

Abstract
Background
Caenorhabditis elegans has emerged over the last decade as a useful model for the study of innate immunity. Its infection with the pathogenic fungus Drechmeria coniospora leads to the rapid up-regulation in the epidermis of genes encoding antimicrobial peptides. The molecular basis of antimicrobial peptide gene regulation has been previously characterized through forward genetic screens. Reverse genetics, based on RNAi, provide a complementary approach to dissect the worm’s immune defenses.
Results
We report here the full results of a quantitative whole-genome RNAi screen in C. elegans for genes involved in regulating antimicrobial peptide gene expression. The results will be a valuable resource for those contemplating similar RNAi-based screens and also reveal the limitations of such an approach. We present several strategies, including a comprehensive class clustering method, to overcome these limitations and which allowed us to characterize the different steps of the interaction between C. elegans and the fungus D. coniospora, leading to a complete description of the MAPK pathway central to innate immunity in C. elegans. The results further revealed a cross-tissue signaling, triggered by mitochondrial dysfunction in the intestine, that suppresses antimicrobial peptide gene expression in the nematode epidermis.
Conclusions
Overall, our results provide an unprecedented system’s level insight into the regulation of C. elegans innate immunity. They represent a significant contribution to our understanding of host defenses and will lead to a better comprehension of the function and evolution of animal innate immunity.
Electronic supplementary material
The online version of this article (doi:10.1186/s12915-016-0256-3) contains supplementary material, which is available to authorized users.
Keywords: Fungal pathogen, Functional genomics, High-throughput screening, Signal transduction, Networks, Osmotic stress, Epistasis, Bioinformatics, Databases, Mitochondrial unfolded protein response
Background
Infection of Caenorhabditis elegans by its natural fungal pathogen Drechmeria coniospora provokes an innate immune response characterized by the expression of antimicrobial peptide (AMP) genes in the worm epidermis [1]. We have focused our attention on the regulation of one group of six AMP genes of the “Neuropeptide-Like Protein” class, nlp-27–nlp-31 and nlp-34, found together in a short genomic interval of less than 12 kb [2], which we call the “nlp-29 cluster”, after the best-studied member of the family. Many genes that play an essential role in controlling nlp-29 AMP gene expression have been defined, acting together in a relatively complex genetic network. Central to this regulation is a conserved p38 MAPK cascade [3], also required for resistance to intestinal bacterial pathogens [4]. Loss of function of any one of the many genes involved provokes a “No Induction of Peptide after Drechmeria Infection” (Nipi) phenotype. After small- and large-scale genetic screens for Nipi mutants [3, 5], our knowledge of anti-fungal innate immunity in C. elegans remains, however, fragmentary. Not only are there missing elements from the associated signal transduction pathways, but how these pathways cross-talk with each other and with the mechanisms involved in general homeostatic regulation is currently unclear [4]. Another largely unexplored aspect of the worm’s antifungal innate defenses relates to the potential for cross-tissue communication. We have demonstrated that a second family of AMP genes, called caenacins (cnc), including cnc-2, are controlled by a cell non-autonomous signal transduction pathway, wherein the nematode TGF-ß, DBL-1, produced in neurons, modulates cnc-2 expression in the epidermis following D. coniospora infection. This pathway does not, however, influence nlp-29 expression [6], which up until now has been found to be controlled cell-autonomously in the epidermis [3, 5, 7–9]. On the other hand, in C. elegans, the disruption of cellular homeostasis in one tissue can influence a stress response in a distant tissue (reviewed in [10–12]); whether this is also the case for nlp-29 remains an open question.
To address these lacunae, since C. elegans lends itself to large-scale functional genomic analyses [13, 14], we undertook a genome-wide RNAi screen for genes involved in the regulation of the AMP gene nlp-29, with a well-characterized reporter gene system used in our previous studies [3]. Many pathogens can infect C. elegans when cultured in liquid in 96 or 384-well plates (reviewed in [15–17]). Since D. coniospora cannot infect worms in liquid, however, we developed a novel solid-based high-throughput assay, using the COPAS Biosort [18] to obtain a quantitative measure of reporter gene expression [19]. In a previous report, we focused on the large family of worm G-protein coupled receptor (GPCR) genes and defined a key role for DCAR-1 that acts as a “damage-associated molecular pattern” receptor, acting upstream of the p38 MAPK cascade [20]. This clearly validated the experimental approach and illustrated the utility of this large-scale reverse genetic screen for identifying individual genes.
Here, we present the full results of the screen, which led to the identification of more than 250 candidate genes. Perhaps surprisingly for such a well-studied organism, there is a relative paucity of functional information available for nematode genes, which stands as a barrier to the interpretation of large-scale studies in C. elegans. For example, in the recent WormBase release (WS250), only a quarter of protein-coding genes (5162/20,362) are associated with a concise description, a similar proportion (27 %) has a UniprotKB gene ontology (GO) annotation, and 57 % of them (11,970/20,362) have any type of GO annotation in WormBase. We have therefore attempted to couple several broad in silico analytical methods with targeted secondary screening to define groups of genes that potentially act together. In doing so, we have been able to identify several distinct biological processes that play an important role in the antifungal response and obtain, for the first time, a comprehensive view of the regulation of AMP gene expression.
Results
A quantitative genome-wide RNAi screen for innate immunity genes
To identify, in an unbiased way, genes potentially involved in the regulation of the induction of antimicrobial peptide genes, we conducted a genome-wide RNAi screen. We first assembled a non-redundant collection of 21,223 RNAi clones from the Ahringer [21] and Vidal [22] libraries (Additional file 1: Table S1). Together, these clones are predicted to target 85 % of the protein coding genes in C. elegans. Using an automated method [19], we screened this library twice and quantified the infection-induced expression of the nlp-29p::gfp reporter gene in young adult worms (Fig. 1a). The entire set of results, a total of more than 46.8 million data points, including measures of body length (time of flight; TOF), optical density, and reporter gene expression (green (GFP) and red (dsRed)) from the analysis of more than 3.9 million individual worms, is publicly available and can be queried via a dedicated web interface (http://bioinformatics.lif.univ-mrs.fr/RNAiScreen; Fig. 1b). The overall continuous, but far from normal, distribution of the results from the first round of screening (Fig. 1c) is very much in line with previous quantitative large-scale screens in other organisms [23].
Fig. 1.
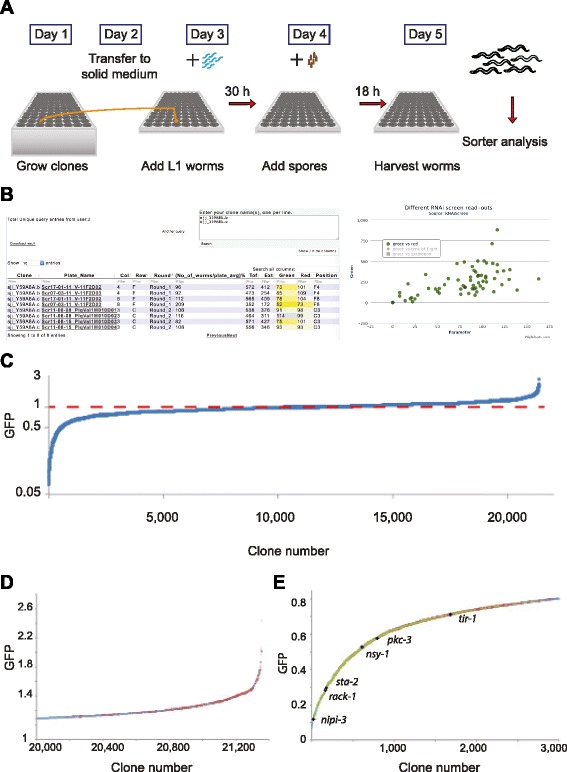
A quantitative genome-wide screen for regulators of AMP gene expression. a Simplified overview of the RNAi screen protocol, adapted from [19]. b Screenshots from the RNAi screen web interface. Left panel: example of results for two clones (insert at top right: the query box) that target the gene gck-3. Contrary to clone sjj_Y59A8A.c that passed the first round of duplicate screening, and for which the results of the second (quadruplicate) round are also displayed, sjj_Y59A8A.b only provoked a 15 % reduction in normalized GFP expression in one of the two first-round tests and so was not retained for the second round. The results for each test are linked to the primary data, which is displayed in the right panel for a single experiment. Users have the option of plotting GFP fluorescence against any or all of three parameters; shown here is GFP versus dsRed expression (in arbitrary units). c The ranked averages of the two values for normalized GFP expression for each of the 21,355 RNAi clones tested (21,223 unique clones, 132 present in duplicate), on a log scale. d The averages, on a linear scale, of the two values for normalized GFP expression for the last 1,355 RNAi clones. The 295 clones that were retested in a second round are indicated in red. e The averages, on a linear scale, of the two values for normalized GFP expression for the first 3000 RNAi clones. The 966 and 360 clones that passed first and second round screening are indicated in red and green, respectively. The results for selected known signaling components are indicated in black
Identification of clones that provoke an exaggerated response
Innate immune responses are limited by negative regulators that contribute to protecting hosts from the collateral damage of their own effector mechanisms [24, 25]. There is emerging evidence that excess NLP-29 can damage host tissue (Dong Yan, Duke University, personal communication). With the aim of identifying negative regulators of the response, in a first step, we retained 295 clones that provoked an average increase of nlp-29p::gfp expression of 30 % or more, but that either did not increase the expression of a control transgene, the constitutive epidermal reporter col-12p::dsRed, nor the average size of the worms, or if they did, the increase was less than 30 % (Additional file 2: Table S2; Fig. 1d). Inactivation of numerous genes that affect molting, such as pan-1 [26], the integrity of the cuticle, including dpy-9, osm-11 [2], and acs-3 [27], or fatty acid metabolism (e.g., fasn-1), is known to provoke the “peptide expression no infection”, or Peni phenotype: an elevation of nlp-29p::gfp expression in the absence of infection [8]. This is associated with an exaggeratedly high expression after infection too [8], which we call here the Hipi phenotype (for hyper-induction of peptide expression after infection). To identify clones that caused only a Hipi phenotype, the 295 clones were retested in quadruplicate for their effect on nlp-29p::gfp expression, both with and without infection. Using cut-offs that captured all the positive controls (fasn-1 and pan-1) but none of the negative controls (sta-1 and K04G11.4 [7]), we removed 21 clones that robustly caused a Peni phenotype (Additional file 3: Table S3). Their characterization will be the subject of a future study.
We then used a simple cut-off to classify 28 clones as being capable of causing a strong Hipi phenotype (termed, “Hipi clones”; Additional file 3: Table S3). We used sequencing and Clone Mapper [28] to verify the identity of the Hipi clones and determine their putative target genes (Additional file 3: Table S3). These included bus-2 and bus-12, which respectively encode a galactosyltransferase and a sugar transporter required for the post-translational modification of surface-exposed proteins [29, 30]. In a detailed analysis, we previously demonstrated that abrogating bus-2 or bus-12 function increases spore binding to the nematode cuticle [31]. To address the question of whether the infectious burden of spores affected the strength of reporter gene expression, we exposed wild-type worms carrying nlp-29p::gfp to varying doses of D. coniospora spores. There was a clear relationship between the concentration of spores and the level of GFP expression (Fig. 2a).
Fig. 2.
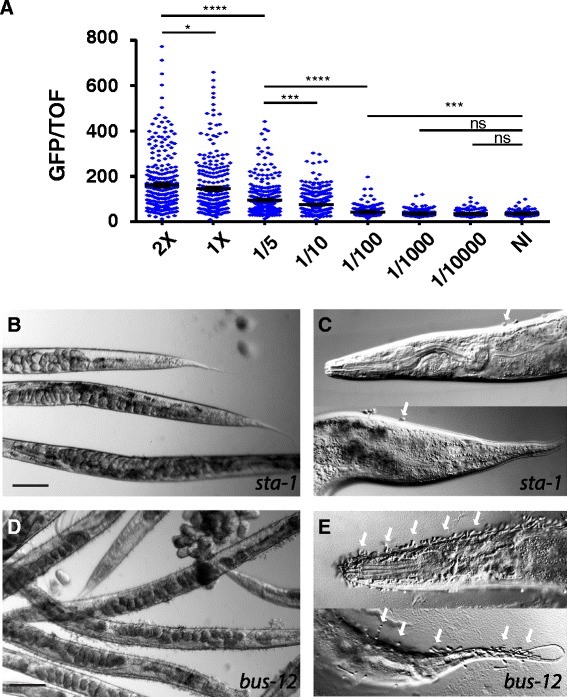
Infection burden affects the strength of the innate immune response. a Normalized fluorescence ratio for worms infected for 18 h with the indicated dilutions of a solution of fresh D. coniospora spores, compared to non-infected (NI) worms. In each sample, a minimum of 230 worms was analyzed. The bar indicates the mean value. Since spore virulence depends on the age of the spores and of the plate from which they were harvested [121], the absolute spore concentration is not an informative measure and is not shown here. Comparisons between selected conditions are shown (Mann–Whitney test); ns, not significant; * P < 0.05; *** P < 0.001; **** P < 0.0001. b–e. Comparison at lower (b, d) and higher (c, e) magnification between worms treated with a control RNAi (sta-1; b, c) or RNAi against bus-12 (d, e). In contrast to the control worms, bus-12(RNAi) animals exhibited a very markedly increased adhesion of spores (white arrows) over the entire body (c), prominently at the head and tail (e). Scale bar in b and d: 50 μm
We therefore conducted a third round of screening, this time directly assessing the adhesion of spores to worms treated with the 28 candidate Hipi clones and, in parallel, the degree of expression of nlp-29p::gfp relative to worms treated with a control RNAi clone targeting sta-1. Half of the clones were again scored as provoking a Hipi phenotype and in each case this was associated with a clear increase in spore binding (Additional file 3: Table S3, Fig. 2b–e). Among the predicted targets of these 14 clones, in addition to bus-2 and bus-12, three other genes are putatively involved in the modification of surface glycans (Table 1). The clone mv_Y38C1AB.5 potentially targets two paralogous genes encoding glycosyltransferases, and is also likely to affect the properties of the cuticle via an effect on surface glycoprotein biosynthesis. Similarly, another predicted target gene, K08E3.5, encodes a uridine triphosphate-glucose-1-phosphate uridylyltransferase, expected to be involved in glycoprotein and glycolipid synthesis. While the connection between spore binding and the remaining target genes is less evident and will require further investigation, these results advance our understanding of the interaction between fungal spores and nematode cuticle and emphasize the cardinal importance of the infection burden in determining the strength of the innate immune response.
Table 1.
Targets of robust Hipi clones
| Gene/sequence name | Brief description |
|---|---|
| bus-2 | Core-1 beta1,3 galactosyltransferaseb |
| bus-12 | Nucleotide-sugar transporterb |
| cpt-6 | Carnitine palmitoyltransferase |
| sdc-2 | Nematode-specific; required for dosage compensation |
| snf-9 | Solute carrier family 6 (SLC6) |
| tkt-1 | Transketolase |
| ykt-6 | v-SNARE |
| C14H10.3 | Pyridoxal-dependent decarboxylase |
| K08E3.5 | Uridine triphosphate:glucose-1-phosphate uridylyltransferaseb |
| F35H12.5 | Epoxide/serine hydrolase |
| K06A9.1 | Nematode-specific; limited similarity to mucin |
| T04G9.4 | Aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl transferase |
| Y38C1AB.1a | Core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferaseb |
| Y38C1AB.5a | Core 1 synthase, glycoprotein-N-acetylgalactosamine 3-beta-galactosyltransferaseb |
| C26B9.3 | Nematode-specific |
aTargeted by a single RNAi clone
bInvolved in carbohydrate metabolism
Identification of clones that abrogate the response
To identify positive regulators of the response, following the first round of screening, we retained clones that reduced the infection-induced expression of nlp-29p::gfp by 20 % or more in both of the tests (i.e., provoked a Nipi phenotype), but excluded those that altered the expression of the control col-12p::dsRed transgene or reduced the average size of the worms more than when we knocked-down the known signaling component rack-1 [9]. The selected 966 Nipi clones were then tested in quadruplicate, and 360 clones giving a robust Nipi phenotype (Additional file 4: Supplementary Methods) were chosen for further study (Fig. 1e and Additional file 5: Table S5; full results available at http://bioinformatics.lif.univ-mrs.fr/RNAiScreen). Among them, 314 clones were predicted to target a single gene, 22 to target two genes each, two to target three genes each, and eight clones an average of 10 each (due to targeting of multigene families of very similar sequence, e.g., the his histone genes; see below). For the remaining 14 clones, no target could be defined because of sequence ambiguity (Additional file 5: Table S5). A total of 404 genes were thus identified as potential targets for these clones, with 15 genes, not counting the many his genes, satisfyingly, being hit by more than one clone. The complete RNAi library contained a further 155 clones that potentially hit the same 404 targets (Additional file 5: Table S5). If the clones we have identified are true positives, then these 155 clones should also have been retained. This gives a measure of the efficiency of the screen (360/(360 + 155), 70 %); in common with other RNAi screens (e.g., [32, 33]), false negatives are thus a notable limitation here. Among the identified targets, in addition to rack-1, we noted the presence of nipi-3, nsy-1, pkc-3, sta-2 and tir-1, all previously characterized for their role in regulating antimicrobial peptide gene expression [1, 3, 7, 9]. Further, among the clones, we identified one targeting dcar-1, encoding a GPCR that we demonstrated to be required for the induction of the innate immune response upon infection [20]. We consider this to be a validation of the screening and selection method.
Going from a genome-wide approach to a more focused analysis allowed attention to be paid to the phenotypes of the worms that had been treated with each of the 360 RNAi clones. A number of clones provoked developmental delays and/or lethality under our experimental protocol. We used the quantitative data from the second round of screening to identify 63 clones associated with pronounced phenotypes (see Additional file 6: Table S4 for criteria). Of these 63 clones, 42 had been associated with severe developmental phenotypes in previous RNAi studies (Additional file 6: Table S4). While we cannot formally exclude the possibility that they exercise both an essential developmental role and a specific role in regulating innate immune defenses, because of their pleiotropic effects they were not included in subsequent analyses. This left us with a list of 297 clones, predicted to target 338 genes (Additional file 5: Table S5), including all the previously characterized genes mentioned above. This list contains many potentially interesting genes, such as akir-1, which encodes the worm ortholog of Akirin, a known regulator of innate immunity in flies and mammals [34, 35], the claudin/calcium channel gamma subunit family gene nsy-4, known to act upstream of nsy-1 during neuronal development [36], as well as several genes encoding transcription factors.
Inspection of this list also revealed a potential confounding factor for subsequent analyses. The prediction of targets for an RNAi clone is based on sequence. Several C. elegans gene families contain multiple members with highly similar nucleotide sequences, so that a single RNAi clone can have many potential targets. In addition to the clone sjj_K07F5.1, predicted to hit 15 msp genes, this principally concerned clones targeting histone genes; a total of 45 his genes were identified as potential targets for just eight RNAi clones (Additional file 5: Table S5). In the absence of functional analyses at the single gene level, it is not possible to ascribe the effect of a given RNAi clone to one or multiple targets. Many published genome-wide RNAi screens in C. elegans have reported target genes but not RNAi clones, and therefore potentially suffer from this confounding factor that can lead to biases in analyses. For some of our subsequent analyses, we removed these two gene classes, giving a set of 288 clones potentially targeting 278 (Nipi non-his non-msp) genes (Tables 2 and 3).
Table 2.
Overview of screen results
| Step | Type of clone | Number of clones | Source |
|---|---|---|---|
| Primary screen | Library | 21,223 | Additional file 1: Table S1 |
| Secondary screen for increased reporter gene expression | Candidate | 295 | Additional file 2: Table S2 |
| Peni | 21 | Additional file 3: Table S3 | |
| Candidate Hipi | 28 | Additional file 3: Table S3 | |
| Tertiary screen for increased reporter gene expression | Hipi | 14 | Additional file 3: Table S3 |
| Secondary screen for decreased reporter gene expression | Candidate Nipi | 966 | http://bioinformatics.lif.univ-mrs.fr/RNAiScreen |
| Tertiary screen for decreased reporter gene expression | Nipi | 360 | Additional file 6: Table S4 |
| With pronounced developmental phenotype | 63 | Additional file 6: Table S4 | |
| Retained | Nipi | 297 | Additional file 5: Table S5 |
| Clones targeting his or msp genes | 9 | Additional file 5: Table S5 | |
| Remainder | Nipi -his -msp | 288 | Additional file 5: Table S5 |
Table 3.
Enrichment of functional classes among 278 Nipi genes
| Category/Phenotype upon RNAia | Number over-lapping genes | Total number genes in class | Percentage of MAPK signaling genes in class | Probabilityb |
|---|---|---|---|---|
| 1. Conserved in D. melanogaster c as described in [124] | 169 | 3710 | 84.8 | 3.77 × 10−54 |
| 2. Stimulate microbial aversion behaviorc [26] | 65 | 374 | 33.3 | 1.39 × 10−50 |
| 3. Suppress over-expression of gpdh-1p::gfp seen in osm-8 mutantc,d [72] | 52 | 252 | 24.2 | 7.87 × 10−44 |
| 4. Decrease acdh-1p::gfp expression [73] | 35 | 146 | 6.1 | 3.33 × 10−31 |
| 5. Induce hsp-6p::gfp (mitochondrial UPR) [66] | 30 | 95 | 0.0 | 1.60 × 10−30 |
| 6. Protein expression [125] | 43 | 446 | 9.1 | 1.18 × 10−21 |
| 7. Required for cytoprotective response – four different reporter genesc [49] | 21 | 71 | 24.2 | 3.08 × 10−20 |
| 8. Required for transgene silencing [57] | 50 | 823 | 24.2 | 1.25 × 10−16 |
| 10. Alter RAB-11 sub-cellular localization and transport of the apical membrane protein PEPT-1 [79] | 34 | 426 | 15.2 | 3.40 × 10−14 |
| 11. Synthetic phenotype with lin-35 [126] | 27 | 252 | 6.1 | 3.95 × 10−14 |
| 12. Required for mitochondrial surveillance and response [74] | 14 | 45 | 9.1 | 2.08 × 10−13 |
| 13. Regulators of gpdh-1 expressiond [75] | 18 | 123 | 6.1 | 2.45 × 10−11 |
| 15. Aberrant GFP::PGL-1 phenotypes [76] | 20 | 170 | 12.1 | 6.87 × 10−11 |
| 16. Suppressors of polyglutamine aggregation [77] | 19 | 173 | 9.1 | 9.65 × 10−10 |
| 17. Mitochondrial [125] | 17 | 160 | 0.0 | 2.35 × 10−8 |
| 18. Upregulated after 12 h of dauer recovery [127] | 44 | 1175 | 12.1 | 3.31 × 10−7 |
| 20. Altered expression in zpf-1 mutant [128] | 21 | 333 | 3.0 | 2.24 × 10−6 |
| 21. Down regulated in an ogt-1 mutant [129] | 30 | 670 | 9.1 | 4.02 × 10−6 |
| 22. Altered expression in rde-4 mutant [128] | 17 | 232 | 6.1 | 7.17 × 10−6 |
| 23. Induce hsp-70p::gfp [130] | 9 | 52 | 9.1 | 1.03 × 10−5 |
| 24. Genes connected to miRNA function [131, 132] | 8 | 42 | 9.1 | 2.79 × 10−5 |
| 25. Increase longevity [133] | 9 | 66 | 3.0 | 8.78 × 10−5 |
| 26. Induced after 24 h of D. coniospora infection (cDNA microarrays) [2] | 21 | 419 | 12.1 | 1.12 × 10−4 |
| 27. Confer hypoxia-resistance [134] | 14 | 198 | 9.1 | 1.98 × 10−4 |
| 29. Induce irg-1p::gfp [59] | 10 | 102 | 0.0 | 4.37 × 10−4 |
| 31. Energy generation [125] | 10 | 104 | 3.0 | 5.23 × 10−4 |
aThe numbers refer to the order of the classes in the complete analysis; some redundant or similar classes have been removed (see Additional file 7: Table S6 for complete data)
bBonferroni-corrected Fischer exact score
cClass significantly enriched in the group of 33 “non-his non-msp MAPK pathway genes”
dClass related to osmotic stress response
The central role of MAPK signaling
The main signaling pathway known to regulate nlp-29 expression has at its core a conserved p38 MAPK cassette involving tir-1, nsy-1, sek-1, and pmk-1 [2, 3]. The identification in the screen of tir-1 and nsy-1 was an important validation of the approach. The failure to identify sek-1 represents a clear example of a false negative arising because of our deliberate selection strategy. The corresponding clone (sjj_R03G5.2) did not pass the first round of screening since it only abrogated reporter gene expression in one of the two trials. For pmk-1, as the corresponding clone (mv_B0218.3) surprisingly did not provoke a Nipi phenotype, we sequenced it. In common with the 54/388 candidate clones that we sequenced (4/28 Hipi and 50/360 Nipi clones, respectively; Additional file 3: Tables S3 and Additional file 5: Table S5), in our cherry-picked library, the clone annotated as mv_B0218.3 did not contain the expected insert. We returned to our original copy of the Vidal RNAi collection; the corresponding clone there was also incorrect. This is another of the known drawbacks in large-scale RNAi screens: the impossibility of being certain of the identity of every clone.
MAPK signaling is central to innate immune defense in many species, including C. elegans and vertebrates [25, 37]. Catalogs of proteins and genes involved in the regulation of MAPK pathways are available for yeast [38], flies [39], and human [40]. Using the Drosophila RNAi Screening Center (DRSC) Integrative Ortholog Prediction Tool [41], we compared the worm orthologs of the members of these lists with our hits. More than 1/5 of the candidates (76/338 Nipi genes; 22 %) had previously been associated with MAPK signaling in at least one other species. The constitution of the list of potential components of MAPK signaling was skewed by the inclusion of multiple histone genes (43/76; Additional file 5: Table S5). Nevertheless, the identification of 33/278 (non-his non-msp) MAPK-related genes (Table 4) reinforces the idea that MAPK signaling is central to the regulation of AMP gene expression in C. elegans epidermis and underscores the conserved nature of this core signaling process.
Table 4.
Nipi genes linked to MAPK signaling
| Gene/sequence name | Brief description |
|---|---|
| ccf-1 | Subunit 7 of CCR4-NOT transcription complex |
| cct-3 | Gamma subunit of eukaryotic cytosolic (‘T complex’) chaperonin |
| cic-1 | Cyclin C |
| cyl-1 | Cyclin L |
| dic-1 | DEAD/H BOX 26; mitochondrial |
| dnc-1 | Dynactin complex subunit p150; DNC-1 is located at cortical microtubule attachment sites |
| ego-2 | Bro1 domain-containing protein; positive regulator Notch signaling |
| exos-9 | Exosome component 9 |
| ftt-2 | 14-3-3 protein |
| hda-1 | Histone deacetylase 1 |
| ima-3 | Importin alpha nuclear transport factor |
| kin-20 | Casein kinase |
| let-92 | Catalytic subunit of PP2A (protein phosphatase 2A) |
| nap-1 | NAP (Nucleosome Assembly Protein) family |
| npp-1 | Nucleoporin |
| npp-10 | Nucleoporin |
| ogdh-1 | 2-oxoglutarate dehydrogenase, mitochondrial |
| puf-9 | PUMILIO RNA-binding protein |
| pyp-1 | Inorganic pyrophosphatase; predicted to participate in nucleosome remodeling |
| rack-1 | Receptor for Activated C Kinase; homolog of G beta [9] |
| rbpl-1 | E3 ubiquitin-protein ligase RBBP6 (Retinoblastoma binding protein 6) |
| rps-26 | Small ribosomal subunit S26 |
| tba-2 | Alpha-tubulin |
| tba-4 | Alpha-tubulin |
| unc-37 | Gro/TLE (Groucho/transducin-like enhancer) |
| vhp-1 | MAP kinase phosphatase |
| wnk-1 | With no lysine kinase [8] |
| xpo-2 | Importin-beta |
| F19F109 | U4/U6U5 tri-snRNP-associated protein 1 |
| K12H44 | Signal peptidase complex subunit 3 |
| R1863 | Signal recognition particle receptor subunit beta |
| F20D122 | Germinal-center associated nuclear protein; required for mRNA export |
References are given for genes previously connected to the regulation of nlp-29 expression. Each gene is targeted by a different RNAi clone
To explore the functional relationship of the non-histone candidate genes potentially involved in MAPK signaling, we submitted them to an analysis using WormNet, a phenotype-centric tool that represents known interactions between genes in a list [42]. Of the 33 genes entered, 28 formed a well-connected network (Fig. 3a). On the basis of an input list of genes, WormNet predicts other genes that could be functionally related to them, ranked by probability. Among the top 200 WormNet candidates, there were 53 genes that were found as candidate Nipi genes in our screen but that had not been included in the original query of 33 genes. At first sight, this remarkable enrichment would appear to be a testament to the predictive power of WormNet. Inspection of the results, however, showed that 38 of these 53 genes encode histones (Additional file 7: Table S6), which generally share functional annotations. Nevertheless, 15 non-histone genes that had been found in our screen were identified as potentially linked to the MAPK network (Additional file 7: Table S6), suggesting that other WormNet candidates could also be involved in the regulation of nlp-29p::gfp expression. Further, this analysis underlines the idea that the genes involved in modulating MAPK signaling are embedded within a broader cellular signaling network.
Fig. 3.

MAPK pathway genes involved in regulation of AMP expression. a Interaction network predicted by WormNet for 33 MAPK pathway-related Nipi genes (Additional file 7: Table S6). The genes ego-2, tag-214, vhp-1, and wnk-1 are not connected to any other of the genes; Y73B3A.18 is not included in the WormNet set of genes. These five genes are not shown here. The remaining 28 genes are connected by 77 edges. As for all large-scale data mining, there are obvious omissions, due to incomplete coverage in databases. One example is nsy-1 that encodes a MAP3K [122] but does not appear here. b Knocking down vhp-1 by RNAi provokes a marked reduction of nlp-29p::gfp reporter gene expression in IG274 worms carrying the frIs7 transgene infected by D. coniospora. The graph shows the quantification of fluorescence of worms treated with control (CT: K04G11.4; blue; n = 296) or vhp-1 (red; n = 258) RNAi. The green fluorescence and length are plotted in arbitrary, but constant units. c–g A cell-non-autonomous regulatory role for vhp-1. Whereas following systemic knock-down of sta-1 (c) or vhp-1 (d), there was no detectable expression of the nlp-29p::gfp reporter gene in IG274 worms carrying the frIs7 transgene in the absence of infection, knocking-down vhp-1 (f) but not sta-1 (e) specifically in the epidermis in strain IG1502 led to ectopic expression of GFP in the nematode intestine. This expression was most pronounced in the posterior intestinal cells (g). The red fluorescence in the pharynx in (e–g) reflects the presence of an additional transgenic marker. Worms were observed at the L4 stage in all cases. Green and red fluorescence are visualized simultaneously with a long pass GFP filter. Scale bar: 50 μm
One of the five MAPK-related candidate target genes that was not part of the MAPK network predicted by WormNet (Fig. 3a) was vhp-1, which encodes a member of the VH1 dual-specificity phosphatase family. Since vhp-1 has been described as a negative regulator of the p38 pathway [43], we would not have expected to have found it as a Nipi gene. The effect of vhp-1(RNAi) on nlp-29p::gfp expression after D. coniospora infection was very pronounced (Fig. 3b). To determine whether this effect was cell-autonomous, we made use of an epidermis-specific RNAi strain, IG1502 [20]. To our surprise, vhp-1(RNAi) provoked a substantial ectopic expression of gfp in the intestine in this strain, even in the absence of infection (Fig. 3c–f). The intestine of C. elegans is functionally regionalized [44]; vhp-1(RNAi)-induced nlp-29p::gfp expression was strongest in the posterior intestinal cells (Fig. 3g). Thus, reducing the activity, specifically in the epidermis, of a phosphatase previously shown to down-regulate p38 MAPK signaling leads to ectopic gene expression of a p38 MAPK target in a distant tissue.
Global functional analyses
Returning to a more global analysis, when we submitted the list of Nipi genes (except his and msp genes) to a WormNet analysis, 231 formed an intensely interconnected network with an average of 11.1 edges/node (Fig. 4). To characterize this broader cellular signaling network, we ran the lists of candidate targets through an Expression Analysis Systematic Explorer (EASE) analysis [45], using our in-house database of functional annotations [46]. A number of classes were significantly enriched (P < 10–3; Additional file 7: Table S6). Several were derived from early genome-wide ChIP-seq studies produced by the model organism encyclopedia of DNA elements (modENCODE) consortium. We did not exploit this data further since its reliability has recently been questioned by the consortium itself [47].
Fig. 4.

Nipi genes are connected by a dense network of interactions. a Interaction network predicted by WormNet for 233 Nipi genes, with 33 MAPK pathway-related genes shown in yellow. b Close-up view of one part of the network shown in b, highlighting two interconnected genes that are apart from the main network and illustrating the relative lack of connections for the known signaling components nsy-1 and nipi-3, which are connected to tir-1. The gene nsy-4 is partially obscured; its position is indicated with an asterisk
Most of the genes (245/278) were found in at least one significantly enriched class (Additional file 7: Table S6). The different classes were more or less related (Fig. 5); for example, genes associated with the stability, localization, and function of P granules (class 15 in Table 3) clustered with those associated with Rab11-positive recycling endosome-linked transport (class 10). Many genes belonged to several functional classes; the most frequently found (in 12/34 classes, Additional file 7: Table S6) encodes the E2 ubiquitin-conjugating enzyme LET-70. The most significantly enriched class was for genes defined as being conserved in Drosophila (through a pairwise comparison with C. elegans), followed by those previously described as stimulating microbial aversion behavior when knocked down by RNAi (classes 1 and 2 in Table 3, respectively). The latter includes genes involved in diverse essential cellular functions [26]. Several other classes linked to stress responses were also highly enriched. One of the most populated classes (50/278) was of genes previously characterized as being necessary for RNAi (class 8 in Table 3). This surprising result was corroborated by an analysis of enriched GO terms using GOrilla [48] (Fig. 6; Additional file 7: Table S6), and is discussed below. The proportion of non-histone MAPK signaling genes present in each class varied widely; for “microbial aversion” (class 2) it was 11/33, but was 0/33 for three classes (classes 5, 17 and 29; Table 3). This list of 33 MAPK-related genes overlapped well (8/33) with the list of genes in class 7, reported to be required for multiple cytoprotective responses (i.e., regulators of gst-4 (detoxification), hsp-4 (endoplasmic reticulum unfolded protein response (UPR)), hsp-6 (mitochondrial UPR,UPRmt) and sod-3 (reactive oxygen species (ROS) response) [49]). There was an equivalent overlap (8/33) with the targets of clones able to suppress over-expression of gpdh-1p::gfp seen in osm-8 mutant worms (class 3). As discussed further below, this gives a further indication of the degree of imbrication of MAPK signaling with different cellular homeostatic processes.
Fig. 5.

Relationship between different functional classes. Hierarchical clustering of genes and functional classes (see Table 3 for class labels; full data in Additional file 7: Table S6; 15 genes from the 245 candidates, present only in one or more of the classes 1, 9, 14, and 31 are not shown). The presence of a gene in a class is represented by a red rectangle, its absence in blue
Fig. 6.

Enrichment of gene ontology terms for Nipi genes. Overview (a) and close-up views (b) of GOrilla analysis of Nipi genes. The color indicates the degree of enrichment, from red (very significantly enriched) to white (not enriched). The regions enlarged in b are indicated by the boxes in a. In addition to the three categories shown in b, in a, the other very significantly enriched processes, between “RNAi” and “Electron transport chain” are generic (“regulation of multicellular organismal process” and “positive regulation of multicellular organismal process”); those on the right are related to development. See also Additional file 7: Table S6
We previously reported a potential role for endocytosis in the induction of nlp-29 expression provoked by fungal infection [7]. Consistent with this, the GOrilla analysis (Fig. 6a; Additional file 7: Table S6), in common with EASE, also highlighted the role of endocytosis in the regulation of nlp-29p::gfp reporter gene expression. They also both drew attention to the potential role of mitochondria in regulating the innate immune response (Fig. 6 and Table 3). For example, all the genes present in at least three of the top four EASE functional classes encode mitochondrial proteins (Additional file 7: Table S6). A Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis [50] of the targets of the 297 Nipi clones (Additional file 5: Table S5) assigned 22 to the category oxidative phosphorylation, corresponding to proteins present in four of the five complexes of the mitochondrial electron transport chain. Extending the analysis to include the targets of the 63 clones that provoked a severe developmental phenotype increased the total number of mitochondrial proteins to 27, covering all five electron transport chain complexes (Fig. 7). The role of mitochondria in the regulation of nlp-29 expression is explored further below.
Fig. 7.
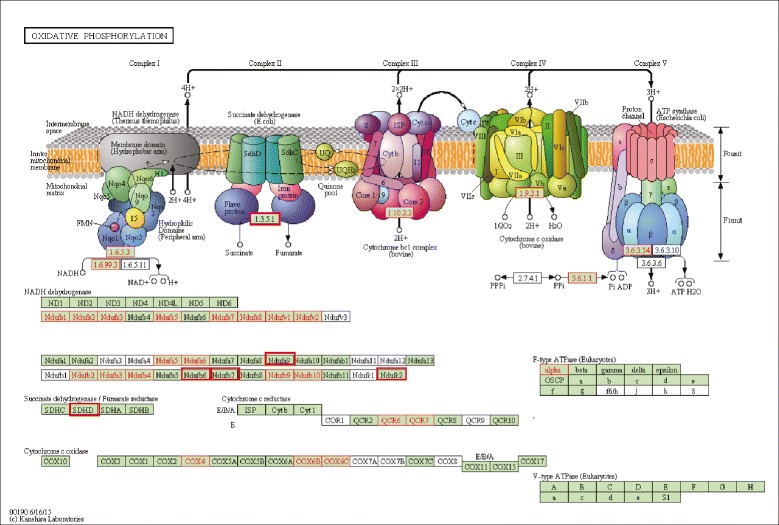
Participation of Nipi gene products in oxidative phosphorylation. The upper part of the figure shows a KEGG-derived schematic representation of the successive complexes that make up the mitochondrial electron transport chain (KEGG pathway cel00190) on which the complexes that include any proteins corresponding to Nipi clone targets are highlighted. For any targets of the 297 clones that did not provoke a strong developmental phenotype, the E.C. name for each complex is boxed in red with red text; for the targets of the remaining 63 clones (Table 2; Additional file 5: Table S5) there is one additional complex (E.C. 1.3.5.1, succinate dehydrogenase), boxed in red with black text. The lower part of the figure shows the individual protein components of the different complexes, annotated as above. Proteins for which there is no KEGG-assigned ortholog in C. elegans are uncolored. A full explanation of the symbols can be found at http://www.kegg.jp/kegg/document/help_pathway.html
Foundling and orphan genes
A total of 33 genes were not found to be associated with any EASE class (Table 5; Additional file 7: Table S6). A WormNet analysis failed to reveal any significant connection between the members of this group (area under the curve = 0.5; P = 0.3). Inspection of this list, however, revealed it to include two genes known to play specific and important roles in the regulation of antimicrobial peptide gene expression, namely dcar-1 and sta-2, which encode, respectively, a DAMP receptor [20] and a STAT-like transcription factor [7]. Our EASE database [46], which currently contains more than 500 classes, has been built up by manual annotation and is necessarily biased to categories that we expect to be of interest in our studies. In an attempt to overcome this limitation, we assembled a far more complete and unbiased collection of functional classes, extracting data automatically from multiple sources including WormBase, FlyBase [51], KEGG [52], and the relevant RNAi databases [53, 54], and combined this with our EASE database to give a collection of more than 3700 classes of genes. Even using this collection, we failed to find any significant enrichment for the group of 33 genes.
Table 5.
Foundling Nipi genes
| Gene/sequence name | Brief description |
|---|---|
| dcar-1 | GPCR receptor; activated by HPLA [20] |
| elo-2 | Palmitic acid elongase; ELO-2 is required with ELO-1 for 20-carbon PUFA production |
| frpr-11 | GPCR of the FMRFamide Peptide Receptor family; unlike dcar-1, acts downstream of gpa-12 and blocks osmotic induction of nlp-29 [20] |
| ins-6 | Predicted type-beta insulin-like peptide |
| mltn-12 | MLt-TeN (mlt-10) related; MLT-10 is a nematode-specific protein required for ecdysis |
| nas-37 | Astacin-class metalloprotease required for ecdysis; N-terminal signal sequence followed by an Astacin protease domain and three protein-binding domains (EGF-like, CUB, and thrombospondin) |
| srsx-25 | GPCR of the serpentine receptor class SX |
| srv-21 | GPCR of the serpentine receptor class V; unlike dcar-1, acts downstream of gpa-12 and blocks osmotic induction of nlp-29 [20] |
| sta-2 | STAT family of transcription factor [7] |
| F56A8.5 | Protein containing an F-box |
| K08C9.5 | Protein containing an F-box |
| C33D9.3 | Nematode-specific |
| F27C8.2a | Nematode-specific |
| F27C8.3a | Nematode-specific |
| F34H10.1 | Ubiquitin/40S ribosomal protein S27a fusion protein |
| K08C9.7 | Ubiquitin/40S ribosomal protein S27a fusion protein |
| R186.8 | 39S ribosomal protein L33, mitochondrial |
| T08D2.2b | Similar to C-terminal half of UDP-N-acetylglucosamine-dolichyl-phosphate N-acetylglucosaminephosphotransferase |
| T08D2.6b | YIPF4-like; YIPF4, poorly characterized membrane spanning protein; in yeast, interacts with Rab GTPases |
| Y60A3A.19 | YIPF4-like.;YIPF4, poorly characterized membrane spanning protein; in yeast, interacts with Rab GTPases |
| C42C1.3 | Nematode-specific |
| F35F10.14 | Nematode-specific |
| F41H8.1c | Nematode-specific domain: GPCR of the serpentine receptor class BC |
| K09C6.6c | Nematode-specific domain: GPCR of the serpentine receptor class BC |
| K09C6.10c | Nematode-specific |
| F42C5.9 | Actin |
| F45E4.5 | Nematode-specific |
| F49H12.5 | Thioredoxin domain-containing protein 12-like |
| F52C6.13 | Nematode-specific |
| tsen-54 | N-terminal half similar to that of tRNA-splicing endonuclease subunit Sen54 |
| Y39G10AR.7 | Nematode-specific |
| Y51H7BR.7 | Contains Spec3 domain, like Drosophila Stumbled |
| Y67D8C.22 | Clarin-like |
a,bTargeted by a single mv clone
cPotential targets of the same sjj clone
References are given for genes previous connected to the regulation of nlp-29 expression; see Additional file 7: Table S6
Genes for which homologues are found only in a specific taxonomic group, irrespective of the level (e.g., animals, nematodes, or Caenorhabditis), are called taxonomically-restricted genes (TRGs). TRGs that are restricted to a very narrow taxonomic group, generally a species, can be called orphan genes [55]. Here, we have described a group of genes for which there is no pertinent functional data in a wide range of publically available databases. In a sense, it is as if these genes have been abandoned. By analogy with the term orphan gene, we apply here the term “foundling gene” to them.
The failure to connect the foundling genes could be because they are not in reality linked to each other in any way, or, the explanation we favor, they collectively play specific roles in nematode epidermal defense against fungal infection, which has not hitherto been sufficiently completely described. Such an idea is in line with the pattern of conservation of the 33 foundling genes; more than half are TRGs and encode proteins that are essentially restricted to nematodes. Others are present in a broad range of invertebrate and vertebrate species, while two (WBGene00018063 and WBGene00018670) are currently orphan genes, with no homologs outside C. elegans (Fig. 8; Table S7 ). Genes with similar phylogenetic profiles are more likely to function together in a common biological process [56, 57]. Thus, these diverse patterns of conservation will contribute to elucidating the function of these foundling genes.
Fig. 8.
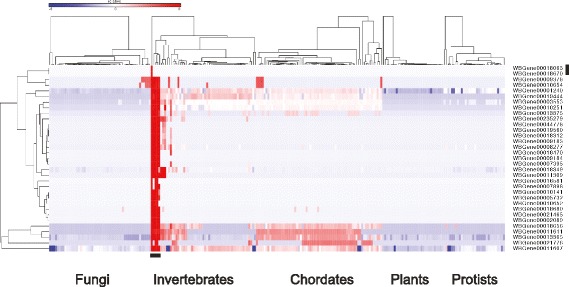
Phylogenetic profiles for 33 foundling genes. Hierarchical clustering of the normalized bit scores for homologs for 33 foundling Nipi genes across the genomes of 238 species present in Ensembl (see Additional file 8: Table S7 for the list of species in the order they appear here). For each of the groups (fungi, invertebrates, etc.), the species are clustered independently. The colour code reflects the relative normalized bit score, from high (red) to low (blue) across the different species. The horizontal bar at the bottom marks the position of the five Caenorhabditis species, from the left to right, C. elegans, C. brenneri, C. briggsae, C. remanei, and C. japonica. Several distinct groups of genes can be discerned, including genes unique to C. elegans (i.e., orphan genes [123]), indicated by the vertical bar on the right
Epistasis and functional analyses with candidate clones
The enriched gene classes included four related to osmotic stress, corresponding to a total of 71 genes (Table 3; Additional file 7: Table S6), consistent with the previously established connection between osmotic stress and antimicrobial peptide gene expression [2, 8, 58]. To investigate this link further, we complemented our in silico analyses with direct assays to test the capacity of the 297 RNAi clones (Table 2) to block the increase in nlp-29p::gfp expression provoked by osmotic stress. Another enriched class was of genes that, when knocked down, provoke the expression of an irg-1p::gfp reporter in the nematode intestine [59]. This category is linked to innate immunity since irg-1 encodes a putative antibacterial effector protein, induced in the intestine upon infection with pathogenic Pseudomonas aeruginosa by zip-2, which promotes defense [59]. The 297 clones were therefore assayed for their capacity to induce the expression of irg-1p::gfp. They were also used in a test of epistasis, by quantitating their potential for abrogation of the elevated nlp-29p::gfp expression associated with a constitutively active form of GPA-12 (GPA-12*), the alpha subunit of a heterotrimeric G protein that acts between DCAR-1 and TIR-1 [5].
More than one third of the clones tested (100/297, 34 %; termed here, “I-clones”) provoked a marked increase in irg-1p::gfp. When compared to the results of Dunbar et al. [59], there was a very satisfactory overlap, with identification of 31 of a possible 44 genes previously associated with the phenotype. This figure is similar to that reported by others when comparing screens performed in different laboratories (75 %; [60]). Importantly, many additional candidate negative regulators of irg-1p::gfp expression were identified (Additional file 5: Table S5). This is one of the positive consequences of our having conducted a quantitative screen. These results illustrate how the targets of the Nipi clones can have pleiotropic roles, being positive regulators of an epidermal AMP gene, but negative regulators of an intestinal defense gene. They also suggest that the reciprocal relationship between gene regulation in these two tissues that we found for vhp-1 may reflect a more general phenomenon.
A high proportion (153/297; 51.5 %) of clones abrogated the high constitutive expression of nlp-29p::gfp seen in a strain expressing GPA-12* (“G-clones”). Since the p38 MAPK cassette functions downstream of gpa-12 in the regulation of nlp-29p::gfp [7, 9], clones targeting components of the MAPK signaling cascade would be expected to be especially well-represented in this category. In fact, this was not the case, as only 18 of the 43 clones (42 %) were G-clones (Additional file 5: Table S5). This could be interpreted to mean that a substantial number of G-clones provoked the phenotype for relatively non-specific reasons. Indeed, of the 153 G-clones, 104 (68 %) were found to block the induction of nlp-29p::gfp normally provoked by osmotic stress, a markedly higher number than expected, since, in the complete set of 297 clones, there were 131 (44 %) in this category (“O-clones”; Additional file 5: Table S5). Further, 21 of the G-clones targeted genes required for the expression of an acdh-1p::gfp reporter gene (class 4, Table 3); acdh-1 encodes a key enzyme in fatty acid metabolism. Finally, a quarter of the targets of the O-clones (37/147) had previously been associated with the response of C. elegans to osmotic stress (Additional file 7: Table S6). The significance of these different overlaps is discussed below.
As described above, an alteration of fungal spore adhesion can lead to a change in defense gene expression. Assaying spore adhesion to worms cultured on RNAi clones proved experimentally challenging because of the variable phenotypes routinely seen with RNAi and especially since there were so many clones to test. We did, however, identify 12 clones that appeared to affect, to a greater or lesser degree, this initial step of the infection process. We were surprised to discover that six were G-clones and five were O-clones, suggesting that the target genes might well play an additional role in governing nlp-29p::gfp expression (Additional file 9: Table S8). As a consequence, we did not remove these clones (representing < 5 % of the total) from our lists.
Conserved protein complexes
Functional modules frequently correspond to physical protein complexes. Several studies have defined a variety of protein complexes from different species. One recent report provided more than one million putative high-confidence co-complex interactions present broadly across animal species [61]. Combining this with data from yeast [62–64], and having identified the C. elegans orthologues of the component proteins when necessary, we compiled a collection of 1925 predicted C. elegans protein complexes (Additional file 10: Table S9). We then associated each of the predicted targets of the Nipi RNAi clones with the different complexes. We focused on complexes with at least three components for which we had picked up more than half of the components in our screen (Additional file 10: Table S9). There was an over-representation of the eukaryotic translation initiation factor (eIF) 2B complex and 66S pre-ribosomal particles, suggesting an important role for protein translation. There was also enrichment for components of the carbon catabolite repression 4-negative regulator of transcription (CCR4-NOT) complex, which is a major mRNA deadenylase, linked to mRNA degradation and general transcriptional regulation, among other functions [65]. We discuss these observations below. The analysis also indicated that there was enrichment in several mitochondrial complexes (Fig. 9). This is consistent with the KEGG analysis described above (Fig. 7), and we focused our attention on this class of gene.
Fig. 9.

Enrichment of proteins encoded by Nipi genes in selected protein complexes. The components of several known complexes are shown, namely, clockwise from top left, with the complex ID(s) in brackets (Additional file 10: Table S9), CCR4-NOT (YChrMod1004), eukaryotic translation initiation factor 2B complex (195; AMMC1231), mitochondrial ETC (218; AMMC1876), 66S pre-ribosomal particles (169; AMMC1267), mitochondrial complex I (65; AMMC1203), and in the center, mitochondrial oxoglutarate dehydrogenase complex (OGDC; YPC_CON1369). Proteins in dark green correspond to members of the final set of 338 Nipi genes, those in light green and orange to the presumed targets of clones that passed the first round of selection or that gave a single positive result in the first round, respectively. The clone that potentially knocks down lpd-5 (corresponding protein in red) abrogated nlp-29p::gfp expression but had a marked effect on development and reduced col-12p::dsRed expression. The remaining proteins are shown in blue
Intestinal UPRmt inhibits epidermal AMP expression
A total of 30 genes identified in our screen have been shown to induce a mitochondrial UPR (UPRmt) when inactivated [66]. For example, the well-characterized spg-7 that encodes a mitochondrial metalloprotease, was picked up with two independent RNAi clones in our screen (Additional file 5: Table S5). This suggested that activation of the UPRmt could block the expression of antimicrobial peptide genes in the epidermis. At the same time, in contrast to intestinal infection with P. aeruginosa [67], infection of young adult C. elegans by D. coniospora does not provoke the UPRmt since the expression of the hallmark genes hsp-6 and hsp-60 is unchanged [2, 46].
Given the links that exist between the UPRmt and antibacterial defenses in C. elegans [68], we decided to explore in more depth the relationship between the response to fungal infection and the UPRmt. As a first step, to validate the results of the screen obtained with the reporter construct, we used qRT-PCR to assay the level of the endogenous nlp-29 transcript following knock-down of five candidate genes, all associated with the activation of other stress reporter transgenes (Additional file 7: Table S6), including spg-7, a well-established means of inducing the UPRmt [69]. Inactivation of four of them (ant-1.1, atp-4, spg-7, and ucr-1) abrogated nlp-29 gene expression after infection to the same degree as the positive control, dcar-1, while knocking down gas-1 did not have a statistically significant effect (Fig. 10a).
Fig. 10.

Cell autonomous and non-autonomous mechanisms influence nlp-29 expression. Quantitative RT-PCR analysis of nlp-29 gene expression level showing the infection-associated “fold induction” (infected/non-infected values) in worms treated with positive and negative RNAi control clones (targeting dcar-1 and sta-1, respectively) or clones that provoke a UPRmt (targeting ant-1.1, atp-4, spg-7, ucr-1, and gas-1) in a wild-type (a) or the intestine-specific RNAi strain MGH171 (b) for 30 h before infection with D. coniospora for 18 h. Results are the average (± SD) from four and three experiments, respectively (see Additional file 11: Table S10). The difference between control and gas-1(RNAi) is not significant (ns) in either strain; * P < 0.05 (unpaired t-test). The SD for dcar-1 in (b) is 14. c Simplified model of pathways and processes involved in the regulation of nlp-29. The screen identified Hipi genes that modulate the adhesion of spores to the worm cuticle, and Nipi genes (central box) required for the expression of nlp-29 upon osmotic stress and infection (purple), or only after infection (yellow), acting downstream (below horizontal line) or upstream of or parallel to GPA-12. Only a very limited number of genes are shown; those in bold were identified in the screen. The Nipi genes fall into multiple functional categories; some are listed on the left, positioned arbitrarily; pointed and flat arrows indicate positive and negative regulation, respectively
In C. elegans, the UPRmt can involve trans-tissue signaling (reviewed in [12]). Thus, for example, provoking an UPRmt just in neurons leads to an UPRmt in the intestine [70]. To address the question of whether the inhibitory effect of the UPRmt on nlp-29 gene expression might also be cell non-autonomous, we assayed the effect of knocking down the same five candidate genes by RNAi specifically in the intestine in the strain MGH171 [26]. In this case, in contrast to intestinal knockdown of dcar-1(RNAi), which gave the same average level of expression of nlp-29 as sta-1(RNAi), consistent with dcar-1’s cell autonomously function [20], intestinal RNAi of ant-1.1, atp-4, spg-7, and ucr-1 was associated with an abrogation of nlp-29 gene expression following D. coniospora infection, while gas-1(RNAi) again did not provoke a statistically significant effect (Fig. 10b). Overall, our results indicate that provoking the UPRmt in the intestine reduces the induction of an antimicrobial peptide gene in the epidermis (Fig. 10c).
Discussion
Qualitative versus quantitative RNAi screens
Genome-wide RNAi screens have been performed in C. elegans for more than a decade. Their experimental basis is relatively straightforward, since RNAi by feeding is an effective technique in worms [71]. In a number of cases, the read-out has been the effect of RNAi on the expression of a reporter gene or the localization of a chimeric reporter protein, to address a specific biological question (e.g., [49, 57, 59, 66, 72–77]). Generally, these have been visual screens. Despite certain advantages [78], visual screens include an element of subjective judgment, lack discriminatory power, and are best suited to identifying clones that provoke a marked phenotype. These will generally target genes at the central nodes of a signaling network. Full understanding of regulatory mechanisms also requires, however, the identification of genes that exert only a minor effect [23].
An alternative is to undertake automated quantitative screens. These require specialized equipment and tools for data storage and analysis [18, 19, 28, 79–82] and are thus more difficult to put in place. Further, they also suffer from the intrinsic variability of RNAi, which cannot be adequately accounted for using formal statistical analyses (Thomas Richardson, University of Washington, personal communication). Coupled with the continuous distribution of the results, this renders the definition of candidates somewhat arbitrary. In this study, as discussed in more detail elsewhere [83], we used the results for clones targeting genes known to be important for the regulation of nlp-29 to establish cut-offs, and in the first round of screening for Nipi clones, we privileged those giving a reproducible effect. Similarly, the cut-offs we used to identify Peni and Hipi clones were based more on criteria of reproducibility rather than strict statistical criteria. The thresholds we adopted will necessarily determine the candidate genes identified, and could bias our global analyses. We have provided, however, for the first time, via a dedicated web interface, the complete set of results for the two rounds of selective screening, measuring multiple parameters for individual worms from each population. Not every clone in the library will contain the expected insert. Extrapolating from our sequencing of 388 clones, which revealed an error rate of 14 %, around 3000 clones in the complete library might be incorrect. With this caveat in mind, the complete set of results contains a substantial amount of information that we have not attempted to exploit, for example, linked to inter-individual variability of gene expression, terminal epidermal cell fate determination, or simply genes that affect the development and size of C. elegans. It also will be an important resource for those wishing to develop new analytical methods; these would be required to leverage the intrinsically variable quantitative data for subsequent analyses.
Genes affecting spore adhesion
One class of genes to emerge from this screen is the Hipi genes that affect the initial adhesion of D. coniospora spores to C. elegans. This requires contact between the spores’ adhesive bud and the outermost layer of the nematode cuticle, the surface coat. In contrast to the underlying collagen- and cuticlin-rich cuticle, the surface coat is rich in structural glycoproteins, including mucins [84, 85]. Two of the candidate Hipi genes, bus-2 and bus-12, were known to affect the surface coat. Their role in adhesion of D. coniospora has been investigated using the corresponding mutants [31]. These were originally isolated because, unlike wild-type worms, they were not susceptible to infection by the bacterial pathogen Microbacterium nematophilum that normally adheres to specific areas of the worm cuticle [86]. Both genes are important for the post-translational modification of surface-exposed proteins [29, 30]. Several other Hipi genes encode conserved enzymes. Their precise role in mediating spore adhesion will require detailed study but, as mentioned above, they too might alter surface protein maturation. Another candidate, K06A9.1, is nematode-specific. It is predicted to encode several protein isoforms, including one of more than 2200 amino acids, comprising 22 degenerate 81 amino acid repeats. Taken together with its distant similarity to mucins, this suggests that it could be a component of the surface coat.
The gene ykt-6 encodes the worm ortholog of Ykt6p, a v-SNARE essential for endoplasmic reticulum-Golgi transport [87]. It could be required for the correct transport of surface proteins. On the other hand, ykt-6 has been linked to insulin signaling in C. elegans [88] and, interestingly, ins-6, which encodes an insulin-like peptide, was identified as a Nipi gene in the current screen (Table 5). An ins-6 loss-of-function mutant, however, did not display a Nipi phenotype (unpublished results). A lack of concordance between phenotypes observed using RNAi and mutant strains has previously been reported (e.g., [89]). The definitive attribution for a role in spore adhesion for the various Hipi genes must therefore await individual genetic validation.
MAPK signaling and osmotic stress responses
The results of the screen reaffirm the central place of p38 MAPK signaling in the regulation of the nlp-29 AMP gene in the epidermis [90] and substantially expand the catalog of genes involved. Many of the same genes are also required not only for xenobiotic detoxification, the UPR, the UPRmt, and the response to ROS [49], but also for the regulation of gpdh-1, a gene that encodes the rate-limiting enzyme in the biosynthesis of the osmoprotectant glycerol [72, 91]. The expression of gpdh-1 does not change following infection with D. coniospora [2, 46], but is elevated upon exposure to high concentrations of salt, via a mechanism that involves inhibition of translation. This is mediated by the general control non-derepressible (GCN-2) kinase signaling pathway that controls eIF-2α phosphorylation and the activity of the with-no-lysine kinase and Ste20 kinases WNK-1 and GCK-3 [92]. In contrast, the results of our screen amply demonstrated that inhibition of translation does not activate nlp-29 expression. Quite the contrary, many clones targeting genes required for translation, including those encoding aminoacyl-tRNA synthetases (hars-1, lars-1, rars-1, tars-1, wars-1) and eIF subunits, were required for expression of nlp-29 after infection. Although we have not yet determined whether these effects are cell autonomous, this AMP gene therefore distinguishes itself from effectors of other stress responses, such as irg-1 and gpdh-1. On the other hand, its expression upon osmotic stress does require wnk-1 and gck-3 [8], which is also the case for gpdh-1 [92]. We show here that the induction of nlp-29 expression after infection also requires wnk-1, acting upstream or in parallel to gpa-12 (Additional file 5: Table S5) and to a lesser extent gck-3 (Fig. 1b). Further, in common with two thirds of the genes that act downstream of gpa-12 (the targets of the G-clones, including hars-1, lars-1, rars-1 and tars-1), we found that nipi-3, which encodes a homolog of Tribbles required for the response to infection [3], is also required for the expression of nlp-29 upon osmotic stress. These results lead to a revision of the infection/osmotic stress dichotomy [8] of our previous models for the regulation of nlp-29 (Fig. 10c).
Cross-tissue communication
One unexpected finding regarding MAPK signaling was that vhp-1(RNAi) abrogates nlp-29 expression, since VHP-1 has been described as a negative regulator of p38 PMK-1 in the nematode intestine [43, 93]. There, the p38 MAPK has a well-characterized role in defense against bacterial pathogens that colonize the gut lumen. We have previously observed that there is an overrepresentation among the genes induced by D. coniospora of genes repressed after infection by the bacteria S. marcescens, E. faecalis, and P. luminescens. This enrichment includes numerous antimicrobial peptide genes of the nlp and cnc classes [46]. In other words, bacterial infection of the gut, which switches on the p38 MAPK pathway, implicating a decrease in VHP-1 activity, abrogates epidermal antimicrobial peptide gene expression. Our current hypothesis is that vhp-1(RNAi) activates the p38 MAPK pathway in the intestine, and that this has the paradoxical consequence of reducing p38 MAPK activity in the epidermis despite a reduction of vhp-1 expression in that tissue too. We currently have no plausible explanation for the observation of ectopic, intestinal expression of nlp-29 following knockdown of vhp-1 only in the epidermis, but note that other analogous examples of cell non-autonomous regulation have been recently reported [78].
A further example of communication between tissues was revealed in our investigation of the impact of the UPRmt on AMP gene expression. Knocking-down, specifically in the intestine, one of several genes known to trigger an UPRmt caused a reduction of nlp gene expression following D. coniospora infection. Recent studies have suggested that intestinal pathogens can provoke the UPRmt in C. elegans and that this switches on defense gene expression in the intestine [67, 74]. The UPRmt is negatively regulated by the Jun kinase KGB-1 [94], which in turn is negatively regulated by VHP-1 [43, 93]. While this mechanism is complemented by another pathway involving ROS-stimulated eIF2α kinase that leads to a reduction in protein translation [95], compromising overall translatory capacity can by itself cause the expression of defense genes such as irg-1 [59, 96] and gpdh-1 [92], but prevents ROS-induced UPRmt [94]. While the precise interplay between this complex series of homeostatic and cellular defense mechanisms is far from being understood [12, 97], these different findings are compatible with a model wherein activation of anti-bacterial defense mechanisms in the intestine, whether directly upon infection with bacterial pathogens, by reducing VHP-1 activity, by reducing protein translation, or following a UPRmt, leads to a suppression of the capacity of the epidermis to express antifungal defense genes. As such, this could constitute a mechanism to ensure an appropriate allocation of resources within the organism, with the aim of concentrating energy to defend one tissue, to the detriment of the capacity of the epidermis to express AMPs.
Fatty acid metabolism and AMP gene expression
Previous studies have suggested a possible link between fatty acid metabolism and innate immunity in C. elegans [8, 27, 98]. This is further reinforced by the fact that both dld-1 and elo-2, respectively encoding a dihydrolipoamide dehydrogenase and a palmitic acid elongase, were identified as Nipi genes in our screen. Further, the expression of acdh-1 and acdh-2, which encode mitochondrial short-chain acyl-CoA dehydrogenases that catalyze the first step of fatty acid beta-oxidation, is markedly reduced when C. elegans is infected either with D. coniospora or with a number of different bacterial intestinal pathogens [46, 99]. The elo-2 paralog elo-3 was previously found to be required for the expression of acdh-1p::gfp [73]. A total of 35 Nipi genes are also regulators of acdh-1p::gfp expression (Table 3). These include the mediator complex gene mdt-15, a major regulator of fatty acid metabolism and longevity [100, 101]. MDT-15 is also required for oxidative stress responses and the induction of specific detoxification genes in response to xenobiotics or heavy metals [102–104]. MDT-15 was recently shown to have a more direct role in innate immunity since it regulates the expression of p38 MAP kinase PMK-1-dependent immune genes and resistance to P. aeruginosa infection [105]. The links between fatty acid metabolism and host defense in C. elegans clearly merit more detailed investigation.
Further functional groups involved in AMP gene expression
Several other groups of functionally related genes were also identified among the Nipi genes. Almost 50 genes had previously been characterized as being necessary for transgene silencing [57]. This is paradoxical since, if RNAi were not efficient in our system, we would not expect an RNAi-dependent reduction in reporter gene expression. Many of the genes in this category clearly play an indirect role in transgene silencing. To give just one example, dpy-4 encodes a cuticle collagen and is required for normal morphology. Whether they play direct roles in modulating nlp-29p::gfp expression remains to be established.
There was a similar overlap of Nipi genes with genes required for the correct sub-cellular localization of the RAB-11, a small GTPase involved in endocytosis, and for transport of the apical membrane protein PEPT-1 [79]. This is consistent with our observation that knocking down dynamin (encoded bydyn-1), which is involved in the scission of newly formed clathrin-coated endocytic vesicle from the cell membrane, or the small GTPase Rab5 (rab-5), which characterizes early endosomes derived from dynamin-dependent and independent endocytosis, abrogates nlp-29 gene expression after infection [7]. Endosomal membranes may function as important platforms for innate immune signaling in C. elegans as in other species [106, 107].
Among the three Nipi genes (icd-1, let-70, and let-92) required both for transgene silencing [57] and endocytosis [79], as mentioned above, the E2 ubiquitin conjugating enzyme gene let-70 is linked to a broad range of cellular and organismal functions. It is noteworthy that we found nine other genes involved in ubiquitination and proteasome-mediated protein catabolism (dcaf-1, hecd-1, pas-3, pas-5, pbs-2, prp-19, rbpl-1, skr-1, usp-39) in the present screen. It is likely therefore that, in C. elegans, ubiquitination plays an important role in regulating innate immune responses, as it does in many species by governing the stability of key signaling molecules [108–111].
Finally, there was enrichment for components of the CCR4-NOT complex. This complex coordinates a variety of cellular processes, acting at all levels of gene expression, including transcription and mRNA or protein stability. It is involved in cellular adaptation to external stress, including the control of the vertebrate innate immune response through the regulation of STAT1 [112]. It may act in a similar manner to influence the activity of the STAT-like transcription factor STA-2 and thereby the expression of AMP genes in C. elegans.
Conservation and innovation in innate immune defenses
In nature, infection represents an extremely strong selection pressure. This is reflected by the evolution of sophisticated host defense mechanisms, driven by the different pathogens that exercise a negative impact on fitness and survival in the environment. In jawed vertebrates, this has led to the emergence of the adaptive immune system, based on a specific collection of genes and mechanisms not found outside the infraphylum [113–115]. Similar specialization involving groups of TRGs involved in immunity is observed in other branches of the animal kingdom [116]. Here, we identified a number of genes required for the expression of an antimicrobial peptide in C. elegans that are restricted to nematodes. They are expected to be part of a lineage-specific defensive innovation. Their further study will contribute to our understanding of the evolution of immunity in C. elegans [117, 118]. Our results also highlighted the links that exist between antimicrobial defenses and the homeostatic mechanisms that counter abiotic stress. This supports an ancient origin for the co-adaptive evolution of stress and innate immune responses (e.g., [119]).
Conclusions
In conclusion, this genome-wide study has allowed the identification of hundreds of genes that modulate the capacity of C. elegans to express the AMP gene nlp-29 following infection with D. coniospora. Not only has it greatly expanded the number of such Nipi genes, but it has also revealed multiple interwoven cellular regulatory mechanisms that impinge on AMP gene expression. Understanding the precise nature of the regulatory activity exercised by the Nipi genes in each of these different functional classes, as well as the many individual genes, will require focused study in the future.
Methods
Nematode strains
All strains were maintained on nematode growth media and fed with E. coli strain OP50. The strain for intestine-specific RNAi, MGH171 sid-1(qt9); alxIs7[vha-6p::SID-1::SL2::gfp] [26] was kindly provided by Justine Melo and the strain AU133 wt; agIs17[irg-1p::gfp;myo-2p::mCherry] [96] by Emily Troemel. Details about the constructions of the strains IG274 (frIs7[nlp-29p::gfp, col-12p::DsRed] IV), IG1389 (frIs7; frIs30[col-19p::gpa-12*,unc-53pB::gfp] I), and the epidermis-specific RNAi strain IG1502 (rde-1(ne219) V; ls[wrt-2p::RDE-1::unc-54 3’utr; myo-2p::RFP3] III; frIs7 IV) are provided elsewhere [3, 5, 20].
A genome-wide RNAi library
In order to cover the maximum number of target genes, as specifically as possible, we combined RNAi clones from the Ahringer genomic [21] and Vidal cDNA [22] libraries. The constitution of the RNAi library was based on the data and tools for target prediction available at the time (WormMart WS220; now retired). If an Ahringer library clone was predicted by WormMart to have more than one primary target, when possible, we added Vidal library clones predicted to target individually any or all of the multiple primary targets. The Ahringer clones were directly redistributed from each 384-well library plate among four daughter 96-well plates. Of the 16,744 clones in our copy of the Ahringer library, 625 failed to grow. We equally sought to replace them with clones from the Vidal library and cherry-picked a total of 5136 clones. Of these, 32 failed to grow, leaving us with a collection of 21,223 clones (Additional file 1: Table S1). Among them, 132 Ahringer clones were present in two wells, so that the combined library included clones in 21,355 wells. This library of 21,355 wells was used in the first round of screening.
Target prediction
Because of limitations in the method used by WormBase to predict the targets of an RNAi clone, as part of this project, we developed the tool CloneMapper [28]. Out of the 21,223 clones, 20,025 were present in CloneMapper, and were predicted to target 16,565 genes (score ≥ 1). For the remaining 1198 clones, despite the known shortcomings [28], we used WormMart WS220 and WormBaseConverter [46] (WS220 to WS240) to identify a further 1304 targets. Combined, the clones are predicted to target 17,415 of the 20,540 protein coding genes (84.8 %) in WS240.
High-throughput RNAi screen
The RNAi screen was performed as previously described in detail [19, 83]. Briefly, synchronized L1 larvae were deposited in 96-well plates containing nematode growth media agar, with a different RNAi clone in each well. After 30 hours at 25 °C, when worms had reached the L3-L4 stage, a fresh solution of D. coniospora spores was added to each well, and worms were harvested 18 hours later for analysis using the COPAS Biosort. All data was stored in a custom-made database (Modul-Bio, Marseille, France) for subsequent analysis. Evaluation of the capacity of RNAi clones to block the increase in nlp-29p::gfp expression provoked by osmotic stress was performed as described [9]. Briefly, following culture on RNAi clones for 48 h, young adult worms were transferred into 96-well U-bottom plates containing 200 μL of 300 mM NaCl and gently agitated for 3 hours at 25 °C before Biosort analysis. Generally, a minimum of 80 synchronized worms were analyzed for size (TOF), extension, and green (GFP) and/or red (dsRed) fluorescence [18]. The inserts of candidate clones were sequenced to establish their identity.
Data analysis and clone selection
Data analysis was performed as previously described in detail [19, 83]. Briefly, in the first round (whole genome) screen, for each well, a mean value for the GFP/TOF ratios for each worm was calculated. From these values, for each plate, a truncated mean (discarding the 25 % lowest and the 25 % highest values) was calculated and used to normalize the average GFP/TOF values for the individual wells, to allow across-plate comparison. Normalized values for TOF (TOF/[truncated mean of TOF]) and dsRed ((dsRed/TOF)/[truncated mean of dsRed/TOF]) were similarly calculated. Details of Nipi clone selection after the second round of screening are given in Supplementary Methods.
Validation of the RNAi screening approach
A full description of the experimental validation of the screening approach can be found in a publicly available PhD thesis [83]. Of note, using the standard feeding protocol, a substantial number of RNAi clones can provoke severe developmental delays and/or larval lethality [21]. In an attempt to circumvent this, we transferred worms from their standard E. coli OP50 diet to RNAi bacteria at the early L3 stage and assayed the same worms when they were adults. Unfortunately, this was not a sufficiently robust method since, of the sequence-verified positive controls we tested, namely pkc-3, rack-1, and sta-2, only sta-2 gave a phenotype [83]. Trying to increase the efficiency of the RNAi by using the RNAi sensitive strain rrf-3 was also unsuccessful [83] since expression from high-copy transgenes is compromised in this background [120].
Analysis of spore adhesion
Worms treated with each of the 28 Hipi clones from the L1 stage were infected as L4s with D. coniospora. To directly correlate spore adhesion and reporter gene expression, worms in the population (n ≥ 30) were visually inspected for their GFP expression, before accessing the adhesion of spores. A score was assigned, taking into account the intrinsic variability in GFP expression associated with infection (see, for example, Fig. 10b). Clones associated with a very high and homogenous induction were assigned a score of 2, clones associated with an induction similar to wild type were assigned a score of 0, and clones associated with an intermediate phenotype assigned a score of 1. Worms were then harvested in 50 mM NaCl, 0.05 % Triton, transferred to 96-well round-bottom well plates, and frozen at –80 °C. Plates were subsequently thawed and the number of spores attached to worms were counted at 230× using a Leica MZ16 stereomicroscope. Clones were assigned to three broad categories, relative to sta-1(RNAi)-treated control worms: 0 = 1–10 spores/worm (same as control); 1 = 10–25 spores; 2 = > 25 spores. A minimum of 30 animals were scored for each clone. Worms treated with the candidate Nipi clones from the L1 stage and infected as L4s with D. coniospora, as above, were analyzed slightly differently. The major part of each sample was analyzed with the Biosort, as above, and for the remainder, the number of spores attached to worms, at the head and vulva, were counted. An adhesion index was calculated: ((number of worms with n > 1 spores at the mouth) + (number of worms with n > 1 spores at the vulva))/(2 × total number worms). Clones associated with a score inferior to that of all control clones, and with a reduction of reporter gene expression greater than 50 % (i.e., loss of spore adhesion was accompanied by a reduction in the observed innate immune response) were selected. Clones selected in both duplicate tests were retained.
Bioinformatic analyses
All analyses used the WS240 WormBase release, unless otherwise stated. Programs were written in Perl and the user interface was developed using HTML, PHP, JavaScript, and MySQL. We used WormNet v3 [42], EASE 2.0 [45] with an in-house database of functional annotations [46], GOrilla [48] with the November 2015 data update, and KEGG [52] release 77.1. For clustering, we used “One minus Pearson correlation” distance matrices within GENE-E (www.broadinstitute.org/cancer/software/GENE-E/).
Data collection
In addition to the previously collected datasets for C. elegans functional classes used in EASE analyses and manually assembled from the literature, including differential transcriptomic and proteomic data, miRNA targets, TF targets etc., further classes were defined using data from a variety of resources.
We extracted all 1094 phenotypes available in WormBase WS246 from ftp://ftp.wormbase.org/pub/wormbase/.
A total of 1221 expression cluster datasets (WS246) were downloaded from ftp://caltech.wormbase.org/pub/wormbase/spell_download/.
We extracted the full list of Drosophila phenotypes from FlyBase release FB2014_06 via http://flybase.org/.bin/cvreport.html?cvterm=FBcv:0000347+childdepth=2, and manually collated closely related classes to give a list of 145 to which we matched the corresponding FlyBase Gene IDs. Using the DRSC Integrative Ortholog Prediction Tool (DIOPT) (http://www.flyrnai.org/cgi-bin/DRSC_orthologs.pl), we identified the C. elegans orthologs for these genes. If more than one ortholog for a given fly gene was predicted, using an in-house perl script, we selected a best hit if the difference in the DIOPT score was greater than or equal to 2 (maximum score 10), but otherwise did not retain a worm ortholog.
Fly RNAi screen data was taken from http://www.flyrnai.org/RNAi_all_hits.txt (downloaded 14-11-14). RNAi clones for which no target gene was listed were excluded. Prediction of C. elegans orthologs was as above.
We downloaded all RNAi screen datasets for Drosophila from version 13 of GenomeRNAi.org [54] and manually collated very similar classes to give 110 datasets. Prediction of C. elegans orthologs was as above.
We extracted the genes corresponding to 136 KEGG_pathways (July 2015) and then converted identifiers to WormBase GeneIDs.
The resulting datasets are available on request.
A separate collection of predicted C. elegans protein-protein complexes was also assembled using experimentally determined protein complexes from other species [61–64]. Prediction of C. elegans orthologs was as above except that, if the difference in the DIOPT score was less than or equal to 2, the top two putative orthologs were retained.
Phylogenetic profiling
To construct phylogenetic profiles, we followed an approach somewhat similar to that of Tabach et al. [57]. We collected data for a wide range of eukaryotic species. We downloaded the complete set of predicted proteins for 66 vertebrates from Ensembl (release 78) and for 55 invertebrates, 53 fungi, 32 plants, and 32 protists from Ensembl genomes (release 25). As many genes have multiple isoforms (e.g., 30,939 for 20,493 protein-coding genes in C. elegans), we chose the longest transcript for each gene. We used BLASTP to compare the proteins predicted for the 33 C. elegans foundling genes against all 238 proteomes and we chose the best hit for each. From this, we generated a “BestHit” matrix (33 × 238), where each entry Cij is the best BLAST bit score of the top hit in species “j” for C. elegans protein “i”. As BLAST bit score depends on protein length, we normalized each bit score by calculating a self-similarity score Cii (by BLASTing each C. elegans protein Ci against itself). We generated a normalized matrix by replacing each Cij by Cij/Cii.
RNA preparation and quantitative RT-PCR
RNA preparation and quantitative RT-PCR were as described [3]. Results were normalized to those of act-1 and were analyzed by the cycling threshold method. Control and experimental conditions were tested in the same ‘run’. Each sample was normalized to its own act-1 control to take into account age-specific changes in gene expression. Primers used for qRT-PCR are for:
act-1: JEP538 ccatcatgaagtgcgacattg JEP539 catggttgatggggcaagag;
nlp-29: JEP952 tatggaagaggatatggaggatatg JEP848 tccatgtatttactttccccatcc.
Availability of data and material
The entire dataset for the first and second rounds of the RNAi screen are publically available at http://bioinformatics.lif.univ-mrs.fr/RNAiScreen/. Data from qRT-PCR experiments are provided in Additional file 11: Table S10. Custom programming scripts are available on request.
Acknowledgments
We thank Ben Lehner, Mirko Francesconi, Nobert Perrimon, Thomas Richardson, Robin Evans, Jerome Reboul, Peter Sarkies, Yanhui Hu, and staff at the Drosophila RNAi Screening Center; Todd Harris, Kimberly van Auken, and staff at WormBase, for discussion and advice; Justine Melo and Emily Troemel for the kind gift of strains; Clement Ghigo and Sophie Cypowyj for their contributions; Pierre Golstein and Yishi Jin for critical reading of the manuscript; and the bioinformatics platforms of the CIML and the Laboratoire d’Informatique Fondamentale de Marseille for providing computing resources, database, and web servers. We are grateful to system administrators Manuel Bertrand and Kai Poutrain for their support and input, and the anonymous reviewers for their very constructive criticisms. Some nematode strains were provided by the Caenorhabditis Genetics Center, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440), or by the National Bioresource Project coordinated by S. Mitani.
Funding
This work was funded by institutional grants from AMU, INSERM and CNRS, program grants from the PACA Regional Council, the ANR (MIME-2007, ANR-12-BSV3-0001-01, ANR-11-LABX-0054 (Investissements d’Avenir–Labex INFORM) and ANR-11-IDEX-0001-02 (Investissements d’Avenir–A*MIDEX)). BS was supported in part by the Fondation ARC.
Abbreviations
- AMP
Antimicrobial peptide
- CCR4-NOT
Carbon catabolite repression 4-negative regulator of transcription
- cnc
Caenacin (Caenorhabditis bacteriocin)
- DIOPT
DRSC integrative ortholog prediction tool
- DRSC
Drosophila RNAi screening center
- DsRed
Red fluorescent protein from Discosoma sp.
- EASE
Expression analysis systematic explorer
- eIF
Eukaryotic translation initiation factor
- GFP
Green fluorescent protein
- GO
Gene ontology
- GPCR
G-protein coupled receptor
- GTP
Guanosine triphosphate
- Hipi
Hyper-induction of peptide expression after infection
- IRG
Infection response gene
- KEGG
Kyoto encyclopedia of genes and genomes
- Nipi
No induction of peptide after Drechmeria infection
- Peni
Peptide expression no infection
- ROS
Reactive oxygen species
- TOF
Time of flight
- TRG
Taxonomically-restricted gene
- UPR
Unfolded protein response
- UPRmt
Mitochondrial unfolded protein response
Additional files
The list of 21,223 clones used in this study. (XLSX 355 kb)
Results for Hipi clone candidates retained in the first round of screening and identification of potential target genes. (XLSX 134 kb)
Results for Hipi clone candidates in the second and third round of screening; categorization of Hipi and Peni clones; clone verification and identification of potential target genes. (XLSX 112 kb)
Details of how Nipi clones where selected after the second round of screening. (PDF 60 kb)
Clone verification and the identification of potential target genes for 360 Nipi clones; genes that are potentially targeted by more than one clone; identification of clones potentially targeting histone genes; phenotypic characterization; comparison with published data. (XLSX 211 kb)
Results for final 360 Nipi clone candidates in the first and second round of screening; identification of clones provoking severe developmental effect. (XLSX 1068 kb)
WormNet and expression analysis systematic explorer (EASE) analyses. (XLSX 131 kb)
Species used in phylogenetic clustering listed in the same order as in Fig. 8. (XLSX 47 kb)
Analysis of spore adhesion following RNAi treatment with Nipi clones; identification of genes potentially involved in spore binding. (XLSX 132 kb)
Analysis of enrichment of predicted Nipi proteins in conserved protein complexes. (XLSX 136 kb)
Data from qRT-PCR experiments. (XLSX 22 kb)
Footnotes
Olivier Zugasti and Nishant Thakur are co-first authors.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
Conceptualization: NP, JJE. Methodology: OZ, NT, JB, BS, JJE. Software: NT, JB, LT. Validation: OZ, NT, BS, JJE. Formal analysis: NT, JJE. Investigation: OZ, JB, BS, CLK, JS, SO, NP. Resources: LT. Data curation: LT, JJE. Writing – original draft: OZ, JJE. Writing – review & editing: OZ, NT, BS, JS, NP, JJE. Visualization: OZ, NT, NP, JJE. Supervision: NP, JJE. Project administration: JJE. Funding acquisition: NP, JJE. All authors read and approved the final manuscript.
Contributor Information
Nathalie Pujol, Email: pujol@ciml.univ-mrs.fr.
Jonathan J. Ewbank, Email: ewbank@ciml.univ-mrs.fr
References
- 1.Couillault C, Pujol N, Reboul J, Sabatier L, Guichou JF, Kohara Y, et al. TLR-independent control of innate immunity in Caenorhabditis elegans by the TIR domain adaptor protein TIR-1, an ortholog of human SARM. Nat Immunol. 2004;5:488–94. doi: 10.1038/ni1060. [DOI] [PubMed] [Google Scholar]
- 2.Pujol N, Zugasti O, Wong D, Couillault C, Kurz CL, Schulenburg H, et al. Anti-fungal innate immunity in C. elegans is enhanced by evolutionary diversification of antimicrobial peptides. PLoS Pathog. 2008;4(7):e1000105. doi: 10.1371/journal.ppat.1000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pujol N, Cypowyj S, Ziegler K, Millet A, Astrain A, Goncharov A, et al. Distinct innate immune responses to infection and wounding in the C. elegans epidermis. Curr Biol. 2008;18(7):481–9. doi: 10.1016/j.cub.2008.02.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim DH, Ewbank JJ. Signaling in the innate immune response. In: The C. elegans Research Community, Editor. WormBook. 2015. p. 1–51. http://www.wormbook.org. Accessed 01 May 2016. [DOI] [PMC free article] [PubMed]
- 5.Labed SA, Omi S, Gut M, Ewbank JJ, Pujol N. The pseudokinase NIPI-4 is a novel regulator of antimicrobial peptide gene expression. PLoS One. 2012;7(3):e33887. doi: 10.1371/journal.pone.0033887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zugasti O, Ewbank JJ. Neuroimmune regulation of antimicrobial peptide expression by a noncanonical TGF-beta signaling pathway in Caenorhabditis elegans epidermis. Nat Immunol. 2009;10(3):249–56. doi: 10.1038/ni.1700. [DOI] [PubMed] [Google Scholar]
- 7.Dierking K, Polanowska J, Omi S, Engelmann I, Gut M, Lembo F, et al. Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe. 2011;9(5):425–35. doi: 10.1016/j.chom.2011.04.011. [DOI] [PubMed] [Google Scholar]
- 8.Lee KZ, Kniazeva M, Han M, Pujol N, Ewbank JJ. The fatty acid synthase fasn-1 acts upstream of WNK and Ste20/GCK-VI kinases to modulate antimicrobial peptide expression in C. elegans epidermis. Virulence. 2010;1(3):113–22. doi: 10.4161/viru.1.3.10974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ziegler K, Kurz CL, Cypowyj S, Couillault C, Pophillat M, Pujol N, et al. Antifungal innate immunity in C. elegans: PKCdelta links G protein signaling and a conserved p38 MAPK cascade. Cell Host Microbe. 2009;5(4):341–52. doi: 10.1016/j.chom.2009.03.006. [DOI] [PubMed] [Google Scholar]
- 10.van Oosten-Hawle P, Morimoto RI. Transcellular chaperone signaling: an organismal strategy for integrated cell stress responses. J Exp Biol. 2014;217(Pt 1):129–36. doi: 10.1242/jeb.091249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taylor RC, Berendzen KM, Dillin A. Systemic stress signalling: understanding the cell non-autonomous control of proteostasis. Nat Rev Mol Cell Biol. 2014;15(3):211–7. doi: 10.1038/nrm3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ewbank JJ, Pujol N. Local and long-range activation of innate immunity by infection and damage in C. elegans. Curr Opin Immunol. 2015;38:1–7. doi: 10.1016/j.coi.2015.09.005. [DOI] [PubMed] [Google Scholar]
- 13.Ahringer J. Reverse genetics. In: The C. elegans Research Community, Editor. WormBook. 2006. doi:10.1895/wormbook.1.47.1. http://www.wormbook.org. Accesse 01 May 2016.
- 14.Boutros M, Ahringer J. The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008;9(7):554–66. doi: 10.1038/nrg2364. [DOI] [PubMed] [Google Scholar]
- 15.Kurz CL, Ewbank JJ. Infection in a dish: high-throughput analyses of bacterial pathogenesis. Curr Opin Microbiol. 2007;10(1):10–6. doi: 10.1016/j.mib.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 16.Pukkila-Worley R, Holson E, Wagner F, Mylonakis E. Antifungal drug discovery through the study of invertebrate model hosts. Curr Med Chem. 2009;16(13):1588–95. doi: 10.2174/092986709788186237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ewbank JJ, Zugasti O. C. elegans: model host and tool for antimicrobial drug discovery. Dis Model Mech. 2011;4(3):300–4. doi: 10.1242/dmm.006684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pulak R. Techniques for analysis, sorting, and dispensing of C. elegans on the COPAS flow-sorting system. Methods Mol Biol. 2006;351:275–86. doi: 10.1385/1-59745-151-7:275. [DOI] [PubMed] [Google Scholar]
- 19.Squiban B, Belougne J, Ewbank J, Zugasti O. Quantitative and automated high-throughput genome-wide RNAi screens in C. elegans. J Vis Exp. 2012;60:e3448. doi: 10.3791/3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zugasti O, Bose N, Squiban B, Belougne J, Kurz CL, Schroeder FC, et al. Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of Caenorhabditis elegans. Nat Immunol. 2014;15(9):833–8. doi: 10.1038/ni.2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421(6920):231–7. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
- 22.Rual JF, Ceron J, Koreth J, Hao T, Nicot AS, Hirozane-Kishikawa T, et al. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 2004;14(10B):2162–8. doi: 10.1101/gr.2505604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman A, Perrimon N. Genetic screening for signal transduction in the era of network biology. Cell. 2007;128(2):225–31. doi: 10.1016/j.cell.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 24.Murray PJ, Smale ST. Restraint of inflammatory signaling by interdependent strata of negative regulatory pathways. Nat Immunol. 2012;13(10):916–24. doi: 10.1038/ni.2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arthur JS, Ley SC. Mitogen-activated protein kinases in innate immunity. Nat Rev Immunol. 2013;13(9):679–92. doi: 10.1038/nri3495. [DOI] [PubMed] [Google Scholar]
- 26.Melo JA, Ruvkun G. Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell. 2012;149(2):452–66. doi: 10.1016/j.cell.2012.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward JD, Mullaney B, Schiller BJ, le He D, Petnic SE, Couillault C, et al. Defects in the C. elegans acyl-CoA synthase, acs-3, and nuclear hormone receptor, nhr-25, cause sensitivity to distinct, but overlapping stresses. PLoS One. 2014;9(3):e92552. doi: 10.1371/journal.pone.0092552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Thakur N, Pujol N, Tichit L, Ewbank JJ. Clone mapper: an online suite of tools for RNAi experiments in Caenorhabditis elegans. G3. 2014;4(11):2137–45. doi: 10.1534/g3.114.013052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palaima E, Leymarie N, Stroud D, Mizanur RM, Hodgkin J, Gravato-Nobre MJ, et al. The Caenorhabditis elegans bus-2 mutant reveals a new class of O-glycans affecting bacterial resistance. J Biol Chem. 2010;285(23):17662–72. [DOI] [PMC free article] [PubMed]
- 30.Gravato-Nobre MJ, Stroud D, O'Rourke D, Darby C, Hodgkin J. Glycosylation genes expressed in seam cells determine complex surface properties and bacterial adhesion to the cuticle of Caenorhabditis elegans. Genetics. 2011;187(1):141–55. doi: 10.1534/genetics.110.122002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rouger V, Bordet G, Couillault C, Monneret S, Mailfert S, Ewbank JJ, et al. Independent synchronized control and visualization of interactions between living cells and organisms. Biophys J. 2014;106(10):2096–104. doi: 10.1016/j.bpj.2014.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hansen M, Hsu AL, Dillin A, Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 2005;1(1):119–28. doi: 10.1371/journal.pgen.0010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lehmann S, Shephard F, Jacobson LA, Szewczyk NJ. Using multiple phenotype assays and epistasis testing to enhance the reliability of RNAi screening and identify regulators of muscle protein degradation. Genes (Basel) 2012;3(4):686–701. doi: 10.3390/genes3040686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goto A, Matsushita K, Gesellchen V, El Chamy L, Kuttenkeuler D, Takeuchi O, et al. Akirins are highly conserved nuclear proteins required for NF-kappaB-dependent gene expression in drosophila and mice. Nat Immunol. 2008;9(1):97–104. doi: 10.1038/ni1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonnay F, Nguyen XH, Cohen-Berros E, Troxler L, Batsche E, Camonis J, et al. Akirin specifies NF-kappaB selectivity of Drosophila innate immune response via chromatin remodeling. Embo J. 2014;33(20):2349–62. doi: 10.15252/embj.201488456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vanhoven MK, Bauer Huang SL, Albin SD, Bargmann CI. The claudin superfamily protein nsy-4 biases lateral signaling to generate left-right asymmetry in C. elegans olfactory neurons. Neuron. 2006;51(3):291–302. doi: 10.1016/j.neuron.2006.06.029. [DOI] [PubMed] [Google Scholar]
- 37.Andrusiak MG, Jin Y. Context specificity of stress-activated MAP Kinase signaling: the story as told by C. elegans. J Biol Chem. 2016;291:7796–804. doi: 10.1074/jbc.R115.711101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chavel CA, Caccamise LM, Li B, Cullen PJ. Global regulation of a differentiation MAPK pathway in yeast. Genetics. 2014;198(3):1309–28. doi: 10.1534/genetics.114.168252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Friedman AA, Tucker G, Singh R, Yan D, Vinayagam A, Hu Y, et al. Proteomic and functional genomic landscape of receptor tyrosine kinase and ras to extracellular signal-regulated kinase signaling. Sci Signal. 2011;4(196):rs10. doi: 10.1126/scisignal.2002029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bandyopadhyay S, Chiang CY, Srivastava J, Gersten M, White S, Bell R, et al. A human MAP kinase interactome. Nat Methods. 2010;7(10):801–5. doi: 10.1038/nmeth.1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hu Y, Flockhart I, Vinayagam A, Bergwitz C, Berger B, Perrimon N, et al. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cho A, Shin J, Hwang S, Kim C, Shim H, Kim H, et al. WormNet v3: a network-assisted hypothesis-generating server for Caenorhabditis elegans. Nucleic Acids Res. 2014;42(Web Server issue):W76–82. doi: 10.1093/nar/gku367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim DH, Liberati NT, Mizuno T, Inoue H, Hisamoto N, Matsumoto K, et al. Integration of Caenorhabditis elegans MAPK pathways mediating immunity and stress resistance by MEK-1 MAPK kinase and VHP-1 MAPK phosphatase. Proc Natl Acad Sci U S A. 2004;101(30):10990–4. doi: 10.1073/pnas.0403546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chauhan VM, Orsi G, Brown A, Pritchard DI, Aylott JW. Mapping the pharyngeal and intestinal pH of Caenorhabditis elegans and real-time luminal pH oscillations using extended dynamic range pH-sensitive nanosensors. ACS Nano. 2013;7(6):5577–87. doi: 10.1021/nn401856u. [DOI] [PubMed] [Google Scholar]
- 45.Hosack DA, Dennis G, Jr, Sherman BT, Lane HC, Lempicki RA. Identifying biological themes within lists of genes with EASE. Genome Biol. 2003;4(10):R70. doi: 10.1186/gb-2003-4-10-r70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Engelmann I, Griffon A, Tichit L, Montanana-Sanchis F, Wang G, Reinke V. A comprehensive analysis of gene expression changes provoked by bacterial and fungal infection in C. elegans. PLoS One. 2011;6(5):e19055. doi: 10.1371/journal.pone.0019055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Araya CL, Kawli T, Kundaje A, Jiang L, Wu B, Vafeados D, et al. Regulatory analysis of the C. elegans genome with spatiotemporal resolution. Nature. 2014;512(7515):400–5. doi: 10.1038/nature13497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shore DE, Carr CE, Ruvkun G. Induction of cytoprotective pathways is central to the extension of lifespan conferred by multiple longevity pathways. PLoS Genet. 2012;8(7):e1002792. doi: 10.1371/journal.pgen.1002792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res. 2010;38(Database issue):D355–60. doi: 10.1093/nar/gkp896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.dos Santos G, Schroeder AJ, Goodman JL, Strelets VB, Crosby MA, Thurmond J, et al. FlyBase: introduction of the Drosophila melanogaster Release 6 reference genome assembly and large-scale migration of genome annotations. Nucleic Acids Res. 2015;43(Database issue):D690–7. doi: 10.1093/nar/gku1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tanabe M, Kanehisa M. Using the KEGG database resource. Curr Protoc Bioinformatics. 2012; Chapter 1:Unit1 12. doi: 10.1002/0471250953.bi0112s38. [DOI] [PubMed]
- 53.Flockhart IT, Booker M, Hu Y, McElvany B, Gilly Q, Mathey-Prevot B, et al. FlyRNAi.org--the database of the Drosophila RNAi screening center: 2012 update. Nucleic Acids Res. 2012;40(Database issue):D715–9. doi: 10.1093/nar/gkr953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmidt EE, Pelz O, Buhlmann S, Kerr G, Horn T, Boutros M. GenomeRNAi: a database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 2013;41(Database issue):D1021–6. doi: 10.1093/nar/gks1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Khalturin K, Hemmrich G, Fraune S, Augustin R, Bosch TC. More than just orphans: are taxonomically-restricted genes important in evolution? Trends Genet. 2009;25(9):404–13. doi: 10.1016/j.tig.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 56.Pellegrini M, Marcotte EM, Thompson MJ, Eisenberg D, Yeates TO. Assigning protein functions by comparative genome analysis: protein phylogenetic profiles. Proc Natl Acad Sci U S A. 1999;96(8):4285–8. doi: 10.1073/pnas.96.8.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tabach Y, Billi AC, Hayes GD, Newman MA, Zuk O, Gabel H, et al. Identification of small RNA pathway genes using patterns of phylogenetic conservation and divergence. Nature. 2013;493(7434):694–8. doi: 10.1038/nature11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rohlfing AK, Miteva Y, Hannenhalli S, Lamitina T. Genetic and physiological activation of osmosensitive gene expression mimics transcriptional signatures of pathogen infection in C. elegans. PLoS One. 2010;5(2):e9010. doi: 10.1371/journal.pone.0009010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dunbar TL, Yan Z, Balla KM, Smelkinson MG, Troemel ER. C. elegans detects pathogen-induced translational inhibition to activate immune signaling. Cell Host Microbe. 2012;11(4):375–86. [DOI] [PMC free article] [PubMed]
- 60.Simmer F, Moorman C, Van Der Linden AM, Kuijk E, Van Den Berghe PV, Kamath R, et al. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. PLoS Biol. 2003;1(1):E12. doi: 10.1371/journal.pbio.0000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wan C, Borgeson B, Phanse S, Tu F, Drew K, Clark G, et al. Panorama of ancient metazoan macromolecular complexes. Nature. 2015;525(7569):339–44. doi: 10.1038/nature14877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gavin AC, Aloy P, Grandi P, Krause R, Boesche M, Marzioch M, et al. Proteome survey reveals modularity of the yeast cell machinery. Nature. 2006;440(7084):631–6. doi: 10.1038/nature04532. [DOI] [PubMed] [Google Scholar]
- 63.Lenstra TL, Benschop JJ, Kim T, Schulze JM, Brabers NA, Margaritis T, et al. The specificity and topology of chromatin interaction pathways in yeast. Mol Cell. 2011;42(4):536–49. doi: 10.1016/j.molcel.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benschop JJ, Brabers N, van Leenen D, Bakker LV, van Deutekom HW, van Berkum NL, et al. A consensus of core protein complex compositions for Saccharomyces cerevisiae. Mol Cell. 2010;38(6):916–28. doi: 10.1016/j.molcel.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 65.Inada T, Makino S. Novel roles of the multi-functional CCR4-NOT complex in post-transcriptional regulation. Front Genet. 2014;5:135. doi: 10.3389/fgene.2014.00135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bennett CF, Vander Wende H, Simko M, Klum S, Barfield S, Choi H, et al. Activation of the mitochondrial unfolded protein response does not predict longevity in Caenorhabditis elegans. Nat Commun. 2014;5:3483. doi: 10.1038/ncomms4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pellegrino MW, Nargund AM, Kirienko NV, Gillis R, Fiorese CJ, Haynes CM. Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature. 2014;516(7531):414–7. doi: 10.1038/nature13818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pellegrino MW, Haynes CM. Mitophagy and the mitochondrial unfolded protein response in neurodegeneration and bacterial infection. BMC Biol. 2015;13:22. doi: 10.1186/s12915-015-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nargund AM, Pellegrino MW, Fiorese CJ, Baker BM, Haynes CM. Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science. 2012;337(6094):587–90. doi: 10.1126/science.1223560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Durieux J, Wolff S, Dillin A. The cell-non-autonomous nature of electron transport chain-mediated longevity. Cell. 2011;144(1):79–91. doi: 10.1016/j.cell.2010.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30(4):313–21. doi: 10.1016/S1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 72.Rohlfing AK, Miteva Y, Moronetti L, He L, Lamitina T. The Caenorhabditis elegans mucin-like protein OSM-8 negatively regulates osmosensitive physiology via the transmembrane protein PTR-23. PLoS Genet. 2011;7(1):e1001267. doi: 10.1371/journal.pgen.1001267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Watson E, MacNeil LT, Arda HE, Zhu LJ, Walhout AJ. Integration of metabolic and gene regulatory networks modulates the C. elegans dietary response. Cell. 2013;153(1):253–66. doi: 10.1016/j.cell.2013.02.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu Y, Samuel BS, Breen PC, Ruvkun G. Caenorhabditis elegans pathways that surveil and defend mitochondria. Nature. 2014;508(7496):406–10. doi: 10.1038/nature13204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lamitina T, Huang CG, Strange K. Genome-wide RNAi screening identifies protein damage as a regulator of osmoprotective gene expression. Proc Natl Acad Sci U S A. 2006;103(32):12173–8. doi: 10.1073/pnas.0602987103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Updike DL, Strome S. A genomewide RNAi screen for genes that affect the stability, distribution and function of P granules in Caenorhabditis elegans. Genetics. 2009;183(4):1397–419. doi: 10.1534/genetics.109.110171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nollen EA, Garcia SM, van Haaften G, Kim S, Chavez A, Morimoto RI, et al. Genome-wide RNA interference screen identifies previously undescribed regulators of polyglutamine aggregation. Proc Natl Acad Sci USA. 2004;101(17):6403–8. doi: 10.1073/pnas.0307697101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.MacNeil LT, Pons C, Arda HE, Giese GE, Myers CL, Walhout AJ. Transcription factor activity mapping of a tissue-specific gene regulatory network. Cell Syst. 2015;1(2):152–62. doi: 10.1016/j.cels.2015.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Winter JF, Hopfner S, Korn K, Farnung BO, Bradshaw CR, Marsico G, et al. Caenorhabditis elegans screen reveals role of PAR-5 in RAB-11-recycling endosome positioning and apicobasal cell polarity. Nat Cell Biol. 2012;14(7):666–76. doi: 10.1038/ncb2508. [DOI] [PubMed] [Google Scholar]
- 80.Morton E, Lamitina T. A suite of MATLAB-based computational tools for automated analysis of COPAS Biosort data. Biotechniques. 2010;48(6):xxv–x. doi: 10.2144/000113427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Montanana F, Julien RA, Vaglio P, Matthews LR, Tichit L, Ewbank JJ. ICeE: an interface for C. elegans experiments. Worm. 2014;3(3):e959420. doi: 10.4161/21624046.2014.959420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marza E, Taouji S, Barroso K, Raymond AA, Guignard L, Bonneu M, et al. Genome-wide screen identifies a novel p97/CDC-48-dependent pathway regulating ER-stress-induced gene transcription. EMBO Rep. 2015;16(3):332–40. doi: 10.15252/embr.201439123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Squiban B. Criblage par ARN interférence du génome complet de C. elegans pour l' identification de nouveaux gènes impliqués dans l' immunité innée. Marseille: Aix-Marseille Université; 2012. [Google Scholar]
- 84.Davies KG, Curtis RH. Cuticle surface coat of plant-parasitic nematodes. Annu Rev Phytopathol. 2011;49:135–56. doi: 10.1146/annurev-phyto-121310-111406. [DOI] [PubMed] [Google Scholar]
- 85.Page AP, Johnstone IL. The cuticle. In: The C. elegans Research Community, Editor. WormBook. 2007. doi:10.1895/wormbook.1.138.1:1-15. Accessed 01 May 2016.
- 86.Gravato-Nobre MJ, Nicholas HR, Nijland R, O'Rourke D, Whittington DE, Yook KJ, et al. Multiple genes affect sensitivity of Caenorhabditis elegans to the bacterial pathogen Microbacterium nematophilum. Genetics. 2005;171(3):1033–45. [DOI] [PMC free article] [PubMed]
- 87.McNew JA, Sogaard M, Lampen NM, Machida S, Ye RR, Lacomis L, et al. Ykt6p, a prenylated SNARE essential for endoplasmic reticulum-Golgi transport. J Biol Chem. 1997;272(28):17776–83. doi: 10.1074/jbc.272.28.17776. [DOI] [PubMed] [Google Scholar]
- 88.Billing O, Natarajan B, Mohammed A, Naredi P, Kao G. A directed RNAi screen based on larval growth arrest reveals new modifiers of C. elegans insulin signaling. PLoS One. 2012;7(4):e34507. doi: 10.1371/journal.pone.0034507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luallen RJ, Bakowski MA, Troemel ER. Characterization of microsporidia-induced developmental arrest and a transmembrane leucine-rich repeat protein in Caenorhabditis elegans. PLoS One. 2015;10(4):e0124065. doi: 10.1371/journal.pone.0124065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Engelmann I, Pujol N. Innate Immunity in C. elegans. In: Söderhäll K, editor. Invertebrate Immunity. Austin, TX: Landes Bioscience; 2010. pp. 1–17. [Google Scholar]
- 91.Lamitina ST, Morrison R, Moeckel GW, Strange K. Adaptation of the nematode Caenorhabditis elegans to extreme osmotic stress. Am J Physiol Cell Physiol. 2004;286(4):C785–91. doi: 10.1152/ajpcell.00381.2003. [DOI] [PubMed] [Google Scholar]
- 92.Lee EC, Strange K. GCN-2 dependent inhibition of protein synthesis activates osmosensitive gene transcription via WNK and Ste20 kinase signaling. Am J Physiol Cell Physiol. 2012;303(12):C1269–77. doi: 10.1152/ajpcell.00294.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mizuno T, Hisamoto N, Terada T, Kondo T, Adachi M, Nishida E, et al. The Caenorhabditis elegans MAPK phosphatase VHP-1 mediates a novel JNK-like signaling pathway in stress response. Embo J. 2004;23(11):2226–34. doi: 10.1038/sj.emboj.7600226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Runkel ED, Liu S, Baumeister R, Schulze E. Surveillance-activated defenses block the ROS-induced mitochondrial unfolded protein response. PLoS Genet. 2013;9(3):e1003346. doi: 10.1371/journal.pgen.1003346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Baker BM, Nargund AM, Sun T, Haynes CM. Protective coupling of mitochondrial function and protein synthesis via the eIF2alpha kinase GCN-2. PLoS Genet. 2012;8(6):e1002760. doi: 10.1371/journal.pgen.1002760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McEwan DL, Kirienko NV, Ausubel FM. Host translational inhibition by Pseudomonas aeruginosa Exotoxin A Triggers an immune response in Caenorhabditis elegans. Cell Host Microbe. 2012;11(4):364–74. [DOI] [PMC free article] [PubMed]
- 97.Cohen LB, Troemel ER. Microbial pathogenesis and host defense in the nematode C. elegans. Curr Opin Microbiol. 2015;23C:94–101. doi: 10.1016/j.mib.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nandakumar M, Tan MW. Gamma-linolenic and stearidonic acids are required for basal immunity in Caenorhabditis elegans through their effects on p38 MAP kinase activity. PLoS Genet. 2008;4(11):e1000273. doi: 10.1371/journal.pgen.1000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Wong D, Bazopoulou D, Pujol N, Tavernarakis N, Ewbank JJ. Genome-wide investigation reveals pathogen-specific and shared signatures in the response of Caenorhabditis elegans to infection. Genome Biol. 2007;8(9):R194. doi: 10.1186/gb-2007-8-9-r194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Taubert S, Van Gilst MR, Hansen M, Yamamoto KR. A Mediator subunit, MDT-15, integrates regulation of fatty acid metabolism by NHR-49-dependent and -independent pathways in C. elegans. Genes Dev. 2006;20(9):1137–49. doi: 10.1101/gad.1395406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang P, Judy M, Lee SJ, Kenyon C. Direct and indirect gene regulation by a life-extending FOXO protein in C. elegans: roles for GATA factors and lipid gene regulators. Cell Metab. 2013;17(1):85–100. doi: 10.1016/j.cmet.2012.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taubert S, Hansen M, Van Gilst MR, Cooper SB, Yamamoto KR. The Mediator subunit MDT-15 confers metabolic adaptation to ingested material. PLoS Genet. 2008;4(2):e1000021. doi: 10.1371/journal.pgen.1000021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, et al. Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell. 2009;8(5):524–41. doi: 10.1111/j.1474-9726.2009.00501.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Goh GY, Martelli KL, Parhar KS, Kwong AW, Wong MA, Mah A, et al. The conserved Mediator subunit MDT-15 is required for oxidative stress responses in Caenorhabditis elegans. Aging Cell. 2014;13(1):70–9. doi: 10.1111/acel.12154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pukkila-Worley R, Feinbaum RL, McEwan DL, Conery AL, Ausubel FM. The evolutionarily conserved Mediator subunit MDT-15/MED15 links protective innate immune responses and xenobiotic detoxification. PLoS Pathog. 2014;10(5):e1004143. doi: 10.1371/journal.ppat.1004143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Blasius AL, Beutler B. Intracellular toll-like receptors. Immunity. 2010;32(3):305–15. doi: 10.1016/j.immuni.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 107.Huang HR, Chen ZJ, Kunes S, Chang GD, Maniatis T. Endocytic pathway is required for Drosophila Toll innate immune signaling. Proc Natl Acad Sci U S A. 2010;107(18):8322–7. doi: 10.1073/pnas.1004031107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vandenabeele P, Bertrand MJ. The role of the IAP E3 ubiquitin ligases in regulating pattern-recognition receptor signalling. Nat Rev Immunol. 2012;12(12):833–44. doi: 10.1038/nri3325. [DOI] [PubMed] [Google Scholar]
- 109.Zinngrebe J, Montinaro A, Peltzer N, Walczak H. Ubiquitin in the immune system. EMBO Rep. 2014;15(1):28–45. doi: 10.1002/embr.201338025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Moynagh PN. The roles of Pellino E3 ubiquitin ligases in immunity. Nat Rev Immunol. 2014;14(2):122–31. doi: 10.1038/nri3599. [DOI] [PubMed] [Google Scholar]
- 111.Davis ME, Gack MU. Ubiquitination in the antiviral immune response. Virology. 2015;479–480:52–65. doi: 10.1016/j.virol.2015.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Chapat C, Corbo L. Novel roles of the CCR4-NOT complex. Wiley Interdiscip Rev RNA. 2014;5(6):883–901. doi: 10.1002/wrna.1254. [DOI] [PubMed] [Google Scholar]
- 113.Boehm T. Evolution of vertebrate immunity. Curr Biol. 2012;22(17):R722–32. doi: 10.1016/j.cub.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 114.Boehm T, McCurley N, Sutoh Y, Schorpp M, Kasahara M, Cooper MD. VLR-based adaptive immunity. Annu Rev Immunol. 2012;30:203–20. doi: 10.1146/annurev-immunol-020711-075038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Flajnik MF. Re-evaluation of the immunological Big Bang. Curr Biol. 2014;24(21):R1060–5. doi: 10.1016/j.cub.2014.09.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Du Pasquier L. Metazoa immune receptors diversification during evolution. Med Sci (Paris) 2009;25(3):273–80. doi: 10.1051/medsci/2009253273. [DOI] [PubMed] [Google Scholar]
- 117.Schulenburg H, Kurz CL, Ewbank JJ. Evolution of the innate immune system: the worm perspective. Immunol Rev. 2004;198:36–58. doi: 10.1111/j.0105-2896.2004.0125.x. [DOI] [PubMed] [Google Scholar]
- 118.Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10(1):47–58. doi: 10.1038/nri2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang L, Li L, Guo X, Litman GW, Dishaw LJ, Zhang G. Massive expansion and functional divergence of innate immune genes in a protostome. Sci Rep. 2015;5:8693. doi: 10.1038/srep08693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Simmer F, Tijsterman M, Parrish S, Koushika SP, Nonet ML, Fire A, et al. Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol. 2002;12(15):1317–9. doi: 10.1016/S0960-9822(02)01041-2. [DOI] [PubMed] [Google Scholar]
- 121.Powell JR, Ausubel FM. Models of Caenorhabditis elegans infection by bacterial and fungal pathogens. In: Ewbank J, Vivier E, editors. Methods Mol Biol, vol. 415. New York, NY: Humana Press; 2008. p. 403–27. [DOI] [PubMed]
- 122.Sagasti A, Hisamoto N, Hyodo J, Tanaka-Hino M, Matsumoto K, Bargmann CI. The CaMKII UNC-43 activates the MAPKKK NSY-1 to execute a lateral signaling decision required for asymmetric olfactory neuron fates. Cell. 2001;105(2):221–32. doi: 10.1016/S0092-8674(01)00313-0. [DOI] [PubMed] [Google Scholar]
- 123.Fischer D, Eisenberg D. Finding families for genomic ORFans. Bioinformatics. 1999;15(9):759–62. doi: 10.1093/bioinformatics/15.9.759. [DOI] [PubMed] [Google Scholar]
- 124.McCarroll SA, Murphy CT, Zou S, Pletcher SD, Chin CS, Jan YN, et al. Comparing genomic expression patterns across species identifies shared transcriptional profile in aging. Nat Genet. 2004;36(2):197–204. doi: 10.1038/ng1291. [DOI] [PubMed] [Google Scholar]
- 125.Kim SK, Lund J, Kiraly M, Duke K, Jiang M, Stuart JM, et al. A gene expression map for Caenorhabditis elegans. Science. 2001;293(5537):2087–92. doi: 10.1126/science.1061603. [DOI] [PubMed] [Google Scholar]
- 126.Ceron J, Rual JF, Chandra A, Dupuy D, Vidal M, van den Heuvel S. Large-scale RNAi screens identify novel genes that interact with the C. elegans retinoblastoma pathway as well as splicing-related components with synMuv B activity. BMC Dev Biol. 2007;7:30. doi: 10.1186/1471-213X-7-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McElwee JJ, Schuster E, Blanc E, Thomas JH, Gems D. Shared transcriptional signature in Caenorhabditis elegans Dauer larvae and long-lived daf-2 mutants implicates detoxification system in longevity assurance. J Biol Chem. 2004;279(43):44533–43. doi: 10.1074/jbc.M406207200. [DOI] [PubMed] [Google Scholar]
- 128.Mansisidor AR, Cecere G, Hoersch S, Jensen MB, Kawli T, Kennedy LM, et al. A conserved PHD finger protein and endogenous RNAi modulate insulin signaling in Caenorhabditis elegans. PLoS Genet. 2011;7(9):e1002299. doi: 10.1371/journal.pgen.1002299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Love DC, Ghosh S, Mondoux MA, Fukushige T, Wang P, Wilson MA, et al. Dynamic O-GlcNAc cycling at promoters of Caenorhabditis elegans genes regulating longevity, stress, and immunity. Proc Natl Acad Sci U S A. 2010;107(16):7413–8. doi: 10.1073/pnas.0911857107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Guisbert E, Czyz DM, Richter K, McMullen PD, Morimoto RI. Identification of a tissue-selective heat shock response regulatory network. PLoS Genet. 2013;9(4):e1003466. doi: 10.1371/journal.pgen.1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Parry DH, Xu J, Ruvkun G. A whole-genome RNAi Screen for C. elegans miRNA pathway genes. Curr Biol. 2007;17(23):2013–22. doi: 10.1016/j.cub.2007.10.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zisoulis DG, Lovci MT, Wilbert ML, Hutt KR, Liang TY, Pasquinelli AE, et al. Comprehensive discovery of endogenous Argonaute binding sites in Caenorhabditis elegans. Nat Struct Mol Biol. 2010;17(2):173–9. doi: 10.1038/nsmb.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet. 2007;3(4):e56. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mabon ME, Mao X, Jiao Y, Scott BA, Crowder CM. Systematic identification of gene activities promoting hypoxic death. Genetics. 2009;181(2):483–96. doi: 10.1534/genetics.108.097188. [DOI] [PMC free article] [PubMed] [Google Scholar]