

Abstract
Transparent films were prepared by cross‐linking polyunsaturated poly(ether carbonate)s obtained by the multicomponent polymerization of CO2, propylene oxide, maleic anhydride, and allyl glycidyl ether. Poly(ether carbonate)s with ABXBA multiblock structures were obtained by sequential addition of mixtures of propylene oxide/maleic anhydride and propylene oxide/allyl glycidyl ether during the polymerization. The simultaneous addition of both monomer mixtures provided poly(ether carbonate)s with AXA triblock structures. Both types of polyunsaturated poly(ether carbonate)s are characterized by diverse functional groups, that is, terminal hydroxy groups, maleate moieties along the polymer backbone, and pendant allyl groups that allow for versatile polymer chemistry. The combination of double bonds substituted with electron‐acceptor and electron‐donor groups enables particularly facile UV‐ or redox‐initiated free‐radical curing. The resulting materials are transparent and highly interesting for coating applications.
Keywords: CO2 utilization, copolymerization, multicomponent reactions, poly(ether carbonate), transparent materials
The utilization of carbon dioxide (CO2) as an inexpensive and renewable C1 feedstock1 is of strategic importance for decreasing our dependence on petroleum‐derived raw materials.2 The direct build‐up of polymers, at least partially from CO2, is a particularly interesting approach.3 In this context, the catalytic copolymerization of CO2 and epoxides to form polycarbonates has proved promising.4 Aliphatic polycarbonates have high transparency5 and, thus, a high potential for coating applications. Alternating polycarbonates are amorphous with a glass transition temperature (T g) of 25 to 45 °C.6 Good resistance to both oxygen and water make them auspicious barrier materials.7 The terminal hydroxy groups play a role in their excellent adhesive properties.7 The high reactivity of these hydroxy groups makes non‐alternating poly(ether carbonate polyol)s, which are characterized by a lower T g value of −40 to −60 °C, promising intermediates for the production of polyurethanes.8 This novel utilization of CO2 exemplifies the industrial potential of the reaction.9
Motivated by the remarkable combination of properties and the goal of finding new materials for the future, the focus of this study is the synthesis and application of tailored polyunsaturated poly(ether carbonate)s, obtained by multicomponent polymerization of CO2 and propylene oxide (PO) with suitable co‐monomers (Scheme 1). Maleic anhydride (MA) and allyl glycidyl ether (AGE) were used as co‐monomers to incorporate double bonds with electron‐acceptor and electron‐donor properties, respectively, into the poly(ether carbonate) backbone. The concurrent presence of such moieties in the prepolymers ought to facilitate UV‐10 or redox‐initiated11 cross‐linking of the poly(ether carbonate) chains into robust materials.
Scheme 1.
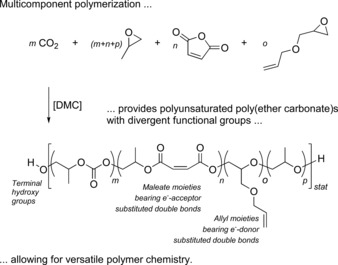
Polyunsaturated poly(ether carbonate)s obtained by multicomponent polymerization are characterized by distinct functional groups.
A double metal cyanide (DMC) based on zinc hexacyanocobaltate was used as the polymerization catalyst.12 DMC catalysts13 are known to be active for the homopolymerization of epoxides14 as well as for the copolymerization of CO2 with epoxides13a, 15 and other co‐monomers, such as cyclic anhydrides16 and lactones.17 The catalytically active metal centers are tetrahedral Zn atoms with nearby Cl atoms, as shown by extended X‐ray absorption fine‐structure analysis.18 The propensity of DMC catalysts to copolymerize various substrates inspired us to incorporate functional co‐monomers (MA and AGE) into a multicomponent polymerization. The molecular weight of the polyunsaturated poly(ether carbonate)s was controlled by using a bifunctional α,ω‐dihydroxypolypropylene oxide as chain‐transfer reagent.19 Since the copolymerization reaction is highly exothermal, monomer mixtures of MA/PO and/or AGE/PO were added in semibatch operation over an extended period of time, thereby maintaining a low concentration of epoxide in the reaction mixture. Simultaneously, the reaction mixture was cooled to remove the heat generated during the chemical reaction. The pressure in the reactor was kept constant by replacing the CO2 consumed during the reaction.
The architecture of the polyunsaturated poly(ether carbonate)s was tailored by adjusting the sequence of monomer addition. A sequential addition procedure, whereby the addition of MA/PO to the reaction mixture was followed by the addition of AGE/PO, provided the polyunsaturated poly(ether carbonate)s 3 with an ABXBA multiblock structure (Figure 1). In a series of experiments, the loading of MA and AGE was varied systematically to obtain polyunsaturated poly(ether carbonate)s 3 a–c with 3, 6, and 12 mol % double bonds (Table 1). The ratio of electron‐acceptor to electron‐donor moieties was fixed at 1:1. As a consequence of the immortal nature of DMC‐catalyzed polymerization reactions,15b, 16a, 17a, 20 we anticipated that all the chain ends grow at approximately the same rate, and that a random incorporation of the co‐monomers takes place (see below).
Figure 1.
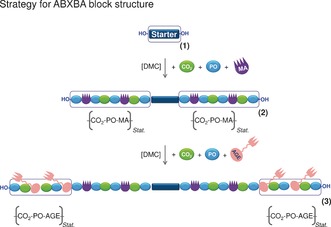
Preparation of polyunsaturated poly(ether carbonate)s 3 a–c with an ABXBA multiblock structure by sequential addition of the co‐monomers.
Table 1.
Chemical composition and molecular weight of polyunsaturated poly(ether carbonate)s 3 a–c and 4.[a]
| Sample | SPolymer [b,c] | CO2 content[c,e] | Composition [mol %] | M n [f] | PDI[f] |
|---|---|---|---|---|---|
| [wt %] | [wt %][b] | m/n/o/p [c,d,e] | [kg mol−1] | [–] | |
| 3 a | 98.2 | 19.1 | 31.8/1.6/1.4/65.2 | 4.7 | 1.3 |
| (23.7) | (42.4/2.1/1.9/53.6) | ||||
| 3 b | 98.3 | 15.9 | 26.1/3.3/2.8/67.8 | 4.5 | 1.8 |
| (19.8) | (34.8/4.4/3.7/57.1) | ||||
| 3 c | 99.1 | 13.1 | 21.8/5.9/5.9/66.4 | 4.0 | 1.3 |
| (16.3) | (29.1/7.9/7.9/55.2) | ||||
| 4 | 99.3 | 14.3 | 24.2/6.0/5.9/63.9 | 4.1 | 1.5 |
| (17.8) | (32.3/8.0/7.9/51.9) |
[a] Reaction conditions: neat, 100 °C, p(CO2)=15 bar, 200 ppm DMC catalyst. [b] SPolymer: Selectivity to polyunsaturated poly(ether carbonate) in weight percent; the only by‐product were traces of cyclic propylene carbonate. [c] Determined by 1H NMR spectroscopy. [d] m=carbonate moiety, n=maleic ester moiety, o=allylether moiety, p=ether moiety. [e] Values in brackets refer to newly formed polymer segments. [f] Determined by gel‐permeation chromatography.
Alternatively, the two monomer mixtures (MA/PO and AGE/PO) were added simultaneously to the reaction mixture to give polyunsaturated poly(ether carbonate)s 4 with an AXA triblock structure (Figure 2). A similar reactivity of the co‐monomers MA and AGE relative to that of PO21 results in an even distribution of the monomers bearing double bonds along the polymer backbone of the newly formed polymer blocks. In the semibatch procedure employed in this study, the slow addition of the monomer mixture(s) led to a small steady‐state concentration of unreacted monomers. Consequently, the composition of the forming polyunsaturated poly(ether carbonate) block are expected to be similar to the composition of the monomer mixture fed into the reactor, irrespective of the actual copolymerization parameters.22 Accordingly, the copolymer blocks ought to have compositions close to the ideal of an aceotropic polymerization reaction. The co‐monomer CO2 can be neglected in this analysis, as it was resupplied to keep the pressure constant. Incorporation of CO2 or MA at the chain end results in the formation of a terminal carbonate or carboxylate group. Consequently, the subsequent monomer has to be an epoxide since homopolymerization of acid anhydrides is not possible under our reaction conditions.
Figure 2.
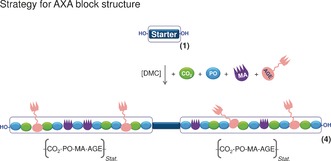
Preparation of polyunsaturated poly(ether carbonate) 4 with an AXA triblock structure by simultaneous addition of the co‐monomers.
Analysis of the product mixture by 1H NMR spectroscopy confirmed that the monomers PO, MA, and AGE had been fully converted (>99 %). The resulting polyunsaturated poly(ether carbonate)s 3 a–c with an ABXBA block structure and 4 with an AXA block structure were obtained with excellent selectivity (98.2–99.3 %). At a pressure as low as 15 bar, the content of CO2 incorporated into the polyunsaturated poly(ether carbonate)s 3 a–c and 4 was 14.3–19.1 wt %. This corresponds to a CO2 content in the newly formed polymer blocks of 16.3–23.7 wt %. Thus, approximately every third repetition unit along the polymer chain was a carbonate moiety. Such a CO2 content is an optimum compromise between the flexibility of the polymer chain, and accordingly a low glass transition temperature (see below), and enhanced sustainability. In all cases, the polyunsaturated poly(ether carbonate)s were obtained with the anticipated molecular weight (M n=4.0–4.7 kg mol−1) and narrow molecular weight distribution (polydispersity index (PDI)=1.3–1.8). No cross‐linking of double bonds was observed at this stage, thereby indicating that the maleate and allyl functionalities are stable under our reaction conditions.
The 1H NMR spectrum of polyunsaturated poly(ether carbonate) 3 c (see Figure S223) is representative of the series of polyunsaturated poly(ether carbonate)s. Closer inspection revealed a multiplet centered at δ=6.3 ppm, which is characteristic of the methine protons of the maleate moieties in the polymer backbone. Signals at δ=5.2 and 5.9 ppm are characteristic of the methylene and methine groups of the allyl moieties. The relative ratio of the maleate and allyl groups was confirmed to be close to unity. The absence of a signal at δ=6.8 ppm corresponding to the fumarate groups demonstrates that the incorporated MA did not isomerize to the corresponding trans isomer. Thus, the chemical composition of polyunsaturated poly(ether carbonate)s 3 a–c and 4 was as anticipated (Table 1). To the best of our knowledge, this is the first study in which a DMC catalyst was successfully employed in the multicomponent polymerization of four co‐monomers.
To confirm the ABXBA multiblock structure of polyunsaturated poly(ether carbonate)s 3, the polymerization reaction was performed stepwise. Commencing with the starter mixture (i), the PO/MA mixture was added in two portions to the reaction mixture (ii/iii) followed by the addition of two portions of the PO/AGE mixture (iv/v).23 After completion of each monomer addition step, a sample of the reaction mixture containing polyunsaturated poly(ether carbonate) 3 c′′ was collected. In all cases, 1H NMR spectroscopic analysis of the mixtures showed that polyunsaturated poly(ether carbonate) 3 c′′ was obtained with full conversion of the monomers and in high selectivity (>99 %). Detailed inspection of the 1H NMR spectra revealed that the multiplet centered at δ=6.3 ppm and characteristic of the methine protons of the maleate moieties in the polymer backbone appeared after the first addition of PO/MA and its intensity increased upon the second addition of a PO/MA mixture (Figure 3 a, ii,iii). The multiplet centered at δ=5.9 ppm characteristic of the methine protons of the allyl moieties in the polymer backbone was observed after the first addition of PO/AGE and increased in intensity upon the second addition of PO/AGE, while the relative intensity of the maleate moieties (δ=6.3 ppm) remained unchanged (Figure 3 a, iv,v). These findings are fully consistent with the ABXBA multiblock structure of the polyunsaturated poly(ether carbonate) 3 c′′ and statistic incorporation of maleate and allyl moieties along with ether and ether carbonate moieties in distinct blocks B and A, respectively.
Figure 3.
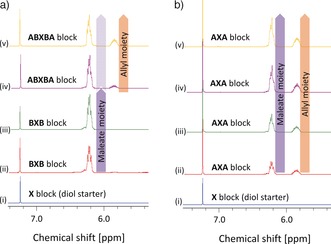
Selected region of the 1H NMR spectra of samples of a) polyunsaturated poly(ether carbonate) 3 a′′ with ABXBA multiblock structures obtained by sequential addition of the co‐monomers (left); i) starter mixture, ii/iii) after stepwise addition of PO/MA, iv/v) after stepwise addition of PO/AGE; b) polyunsaturated poly(ether carbonate) 4′ with an AXA triblock structure obtained by simultaneous addition of the co‐monomers (right); i) starter mixture, ii–v) after stepwise addition of PO/MA and PO/AGE. For the full spectra see the Supporting Information.23
To verify the AXA triblock structure of polyunsaturated poly(ether carbonate)s 4, an analogous experiment was performed in which the PO/MA and the PO/AGE mixtures were added simultaneously to the reaction mixture in four portions. After completion of each monomer addition step, a sample of the reaction mixture was collected and analyzed by 1H NMR spectroscopy and gel‐permeation chromatography. The 1H NMR spectroscopic analysis revealed that the relative intensity of ether (CH 3, δ=1.10 ppm), ether carbonate (CH 3, δ=1.25 ppm), allyl (CH, δ=5.9 ppm), and maleate (CH, δ=6.3 ppm) moieties increased simultaneously after each monomer addition step (Figure 3 b, ii–v).23 These results are fully consistent with the AXA triblock structure of polyunsaturated poly(ether carbonate) 4′ and statistical incorporation of the four monomers CO2, PO, MA, and AGE into block A.
The molecular weight (of the polymer chains in 3 c′′ determined by gel‐permeation chromatography increased stepwise from 1.5 (corresponding to the starter block) to 2.1, 3.0, 3.9, and 5.0 kg mol−1 after stepwise addition of the monomer mixture (Figure 4). The molecular weights in the oligomer range, which correspond well with the respective expected molecular weights (2.0, 2.8, 3.7, and 4.5 kg mol−1, assuming 13.1 wt % incorporated CO2 and systematic error through use of an external reference), and the narrow PDI of 1.06–1.27, indicate that the molecular weight was indeed regulated by the diol starter. This is in excellent agreement with the concept of immortal polymerization. Furthermore, the controlled stepwise growth of the polymer chains and the option to restart the reaction provide strong evidence that the DMC‐catalyzed polymerization follows an immortal polymerization mechanism.
Figure 4.
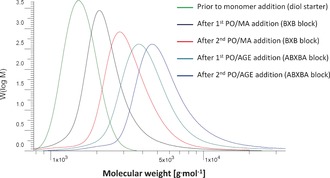
GPC traces for polyunsaturated poly(ether carbonate) 3 c′′ after steps i–v (see Figure 3 a, left and caption).
The polyunsaturated poly(ether carbonate)s 3 a–c and 4 are colorless liquids with a low viscosity (from 17.0 to 22.0 Pa s).23 The viscosity increased slightly as the content of carbonate linkages increased, consistent with the incorporation of less flexible carbonate moieties into the polymer backbone. Likewise, a low glass transition temperature of −43.1 to −47.4 °C is characteristic of the soft segments formed by the ether moieties in the backbone polymer.23
The low viscosity and the low glass transition temperature of these polyunsaturated poly(ether carbonate)s make them ideally suited prepolymers for further processing. To prepare a poly(ether carbonate) film, the double bonds of the polyunsaturated poly(ether carbonate)s were cross‐linked by UV‐initiated curing. A sample of polyunsaturated poly(ether carbonate) 3 a was mixed with dibenzoyl peroxide as a photoinitator (PI), and the time‐dependent viscoelastic behavior during curing was monitored with a rheometer under isothermal and low shear conditions (40 °C, 1 Hz, 10 % shear). Upon exposure to UV light (λ=300–500 nm, I UV=22.5 W c−2), the storage modulus (G′), which was initially lower than the loss modulus (G′′), and consistent with the liquid state of the poly(ether carbonate), started to increase after 3.6 min (t onset; Figure 5). The onset of the curing process is characteristic of the formation of a three‐dimensional network through cross‐linking of the polymer chains by free‐radical polymerization of the double bonds. The gel time (t g), at which G′ and G′′ are equal,24 was attained after 9.3 min. The final value of G′ was higher than that of G′′ of poly(ether carbonate) 3 a, thus confirming the successful cross‐linking of poly(ether carbonate) 3 a. Similarly, the cross‐linking of the polyunsaturated poly(ether carbonate)s 3 b and 3 c revealed the onset of the curing process after 2.8 min and 1.2 min, respectively. The respective gel points were achieved after 6.6 and 5.3 min. Thus, the greater the number of double bonds in the poly(ether carbonate), the faster the curing. For the actual application of the material, the curing time can be readily adjusted by tailoring the intensity of the UV light, the concentration of the initiator, and the temperature.24
Figure 5.
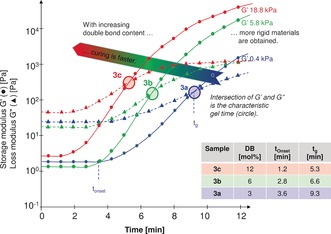
Influence of double‐bond loading on the viscoelastic behavior of the polyunsaturated poly(ether carbonate)s 3 a–c during irradiation with UV light.
The importance of the concurrent presence of maleate and allyl groups with electron‐acceptor and electron‐donor properties was explored in a set of reference experiments. No change in the values of G′ and G′′ was observed upon irradiation of polyunsaturated poly(ether carbonate) 5, which contains only maleate groups (6 mol %), with UV light for 60 min under otherwise identical reaction conditions.23 Similarly, irradiation of polyunsaturated poly(ether carbonate) 6, which contains only allyl moieties (6 mol %), with UV light did not give rise to curing. In contrast, the gel point with a mixture of polyunsaturated poly(ether carbonate)s 5 and 6 at a ratio of 1:1 was reached within 6 min. Thus, the presence of both electron‐acceptor and electron‐donor moieties is essential for the facile cross‐linking of the polymer chains. This is consistent with the formation of electron donor/acceptor complexes by the partial transfer of a π electron from the allyl moiety to the π orbital of the maleate group, which initiates the cross‐linking of the polymer chains. Furthermore, the propagating species is most likely an electron donor/acceptor complex rather than a discrete allyl or maleate moiety.25 The rate of a diffusion‐controlled reaction of the double bonds, without prior formation of donor/acceptor complexes, would decrease significantly as the viscosity of the medium increases during curing.
The curing of polyunsaturated poly(ether carbonate) 3 c was compared with that of 4 to study the influence of the polymer architecture on the viscoelastic behavior. Compared to 3 c with an ABXBA multiblock structure (t g=5.3 min; Table 2, entry 3), the gel time was much shorter for 4 with an AXA triblock structure (t g=4.3 min; Table 2, entry 4). The faster curing of polyunsaturated poly(ether carbonate) 4 is attributed to the better local dispersion of the electron‐acceptor and electron‐donor double bonds, which facilitates an efficient cross‐linking of the polymer chains.
Table 2.
Concentration of unsaturated moieties in poly(ether carbonate)s 3 a–c, 4–6 and viscoelastic behavior upon exposure to UV light.
| Entry | Sample | Unsaturated moieties[a] | t onset [b] | t g [b] | G′12min | |
|---|---|---|---|---|---|---|
| maleate [mol %] | allyl [mol %] | [min] | [min] | [kPa] | ||
| 1 | 3 a | 1.6 | 1.4 | 3.6 | 9.3 | 0.4 |
| 2 | 3 b | 3.3 | 2.8 | 2.8 | 6.6 | 5.8 |
| 3 | 3 c | 5.9 | 5.9 | 1.2 | 5.3 | 18.8 |
| 4 | 4 | 6.0 | 5.8 | 0.8 | 4.3 | 41.8 |
| 5 | 5 | 6.0 | – | >60 | n.o. | n.o. |
| 6 | 6 | – | 6.0 | >60 | n.o. | n.o. |
| 7 | 5 + 6 | 3.0 | 3.0 | 2.0 | 6 | 6.0 |
[a] Determined by 1H NMR spectroscopy. [b] 40 °C, determined with a rheometer equipped with a plate–plate measuring system; t onset=curing onset, t g=gel time; G′12min=storage modulus after 12 min exposure to UV light; n.o. not observed.
To explore the effect of the cross‐linking density on the hardness of the film, the value of the storage modulus G′ of the polyunsaturated poly(ether carbonate)s 3 a–c was compared after exposure of the sample to UV light for 12 min (Figure 5 and Table 2, entries 1–3). The storage modulus G′ of the cross‐linked film increased as the number of double bonds in the polymer chains increased. Furthermore, the hardened material was noticeably more rigid as the number of double bonds of the polyol and, thus, the density of cross‐linking increased. Compared to a film prepared with poly(ether carbonate) 3 c, the film prepared with poly(ether carbonate) 4 (Table 2, entry 4) was much harder and had a significantly higher storage modulus G′ (18.8 versus 41.8 kPa). Similar to the shorter gel time, this increased hardness of the film prepared with poly(ether carbonate) 4 is attributed to the AXA architecture, which allows for better dispersion of the cross‐linking points. The values of the storage modulus of the cured films are ideal for the application of these materials as soft coatings24, 26 which feel warm to the touch.
In conclusion, the synthesis of transparent films from CO2‐based, environmentally sustainable, polyunsaturated poly(ether carbonate)s was achieved by the DMC‐catalyzed multicomponent polymerization of CO2 with propylene oxide, maleic anhydride, and allyl glycidyl ether. Although characterized by divergent functional groups, the polyunsaturated poly(ether carbonate)s showed remarkable stability, both during and after polymerization, which is highly desirable for the preparation of advanced materials and for subsequent applications. Specific incorporation of unsaturated co‐monomers along the poly(ether carbonate) backbone led to tailored polyunsaturated poly(ether carbonate)s with clearly defined compositions and architectures. The sequential addition of the co‐monomers resulted in polyunsaturated poly(ether carbonate)s with an ABXBA multiblock structure; the simultaneous addition of maleic anhydride and allyl glycidyl ether led to polyunsaturated poly(ether carbonate)s with an AXA triblock structure. This enables the adjustment of the physicochemical and macroscopic properties, such as viscosity, gel time, and hardness. This novel method will be promising for the synthesis of advanced sustainable materials.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The experiments were performed by Dario Gottschalk and Sibel Ciftci. We thank V. Marker and A. Keldenich for experimental support. M.A.S. acknowledges funding as an Alexander von Humboldt Fellow.
M. A. Subhani, B. Köhler, C. Gürtler, W. Leitner, T. E. Müller, Angew. Chem. Int. Ed. 2016, 55, 5591.
References
- 1. Seong J. E., Na S. J., Cyriac A., Kim B. W., Lee B. Y., Macromolecules 2010, 43, 903–908. [Google Scholar]
- 2.
- 2a. Markewitz P., Kuckshinrichs W., Leitner W., Linssen J., Zapp P., Bongartz R., Schreiber A., Müller T. E., Energy Environ. Sci. 2012, 5, 7281–7305; [Google Scholar]
- 2b. Peters M., Köhler B., Kuckshinrichs W., Leitner W., Markewitz P., Müller T. E., ChemSusChem 2011, 4, 1216–1240; [DOI] [PubMed] [Google Scholar]
- 2c. Cokoja M., Bruckmeier C., Rieger B., Herrmann W. A., Kühn F. E., Angew. Chem. Int. Ed. 2011, 50, 8510–8537; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8662–8690; [Google Scholar]
- 2d. Sakakura T., Choi J. C., Yasuda H., Chem. Rev. 2007, 107, 2365–2387; [DOI] [PubMed] [Google Scholar]
- 2e. Quadrelli E. A., Centi G., Duplan J. L., Perathoner S., ChemSusChem 2011, 4, 1194–1215; [DOI] [PubMed] [Google Scholar]
- 2f. Aresta M., Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010; [Google Scholar]
- 2g. Darensbourg D. J., Inorg. Chem. 2010, 49, 10765–10780. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Darensbourg D. J., Andreatta J. R., Moncada A. I., Carbon Dioxide as Chemical Feedstock, Wiley-VCH, Weinheim, 2010; [Google Scholar]
- 3b. Müller T. E., Prepr. Am. Chem. Soc. Div. Fuel Chem. 2008, 53, 317; [Google Scholar]
- 3c. Darensbourg D. J., Holtcamp M. W., Coord. Chem. Rev. 1996, 153, 155–174. [Google Scholar]
- 4.
- 4a. Darensbourg D. J., Wilson S. J., Green Chem. 2012, 14, 2665–2671; [Google Scholar]
- 4b. Inoue S., Koinuma H., Tsuruta T., Makromol. Chem. 1969, 130, 210; [Google Scholar]
- 4c. Rokicki A., Kuran W., J. Macromol. Sci. Rev. Macromol. Chem. Phys. 1981, C21, 135–186; [Google Scholar]
- 4d. Elmas S., Subhani M. A., Harrer M., Leitner W., Sundermeyer J., Müller T. E., Catal. Sci. Technol. 2014, 4, 1652–1657. [Google Scholar]
- 5. Wu G. P., Wei S. H., Ren W. M., Lu X. B., Xu T. Q., Darensbourg D. J., J. Am. Chem. Soc. 2011, 133, 15191–15199. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Klaus S., Lehenmeier M. W., Anderson C. E., Rieger B., Coord. Chem. Rev. 2011, 255, 1460–1479; [Google Scholar]
- 6b. Luinstra G. A., Borchardt E. in Synthetic Biodegradable Polymers, Vol. 245 (Eds.: B. Rieger, A. Künkel, G. W. Coates, R. Reichardt, E. Dinjus, T. A. Zevaco), Springer, Berlin, 2012, pp. 29–48. [Google Scholar]
- 7.
- 7a. Cyriac A., Lee S. H., Lee B. Y., Polym. Chem. 2011, 2, 950–956; [Google Scholar]
- 7b. Qin Y. S., Wang X. H., Biotechnol. J. 2010, 5, 1164–1180; [DOI] [PubMed] [Google Scholar]
- 7c. Eun-Sung L., Choi J.-Y., Seul-Ki K., Seon-Mi Y., Baik N.-S., Yong-Jun J., US 2007/0001608, 2007;
- 7d. Luinstra G. A., Polym. Rev. 2008, 48, 192–219. [Google Scholar]
- 8. Langanke J., Wolf A., Hofmann J., Böhm K., Subhani M. A., Müller T. E., Leitner W., Gürtler C., Green Chem. 2014, 16, 1865–1870. [Google Scholar]
- 9. von der Assen N., Bardow A., Green Chem. 2014, 16, 3272–3280. [Google Scholar]
- 10. Faure S., PivaLeBlanc S., Piva O., Pete J. P., Tetrahedron Lett. 1997, 38, 1045–1048. [Google Scholar]
- 11. Sideridou I. D., Achilias D. S., Karava O., Macromolecules 2006, 39, 2072–2080. [Google Scholar]
- 12. Ehlers S., Esser U., Fechtel T., Foehles F., Hofmann J., Klinksiek B., Ruhland M., Scholz J. (BAYER AG), WO-A 01/80994, 2001.
- 13.
- 13a. Robertson N. J., Qin Z. Q., Dallinger G. C., Lobkovsky E. B., Lee S., Coates G. W., Dalton Trans. 2006, 5390–5395; [DOI] [PubMed] [Google Scholar]
- 13b. Chen S., Hua Z. J., Fang Z., Qi G. R., Polymer 2004, 45, 6519–6524; [Google Scholar]
- 13c. Chen S., Chen L., Colloid Polym. Sci. 2003, 281, 288–291; [Google Scholar]
- 13d. Wu L. C., Yu A. F., Zhang M., Liu B. H., Chen L. B., J. Appl. Polym. Sci. 2004, 92, 1302–1309; [Google Scholar]
- 13e. Kim I., Byun S. H., Ha C. S., J. Polym. Sci. Part A 2005, 43, 4393–4404; [Google Scholar]
- 13f. Zhang W. Y., Lu L. B., Cheng Y., Xu N., Pan L. S., Lin Q., Wang Y. Y., Green Chem. 2011, 13, 2701–2703; [Google Scholar]
- 13g. Zhang X. H., Chen S., Wu X. M., Sun X. K., Liu F., Qi G. R., Chin. Chem. Lett. 2007, 18, 887–890; [Google Scholar]
- 13h. Klaus S., Lehenmeier M. W., Herdtweck E., Deglmann P., Ott A. K., Rieger B., J. Am. Chem. Soc. 2011, 133, 13151–13161. [DOI] [PubMed] [Google Scholar]
- 14. Le-Khac B. (Arco Chemical Technology, L.P.), US 08/302,296, 1996.
- 15.
- 15a. Wegener G., Brandt M., Duda L., Hofmann J., Klesczewski B., Koch D., Kumpf R.-J., Orzesek H., Pirkl H.-G., Six C., Steinlein C., Weisbeck M., Appl. Catal. A 2001, 221, 303–335; [Google Scholar]
- 15b. Dienes Y., Leitner W., Müller M. G. J., Offermans W. K., Reier T., Reinholdt A., Weirich T. E., Müller T. E., Green Chem. 2012, 14, 1168–1177. [Google Scholar]
- 16.
- 16a. Liu Y., Huang K., Peng D., Wu H., Polymer 2006, 47, 8453–8461; [Google Scholar]
- 16b. Gürtler C., Müller T. E., Kermagoret A., Dienes Y., Barruet J., Wolf A., Grasser S. (Bayer Intellectual Property GmbH), WO2013087582, 2013.
- 17.
- 17a. Liu S., Xiao H., Huang K., Lu L., Huang Q., Polym. Bull. 2006, 56, 53–62; [Google Scholar]
- 17b. Müller T. E., Gürtler C., Wohak M., Hofmann J., Subhani A. M., Leitner W., Peckermann I., Wolf A. (Bayer MaterialScience AG, Germany), WO2014033070, 2014.
- 18. Lawniczak-Jablonska K., Dynowska E., Lisowski W., Sobczak J. W., Chruściel A., Hreczuch A., Libera J., Reszka A., X-Ray Spectrom. 2015, 44, 330–338. [Google Scholar]
- 19. Müller T. E., Gürtler C., Wohak M., Hofmann J., Subhani M. A., Cosemans M., Leitner W. (Bayer Intellectual Property GmbH, Germany), WO2013011014, 2013.
- 20. Müller T. E., Gürtler C., Kermagoret A., Dienes Y., Busygin I., Köhler B., Leitner W. (Bayer MaterialScience AG, Germany), WO2012059550, 2012.
- 21. Müller T. E., Subhani A., Köhler B., Leitner W., Gürtler C. (Bayer MaterialScience AG), EP20130183208, 2015.
- 22.T. Cherubin, Universität-Gesamthochschule-Essen 1999.
- 23.See the Supporting Information.
- 24. Mezger T. G., The Rheology Handbook, Vincentz Network, Hannover, 2011. [Google Scholar]
- 25.
- 25a. Auvergne R., Colomines G., Robin J. J., Boutevin B., Macromol. Chem. Phys. 2007, 208, 690–701; [Google Scholar]
- 25b. Klumperman B., Polym. Chem. 2010, 1, 558–562. [Google Scholar]
- 26. Zeng H., Polymer Adhesion, Friction, and Lubrication, Wiley, Hoboken, 2013. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary