

Abstract
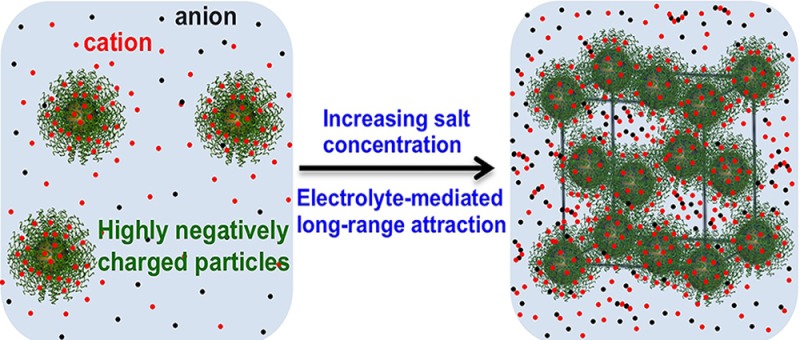
Solutions at high salt concentrations are used to crystallize or segregate charged colloids, including proteins and polyelectrolytes via a complex mechanism referred to as “salting-out”. Here, we combine small-angle X-ray scattering (SAXS), molecular dynamics (MD) simulations, and liquid-state theory to show that salting-out is a long-range interaction, which is controlled by electrolyte concentration and colloid charge density. As a model system, we analyze Au nanoparticles coated with noncomplementary DNA designed to prevent interparticle assembly via Watson–Crick hybridization. SAXS shows that these highly charged nanoparticles undergo “gas” to face-centered cubic (FCC) to “glass-like” transitions with increasing NaCl or CaCl2 concentration. MD simulations reveal that the crystallization is concomitant with interparticle interactions changing from purely repulsive to a “long-range potential well” condition. Liquid-state theory explains this attraction as a sum of cohesive and depletion forces that originate from the interelectrolyte ion and electrolyte–ion–nanoparticle positional correlations. Our work provides fundamental insights into the effect of ionic correlations in the salting-out mechanism and suggests new routes for the crystallization of colloids and proteins using concentrated salts.
Short abstract
The electrolyte-mediated crystallization of highly charged nanoparticles is driven by long-range attractions with a spatial extension of ∼4 nm from the nanoparticle surface.
Introduction
Controlling the crystallization of colloids, including proteins, from solutions has been a scientific goal for decades.1−7 The crystallization of charged colloids is often induced by using high salt concentrations, a process referred to as “salting-out”.7 Colloids can also be concentrated and crystallized via the well-understood depletion forces induced by the addition of polymers5,8 or micelle forming surfactants.9 However, colloidal crystallization in high ionic strength solutions is subtle and not understood. Crystallization via “salting-out” is observed for specific salts in a narrow range of salt concentrations, when the interparticle interactions are weakly attractive.10 Above this salt concentration range, in the regime of stronger attractive interactions, amorphous precipitates are observed. It is generally believed that short-ranged attractions due to ionic correlations and solvation effects drive the colloidal assembly.11 By contrast, the present study reveals that, in high ionic strength solutions, the interparticle attraction between like-charged nanoparticles extends a few nm from the colloidal surface. This “long-range” attraction is induced by the electrolyte ions, and is not an effect of van der Waals forces.
Long range interactions between like-charged colloids near surfaces12,13 have been explored for decades. These interactions are attractive near surfaces due to hydrodynamic effects,14 but in bulk solutions they are found to be purely repulsive.15 Here, we show that electrolyte-mediated long-range interparticle attractions are possible in bulk solutions in the regime of high ionic strength. To enhance the electrostatic coupling between the nanoparticles and the electrolyte ions, our experimental design used highly charged (>2000 e–/nanoparticle) DNA coated spherical gold nanoparticles (AuNPs) in solutions containing high concentrations of NaCl or CaCl2. To avoid interparticle assembly via Watson–Crick hybridization,16,17 we used DNAs that lacked self-complementary single-stranded sticky ends. Naively, one might expect that, in the absence of hybridization, the interactions between DNA coated AuNPs are purely repulsive. Here, small-angle X-ray scattering (SAXS) shows that, depending on the salt concentration and the DNA, FCC crystals are formed with nearest-neighbor distances (dNN) that are comparable with twice the nanoparticle hydrodynamic radius R. This demonstrates the emergence of concentrated electrolyte-mediated attractions.
Various mean field theories have been developed to compute the effective interactions between charged colloids, for example, the Derjaguin–Landau–Verwey–Overbeek (DLVO)18 theory and its extensions that include the renormalized charges of the colloids. However, at high ionic strengths these models cannot account for the correlations among ions surrounding strongly charged colloids. Recently, numerical techniques have elucidated that ionic correlations in confined concentrated electrolytes can induce attractions between like-charged surfaces at concentrations larger than 300 mM NaCl.19 These attractions are distinct from the multivalent (Z ≥ 3) counterion-mediated attractions in DNA and other polyelectrolytes,20−22 which are observed at low ionic strengths (μM–mM), are short-ranged (a few Å corresponding to the multivalent ion diameter), and lead to unstable precipitates in the absence of specific short-range attractions as the salt concentration increases. Here, we find attractions at high ionic strengths (>100 mM) and even in monovalent salts, resembling the “salting-out” effect. By molecular dynamics (MD) simulations and liquid-state theory we provide evidence that the ionic correlations in the concentrated electrolyte induce interparticle long-range attractions and drive the assembly.
Results and Discussion
SAXS Studies of DNA Coated AuNP Assembly
To analyze the effect of charge density, DNA rigidity, and electrolyte concentration, we studied four sample sets. These sets correspond to two nanoparticle types: AuNPs (nominally, 10 nm diameter) functionalized with single-stranded (ss)-DNA (ss-DNA-AuNP) or double-stranded (ds)-DNA (ds-DNA-AuNP) (insets, Figures 1A and 1C), each dispersed in two solution types, NaCl and CaCl2. For all samples, the nanoparticle concentration was ∼50 nM, corresponding to an average center-to-center interparticle distance of ∼400 nm in the gas phase. For each set, ionic strengths (μs) in the range ∼30–2000 mM were examined (Tables S1 and S2). By definition, for NaCl solutions, μs = [NaCl] and for CaCl2 solutions, μs = 3 × [CaCl2]. In salt free solutions, dynamic light scattering (DLS) yield hydrodynamic radii of R ∼ 19 nm and R ∼ 13 nm for ss-DNA-AuNP and ds-DNA-AuNP, respectively (Figure S1), corresponding to volume fractions of ∼8.7 × 10–4 and ∼2.7 × 10–4. These salt free values mark the upper bounds for the volume fractions since the radial extension of the DNA on the nanoparticles, as expected,23 is found (section 4 in the Supporting Information) to decrease with increasing μs due to the enhanced screening of the intra-DNA electrostatic repulsions.
Figure 1.
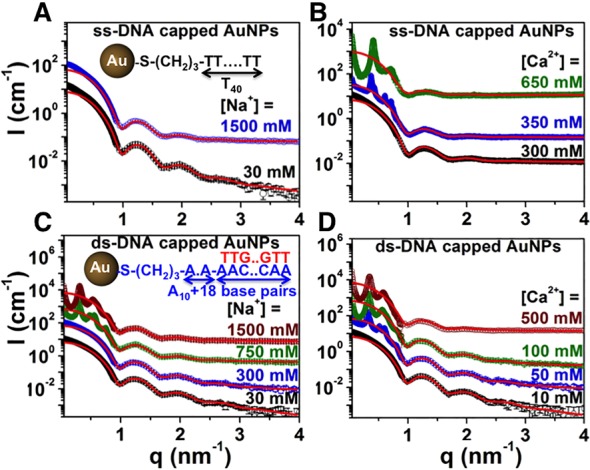
Ionic-strength-dependent assembly behavior of DNA coated AuNPs. 1D SAXS intensity profiles for ss-DNA-AuNP and ds-DNA-AuNP in NaCl (A, C) and CaCl2 (B, D) solutions. The data shown is the scattered intensity above the background scattering from empty capillary and pure water. The insets in panels A and C show the DNA-grafted-AuNP components. There are ∼60 thiolated-DNA tethered to each AuNP. About 40% of the strands on ds-DNA-AuNPs were in duplexed form. The ss-DNA is a T40 strand. The DNA chain in panel C consists of a 10 base long ss-DNA spacer A10 and an 18 base-pair long ds-DNA segment. Therefore, the total charge on the nanoparticles is ∼2400 e–/NP and ∼2100 e–/NP for ss-DNA-AuNP and ds-DNA-AuNP, respectively. Solid red lines are the expected scattered intensities from isolated DNA-grafted-AuNPs.
For all the sample sets, Figure 1 shows representative SAXS intensity profiles (I) as a function of the scattering vector magnitude q (= 4π sin θ/λ). Here, λ is the X-ray wavelength and 2θ is the scattering angle. For ss-DNA-AuNPs in NaCl solutions, the main features of the intensity profiles are μs-independent. To illustrate, two extreme μs cases are shown in Figure 1A. These SAXS profiles exhibit the characteristics of scattering from isolated DNA-coated-AuNPs, which is predominantly due to the electron-dense Au cores.24 Based on SAXS from a solid homogeneous sphere,25 the position of the first minima (qmin ∼ 1 nm–1) corresponds to a Au core radius of RAu ∼ 4.5/qmin = 4.5 nm. Unlike ss-DNA-AuNPs in NaCl solutions, ss-DNA-AuNPs in CaCl2 or ds-DNA-AuNPs in NaCl or CaCl2 solutions aggregate into clusters above a threshold ionic strength μt, as evidenced by the appearance of sharp intensity modulations in the q < 1 nm–1 region (Figures 1B–1D). DLS measurements show that a typical cluster size is ∼1.7 μm (Figure S1C).
Comparison of μt in different sample sets shows that Ca2+ induces aggregation of DNA-coated-AuNPs at much lower μs than Na+ (Figures 1C and 1D). Similarly, ds-DNA-AuNPs form aggregates at a much lower μs than ss-DNA-AuNPs (Figures 1B and 1D). Thus, the DNA-coated-AuNPs form aggregates more readily when the DNA charge density and the counterion valency are increased. These trends indicate that the responsible attractions cannot originate from van der Waals forces.
Figure 1 shows the
simulated intensities P(q) for isolated
DNA-grafted-AuNPs (solid red lines). For all the cases where nanoparticle
aggregation is not observed, the measured I(q) are well described by simulations based on mean Au core
size ⟨RAu⟩ = 4.5 nm and
polydispersity (PD) = 8.5% or ⟨RAu⟩ = 4.4 nm and PD = 7.7%, depending upon the nanoparticle
batch used (section 2.1 in the Supporting Information). This analysis allows extraction of the structure factor [S(q) = 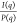 ] for nanoparticle aggregates (Figures 2A and 2B).
] for nanoparticle aggregates (Figures 2A and 2B).
Figure 2.

Structure of DNA coated AuNP assemblies. (A, B) SAXS-derived S(q) for DNA-grafted-AuNP aggregates (circles) along with simulations based on FCC lattices (red lines). For reference, the expected peak positions and relative intensities for Bragg reflections from ideal FCC lattices are shown (A, vertical black lines). The labels ss and ds correspond to ss-DNA-AuNP and ds-DNA-AuNP, respectively. (C) Representative radial distribution functions for ds-DNA-AuNPs in CaCl2 as a function of μs along with the expected positions and relative populations (P/12) for neighbors in a FCC lattice (black lines). For visual comparisons, g(r) is plotted against normalized radial distance r/r1. Here, r1 = dNN represents the nearest-neighbor interparticle distance. Monte Carlo simulations (section 1.4 in the Supporting Information) for g(r) based on random close packing (RCP) of hard spheres (blue lines) reasonably describe the experimental g(r) for μs much higher than μt. (D) Schematic of the observed changes in colloidal packing as a function of ionic strength.
Two types of S(q) profiles are
observed. First, regardless of the DNA-coating and the salt solution, S(q) exhibits similar features at the threshold
ionic strength (μt) for aggregation. These S(q) are plotted against q/q1 (Figure 2A), where, q1 is the position of the principal peak. Similarly, for μs ≫ μt, S(q) vs q/q1 profiles
are nearly identical (Figure 2B), but subtly different from the profiles at μs = μt. The analysis of S(q) based on a formalism by Förster et al.26 shows that, for μs = μt, DNA functionalized AuNPs are arranged on FCC lattices (Figure 2A, and section 2.2
in the Supporting Information). The positions
of the principal FCC (1 1 1) peak yield lattice parameters aFCC (= 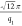 ) = 29.2, 36.7, and 34.4 nm for ss-DNA-AuNPs
in CaCl2, and ds-DNA-AuNPs in NaCl and CaCl2 solutions, respectively (see also Table S1). For the three cases in Figure 2A, the widths of the (hkl) diffraction profiles yield average crystallite
sizes of 200–300 nm. Taken together, the DLS-measured aggregate
size (1.7 μm) and the SAXS-derived crystallite size imply that
the DNA-grafted-AuNPs assemble into polycrystalline aggregates at
ionic strengths equal to or slightly above μt. Therefore,
under appropriate conditions, electrolyte-mediated interactions can
induce crystalline order in DNA functionalized AuNPs even in the absence
of Watson–Crick hybridization.
) = 29.2, 36.7, and 34.4 nm for ss-DNA-AuNPs
in CaCl2, and ds-DNA-AuNPs in NaCl and CaCl2 solutions, respectively (see also Table S1). For the three cases in Figure 2A, the widths of the (hkl) diffraction profiles yield average crystallite
sizes of 200–300 nm. Taken together, the DLS-measured aggregate
size (1.7 μm) and the SAXS-derived crystallite size imply that
the DNA-grafted-AuNPs assemble into polycrystalline aggregates at
ionic strengths equal to or slightly above μt. Therefore,
under appropriate conditions, electrolyte-mediated interactions can
induce crystalline order in DNA functionalized AuNPs even in the absence
of Watson–Crick hybridization.
Figure 2B shows that, for μs ≫ μt, the assembly does not consist of FCC crystallites. More information about the nanoparticle packing in these aggregates is gleaned from radial distribution function g(r) (eq S10 in the Supporting Information). Figure 2C shows the μs-dependence of g(r) for ds-DNA-AuNPs in CaCl2 solutions. For the 50 mM [Ca2+] case (μs = μt), the amplitudes and the positions of maxima in g(r) at r/r1 = 1, √2, √3, √4, √5, etc. are consistent with FCC lattices (Figure 2C, bottom). With increasing μs, the r/r1 = √2 modulation smears out. Further, the g(r) exhibit a slightly split doublet with nearly equal amplitude maxima at r/r1 ∼ √3 and ∼ √4 (Figure 2C, middle and top). This doublet is a signature of a glassy phase.27 Specifically, the g(r) for [Ca2+] = 100 mM (Figure 2C, middle) resembles the g(r) for the “metallic-glass-like” packing of spherical colloids.2 Similarly, the g(r) for [Ca2+] = 250 mM, where the r/r1 = √2 feature is mostly smeared out, is reminiscent of the g(r) for random-close-packed (RCP) spheres.28 These observations imply that the packing of DNA-grafted-AuNPs transforms from isolated particles (gas-like) to face centered cubic (FCC) to “glass-like” arrangement with increasing μs (Figure 2D). The structural phase transition sequence is similar to that observed for protein crystallization.10 Furthermore, similar to the case of proteins, the crystallization of DNA-coated AuNPs occurs in a narrow μs regime, for example, μs ∼ 1050–1500 mM for ss-DNA-AuNP in CaCl2 (Tables S1 and S2). Our results suggest that the electrolyte concentration induced “gas” to “crystalline” to “amorphous” transitions are a general feature of the assembly of charged colloids in high ionic strength solutions.
Some insight into the assembly mechanism of DNA-grafted-AuNPs is obtained from the (nearest-neighbor distance) dNN vs μs trends (Tables S1 and S2 and Figures S2 and S3). First, the dNN continuously decreases with increasing μs to reach a constant value in the glassy state, which is ∼94% of the dNN observed for FCC crystals at μs = μt. Second, the observed dNN are smaller than estimates for 2R that are based on the combination of modified Daoud–Cotton model parameters23 for the ss-DNA radial extension and the experimental values for the average inter-base-pair separation for ds-DNA in Watson–Crick hybridization driven assemblies29 (Figures S2 and S3). Both these observations suggest a dense packing of DNA-grafted-AuNPs that is driven by electrolyte-mediated attractions.
MD Simulations for Potential of Mean Force between DNA-Coated AuNPs
The hypothesis of electrolyte-mediated interparticle attractions was validated by MD simulations (section 1.3 in the Supporting Information). Figure 3A shows the potential of mean force between two ds-DNA-AuNPs as a function of the distance between their centers in the presence of an electrolyte with divalent cations and monovalent anions (2:1 electrolyte). Here, the two DNA-grafted-AuNPs interact only via short-ranged repulsive steric interactions, and long-ranged Coulomb potentials. Two values of μs were simulated: 15 mM (μs ≪ μt) and 150 mM (μs = μt). For the 15 mM case, the interaction is repulsive for all interparticle separations. At the onset of crystallization (150 mM case), the potential barrier at low interparticle separations reflects the steric and electrostatic repulsions arising due to the strong interdigitation of the DNA strands on neighboring nanoparticles. However, the effective potential is clearly attractive over a ∼7 nm wide region. The minima position in the interparticle potential (Figure 3A) corresponds to the case where the DNA coronas of the two nanoparticles are just touching (Figure 3B). Thus, the interparticle interactions are attractive at separation distances where ds-DNA chains with maximum extension can overlap slightly, but also at separation distances that are ∼4 nm larger than the tangential contact distance between the nanoparticles (Figure 3C). The range of attractive interactions is approximately five times higher than the Debye screening length (κ–1 = 0.78 nm) for μs = 150 mM. Attractions between high charge density macromolecules such as DNA in bulk solutions30 and at interfaces31 have been previously observed at or above μs = 150 mM for 2:1 electrolytes. However, these attractions were hypothesized to be short-ranged, with a decay length comparable to the hydrated divalent cation diameter.
Figure 3.
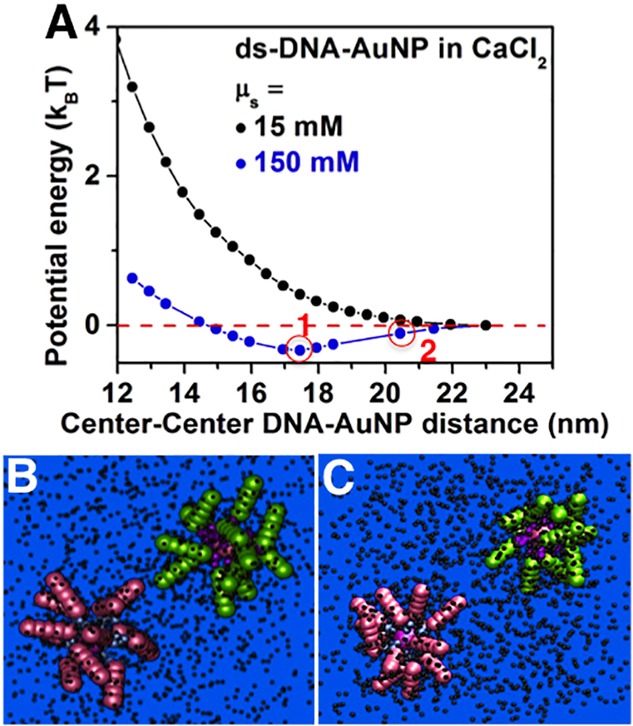
Effective interaction potential between two DNA-grafted-AuNPs. (A) Potential energy as a function of interparticle separation for two ds-DNA-AuNPs in two solutions of different μs. The minima position (circled point 1) corresponds to tangential contact between the two ds-DNA capped AuNPs. Simulation snapshots corresponding to circled points 1 and 2 are shown in panels B and C, respectively.
Due to computational
constraints, the MD simulations were performed
for RAu = 1.5 nm particles with 12 DNA/AuNP
and only for 2:1 electrolytes at the two ionic strengths described
above (section 1.3 in the Supporting Information). Correcting for the radius of the AuNPs, MD simulations show that
the equilibrium inter-ds-DNA-AuNP separation is 23.6 nm, close to
the experimental dNN = ( ) = 24.3 nm for the μs =
150 mM case. The nanoparticle size-correction should also be applied
to the potential well depth (∼0.33kBT, Figure 3A). This is because liquid-state theory (next section) shows that the magnitude of the two-body attraction
depends on the nanoparticle size. The size-corrected potential well
depth is 0.45kBT.
) = 24.3 nm for the μs =
150 mM case. The nanoparticle size-correction should also be applied
to the potential well depth (∼0.33kBT, Figure 3A). This is because liquid-state theory (next section) shows that the magnitude of the two-body attraction
depends on the nanoparticle size. The size-corrected potential well
depth is 0.45kBT.
The interparticle attractive potential well is shallow. However, crystallization is a many-particle collective process. Taking into account only the coordination number of 12 in a FCC lattice, the potential energy/particle becomes ∼5.4kBT. Considerations of DNA-coated nanoparticles at finite concentration could further increase this energy estimate via inclusion of multiparticle effects that are absent in our potential of mean force calculations, due to the assumption of infinite dilution of nanoparticles. We note that the attractive potential well condition coincides with a strong enhancement in the cation–anion positional correlations in the supporting electrolyte and the DNA corona (Figures S4–S6 and accompanying text).
Finally, previous simulation studies that utilized simplifying assumptions of screened Coulomb or Yukawa-like effective potentials32 yielded short-ranged attractions between functionalized nanoparticles. Now, by explicitly considering the positional correlations between electrolyte ions in bulk solutions and between the electrolyte ions and the nanoparticles, our simulations reveal the long-range nature of the observed electrolyte mediated attractions.
Liquid-State Theory for Like-Charged Attraction
Insights into the origin of the attraction between like-charged objects are provided by a liquid-state-theory based analytical approach. Specifically, the interaction potential between the nanoparticles can be derived from first principles (eqs S12–S17) in an algebraic form that distinguishes contributions from ion entropy and ion–nanoparticle and interion correlations. The range of the interaction is connected to the length over which the nanoparticles influence the ionic density profile in the electrolyte. This length typically extends beyond the radial size of the DNA linkers because of electrostatic and steric interactions.24 At low salt concentrations, this extension is well approximated by the Debye length, whereas at high concentrations, it is typically larger than the Debye length, measuring a few hydrated ionic radii.
To illustrate like-charge attraction in a simpler case, we calculate the potential of mean force between like-charged ions in primitive electrolytes. Figure 4A shows that, at sufficiently high concentrations, like charges attract, mediated by opposite charges. These attractions appear roughly above 0.1 M for a 1:1 electrolyte of the primitive model (e.g., NaCl) and a few tens of mM for 2:1 electrolyte (e.g., CaCl2). Furthermore, the range of the interaction is greater than 2.5 nm [6–7 × the hydrated ionic radii33].
Figure 4.
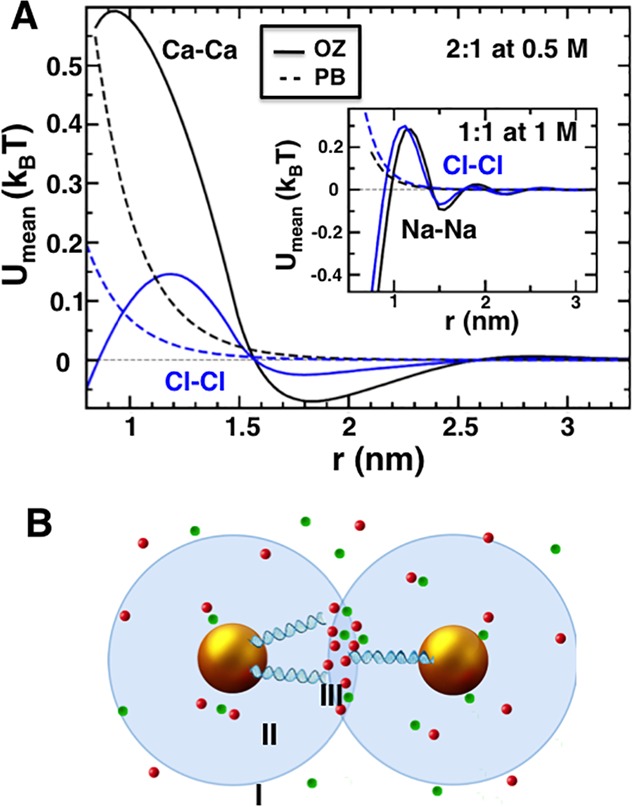
Origin of like-charge attraction at high salt concentration. (A) Calculations of the potentials of mean force in primitive model 2:1 and 1:1 electrolytes. At sufficient concentrations, like charges attract. Mean field theory [Poisson–Boltzmann (PB)] misses these attractions, while liquid-state theory [Ornstein–Zernike equation (OZ)] captures these effects. (B) Schematic of the regions that are influenced by a DNA coated AuNP (region II), and the overlap of spheres of influence of two DNA coated AuNPs (region III).
To extend these conclusions, we calculate the electrolyte-induced interaction between two smooth, parallel, like-charged surfaces, by solving the Ornstein–Zernike equation with the anisotropic hypernetted chain (HNC) closure.19,34,35 The mean potential between two highly charged surfaces (Figure S9) exhibits a qualitatively similar spatial profile as the interaction between electrolyte ions. The induced attraction is strongly amplified by a small dielectric contrast between the surfaces and the solvent, driven by an enhanced depletion of ions caused by polarization charge. Furthermore, at small interplate separations and for high salt concentrations, exclusion of electrolyte ions from the volume confined by the two plates results in very strong interplate attraction due to the osmotic pressure difference. A similar effect for DNA-coated AuNPs could explain the crystal to glass transition observed at high salt concentrations (Figure S9 and accompanying text).
DNA-coated AuNPs should attract in high salt concentrations in a manner analogous to the like-charged ions in primitive model electrolytes and the like-charged surfaces, with differences in the magnitude because of geometric reasons. Additionally, the cohesive forces driven by ion-bridging and ionic correlations are dominant in polyelectrolyte gels and blends if the pair correlation functions and the ionic-interaction potentials of the local salt are oscillatory,36 such as those shown in Figure 4A. Although the mean attraction per charge can be small compared to the thermal energy (Figure 4A), the attractive force between DNA-coated AuNPs should be amplified due to the polyvalency of the nanoparticles and the large number of associated ions in the overlap region of influence between two nanoparticles (region III, Figure 4B). Our MD simulations point to such enhanced correlations between the DNA charges and the electrolyte-ions and between the electrolyte-ions in the DNA corona (regions II and III, Figure 4B). Specifically, in going from μs = 15 mM to μs = 150 mM for a 2:1 electrolyte, the number of cations in the DNA corona increases by 25%, overcompensating the charge on DNA-coated AuNPs by ∼20%. A near electroneutrality condition is achieved by a simultaneous ∼12-fold increase in the number of associated anions (Figure S6 and accompanying text). Second, the enhanced local concentration of cations and anions in the overlap region (region III, Figure 4B) elevates the local activity of the ions, and reduces the excluded volume for the salt. This should induce depletion attractions between nanoparticles due to a locally decreased osmotic pressure. The combined effect of these cohesive forces and depletion-like attractions is calculated by the MD. Interestingly, the total effective potential (eqs S16 and S17 in the Supporting Information), as in the case of the Asakura–Oosawa depletion potential,37 is determined by the number of mediating particles (polymers in Asakura–Oosawa case; ions in the current case) in the overlap volume of the influence spheres. For the typical parameters of DNA-grafted nanoparticles in NaCl and CaCl2 solutions, the effective potential may exceed 1kBT if the concentrations are, roughly, larger than 0.1 M (section 8 in the Supporting Information). On the basis of these rough estimates we expect an attractive interaction between DNA-grafted nanoparticles, induced by the ions, via ion entropy, “ion bridges”, and ionic cohesion. In principle, these contributions can be extracted from an algebraic form for the thermally averaged potential between two nanoparticles (derived in section 8 in the Supporting Information),
| 1 |
where V0 is the overlap volume of region III,
| 2 |
The subindices I, II, and III refer to the regions shown in Figure 4B, Zi is an ion partition sum corresponding to region i at a fixed configuration of the nanoparticles, and zi is the fugacity of species i. For a mixture of hard spheres and small depletants, eq 1 reduces to the Asakura–Oosawa potential, with D being the sum of the hard sphere and the depletant diameters. Ions however interact over long distance and add energetic contributions, which can be quantified by an excess chemical potential (ion cohesion), a local Donnan potential (a mean electrostatic potential), and a direct ion–nanoparticle interaction (ion bridges) (eq S16).
The linear dependence of the interparticle attraction on the overlap volume V0 (eq 1) was used to obtain the size-corrected value for the MD simulations derived interparticle potential energy. Here, the radius of the influence sphere was assumed to be 2 nm greater than that for the DNA-coated AuNP to correspond to the 4 nm range of the attractive interactions. Furthermore, the interparticle attraction also increases exponentially with the counterion valency due to the Boltzmann weight in the partition sums Zi. This correlates well with the SAXS observation that the threshold ionic strength for nanoparticle aggregation is ∼5× lower for ds-DNA-AuNP in CaCl2 than in NaCl solutions. While the effective potential is generally attractive, the nanoparticles are stabilized by the opposing steric and electrostatic repulsions between the DNA chains, which increase sharply if the nanoparticles interdigitate.
Conclusions
We experimentally show that, in the absence of specific short-range interactions, highly charged nanoparticles undergo “gas-like” to crystalline to “glass-like” transformations with increasing salt concentration. MD simulations reveal that crystallization of the highly charged nanoparticles is driven by electrolyte-mediated attraction with a spatial extension of 4 nm from the nanoparticle surface. MD simulations and liquid-state theory suggest that the attractive interactions arise due to enhanced ionic correlations in the concentrated electrolyte and are the sum of cohesive forces and depletion interactions. These results provide fundamental insights into the very commonly observed “salting-out” phenomenon, which is extensively used to crystallize and concentrate colloids, including polyelectrolytes and proteins.
Acknowledgments
S.K., C.A.M., and M.J.B. were funded by AFOSR (FA9550-11-1-0275), L.M.M. was funded by National Defense Science and Engineering Graduate (NDSEG) fellowship, and G.I.G.-G., M.O.d.l.C., and J.W.Z. were funded by Center for Bioinspired Energy Sciences (CBES), which is an Energy Frontier Research Center funded by U.S. Department of Energy, Office of Basic Energy Sciences under Award Number DE-SC0000989. The SAXS experiments were performed at the APS 5ID-D beamline, which is supported through E. I. duPont de Nemours & Co., Northwestern University (NU), The Dow Chemical Co., and the NSF funded MRSEC at NU. The NSF-MRSEC (DMR-1121262) supported the use of J. B. Cohen X-ray diffraction facility at NU. Use of the APS was supported by DOE-BES (DE-AC02-06CH11357). We thank Steven Weigand of DND-CAT for assistance with the SAXS setup and data reduction, and Kurinji Krishnamoorthy at NU for assistance with AuNP functionalization.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00023.
Methods and materials, SAXS details, DLS measurements, and additional discussions (PDF)
Author Contributions
M.J.B., S.K., M.O.d.l.C, and G.I.G.-G. designed research. L.M.M. and C.A.M. synthesized oligonucleotides and DNA-coated AuNPs. S.K. and L.M.M. collected SAXS and DLS data. S.K. and M.J.B. analyzed SAXS data. G.I.G.-G. performed simulations. J.W.Z performed theoretical analysis. S.K., G.I.G.-G., M.O.d.l.C, J.W.Z., and M.J.B. analyzed the results and wrote the paper.
The authors declare no competing financial interest.
Supplementary Material
References
- Pusey P. N.; Vanmegen W. Phase-Behavior of Concentrated Suspensions of Nearly Hard Colloidal Spheres. Nature 1986, 320, 340–342. 10.1038/320340a0. [DOI] [Google Scholar]
- Sirota E. B.; Ouyang H. D.; Sinha S. K.; Chaikin P. M.; Axe J. D.; Fujii Y. Complete Phase-Diagram of a Charged Colloidal System - A Synchrotron X-Ray-Scattering Study. Phys. Rev. Lett. 1989, 62, 1524–1527. 10.1103/PhysRevLett.62.1524. [DOI] [PubMed] [Google Scholar]
- Leunissen M. E.; Christova C. G.; Hynninen A. P.; Royall C. P.; Campbell A. I.; Imhof A.; Dijkstra M.; van Roij R.; van Blaaderen A. Ionic Colloidal Crystals of Oppositely Charged Particles. Nature 2005, 437, 235–240. 10.1038/nature03946. [DOI] [PubMed] [Google Scholar]
- Yethiraj A.; van Blaaderen A. A Colloidal Model System with an Interaction Tunable from Hard Sphere to Soft and Dipolar. Nature 2003, 421, 513–517. 10.1038/nature01328. [DOI] [PubMed] [Google Scholar]
- Lu P. J.; Zaccarelli E.; Ciulla F.; Schofield A. B.; Sciortino F.; Weitz D. A. Gelation of Particles with Short-Range Attraction. Nature 2008, 453, 499–504. 10.1038/nature06931. [DOI] [PubMed] [Google Scholar]
- Mirkin C. A.; Letsinger R. L.; Mucic R. C.; Storhoff J. J. A DNA-Based Method for Rationally Assembling Nanoparticles into Macroscopic Materials. Nature 1996, 382, 607–609. 10.1038/382607a0. [DOI] [PubMed] [Google Scholar]
- McPherson A. Introduction to Protein Crystallization. Methods 2004, 34, 254–265. 10.1016/j.ymeth.2004.03.019. [DOI] [PubMed] [Google Scholar]
- Sacanna S.; Irvine W. T. M.; Chaikin P. M.; Pine D. J. Lock and Key Colloids. Nature 2010, 464, 575–578. 10.1038/nature08906. [DOI] [PubMed] [Google Scholar]
- Young K. L.; Jones M. R.; Zhang J.; Macfarlane R. J.; Esquivel-Sirvent R.; Nap R. J.; Wu J.; Schatz G. C.; Lee B.; Mirkin C. A. Assembly of Reconfigurable One-Dimensional Colloidal Superlattices Due to a Synergy of Fundamental Nanoscale Forces. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 2240–2245. 10.1073/pnas.1119301109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumetz A. C.; Snellinger-O’Brien A. M.; Kaler E. W.; Lenhoff A. M. Patterns of Protein-Protein Interactions in Salt Solutions and Implications for Protein Crystallization. Protein Sci. 2007, 16, 1867–1877. 10.1110/ps.072957907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum D.; Zamora P. C.; Zukoski C. F. Phase Behavior of Small Attractive Colloidal Particles. Phys. Rev. Lett. 1996, 76, 150–153. 10.1103/PhysRevLett.76.150. [DOI] [PubMed] [Google Scholar]
- Larsen A. E.; Grier D. G. Like-Charge Attractions in Metastable Colloidal Crystallites. Nature 1997, 385, 230–233. 10.1038/385230a0. [DOI] [Google Scholar]
- Bowen W. R.; Sharif A. O. Long-Range Electrostatic Attraction Between Like-Charge Spheres in a Charged Pore. Nature 1998, 393, 663–665. 10.1038/31418. [DOI] [Google Scholar]
- Squires T. M.; Brenner M. P. Like-Charge Attraction and Hydrodynamic Interaction. Phys. Rev. Lett. 2000, 85, 4976–4979. 10.1103/PhysRevLett.85.4976. [DOI] [PubMed] [Google Scholar]
- Behrens S. H.; Grier D. G. Pair Interaction of Charged Colloidal Spheres near a Charged Wall. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top. 2001, 64, 050401. 10.1103/PhysRevE.64.050401. [DOI] [PubMed] [Google Scholar]
- Nykypanchuk D.; Maye M. M.; van der Lelie D.; Gang O. DNA-Guided Crystallization of Colloidal Nanoparticles. Nature 2008, 451, 549–552. 10.1038/nature06560. [DOI] [PubMed] [Google Scholar]
- Park S. Y.; Lytton-Jean A. K. R.; Lee B.; Weigand S.; Schatz G. C.; Mirkin C. A. DNA-Programmable Nanoparticle Crystallization. Nature 2008, 451, 553–556. 10.1038/nature06508. [DOI] [PubMed] [Google Scholar]
- Verwey E. J. W.; Overbeek J. T. G.. Theory of the Stability of Lyophobic Colloids; Elsevier: New York, 1948. [Google Scholar]
- Zwanikken J. W.; Olvera de la Cruz M. Tunable Soft Structure in Charged Fluids Confined by Dielectric Interfaces. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 5301–5308. 10.1073/pnas.1302406110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspaud E.; Olvera de la Cruz M.; Sikorav J. L.; Livolant F. Precipitation of DNA by Polyamines: A Polyelectrolyte Behavior. Biophys. J. 1998, 74, 381–393. 10.1016/S0006-3495(98)77795-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong G. C. L.; Pollack L. Electrostatics of Strongly Charged Biological Polymers: Ion-Mediated Interactions and Self-Organization in Nucleic Acids and Proteins. Annu. Rev. Phys. Chem. 2010, 61, 171–189. 10.1146/annurev.physchem.58.032806.104436. [DOI] [PubMed] [Google Scholar]
- Rouzina I.; Bloomfield V. A. Macroion Attraction Due to Electrostatic Correlation Between Screening Counterions 1. Mobile Surface-Adsorbed Ions and Diffuse Ion Cloud. J. Phys. Chem. 1996, 100, 9977–9989. 10.1021/jp960458g. [DOI] [Google Scholar]
- Tan S. J.; Kahn J. S.; Derrien T. L.; Campolongo M. J.; Zhao M.; Smilgies D.-M.; Luo D. Crystallization of DNA-Capped Gold Nanoparticles in High-Concentration, Divalent Salt Environments. Angew. Chem., Int. Ed. 2014, 53, 1316–1319. 10.1002/anie.201307113. [DOI] [PubMed] [Google Scholar]
- Kewalramani S.; Zwanikken J. W.; Macfarlane R. J.; Leung C. Y.; Olvera de la Cruz M.; Mirkin C. A.; Bedzyk M. J. Counterion Distribution Surrounding Spherical Nucleic Acid-Au Nanoparticle Conjugates Probed by Small-Angle X-Ray Scattering. ACS Nano 2013, 7, 11301–11309. 10.1021/nn405109z. [DOI] [PubMed] [Google Scholar]
- Als-Nielsen J.; McMorrow D.. Elements of Modern X-Ray Physics, 2nd ed.; John Wiley: Chichester, U.K., 2011. [Google Scholar]
- Forster S.; Timmann A.; Konrad M.; Schellbach C.; Meyer A.; Funari S. S.; Mulvaney P.; Knott R. Scattering Curves of Ordered Mesoscopic Materials. J. Phys. Chem. B 2005, 109, 1347–1360. 10.1021/jp0467494. [DOI] [PubMed] [Google Scholar]
- Paloli D.; Mohanty P. S.; Crassous J. J.; Zaccarelli E.; Schurtenberger P. Fluid-Solid Transitions in Soft-Repulsive Colloids. Soft Matter 2013, 9, 3000–3004. 10.1039/c2sm27654b. [DOI] [Google Scholar]
- Finney J. L. Random Packings and Structure of Simple Liquids. I. Geometry of Random Close Packing. Proc. R. Soc. London, Ser. A 1970, 319, 479–493. 10.1098/rspa.1970.0189. [DOI] [Google Scholar]
- Hill H. D.; Macfarlane R. J.; Senesi A. J.; Lee B.; Park S. Y.; Mirkin C. A. Controlling the Lattice Parameters of Gold Nanoparticle FCC Crystals with Duplex DNA Linkers. Nano Lett. 2008, 8, 2341–2344. 10.1021/nl8011787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X. Y.; Kwok L. W.; Park H. Y.; Lamb J. S.; Andresen K.; Pollack L. Measuring Inter-DNA Potentials in Solution. Phys. Rev. Lett. 2006, 96, 138101. 10.1103/PhysRevLett.96.138101. [DOI] [PubMed] [Google Scholar]
- Koltover I.; Wagner K.; Safinya C. R. DNA Condensation in Two Dimensions. Proc. Natl. Acad. Sci. U. S. A. 2000, 97, 14046. 10.1073/pnas.97.26.14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truzzolillo D.; Bordi F.; Sciortino F.; Sennato S. Interaction Between Like-Charged Polyelectrolyte- Colloid Complexes in Electrolyte Solutions: A Monte-Carlo Simulation Study in the Debye-Hückel Approximation. J. Chem. Phys. 2010, 133, 024901. 10.1063/1.3459125. [DOI] [PubMed] [Google Scholar]
- Nightingale E. R. Phenomenological Theory of Ion Solvation - Effective Radii of Hydrated Ions. J. Phys. Chem. 1959, 63, 1381–1387. 10.1021/j150579a011. [DOI] [Google Scholar]
- Jing Y. F.; Jadhao V.; Zwanikken J. W.; Olvera de la Cruz M. Ionic Structure in Liquids Confined by Dielectric Interfaces. J. Chem. Phys. 2015, 143, 194508. 10.1063/1.4935704. [DOI] [PubMed] [Google Scholar]
- Kjellander R.; Marcelja S. Correlation and Image Charge Effects in Electric Double Layers. Chem. Phys. Lett. 1984, 112, 49–53. 10.1016/0009-2614(84)87039-6. [DOI] [Google Scholar]
- Sing C. E.; Zwanikken J. W.; Olvera de la Cruz M. Electrostatic Control of Block Copolymer Morphology. Nat. Mater. 2014, 13, 694–698. 10.1038/nmat4001. [DOI] [PubMed] [Google Scholar]
- Asakura S.; Oosawa F. Interaction Between Particles Suspended in Solutions of Macromolecules. J. Polym. Sci. 1958, 33, 183–192. 10.1002/pol.1958.1203312618. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.