

Abstract
Fusobacteria are associated with colorectal cancer (CRC) and are amplified during colorectal carcinogenesis. Compared to the adenoma-carcinoma sequence of carcinogenesis, serrated neoplasm has distinct clinical features and a different molecular background. We aimed to compare the gut microbiome between tubular adenoma (TA) and sessile serrated adenoma/polyp (SSA/P). Patients with TA, SSA/P, or CRC were recruited. Three pieces of colorectal mucosal tissue were obtained from each patient by endoscopic biopsy. 16S rRNA gene pyrosequencing and phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt) were performed. Among 26 enrolled patients, 8, 10, and 8 had TA, SSA/P, and CRC, respectively. The relative abundance of Fusobacteria did not differ significantly between the TA and SSA/P groups (4.3% and 1.9%, P = 0.739) but was higher in the CRC group (33.8%) than in the TA or SSA/P group, respectively (TA vs. CRC, P = 0.002, false discovery rate [FDR] = 0.023; SSA/P vs. CRC, P < 0.001, FDR = 0.001). PICRUSt revealed that most functions in the TA metagenome were similar to those in the SSA/P metagenome. The gut microbiome, including relative abundance of Fusobacteria, did not differ between TA and SSA/P, suggesting that Fusobacteria may contribute to both the serrated pathway and the adenoma-carcinoma sequence.
Most colorectal cancers (CRCs) are preceded by dysplastic adenomas that can progress to malignancy, a process known as the adenoma-carcinoma sequence1. Accumulation of genetic changes, including loss of the tumor suppressor gene adenomatous polyposis coli (APC) followed by activating and inactivating mutations in KRAS, PIK3CA, and TP532, is usually required for the development of CRC. Additionally, it has been suggested that an altered gut microbiome may affect the development of CRC through interaction with the innate immune system and other host factors3,4,5. Fusobacterium species are in the spotlight in the development of CRC because many studies have shown that Fusobacterium is abundant in CRC tissue compared to adjacent normal tissue using next-generation sequencing6,7,8. In addition, Kostic et al. demonstrated that the F. nucleatum induced development of colonic tumor in APCMin/+mice. These results imply that Fusobacteria can affect the early stage of the adenoma-carcinoma sequence, such as APC mutated adenoma.
In addition to TA, serrated lesions are precursors of CRCs that exhibit hypermethylation and may account for one-third of all CRCs9. Sessile serrated adenoma/polyp (SSA/P) has distinct endoscopic features including a mucus cap, a color that is usually similar to normal mucosa, and indistinct edges9. Colorectal carcinogenesis of SSA/P is regarded as the results of epigenetic alterations in genes involving cellular proliferation and differentiation, caused by hypermethylation of promoter DNA, in addition to genetic mutations including BRAF9. The gut microbiome may also contribute to the serrated polyp-carcinoma sequence as well as the adenoma-carcinoma sequence because DNA methylation is closely related to the gut microbiome10,11. Nevertheless, the role of Fusobacteria in the serrated polyp-carcinoma sequence has not been fully evaluated. To identify whether microbial composition, including Fusobacteria, is different between tubular adenoma (TA) and SSA/P, we planned to compare the gut microbiome between those two types of CRC precursor. If Fusobacteria is also associated with the serrated polyp-carcinoma sequence, the microbial composition of SSA/P, including relative abundance of Fusobacteria, may be similar to that of TA. In this study we compared the gut microbiome among three different diseases—TA, SSA/P, and CRC—using 16S rRNA gene pyrosequencing.
Results
Baseline characteristics and microbiome reads
Table 1 shows baseline patient characteristics and microbiome reads. Among 26 enrolled patients, 8 had CRC, 8 had TA, and 10 had SSA/P. Median age was 68, 54, and 63 years in the TA, SSA/P, and CRC group, respectively (P = 0.119). The proportion of males did not differ significantly among the groups (P = 0.244). Five (62.5%) TAs and 7 (70.0%) SSA/Ps were in the proximal colon, whereas all lesions in the CRC group were in the distal colon.
Table 1. Baseline characteristics and microbiome reads.
| Variable | TA | SSA/P | CRC | P-value |
|---|---|---|---|---|
| n | 8 | 10 | 8 | |
| Age, year, median (IQR) | 68 (55–74) | 54 (49–66) | 63 (60–76) | 0.119 |
| Male, n (%) | 6 (75.0) | 5 (50.0) | 7 (87.5) | 0.244 |
| Tumor location, n (%) | 0.006 | |||
| Proximal colon | 5 (62.5) | 7 (70.0) | 0 (0.0) | |
| Distal colon | 3 (37.5) | 3 (30.0) | 8 (100.0) | |
| Read count, median (IQR) | 5,540 (3,150–6,875) | 6,162 (4,483–7,394) | 4,969 (1,642–6,132) | 0.247 |
| OTU, median (IQR) | 45.0 (33.3–71.5) | 44.0 (28.0–74.5) | 55.0 (33.8–80.3) | 0.847 |
| Chao1 estimator, median (IQR) | 46.8 (36.9–88.5) | 48.6 (30.8–81.0) | 65.4 (44.8–88.8) | 0.551 |
| Shannon’s diversity index, median (IQR) | 3.22 (2.89–3.75) | 3.51 (2.55–4.00) | 3.56 (2.52–4.21) | 0.912 |
| Simpson’s diversity index, median (IQR) | 0.82 (0.78–0.86) | 0.84 (0.70–0.90) | 0.84 (0.69–0.91) | 0.872 |
TA, tubular adenoma; SSA/P, sessile serrated adenoma/polyp; CRC, colorectal cancer; OTU, operational taxonomic unit; IQR, interquartile rnage.
A total of 136,414 read were obtained from 26 enrolled patients through 16S rRNA gene pyrosequencing. The median number of reads was 5,540, 6,162 and 4,969 in TA, SSA/P, and CRC groups, respectively (P = 0.247). Median operational taxonomic units (OTUs), Chao1 estimator, and Shannon’s and Simpson’s diversity index did not differ among the groups.
Relative bacterial abundance
Figure 1A shows relative bacterial abundance at the phylum level for each group. The five most abundant phyla were Fusobacteria, Firmicutes, Proteobacteria, Bacteroidetes, and Actinobacteria in all three groups. Relative abundance of Fusobacteria did not differ between the TA and SSA/P group (4.3% and 1.9%, P = 0.739), but was significantly higher in the CRC group (33.8%) than in the TA or SSA/P group, respectively (Table 2; TA vs. CRC, P = 0.002, FDR = 0.023; SSA/P vs. CRC, P < 0.001, FDR = 0.001).
Figure 1.

Relative bacterial abundance at the phylum level for all lesions (A) and only lesions located in the distal colon (B).
Table 2. Relative bacterial abundance of gastric mucosa at the phylum level according to histology.
| TA | SSA/P | CRC | TA vs. SSA/P | TA vs. CRC | SSA/P vs. CRC | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | SD | Mean | SD | Mean | SD | P-value | FDR | P-value | FDR | P-value | FDR | |
| Acidobacteria | 0.10% | 0.31% | 0.01% | 0.04% | 0.00% | 0.00% | 0.747 | >0.999 | >0.999 | 0.867 | >0.999 | >0.999 |
| Armatimonadetes | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | N/A | N/A | N/A | N/A | N/A | N/A |
| Candidate_division_TM7 | 0.02% | 0.07% | 0.00% | 0.00% | 0.00% | 0.00% | 0.409 | >0.999 | >0.999 | 0.867 | N/A | N/A |
| Chlorobi | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | >0.999 | >0.999 | >0.999 | 0.867 | >0.999 | >0.999 |
| Chloroflexi | 0.00% | 0.00% | 1.54% | 5.56% | 0.00% | 0.00% | >0.999 | >0.999 | >0.999 | 0.867 | >0.999 | >0.999 |
| Deinococcus-Thermus | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | N/A | N/A | N/A | N/A | N/A | N/A |
| Gemmatimonadetes | 0.15% | 0.44% | 0.31% | 1.13% | 0.00% | 0.00% | >0.999 | >0.999 | >0.999 | 0.867 | >0.999 | >0.999 |
| Lentisphaerae | 0.00% | 0.00% | 0.02% | 0.07% | 0.00% | 0.00% | >0.999 | >0.999 | >0.999 | 0.867 | >0.999 | >0.999 |
| Nitrospirae | 0.00% | 0.00% | 0.08% | 0.27% | 0.00% | 0.00% | >0.999 | >0.999 | >0.999 | 0.867 | >0.999 | >0.999 |
| Synergistetes | 0.25% | 0.74% | 0.07% | 0.24% | 0.00% | 0.00% | 0.443 | >0.999 | 0.471 | 0.867 | >0.999 | >0.999 |
| TM6 | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | N/A | N/A | N/A | N/A | N/A | N/A |
| Tenericutes | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | 0.00% | N/A | N/A | N/A | N/A | N/A | N/A |
| Verrucomicrobia | 0.03% | 0.06% | 0.43% | 1.21% | 0.00% | 0.01% | 0.844 | >0.999 | 0.594 | 0.867 | 0.566 | >0.999 |
| Spirochaetes | 0.00% | 0.00% | 0.00% | 0.00% | 0.13% | 0.35% | >0.999 | >0.999 | 0.206 | 0.759 | 0.133 | 0.654 |
| Other | 1.71% | 1.72% | 0.66% | 0.97% | 0.33% | 0.57% | 0.113 | >0.999 | 0.029 | 0.141 | 0.419 | >0.999 |
| Cyanobacteria | 0.11% | 0.22% | 0.10% | 0.36% | 0.42% | 1.20% | 0.544 | >0.999 | >0.999 | 0.867 | 0.752 | >0.999 |
| Actinobacteria | 2.34% | 2.61% | 2.75% | 3.65% | 1.46% | 2.65% | 0.830 | >0.999 | 0.417 | 0.867 | 0.184 | 0.654 |
| Bacteroidetes | 16.68% | 13.07% | 12.96% | 11.73% | 19.42% | 13.68% | 0.881 | >0.999 | 0.673 | 0.867 | 0.204 | 0.654 |
| Proteobacteria | 51.25% | 26.71% | 52.34% | 27.80% | 22.02% | 16.24% | 0.896 | >0.999 | 0.027 | 0.141 | 0.013 | 0.101 |
| Firmicutes | 23.10% | 18.23% | 26.87% | 20.50% | 22.46% | 13.29% | 0.794 | >0.999 | 0.815 | 0.867 | 0.804 | >0.999 |
| Fusobacteria | 4.26% | 8.24% | 1.85% | 4.33% | 33.75% | 23.17% | 0.739 | >0.999 | 0.002 | 0.023 | <0.001 | 0.001 |
TA, tubular adenoma; SSA/P, sessile serrated adenoma/polyp; CRC, colorectal cancer; FDR, false discovery rate; SD, standard deviation.
Because all lesions of the CRC group were in the distal colon, we performed a subgroup analysis for lesions located in the distal colon. As shown in Fig. 1B, relative abundance of Fusobacteria in the CRC group was higher than that in the TA or SSA/P group. Bacterial abundance at the phylum level was not associated with tumor location.
Relative abundance of Fusobacteria in each tissue
Figure 2 shows relative abundance of Fusobacteria in each tissue. Although the relative abundance of Fusobacteria varied, 37.5% (3 of 8) and 50.0% (5 of 10) of lesions contained Fusobacteria in the TA and SSA/P group, respectively. In the CRC group, Fusobacteria was identified in all lesions. Interestingly, Fusobacteria was identified not only in the lesions but also in adjacent normal tissues. Four (50.0%), three (30.0%), and eight (100.0%) adjacent normal tissues in the TA, SSA/P, and CRC group, respectively, showed the presence of Fusobacteria.
Figure 2. Relative abundance of Fusobacteria in each sample.
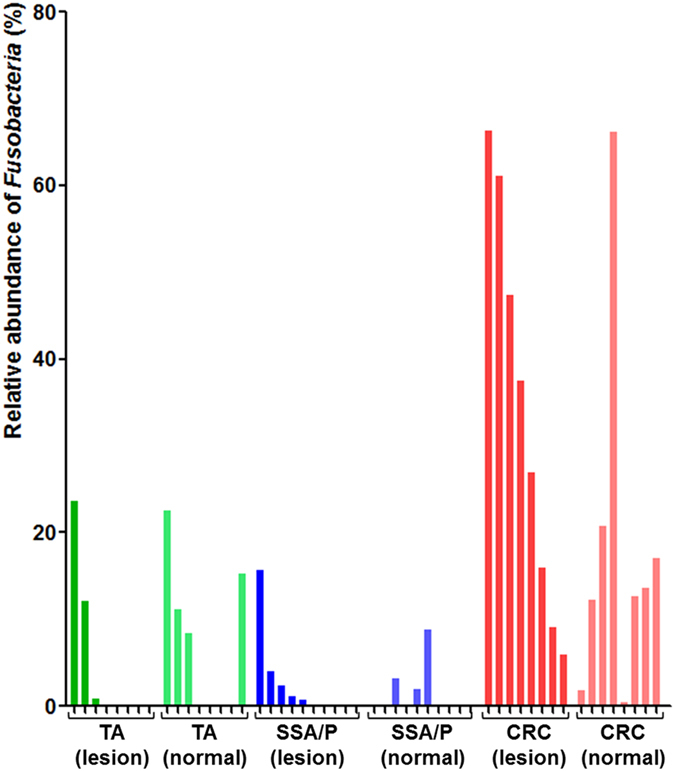
Samples were grouped by disease and biopsy site (lesion vs. adjacent normal tissue). TA, tubular adenoma; SSA/P, sessile serrated adenoma/polyp; CRC, colorectal cancer.
Principal coordinates analysis
PCoA was performed to cluster the communities along axes of maximal variance (Fig. 3). The maximum variations were 35.4% and 18.7% in PCo1 and PCo2, respectively. No difference was observed between TAs and SSA/Ps. Bacterial communities in the TA or SSA/P group showed distinct patterns from those in the CRC group.
Figure 3. Principal coordinate analysis plot.
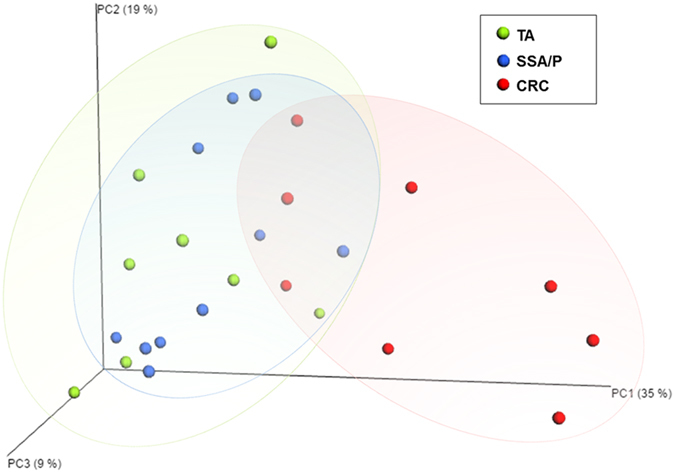
The plots shows the clustering pattern among the TA (green), SSA/P (blue), and CRC (red) groups based on a principal coordinates analysis. TA, tubular adenoma; SSA/P, sessile serrated adenoma/polyp; CRC, colorectal cancer.
Functional composition of the microbiome
After prediction of the microbial metagenome from the results of 16S rRNA gene pyrosequencing using PICRUSt, weighted NSTI was calculated in order to assess the reliability of the predicted metagenome. The predicted metagenome in all three groups showed excellent weighted NSTI (median [IQR]; TA, 0.050 [0.037–0.080]; SSA/P, 0.063 [0.036–0.091]; CRC, 0.063 [0.047–0.079]; P = 0.828)12.
Relative abundance of predicted function in each metagenome was demonstrated according to the level 1 KEGG module (Table 3). Although FDR was not significant, the relative abundance of the cellular processes category was higher in the TA (3.3%) and SSA/P (3.0%) groups than in the CRC group (2.3%) (TA vs. CRC, P = 0.010, FDR = 0.088; SSA/P vs. CRC, P = 0.027, FDR = 0.208). In contrast, the relative abundance of the genetic information processing category tended to be higher in the CRC group (19.4%) than in the TA (17.7%) and SSA/P (17.8%) groups (TA vs. CRC, P = 0.038, FDR = 0.088; SSA/P vs. CRC, P = 0.122, FDR = 0.208).
Table 3. Relative abundance of predicted function for specific KEGG modules (level 1) according to histology.
| KEGG modules Level 1 | Relative abundance for specific KEGG modules (%) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TA |
SSA/P |
CRC |
TA vs. SSA/P | TA vs. CRC | SSA/P vs. CRC | ||||||||
| Mean | SD | Mean | SD | Mean | SD | P-value | FDR | P-value | FDR | P-value | FDR | ||
| Cellular Processes | 3.30 | 0.74 | 3.01 | 0.89 | 2.30 | 0.34 | 0.274 | 0.965 | 0.010 | 0.088 | 0.027 | 0.208 | |
| Environmental Information Processing | 14.81 | 1.39 | 15.69 | 1.99 | 13.77 | 2.04 | 0.408 | 0.965 | 0.442 | 0.124 | 0.068 | 0.208 | |
| Genetic Information Processing | 17.65 | 1.83 | 17.82 | 2.31 | 19.43 | 1.45 | 0.965 | 0.965 | 0.038 | 0.088 | 0.122 | 0.208 | |
| Human Diseases | 1.05 | 0.23 | 0.90 | 0.15 | 0.83 | 0.07 | 0.237 | 0.965 | 0.021 | 0.088 | 0.408 | 0.341 | |
| Metabolism | 47.05 | 1.78 | 46.78 | 1.54 | 48.06 | 1.19 | 0.829 | 0.965 | 0.195 | 0.097 | 0.101 | 0.208 | |
| Organismal Systems | 0.66 | 0.08 | 0.68 | 0.12 | 0.73 | 0.07 | 0.696 | 0.965 | 0.083 | 0.088 | 0.515 | 0.351 | |
| Unclassified | 15.48 | 1.22 | 15.13 | 1.26 | 14.89 | 0.85 | 0.573 | 0.965 | 0.382 | 0.124 | 1.000 | 0.581 | |
KEGG, Kyoto Encyclopedia of Genes and Genomes; TA, tubular adenoma; SSA/P, sessile serrated adenoma/polyp; CRC, colorectal cancer; SD, standard deviation; FDR, false discovery rate; N/A, not applicable.
The relative abundance of functional category according to the level 3 KEGG module is illustrated in Fig. 4. Overall, distribution of each predictive function at the level 3 KEGG module did not differ among the three groups. However, several functions of the cellular process category including bacterial chemotaxis, bacterial motility proteins, and flagella assembly tended to be highly expressed in the TA and SSA/P metagenomes compared to the CRC metagenomes. On the contrary, several functions of the genetic information processing category including RNA degradation, DNA repair and recombination proteins, and mismatch repair tended to be more highly expressed in the CRC metagenome than in the TA and SSA/P metagenomes. Detailed relative abundance of predicted functions for specific level 3 KEGG modules is shown in supplementary Dataset 1.
Figure 4. Relative abundance of functions predicted by phylogenetic investigation of communities by reconstruction of unobserved states.

(A) TA, (B) SSA/P, and (C) CRC. Values represent percentage of relative abundance of each level 1 KEGG module. TA, tubular adenoma; SSA/P, sessile serrated adenoma/polyp; CRC, colorectal cancer; KEGG, Kyoto Encyclopedia of Genes and Genomes.
Discussion
Many studies have revealed that Fusobacteria are prevalent in CRCs compared to adjacent normal tissues6,7,8. It has been shown that Fusobacteria are present in colorectal premalignant lesions including TA and SSA/P although the relative abundance is lower than in CRC tissues13. Moreover, it was shown that abundance of F. nucleatum in CRC tissue was associated with a lower density of CD3+ T cell14. It has been suggested that Gut bacteria including Fusobacteria may have an influence on the development of CRC through interaction with the innate immune system or host factors3. These evidences support that Fusobacteria may have a distinct role in the carcinogenesis of the colorectum.
Recently, Kostic et al. demonstrated that F. nucleatum can induce intestinal tumorigenesis in APCMin/+mice, and showed that Fusobacteria can affect the early stage of the adenoma-carcinoma sequence such as APC-mutated adenoma. However, the influence of Fusobacteria on CRC development through the serrated pathway remained relatively unknown. Compared to the adenoma-carcinoma sequence, sessile serrated adenoma/polyp has an obviously different pathway to the development of CRC1,9,15,16. For example, abnormal hypermethylation of promoter CpG island is common in the serrated pathway to CRC9. In addition, BRAF mutations are strongly associated with CpG island methylator phenotype (CIMP)-high CRCs, acting as an alternative to the KRAS mutations that commonly occur in chromosomal instability cancers9. Moreover, APC mutation is relatively rare in SSA/Ps15. We hypothesized that if Fusobacteria are not associated with SSA/P, the gut microbiome, including relative abundance of Fusobacteria, might differ between TA and SSA/P tissues. In our study, however, the relative abundance of Fusobacteria did not differ between the TA and SSA/P groups. PCoA also showed that the gut microbiome in the TAs was more similar to that of the SSA/Ps than the CRCs. These results suggested that Fusobacteria is a possible contributor to both the adenoma-carcinoma sequence and the serrated polyp-carcinoma sequence.
In this study, the relative abundance of Fusobacteria in the TA or SSA/P groups was lower than in the CRC group. Considering that Fusobacteria are extremely rare in healthy individuals17, their abundance in 37.5–50% of lesions may be higher than the relative abundance of Fusobacteria in normal colorectal mucosal of healthy individuals. These findings were consistent with previous studies. One recent study by Ito et al. using quantitative polymerase chain reaction for F. nucleatum in formalin-fixed paraffin-embedded tumor tissues showed that only 30–40% of serrated adenomas expressed positivity for F. nucleatum13. Additionally, F. nucleatum expression did not differ between serrated and non-serrated adenomas. Moreover, F. nucleatum was more expressed in CRC tissues than in both serrated and non-serrated adenomas. Based on their results, Ito et al. concluded that expression of F. nucleatum may contribute to the progression of colorectal neoplasia.
Differences in the gut microbiome, including the relative abundance of Fusobacteria, between TA or SSA/P and CRC can be explained by the physiologic and metabolic changes that occur during colon carcinogenesis, including changes in colonic barrier function and rupture and bleeding of the colonic epithelium18. For example, Bacteroides fragilis (ETBF) strains produce B. fragilis metalloprotease toxin19,20. This metalloprotease facilitates cleavage of E-cadherin, that is tumor suppressor protein, in intestinal epitheliums, causing cell proliferation and permeabilization of the intestinal barrier21. As a result of changes in colonic barrier permeability and cellular metabolism, Fusobacteria might be more prevalent in CRC tissues than in adenomas18.
One interesting finding of our study is that Fusobacteria was identified in the adjacent normal tissues as well as colorectal neoplasms. The relative abundance of Fusobacteria between lesions and adjacent normal tissues was similar in each group. This finding can be explained by the field cancerization effect, i.e., a field of cellular and molecular alterations that predisposes to the development of neoplasms within that territory22,23. Other studies on gut microbiomes in patients with CRC also showed that Fusobacteria are abundant in adjacent normal tissues as well as CRC tissues24.
An additional merit of our study is functional analysis for the predicted metagenome using PICRUSt in TAs, SSA/Ps, and CRCs. Although statistical power was not achieved because of the small sample size, a tendency for functional differences among the three metagenomes could be identified. Overall, the relative abundance of the cellular processes category including bacterial chemotaxis, bacterial motility proteins, and flagella assembly tended to be higher in the TA and SSA/P groups, whereas that of the genetic information processing category including RNA degradation, DNA repair and recombination proteins, and mismatch repair tended to be higher in the CRC group. These results, however, were metagenomic inferences without statistical significance. Therefore, more experimental studies on these bacterial functions should be required to clarify the role of the gut microbiome in CRC development.
Although this study was the first to compare the gut microbiome between two types of CRC precursor, TA and SSA/P, it has several limitations. The small sample size is the first limitation. Although some significant differences in the gut microbiome were identified at the phylum level, statistical power could not be achieved at a deeper level because of the small sample size. Further large-scaled studies are needed for identifying detailed differences of gut microbiome among disease status. Second, all CRCs in our study were located in the distal colon. Unfortunately, patients with proximal colon cancer could not be recruited during the study period therefore we performed a subgroup analysis for lesions located in the distal colon. Subgroup analysis revealed that the relative abundance of bacteria including Fusobacteria did not differ according to the tumor location in our study. Finally, although our data showed the comparison of gut microbiome between patients with TA and those with SSA/P, we could not make a definitive conclusion about the role of Fusobacteria in serrated pathways. Experimental studies using a SSA/P animal model may provide data supporting our current study.
Despite these limitations, our data provide a better understanding of the role of Fusobacteria in the development of CRC. The relative abundance of Fusobacteria in CRCs was higher than that in the TAs or SSA/Ps. In addition, the gut microbiome, including relative abundance of Fusobacteria, did not differ between the TAs and SSA/Ps. Therefore, Fusobacteria may contribute to the serrated pathway as well as the adenoma-carcinoma sequence.
Methods
Study population
Patients that were pathologically diagnosed with TA, SSA/P, or CRC were recruited for this study. Written informed consent was obtained from all patients. Patients who were diagnosed with inflammatory bowel disease and human immunodeficiency virus enteropathy were excluded. Additionally, patients who had been treated with antibiotics or antidiarrheal agents within 3 months prior to enrollment were excluded. The study protocol was approved by the Institutional Review Board of Hanyang University Guri Hospital (GURI 2014-10-010). All experiments were performed in accordance with relevant guidelines and regulations.
Tissue sampling and bacterial DNA extraction
Three pieces of mucosal tissue were obtained from patients with TA, SSA/P, or CRC by endoscopic biopsy. Additionally, four biopsy samples were taken from adjacent normal tissues of each patient.
Extraction of bacterial DNA was performed from mucosal biopsy samples as we previously reported25. Briefly, 100 mg of frozen gastric mucosal tissues was suspended in 750 μL of sterile bacterial lysis buffer (200 mmol/L NaCl, 100 mmol/L EDTA [pH 8.0], 20 mmol/L Tris base, 20 mg/mL lysozyme) and incubated at 37 °C for 30 minutes. Then, we added 20 μL of proteinase K and 80 μL of 10% SDS to the mixture and incubated it at 65 °C for 30 minutes. Finally, bead beating was performed for 90 seconds at 5,300 rpm (PRECELLYS 24; Bertin Technologies, Le Bretonneux, France) after adding a 300 mg of 0.1-mm zirconium beads (BioSpec Products, Bartlesville, OK, USA) for finishing homogenization. The homogenized mixture was cooled on ice and then centrifuged at 14,000 rpm for 5 minutes. Bacterial DNA was extracted from the supernatant by phenol/chloroform/iso-amyl alcohol (25:24:1) followed by chloroform/iso-amyl alcohol (24:1), and precipitated by absolute ethanol at −20 °C for 1 hour. The precipitated DNA was suspended in DNase-free H2O and cleaned up using a DNA clean-up kit according to the manufacturer’s instructions. Isolated DNA was stored at −80 °C until use in microbial characterization
Amplification of 16S rRNA gene and sequencing
Extracted gDNA was amplified using primers targeting the V1 to V3 regions of the 16S rRNA gene. For bacterial gDNA amplification, barcoded primers of 27F 5′-CCATCTCATCCCTGCGTGTCTCCGACTCAG-MID-GAGTTTGATCMTGGCTCAG-3′, which was consisted of the GS FLX+ adapter sequence A (underlined sequence), linker nucleotides (bolded sequence), multiplex identifiers sequence (MID), and the universal 16S rRNA-specific sequence (italic sequence). The reverse primer 518R 5′-CCTATCCCCTGTGTGCCTTGGCAGTCTCAG-MID-WTTACCGCGGCTGCTGG-3′ consisted of the GS FLX+ adapter sequence B (underlined sequence), linker nucleotides (bolded sequence), MID, and the universal 16S rRNA-specific sequence (italic sequence). The amplifications were performed at the following conditions: initial denaturation at 95 °C for 5 minutes, followed by 35 cycles of denaturation at 95 °C for 40 seconds, primer annealing at 57 °C for 40 seconds, and extension at 72 °C for 60 seconds; followed by a final elongation at 72 °C for 60 seconds. 16S rRNA PCR products were quantified, pooled, and purified for the sequencing reaction. Sequencing was performed using a 454 Life Sciences Genome Sequencer FLX+ machine (Roche, Florence, SC, USA) according to the manufacturer’s instructions.
Analysis of 16S rRNA Sequences
Initially generated partial reads of 16S rRNA were trimmed for quality using the standard SFF software tools from Roche/454. Pre-processing and clustering cluding raw read filtering and trimming, reads picking, and OTU clustering using the CD-HIT-OTU26. In addition, bacterial 16S rRNA sequence data of the gastric mucosal microbiome were processed through the QIIME pipeline27. Reads which showed low quality, contained incorrect primer sequences, or contained more than one ambiguous base were excluded. The remaining reads were classified into CRC, tubular adenoma, and serrated adenoma groups based on their unique nucleotide barcodes. Chimeric sequences were filtered out using the UCHIME algorithm after nearest alignment space termination (NAST) based on the SILVA database28,29. Taxonomic composition from phylum to species levels and bacterial diversity for each sample were evaluated based on 97% similarity using the QIIME30. A UniFrac distance was calculated from representative reads of each OTU using the UniFrac program31. Additionally, Unifrac and weighted Unifrac distances were fed into a principal coordinate analysis (PCoA)31.
DNA sequences obtained from this metagenomic project have been deposited in the NCBI Short Read Archive under the Accession No. SRA306790.
Metagenome prediction
Functional composition of the microbiome was predicted using phylogenetic investigation of communities by reconstruction of unobserved states (PICRUSt)12. PICRUSt uses evolutionary modeling to predict metagenomes from 16S data compared with a reference genome database12. Metagenome prediction in this study was conducted using Galaxy, which is an open, web-based platform for computational biomedical research (http://huttenhower.sph.harvard.edu/galaxy)12. To evaluate dissimilarity between reference genomes and the metagenome, the nearest sequenced taxon index (NSTI), which is the sum of phylogenetic distances for each organism in the OTU table to its nearest relative with a sequenced reference genome, was calculated12. NSTI has a negative correlation with accuracy of PICRUSt. Functions of predicted metagenomes were classified according to the Kyoto Encyclopedia of Genes and Genomes (KEGG).
Statistical analysis
Baseline patient characteristics and results of 16S rRNA gene pyrosequencing were described as median with interquartile range (IQR) or number with proportion. Fisher’s exact test or Kruskal Wallis test was used for group comparisons. Relative abundance of different bacteria phyla in each group was presented as mean with standard deviation. Assigned OTUs were compared individually between study groups using Mann-Whitney U test with false discovery rate (FDR) control to correct for multiple comparisons32. All statistical procedures were conducted using R (version 2.15.3; R Foundation for Statistical Computing, Vienna, Austria).
Additional Information
How to cite this article: Park, C. H. et al. Role of Fusobacteria in the serrated pathway of colorectal carcinogenesis. Sci. Rep. 6, 25271; doi: 10.1038/srep25271 (2016).
Supplementary Material
Acknowledgments
This work was funded by a grant of the Korean Health Technology R&D Project, Ministry of Health & Welfare, Republic of Korea. (A120176)(DSH).
Footnotes
Author Contributions Conception and design: D.S.H. and C.S.E. Acquisition of data: D.S.H. and C.S.E. Laboratory analysis: A.L. and Y.L. Analysis and interpretation of data: C.H.P., D.S.H., Y.O. and C.S.E. Drafting Manuscript: C.H.P. Review of manuscript: C.H.P., D.S.H., A.L., Y.L. and C.S.E. Study Supervision: D.S.H.
References
- Brenner H., Kloor M. & Pox C. P. Colorectal cancer. Lancet 383, 1490–1502, 10.1016/S0140-6736(13)61649-9 (2014). [DOI] [PubMed] [Google Scholar]
- Vogelstein B. & Kinzler K. W. Cancer genes and the pathways they control. Nat Med 10, 789–799, 10.1038/nm1087 (2004). [DOI] [PubMed] [Google Scholar]
- Yamauchi M. et al. Colorectal cancer: a tale of two sides or a continuum? Gut 61, 794–797, 10.1136/gutjnl-2012-302014 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara T. et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res 74, 1311–1318, 10.1158/0008-5472.CAN-13-1865 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mima K. et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut (in press), 10.1136/gutjnl-2015-310101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic A. D. et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res 22, 292–298, 10.1101/gr.126573.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellarin M. et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res 22, 299–306, 10.1101/gr.126516.111 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi J. R. et al. Towards the human colorectal cancer microbiome. PLoS One 6, e20447, 10.1371/journal.pone.0020447 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rex D. K. et al. Serrated lesions of the colorectum: review and recommendations from an expert panel. Am J Gastroenterol 107, 1315–1329; quiz 1314, 1330, 10.1038/ajg.2012.161 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongers G. et al. Interplay of host microbiota, genetic perturbations, and inflammation promotes local development of intestinal neoplasms in mice. J Exp Med 211, 457–472 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar H. et al. Gut microbiota as an epigenetic regulator: pilot study based on whole-genome methylation analysis. MBio 5, e02113–14, 10.1128/mBio.02113-14 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31, 814–821, 10.1038/nbt.2676 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int J Cancer 137, 1258–1268, 10.1002/ijc.29488 (2015). [DOI] [PubMed] [Google Scholar]
- Mima K. et al. Fusobacterium nucleatum and T Cells in Colorectal Carcinoma. JAMA Oncol 1, 653–661, 10.1001/jamaoncol.2015.1377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehari R. Infrequent APC mutations in serrated adenoma. Tohoku J Exp Med 193, 181–186, 10.1620/tjem.193.181 (2001). [DOI] [PubMed] [Google Scholar]
- Rosty C. et al. Serrated polyps of the large intestine: current understanding of diagnosis, pathogenesis, and clinical management. J Gastroenterol 48, 287–302, 10.1007/s00535-012-0720-y (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Q. et al. Gut microbiome development along the colorectal adenoma-carcinoma sequence. Nat Commun 6, 6528, 10.1038/ncomms7528 (2015). [DOI] [PubMed] [Google Scholar]
- Tjalsma H., Boleij A., Marchesi J. R. & Dutilh B. E. A bacterial driver-passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol 10, 575–582, 10.1038/nrmicro2819 (2012). [DOI] [PubMed] [Google Scholar]
- Wu S. et al. The Bacteroides fragilis toxin binds to a specific intestinal epithelial cell receptor. Infect Immun 74, 5382–5390, 10.1128/IAI.00060-06 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toprak N. U. et al. A possible role of Bacteroides fragilis enterotoxin in the aetiology of colorectal cancer. Clin Microbiol Infect 12, 782–786, 10.1111/j.1469-0691.2006.01494.x (2006). [DOI] [PubMed] [Google Scholar]
- Wu S. et al. Bacteroides fragilis toxin stimulates intestinal epithelial cell shedding and gamma-secretase-dependent E-cadherin cleavage. J Cell Sci 120, 1944–1952, 10.1242/jcs.03455 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippman S. M. & Hawk E. T. Cancer prevention: from 1727 to milestones of the past 100 years. Cancer Res 69, 5269–5284, 10.1158/0008-5472.CAN-09-1750 (2009). [DOI] [PubMed] [Google Scholar]
- Lochhead P. et al. Etiologic field effect: reappraisal of the field effect concept in cancer predisposition and progression. Mod Pathol 28, 14–29, 10.1038/modpathol.2014.81 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allali I. et al. Gut microbiome compositional and functional differences between tumor and non-tumor adjacent tissues from cohorts from the US and Spain. Gut Microbes 6, 161–172, 10.1080/19490976.2015.1039223 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eun C. S. et al. Differences in gastric mucosal microbiota profiling in patients with chronic gastritis, intestinal metaplasia, and gastric cancer using pyrosequencing methods. Helicobacter 19, 407–416, 10.1111/hel.12145 (2014). [DOI] [PubMed] [Google Scholar]
- Li W. et al. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief Bioinform 13, 656–668, 10.1093/bib/bbs035 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso J. G. et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7, 335–336, 10.1038/nmeth.f.303 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R. C. et al. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200, 10.1093/bioinformatics/btr381 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeSantis T. Z. et al. NAST: a multiple sequence alignment server for comparative analysis of 16S rRNA genes. Nucleic Acids Res 34, W394–399, 10.1093/nar/gkl244 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Garrity G. M., Tiedje J. M. & Cole J. R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73, 5261–5267, 10.1128/AEM.00062-07 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C. & Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71, 8228–8235, 10.1128/AEM.71.12.8228-8235.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y. et al. Controlling the false discovery rate in behavior genetics research. Behav Brain Res 125, 279–284, 10.1016/s0166-4328(01)00297-2 (2001). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.