561.2101).
− 42.1 (c 0.34, MeOH); UV (MeOH) λmax (log ε) 206.5 (4.76), 231.5 (4.30), 283.0 (3.92) nm; IR vmax 3339, 2932, 1654, 1603, 1515, 1496, 1464, 1374, 1271, 1207, 1123, 1066, 1033, 951, 853 cm−1; 1H and 13C NMR data see Table 2 and 3. HRESIMS: m/z 561.2089 [M + Na]+ (calcd for C21H26O9Na, 561.2101).
(+)-Phyllanglaucin B (4a)
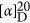 + 62.6 (c 0.06, MeOH); ECD (MeOH) λmax (Δε) 207 (+27.09), 242 (+6.72), 275 (+3.07), 299(+1.29) nm.
+ 62.6 (c 0.06, MeOH); ECD (MeOH) λmax (Δε) 207 (+27.09), 242 (+6.72), 275 (+3.07), 299(+1.29) nm.
(−)-Phyllanglaucin B (4b)
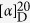 − 64.8 (c 0.25, MeOH); ECD (MeOH) λmax (Δε) 208 (−30.83), 242 (−8.04), 277 (−3.53), 298 (−1.49) nm.
− 64.8 (c 0.25, MeOH); ECD (MeOH) λmax (Δε) 208 (−30.83), 242 (−8.04), 277 (−3.53), 298 (−1.49) nm.
Phyllanglaucin C (5)
colorless gum, UV (MeOH) λmax (log ε) 210.0 (4.70), 230.0 (4.27), 280.5 (3.69) nm; IR vmax 3367, 2939, 2877, 2838, 1677, 1598, 1502, 1460, 1425, 1329, 1277, 1235, 1215, 1125, 1091, 1032, 955, 830 cm−1; 1H and 13C NMR data see Table 2 and 3; HRESIMS: m/z 623.2456 [M + Na]+ (calcd for C21H26O9Na, 623.2468).
(+)-Phyllanglaucin C (5a)
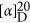 + 7.5 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 209 (+8.91), 238 (−2.06), 294 (−0.65) nm.
+ 7.5 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 209 (+8.91), 238 (−2.06), 294 (−0.65) nm.
(−)-Phyllanglaucin C (5b)
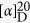 − 7.4 (c 0.24, MeOH); ECD (MeCN) λmax (Δε) 207 (−8.40), 237 (+2.84), 295 (+0.79) nm.
− 7.4 (c 0.24, MeOH); ECD (MeCN) λmax (Δε) 207 (−8.40), 237 (+2.84), 295 (+0.79) nm.
Phyllanglaucin D (6)
colorless gum, UV (MeOH) λmax (log ε) 211.0 (4.60), 230.0 (4.22), 280.5 (3.64) nm; IR vmax 3348, 2937, 2874, 2838, 1598, 1502, 1460, 1424, 1329, 1277, 1235, 1215, 1125, 1090, 1033, 955, 830 cm−1; 1H and 13C NMR data see Table 2 and 3; HRESIMS: m/z 623.2461 [M + Na]+ (calcd for C21H26O9Na, 623.2468).
(+)-Phyllanglaucin D (6a)
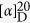 + 14.4 (c 0.24, MeOH); ECD (MeCN) λmax (Δε) 207 (+11.80), 230 (−1.48), 282 (+1.41) nm.
+ 14.4 (c 0.24, MeOH); ECD (MeCN) λmax (Δε) 207 (+11.80), 230 (−1.48), 282 (+1.41) nm.
(−)-Phyllanglaucin D (6b)
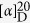 − 13.0 (c 0.18, MeOH); ECD (MeCN) λmax (Δε) 209 (−8.66), 229 (+1.04), 285 (−1.11) nm.
− 13.0 (c 0.18, MeOH); ECD (MeCN) λmax (Δε) 209 (−8.66), 229 (+1.04), 285 (−1.11) nm.
acernikol (7)
colorless gum, UV (MeOH) λmax (log ε) 211.0 (4.71), 230.0 (4.28), 280.5 (3.72) nm; IR vmax 3366, 2937, 2873, 1599, 1501, 1460, 1424, 1382, 1328, 1277, 1236, 1214, 1124, 1031, 953, 831 cm−1; 1H and 13C NMR data see Table 2 and 3; HRESIMS: m/z 609.2294 [M + Na]+ (calcd for C21H26O9Na, 609.2312).
(+)-(7R,8S,7′R,8′R)-acernikol (7a)
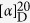 + 22.4 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 206 (+15.48), 244 (+2.32), 294 (−0.79) nm.
+ 22.4 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 206 (+15.48), 244 (+2.32), 294 (−0.79) nm.
(−)-(7S,8R,7′S,8′S)-acernikol (7b)
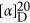 − 20.0 (c 0.12, MeOH); ECD (MeOH) λmax (Δε) 207 (−11.94), 244 (−2.18), 294 (+0.76) nm.
− 20.0 (c 0.12, MeOH); ECD (MeOH) λmax (Δε) 207 (−11.94), 244 (−2.18), 294 (+0.76) nm.
acernikol (8)
colorless gum, UV (MeOH) λmax (log ε) 211.5 (4.72), 230.0 (4.33), 281.0 (3.74) nm; IR vmax 3356, 2923, 2852, 1599, 1501, 1462, 1425, 1382, 1328, 1277, 1236, 1214, 1124, 1030, 954, 830 cm−1; 1H and 13C NMR data see Table 1; HRESIMS: m/z 609.2297 [M + Na]+ (calcd for C21H26O9Na, 609.2312).
(−)-(7R,8S,7′S,8′S)-acernikol (8a)
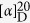 − 10.2 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 207 (−11.60), 243 (+6.51), 281 (+1.76) nm.
− 10.2 (c 0.13, MeOH); ECD (MeOH) λmax (Δε) 207 (−11.60), 243 (+6.51), 281 (+1.76) nm.
(+)-(7S,8R,7′R,8′R)-acernikol (8b)
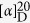 + 10.7 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 206 (+2.98), 243 (−3.76), 281 (−1.32) nm.
+ 10.7 (c 0.11, MeOH); ECD (MeOH) λmax (Δε) 206 (+2.98), 243 (−3.76), 281 (−1.32) nm.
ECD Calculations
Conformational search using molecular mechanics calculations was performed in Discovery Studio 3.5 Client with MMFF force field with 20 kcal/mol upper energy limit51, and nine low energy conformations were chosen as predominant conformers from all the conformations produced by the change of dihedral of the middle four carbons C1-C7-C8-C1′ of 7R,8R-2. The nine predominant conformers were optimized at the B3LYP/6-311++G(2d,p) level in MeOH using the IEFPCM solvation model. The optimized geometries and thermodynamic parameters of all conformations were provided in supplementary information. The calculations were performed using Gaussian 0952, and figured using GaussView 5.053.
The theoretical calculation of ECD was performed using time dependent Density Functional Theory (TDDFT) at LC-wPBE/6-311++G(2d,p)//B3LYP/6-311++G(2d,p) level in MeOH with IEFPCM solvation model. The ECD spectra of compound 2a were obtained by weighting the Boltzmann distribution ratio of each geometric conformation54. The ECD spectra were simulated by overlapping Gaussian functions for each transition according to:
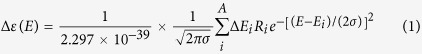 |
The σ represented the width of the band at 1/e height, and ΔEi and Ri were the excitation energies and rotational strengths for transition i, respectively.
Additional ECD calculations were performed with two other functionals, i.e., CAM-B3LYP and WB97XD, to further confirm our calculated results.
DPPH Radical Scavenging Activity
The DPPH radical assay was carried out as previously described with minor modifications50. Each sample (final concentrations of 3.125, 6.25, 12.5, 25, and 50 μg/mL) was mixed with DPPH (final concentration of 100 μM) EtOH (with 5% DMSO) solution in 96-well microplate. The mixtures were deposited in the dark at room temperature for 1 h, and then the absorbance was measured at 515 nm using a microplate reader. An EtOH (with 5% DMSO) was used as negative control, and the positive control was Trolox (10 mM, sigma). All tests were performed triplicate, and the results were averaged. The final results were reported as IC50, a concentration of a sample that scavenged 50% DPPH free radicals in the reaction solution.
Additional Information
How to cite this article: Wu, Z. et al. Enantiomeric Lignans and Neolignans from Phyllanthus glaucus: Enantioseparation and Their Absolute Configurations. Sci. Rep. 6, 24809; doi: 10.1038/srep24809 (2016).
Supplementary Material
Acknowledgments
We thank Prof. Yunhe He of Zhejiang A&F University for identification of the plant. We also thank Prof. Junjun Liu of School of Pharmacy, Tongji Medical College, Huazhong University of Science and Technology, for kindly discussing and analyzing ECD calculation. We greatly acknowledge the financial supports from the National Natural Science Foundation of China (Nos 31200258, 31370372, and 81202423), the New Century Excellent Talents in University, State Education Ministry of China (NCET-2008-0224), and National Science and Technology Project of China (No. 2011ZX09102-004).
Footnotes
Author Contributions Z.W. and Y.L. contributed equally to this work. They conducted the main experiments, data analyzes, and wrote the manuscript; L.Z. and Y.W. assisted the chiral HPLC experiments; J.Y. performed the ECD calculations; H.Z. and J.Z. analyzed the spectroscopic data; Z.H., Z.L. and J.W. revised and polished this manuscript; Y.Z. and Y.X. designed the experiments and commented the manuscript. All authors reviewed the manuscript.
References
- Suzuki S. & Umezawa T. Biosynthesis of lignans and norlignans. J. Wood Sci. 53, 273–284 (2007). [Google Scholar]
- Pan J. Y. et al. An update on lignans: natural products and synthesis. Nat. Prod. Rep. 26, 1251–1292 (2009). [DOI] [PubMed] [Google Scholar]
- Pereira A. C. et al. Schistosomicidal and trypanocidal structure-activity relationships for (±)-licarin A and its (−)-and (+)-enantiomers. Phytochemistry. 72, 1424–1430 (2011). [DOI] [PubMed] [Google Scholar]
- Silva R. D. et al. Trypanocidal structure-activity relationship for cis- and trans-methylpluviatolide. Phytochemistry. 69, 1890–1894 (2008). [DOI] [PubMed] [Google Scholar]
- Lai Y. J. et al. Neolignans with a rare 2-oxaspiro[4.5]deca-6,9-dien-8-one motif from the stem bark of Cinnamomum subavenium. J. Nat. Prod. 78, 1740–1744 (2015). [DOI] [PubMed] [Google Scholar]
- Shi Y. S. et al. Chiral resolution and absolute configuration of a pair of rare racemic spirodienone sesquineolignans from Xanthium sibiricum. Org. Lett. 16, 5406–5409 (2014). [DOI] [PubMed] [Google Scholar]
- Cheng Z. B. et al. (±)-Torreyunlignans A–D, rare 8–9′ linked neolignan enantiomers as phosphodiesterase-9A inhibitors from Torreya yunnanensis. J. Nat. Prod. 77, 2651–2657 (2014). [DOI] [PubMed] [Google Scholar]
- Yang D. T. et al. (+)- and (−)-liriodenol, a pair of novel enantiomeric lignans from Liriodendron hybrid. Bioorg. Med. Chem. Lett. 25, 1976–1978 (2015). [DOI] [PubMed] [Google Scholar]
- Qi W. Y., Hua L. & Gao K. Chemical constituents of the plants from the genus Phyllanthus. Chem. Biodivers. 11, 364–395 (2014). [DOI] [PubMed] [Google Scholar]
- Lv J. J. et al. Stereochemistry of cleistanthane diterpenoid glucosides from Phyllanthus emblica. RSC. Adv. 5, 29098–29107 (2015). [Google Scholar]
- Fan Y. Y. et al. Phainanoids A–F, A new class of potent immunosuppressive triterpenoids with an unprecedented carbon skeleton from Phyllanthus hainanensis. J. Am. Chem. Soc. 137, 138–141 (2014). [DOI] [PubMed] [Google Scholar]
- Lv J. J. et al. Anti-hepatitis B virus norbisabolane sesquiterpenoids from Phyllanthus acidus and the establishment of their absolute configurations using theoretical calculations. J. Org. Chem. 79, 5432–5447 (2014). [DOI] [PubMed] [Google Scholar]
- Zhao J. Q. et al. Phyllanflexoid C: first example of phenylacetylene-bearing 18-nor-diterpenoid glycoside from the roots of Phyllanthus flexuosus. Tetrahedron Lett. 54, 4670–4674 (2013). [Google Scholar]
- Ren Y. L. et al. Potent cytotoxic arylnaphthalene lignan lactones from Phyllanthus poilanei. J. Nat. Prod. 77, 1494–1504 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanh N. V. et al. A new flavone sulfonic acid from Phyllanthus urinaria. Phytochem. Lett. 7, 182–185 (2014). [Google Scholar]
- Mensah J. L., Gleye J., Moulis C. & Fouraste I. Alkaloids from the leaves of Phyllanthus discoideus. J. Nat. Prod. 51, 1113–1115 (1988). [Google Scholar]
- Zhang Y. J., Abe T., Tanaka T., Yang C. R. & Kouno I. Phyllanemblinins A–F, new ellagitannins from Phyllanthus emblica. J. Nat. Prod. 64, 1527–1532 (2001). [DOI] [PubMed] [Google Scholar]
- Hu Z. X. et al. Phytochemical and chemotaxonomic studies on Phyllanthus urinaria. Biochem. Syst. Ecol. 56, 60–64 (2014). [Google Scholar]
- Zhu H. C. et al. Bioactive acylphloroglucinols with adamantyl skeleton from Hypericum sampsonii. Org. Lett. 16, 6322–6325 (2014). [DOI] [PubMed] [Google Scholar]
- Lai Y. J. et al. Scapiformolactones A–I: germacrane sesquiterpenoids with an unusual Δ3-15,6-lactone moiety from Salvia scapiformis. Phytochemistry. 96, 378–388 (2013). [DOI] [PubMed] [Google Scholar]
- Yu S. et al. New cytotoxic lignan glycosides from Phyllanthus glaucus. Nat. Prod. Res. 30, 419–425 (2016). [DOI] [PubMed] [Google Scholar]
- Agrawal P. K., Agarwal S. K. & Rastogi R. P. A new neolignan and other phenolic constituents from Cedrus deodara. Phytochemistry. 19, 1260–1261 (1980). [Google Scholar]
- Liu Q. B. et al. Antioxidant and anti-inflammatory active dihydrobenzofuran neolignans from the seeds of Prunus tomentosa. J. Agric. Food Chem. 62, 7796–7803 (2014). [DOI] [PubMed] [Google Scholar]
- Antus S. et al. Chiroptical properties of 2,3-dihydrobenzo[b]furan and chromane chromophores in naturally occurring O-heterocycles. Chirality. 13, 493–506 (2001). [DOI] [PubMed] [Google Scholar]
- Hsiao J. J. & Chiang H. C. Lignans from the wood of Aralia bipinnata. Phytochemistry. 39, 899–902 (1995). [Google Scholar]
- Zhu J. X. et al. Phenylpropanoids and lignanoids from Euonymus acanthocarpus. Arch. Pharm. Res. 35, 1739–1747 (2012). [DOI] [PubMed] [Google Scholar]
- Zhou C. C. et al. Two new compounds from Crataegus pinnatifida and their antithrombotic activities. J. Asian Nat. Products Res. 16, 169–174 (2014). [DOI] [PubMed] [Google Scholar]
- Zeng Q. et al. Chemical constituents from Metasequoia glyptostroboides Hu et Cheng. Biochem. Syst. Ecol. 50, 406–410 (2013). [Google Scholar]
- Liu Y. B. et al. Chemical constituents of Toona ciliata var. pubescens. Chin. J. Nat. Med. 9, 115–119 (2011). [Google Scholar]
- Rayanil K., Nimnoun C. & Tuntiwachwuttikul P. New phenolics from the wood of Casearia grewiifolia. Phytochem. Lett. 5, 59–62 (2012). [Google Scholar]
- Yoshikawa K., Mimura N. & Arihara S. Isolation and absolute structures of enantiomeric 1,2-bis(4-hydroxy-3-methoxyphenyl)-1,3-propanediol 1-O-glucosides from the bark of Hovenia trichocarpa. J. Nat. Prod. 61, 1137–1139 (1998). [DOI] [PubMed] [Google Scholar]
- Wu W., Bi X. L., Cao J. Q., Zhang K. Q. & Zhao Y. Q. New antitumor compounds from Carya cathayensis. Bioorg. Med. Chem. Lett. 22, 1895–1898 (2012). [DOI] [PubMed] [Google Scholar]
- Crabbé P. & Klyne W. Optical rotatory dispersion and circular dichroism or aromatic compounds: a general survey. Tetrahedron. 23, 3449–3503 (1967). [Google Scholar]
- Yoshinari K., Shimazaki N., Sashida Y. & Mimaki Y. Flavanone xyloside and lignans from Prunus jamasakura bark. Phytochemistry. 29, 1675–1678 (1990). [Google Scholar]
- Xiong L. et al. Lignans and neolignans from Sinocalamus affinis and their absolute configurations. J. Nat. Prod. 74, 1188–1200 (2011). [DOI] [PubMed] [Google Scholar]
- Morikawa T., Tao J., Ueda K., Matsuda H. & Yoshikawa M. Medicinal foodstuffs. XXXI. Structures of new aromatic constituents and inhibitors of degranulation in RBL-2H3 cells from a Japanese folk medicine, the stem bark of Acer nikoense. Chem. Pharm. Bull. 51, 62–67 (2003). [DOI] [PubMed] [Google Scholar]
- Huo C. H., Liang H., Zhao Y. Y., Wang B. & Zhang Q. Y. Neolignan glycosides from Symplocos caudata. Phytochemistry. 69, 788–795 (2008). [DOI] [PubMed] [Google Scholar]
- Fang L. et al. Neolignans and glycosides from the stem bark of Illicium difengpi. J. Nat. Prod. 73, 818–824 (2010). [DOI] [PubMed] [Google Scholar]
- Hanawa F., Shiro M. & Hayashi Y. Heartwood constituents of Betula maximowicziana. Phytochemistry. 45, 589–595 (1997). [Google Scholar]
- Shi S. P., Jiang D., Dong C. X. & Tu P. F. Lignans from the roots and rhizomes of Clematis manshurica. Zeitschrift für Naturforschung B. 61, 1299–1303 (2006). [Google Scholar]
- Liu J. F. et al. Two new lignans and anti-HBV constituents from Illicium henryi. Chem. Biodivers. 8, 692–698 (2011). [DOI] [PubMed] [Google Scholar]
- Liu Q. B. et al. Neolignans from the seeds of Prunus tomentosa (Rosaceae) and their chemotaxonomic interest. Biochem. Syst. Ecol. 55, 236–240 (2014). [Google Scholar]
- Auten R. L. & Davis J. M. Oxygen toxicity and reactive oxygen species: the devil is in the details. Pediatr. Res. 66, 121–127 (2009). [DOI] [PubMed] [Google Scholar]
- Coombes J. S. & Fassett R. G. Antioxidant therapy in hemodialysis patients: a systematic review. Kidney Int. 81, 233–246 (2012). [DOI] [PubMed] [Google Scholar]
- Perl A. Oxidative stress in the pathology and treatment of systemic lupus erythematosus. Nat. Rev. Rheumatol. 9, 674–686 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller J., Fuchs B., Arnhold J. & Arnold K. Contribution of reactive oxygen species to cartilage degradation in rheumatic diseases: molecular pathways, diagnosis and potential therapeutic strategies. Curr. Med. Chem. 10, 2123–2145 (2003). [DOI] [PubMed] [Google Scholar]
- Miller M. W. & Sadeh N. Traumatic stress, oxidative stress and post-traumatic stress disorder: neurodegeneration and the accelerated-aging hypothesis. Mol. Psychiatr. 19, 1156–1162 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorrini C., Harris I. S. & Mak T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947 (2013). [DOI] [PubMed] [Google Scholar]
- Saleem M., Kim H. J., Ali M. S. & Lee Y. S. An update on bioactive plant lignans. Nat. Prod. Rep. 22, 696–716 (2005). [DOI] [PubMed] [Google Scholar]
- Krishnaiah D., Sarbatly R. & Nithyanandam R. A review of the antioxidant potential of medicinal plant species. Food Bioprod. Process. 89, 217–233 (2011). [Google Scholar]
- Smith S. G. & Goodman J. M. Assigning stereochemistry to single diastereoisomers by GIAO NMR calculation: The DP4 probability. J. Am.Chem. Soc. 132, 12946–12959 (2010). [DOI] [PubMed] [Google Scholar]
- Frisch M. J. et al. Gaussian 09. Gaussian Inc. : Wallingford, CT, 2010. [Google Scholar]
- Dennington R., Keith T. & Millam J. GaussView, Version 5.0. Semichem Inc.: Shawnee Mission, KS, 2009
- Tähtinen P., Bagno A., Klika K. D. & Pihlaja K. Modeling NMR parameters by DFT methods as an aid to the conformational analysis of cis-fused 7a(8a)-methyl octa(hexa)hydrocyclopenta [d][1,3] oxazines and [3,1] benzoxazines. J. Am. Chem. Soc. 125, 4609–4618 (2003). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.