

Abstract
Background/Objective
Novel approaches allowing efficient, readily-translatable image-guided drug delivery (IGDD) against solid tumors is needed. The objectives of this study were to: 1) develop echogenic low temperature sensitive liposomes (E-LTSLs) loaded with an ultrasound (US) contrast agent (perfluoropentane, PFP), 2) determine the in vitro and in vivo stability of contrast agent encapsulation, 3) co-encapsulate and characterize doxorubicin (Dox) E-LTSL, and cellular uptake and cytotoxicity in combination with high intensity focused ultrasound (HIFU).
Method
E-LTSLs were loaded passively with PFP and actively with Dox. PFP encapsulation in E-LTSL was determined by transmission electron microscopy (TEM), and US imageability was determined in tissue mimicking phantoms and mouse tumor model. Dox release from E-LTSL in physiological buffer was quantified by fluorescence spectroscopy. Cellular uptake and cytotoxicity of E-LTSL in presence of HIFU-induced mild hyperthermia (~40–42°C) was determined in a 3D-tumor spheroid model.
Results
TEM & US confirmed that the PFP emulsion was contained within LTSLs. Phantom and animal studies showed that the E-LTSLs were echogenic. Temperature versus size increase and Dox release kinetics of E-LTSLs demonstrated no difference compared to LTSL alone. Dox release was < 5% within 1 h at baseline (25 °C) and body (37°C) temperatures, and was > 99% under hyperthermia. E-LTSL plus HIFU achieved significantly greater Dox uptake in spheroids and cytotoxicity compared to body temperature.
Conclusion
A stable US-imageable liposome co-loaded with Dox and PFP for in vivo IGDD was developed. Data suggest that HIFU can induce cellular uptake and toxicity with E-LTSLs.
1. Introduction
Stealth liposomes theoretically can be used to selectively deliver chemotherapeutic agents to tumors cells, and multiple formulations are currently approved for clinical use1,2. However, their full therapeutic potential has not been realized because the tumor-directed dose escalations achieved to date have not yet improved treatment efficacy (measured as minimized post-treatment cancer recurrence)3–5. Overcoming this limitation and capturing the full therapeutic potential of this technology requires improved ability to control and selectively release liposome-borne drug payloads within tumors, and the ability to image liposomes and released drugs in real time at sufficient resolution to reveal the concentrations and dynamics of intra-tumoral drug delivery.
In previous reports, it has been shown that that lysolipid incorporated low temperature sensitive liposomes (LTSLs) can selectively release doxorubicin upon mild, non-destructive elevations of tumor temperature6–10. For example, Kong et al. showed a tight correlation between doxorubicin concentrations and tumor growth in athymic nude mice bearing the hypopharyngeal FaDu human tumor xenograft upon mild hyperthermia (~ 40–42°C). Similarly, we have shown that the intravascular release of drugs from LTSLs induced by mild local hyperthermia (40–42 °C) using magnetic resonance image guided-high intensity focused ultrasound (MR-HIFU) resulted in improved drug accumulation and distribution in tumors6,11. Despite the potential for enhanced tumor drug accumulation from LTSLs, the ability of physicians to accurately implement real-time image guided drug delivery (IGDD) remains an issue. To address this, novel LTSLs containing doxorubicin and an MRI contrast agent (manganese) (Dox/Mn-LTSLs) that can successfully report on the temporal and spatial patterns of drug delivery have been reported12–16. These studies have shown that the enhanced areas in the magnetic resonance images can allow estimation of the mean amount of doxorubicin that are delivered to the tumor and the mean initial rate of drug delivery. More recently, we and others reported an imageable LTSLs containing a combined payload of gadolinium-based MR contrast agents and the antitumor drug doxorubicin (Dox) combined with MR-HIFU to enable in vivo IGDD that can similarly allow chemodosimetry at the targeted tumor site17,18. The MR-HIFU approach is attractive, however, MR-contrast agents may alter the chemical shift thus disrupting the predicted correlation with actual drug distribution, and hence fidelity of temperature measurement19,20. Thus, even when effectively co-loaded with antitumor drugs, MR- contrast agents may impact drug release. To address this, Davis et al. combined the well-established established proton resonance frequency shift (PRFS) method, which provides a time-series of temperature change images with manganese containing low-temperature sensitive liposomes (Mn-LTSL) to provide absolute tissue temperature21. In this study, Mn ions that were released from LTSL had a 2–3 times greater longitudinal relaxivity than encapsulated Mn2+ allowing it to be imaged with MRI. Thus, localized one time measurement of absolute temperature could be achieved using this MRI technique. While MRI continues to be investigated for drug delivery and thermometry development, there is a critical clinical need to develop an alternative, less-constrained technology, such as ultrasound (US) that can reliably and accurately provide IGDD.
As an initial step, the objective of this study was to develop LTSLs containing a less-problematic ultrasound (US)-based contrast agent. US is widely used for diagnostic imaging in clinics and for controlled drug delivery using microbubbles (MBs)22. 23. MBs are encapsulated in a solid lipid or polymeric shell24, but they are limited by a short half-life that is caused by quick destruction by the reticulo-endothelial system due to their large size and instability upon systemic administration. To circumvent these problems, in this study we encapsulated the US contrast agent perfluoropentane (PFP) in a size-controlled manner within LTSLs to create echogenic LTSLs (E-LTSLs). PFP is a very hydrophobic, nontoxic, noncarcinogenic fluoroalkane with a boiling point (29 °C) between room and body temperatures25. The phase transition temperature for PFP is clinically relevant, as it allows PFP to be injected in the form of liquid droplets dispersed in an aqueous medium that then are converted to echogenic bubbles upon warming to body temperature26. The feasibility of incorporating PFP into stealth-liposomes in a size-controlled manner has been reported previously using lipid-based surfactants27,28 (1,2-dihexadecanoyl-sn-glycero-3-phosphocholine and 1,2-dipalmitoyl-sn-glycero-3-phosphate to emulsify perfluorohexane (PFC6) for loading into liposomes. Negatively-stained liposomes synthesized using these surfactants suggested that PFC6 emulsions were effectively loaded. However, to our best knowledge, this approach has not been applied for synthesis of E-LTSL. Additionally, lipid surfactants may have limited abilities to retain PFC6 emulsions within the liposome aqueous core because PFC6 is so hydrophobic that it can associate with the hydrophobic lipid tails in the liposome bilayer. In one study, Ibsen et al. incorporated a water-soluble surfactant 1,3-propanediol (1,3-PD) into the aqueous core of the liposome to entrap PFP and induce emulsion formation in non-thermosensitive liposome29,30. The formulation approach presented herein adapts the 1,3-PD emulsion methodology, and demonstrates its novel utility for synthesis of E-LTSLs.
2. Material and Methods
2.1 Chemicals
Perfluoropentane (99%, Exfluor Research Corporation, Texas, USA) was used as the US contrast agent. Monostearoyl-2-hydroxy-sn-glycero-3-phosphocholine (MSPC), 1,2-dipalmitoylsn-glycero-3-phosphocholine (DPPC), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (Polyethylene Glycol)2000] (DSPEPEG2000) were obtained from Corden Pharma Corporation (Colorado, USA). Dox was obtained from LC laboratory (MA, USA). Agarose and psyllim fiber were purchased from BDH (Pennsylvania, USA) and Konsyl Pharmaceuticals, (Maryland, USA), respectively. Graphite was purchased from Alpha Aesar, Ward Hill, MA. Acetonitrile (HPLC grade) was obtained from Pharmco-AAPER (Connecticut, USA). Ethylene glycol (99%, spectrophotometric grade), phenylboronic acid (98%), and 2,2-dimethoxypropane (98%) were purchased from Alpha Aesar (Massachusetts, USA). C26 cells were kindly provided by the National Cancer Institute. PC3 prostate and A549 lung cancer cells were kindly provided by Dr. Elankumaran Subbiah (Virginia Tech), and Dr. Lin Lin (Oklahoma State University).
2.2 Synthesis of E-LTSLs
E-LTSLs were prepared by hydration of lipid film followed by the extrusion method described previously31. Briefly, three phospholipids (MSPC, DPPC, and DSPE-mPEG2000) were dissolved in a minimum amount of chloroform (CHCl3) at a molar ratio of 85.3:9.7:5.0. The solvent was evaporated and the resulting lipid film was hydrated in citrate buffer (pH 4.0) mixed with 1,3-PD (0.65M) at 55 °C and extruded five times through double-stacked 200 nm polycarbonate filters to yield a final lipid concentration of 50 mg/mL. A PD-10 size-exclusion column equilibrated with 5–10 column volumes of 1× phosphate buffer saline (PBS) was used to remove free 1,3-PD from the outside of the liposomes. PFP-loaded E-LTSLs were prepared using a one-step sonoporation method. Briefly, 2 mL of the liposomal formulations were incubated under continuous sonication (~20 khz) in 3 mL vials along with PFP (boiling point 30°C; 20 µL/100 mg lipid) for 1–2 min. PFP and LTSLs were kept cold prior to being combined, and the sonication bath was kept at 4°C to minimize PFP vaporization. Free PFP was removed by column purification. This method was repeated at least in triplicate (n = 3) for evaluation.
2.3 Gas chromatography-mass spectrometry (GC-MS) validation of 1,3-PD loading in LTSLs
2.3.1 Extraction/derivatization procedure
A 1:1 volume ratio of chloroform and liposome (1 mL, 50 mg lipids) was mixed for 10 min. The mixture then was centrifuged at 3000 × g for 5 min to remove the lipid components which settled at the organic/water interface. Next, 1 mL internal standard (7.5 mg/dL ethylene glycol in acetonitrile) was added to 500 µL of the top aqueous layer containing PD to achieve a single phase. This was mixed and centrifuged at 3000 × g for 5 min. Then, 500 µL of this solution was added to 650 µL of derivatizing agent (0.5 % phenylboronic acid in 2,2-dimethoxypropane) and mixed at room temperature for 15 min. This mixture was then centrifuged at 3000 × g for 5 min, and 400 µL of the top organic layer was removed for gas chromatography analysis (Fig. 1a).
Figure 1.
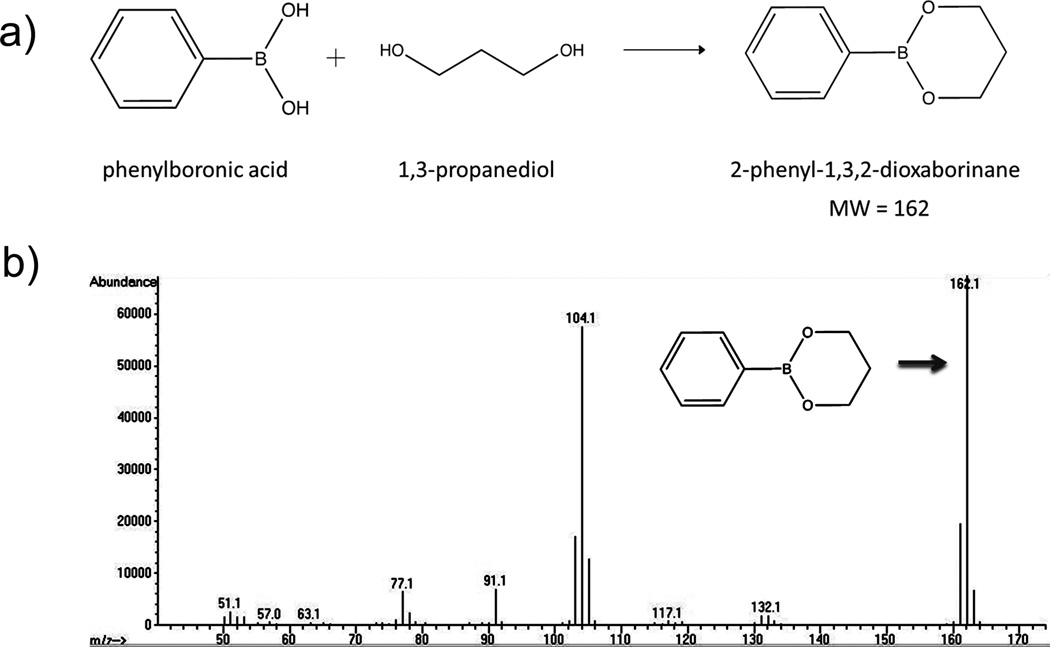
Determination of 1,3-PD in E-LTSL by GC-MS. (A) Derivatized 1,3-PD (MW: 162), (B) Confirmation of presence of derivatized 1,3-PD following two rounds of column purification.
2.3.2 GC-MS analysis
Analysis of PD in liposomes was carried out using an HP5890II gas chromatograph equipped with an FID (Agilent Technologies, Palo Alto, CA, USA). A DB-1column (30 m × 0.25 mm × 0.25 µm) was used. The carrier gas (helium) was delivered at a flow rate of 1 mL/min. Injections (1 µL) were made in the splitless mode with an injector temperature of 250 °C. The detector temperature was set at 300 °C. The temperature program employed was as follows: initial oven temperature at 100 °C for 1 min, increasing 5 °C/min to 150 °C, then 20 °C/min to 290 °C, and holding for 1 min. Data were collected and analyzed using Chemstation software (Agilent Technologies). Peaks were compared to known ethylene glycol and 1,3-PD external standards. Confirmation of the presence of 1,3-PD in E-LTSLs was performed using an HP6890 gas chromatograph coupled with a 5973 mass selective detector (Agilent Technologies). A DB-5MS column (30 m × 0.25 mm × 0.25 µm) was used. The carrier gas (helium) was delivered at a flow rate of 1 mL/min. Injections (1 µL) were made in the splitless mode at an injector temperature of 250 °C. The temperature program employed was as follows: initial oven temperature at 100°C for 2 min, increasing 20°C/min to 280°C, and holding for 3 min. The mass range in the full scan mode covered 50–550 m/z. The transfer line temperature was set at 230 °C, and the solvent delay was 3.10 min. Total ion traces were compared to known ethylene glycol and PD external standards. Mass spectra were deconvoluted using AMDIS 32 software, and the compounds were identified by searching the NIST spectral library.
2.4 TEM imaging of E-LTSL
LTSLs and E-LTSLs were imaged using a negative staining technique and a transmission electron microscope (TEM). Heated (37 and 42°C) LTSLs and E-LTSLs were diluted 500–1000× in PBS. A 10 µL drop of diluted liposomes was pipetted onto a carbon grid (Lacey or Holey grid) and left for 1 min so that the liposomes adsorbed to the grid, then the liquid was wicked away with a piece of filter paper. The grid was allowed to dry for 30 sec. For negative staining, a 9 µL drop of 2% phosphotungstic acid (PTA) was pipetted onto the grid and left for 30 sec, then it was wicked away with a piece of filter paper. Again, the grid was briefly dried before imaging. The imaging was conducted at 200 kV using a JEOL JEM-2100 TEM (JEOL USA, Peabody, MA, USA).
2.5 Monitoring of E-LTSL echogenicity in the agarose phantom model
For US imaging, the phantom recipe (3.0% (w/v) agarose and 3% (w/v) psyllium fiber or 3% (w/v) agarose and 0.25% (w/v) graphite) was optimized to withstand temperatures up to ~70 °C. E-LTSL samples were tested directly after free PFP was removed using the PD-10 column procedure. Prior to imaging, the samples were diluted with PBS to achieve a theoretical concentration of 20–30 mg lipids. Typically, 1 mL of E-LTSL sample was added to the agarose-psyllium phantom wells. For imaging, the phantom well was positioned vertically in an Isotemp water bath such that the lower half of the phantom cell was submerged, and the MS250 US transducer (13–24 MHz) was positioned horizontally just above the water surface. The temperature of each phantom well was recorded via thermocouples positioned at the top of each well (~1 cm below the sample surface). The water bath temperature was set so as to elevate water temperature by ~1 °C/min. Unpurified PBS mixtures containing PFP were used as a positive control. Column-purified PBS mixtures containing PFP alone or 1,3-PD plus PFP were used as additional controls.
2.6 Imageability of E-LTSL in a mouse tumor model
All animal-related procedures were approved and carried out under the guidelines of the Oklahoma State University (OSU) Animal Care and Use Committee. Tumor imaging was performed in six- to 8-week-old male athymic nude mice (BALB/c strain, body weight 20–25 g). Animals were kept as five/cage under specific-pathogen-free conditions with water and food ad-libitum. For tumor initiation, human prostate adenocarcinma cells (PC3) were established as previously described32. PC3 cells at 80–90 % confluence were harvested, washed and diluted with sterile PBS buffer. Three-five million cells were injected subcutaneously, in a total volume of 100 µl, into the rear thigh region of mouse leg using a 27 ga needle. Mice were monitored and weighed daily for tumor growth by serial caliper measurement. Tumor volume was calculated using the formula (length × width2)/2, where length (a) is the largest dimension and width (b) the smallest dimension perpendicular to the length (a × b2)/2. Tumor imaging was initiated when the tumor sizes reached 200–300 mm3. For imaging, the tumors were positioned in specially designed holders that allow the isolated hind flank tumor to be placed in a water bath for 1 hour. The water bath temperature was set to ~43°C; a temperature that had already been calibrated to give tumor temperatures of 41–42°C. All US imaging was conducted using a VisualSonics Vevo 2100 ultrasound MS550D transducer (22–55 MHz).
2.7 Dox loading in E-LTSL
Encapsulation of Dox into the E-LTSLs was carried out using a pH-gradient loading protocol as described by Mayer et al. (21). In general, the outside of the E-LTSLs was adjusted (by column) to about pH 7.4 using PBS, whereas the inside remained acidic at pH 4. Dox was loaded at 2 mg per 100 mg lipid concentration at 37 °C for 1 h. Free Dox and PFP was removed using a PD-10 column. For all in vitro characterization, LTSL was used as a positive control. LTSL synthesis was carried out using our previously published procedure31.
2.8 Size to temperature analysis of E-LTSL
Liposomes (LTSLs and E-LTSLs) were characterized for size (z-average) using a dynamic light scattering (DLS) instrument (Zetapals, Brookhaven Instruments Corporation) by employing a NNLS(non-negatively least squares) algorithm. The calibration standard used was a Thermo scientific 3090A polystyrene nanosphere standard with a particle size of 92+/− 3 nm. During measurement, the filter wheel attenuation for sample optimization was set to between 500kcps – 750kcps for the detectors APD (Avalanche Photo Diode) and PMT (Photo Multiplier Tube) respectively. Briefly, 10–20 µL of LTSLs or E-LTSLs were added to 2 mL of water in a cuvette, and DLS measurements were taken between 25 and 42 °C by increasing the sample holder temperature periodically by 1°C and simultaneously recording the size of the liposomes. For each temperature point, an average of five measurements were taken, and the mean size and standard error of the mean (SEM) were calculated for the LTSL and E-LTSL samples.
2.9 Dox release from LTSLs and E-LTSLs
2.9.1 Thermoscan release assay
Stability at body temperature (37°C) was assessed by measuring release of encapsulated Dox from E-LTSLs as a function of temperature (25–42°C) in vitro in phosphate buffered saline (PBS) and in 10% FBS. For Dox release study, samples (50 mg lipids/1 mg Dox) were diluted 50-fold in either PBS or FBS, and 3 mL of sample was placed in a quartz cuvette equipped with a stopper and magnetic stirrer. A Flurolog 3 spectrofluorometer (HORIBA Jobin Yvon, NJ, USA) equipped with an external temperature controller (Wavelength Electronics, Model LFI-3751, MT, USA) was used to measure Dox release from the E-LTSLs. Dox release was assessed by excitation at 480 nm and fluorescence emission monitored at 590 nm every 1°C from 25–39°C. Each temperature point was held for 3 minutes to ensure the temperature had fully equilibrated. From 39–42°C, fluorescence readings were taken every 0.2°C and each point was equilibrated for 1 minute. LTSL (no PFP) was used under identical conditions as a control.
2.9.2 Dox release kinetics
To measure Dox release as a function of time at a constant temperature, kinetics experiments were performed. For fluorescence measurements, at the same volumes as described in 2.9.1, the samples were equilibrated to the desired temperature (25, 30, 35, and 37–42°C) for 30 min. Baseline fluorescence measurements for each sample were taken at 25°C and complete release was determined by heating the sample for 5 minutes at 45°C. Drug release based on fluorescence quantification at a given time (t) under constant temperature exposure was determined using the equation below:
where Io represented the initial fluorescence intensity of E-LTSL or LTSL suspension at 25°C, and It is its intensity at time (t) at a predetermined temperature. Im represented the fluorescence intensity of completely released Dox at 45°C. Data was obtained as % release of encapsulated Dox at a given temperature.
2.9.3 Dox release as a function of sonication time and US exposure
The effect of co-encapsulation of Dox and PFP on E-LTSL encapsulation or with incubation at 37 °C for > 20 min in the presence of continuous-wave focused US (13–24 MHz VisualSonics transducer) versus its release from conventional LTSLs was studied as follows. E-LTSLs were incubated under continuous sonication (~20 khz, 4 °C) in 3 mL vials for 0, 30, 60, 90,120, 150, 180, and 300 sec. Also, under conditions tested, sample temperature rose by 3°C with longer sonication time (>5min.). This temperature was well below the boiling point of PFP (30°C) and the transition temperature of the LTSL (41–42°C). Similarly, to determine the effect of ultrasound exposure, 1 mL of column-purified E-LTSL sample was added to the agarose-psyllium phantom wells and heated to 37 °C. Ultrasound imaging was performed for > 20 min under constant exposure of 13–24 MHz. During this time, 25–50 µL of sample was collected periodically (0, 3, 6, 9, 12, 15, 18, and 21 min.) from phantom wells. Collected samples were analyzed for Dox fluorescence at 480 nm excitation and 590 nm emissions using a SpectraMax M2e spectrophotometer.
2.10 Cellular uptake of E-LTSL in combination with HIFU
2.10.1. HIFU set-up, 3D-spheroid generation and intracellular Dox estimation
For HIFU treatment, initially A549 human lung adenocarcinoma epithelial cell tumor spheroids were generated as described in our previous publication31. Briefly, uniform sized MCTs were prepared using a liquid overlay technique by coating 96-well plates with 1% agarose (w/v) in phosphate buffered saline (PBS). A549-lung cancer cells were seeded at 1–2 × 104 cells per well in 200 µL of RPMI 1640 supplemented with 10% FBS and 1% penicillin/streptomycin. Cells were incubated at 37 °C with 5% CO2 for 2–3 days. Prior to HIFU treatment, cell morphology was characterized using scanning electron microscopy (SEM). Then, the spheroids were gently transferred in a 0.5 ml thin-walled PCR tube, and placed vertically with its conical bottom aligned within the beam focus of the HIFU transducer as described previously by Hu et al33. 150µL of E-LTSL or LTSL containing 100µM Dox was added to the vials and the hyperthermia treatment of spheroids was performed using an Alpinion wet-type VIFU 2000 HIFU system. Our HIFU transducer has 1.5 MHz central frequency, 45 mm radius and 64 mm aperture diameter with central opening 40 mm in diameter. Prior to actual imaging/hyperthermia, we calibrated the instrument to a temperature of 42°C in PCR tubes by optimizing the HIFU parameters (50% duty cycle, 5 MHz pulse repetitive frequency, 5.5 W electric power and 3.25 W total acoustic power and 60 s time). Temperature elevation in cell suspension was monitored using a thermocouple (Mannix Model DT8852) during the experiment. For drug uptake estimation, B-mode images were collected continuously at optimal HIFU settings at a single hyperthermia point (volume: 1 × 1 × 10 mm). After HIFU treatment, the supernatant and the treated spheroids were collected separately. Spheroid samples (n=10) were rinsed in PBS, and lysed with DMSO. Then, the Dox fluorescence in the lysed sample and supernatant was determined at 480 nm excitation and 590 nm emissions using a SpectraMax M2e spectrophotometer.
2.10.2 Cytotoxicity Assay of E-LTSL and LTSL in combination with HIFU
An in-vitro homogeneous, colorimetric method for determining the number of viable cells using the CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay (Promega, USA) was used to determine any cytotoxic effects of the released Dox from LTSL and E-LTSL upon HIFU in two cell types (Colon C26 and A549 lung cancer cells). Briefly, ~5 × 103 cells suspended in 100 µL of DMEM supplemented with 10% FBS, L-glutamine, NaHCO3, pyridoxine-HCl, and 45,000 mg/L glucose and preserved with 1% penicillin-streptomycin solution were seeded in 96-well plates and incubated for 24 hours at 37 °C in a 5% CO2 atmosphere. Later, the adhered cells were incubated at an equivalent dose of 10µM of Dox containing LTSL and E-LTSL pretreated with HIFU at 4°C for 1h along with untreated control. Following this, the culture media was discarded, and the cells in each well were washed with PBS and re-suspended with 100 µL of cell culture media, and was incubated at 37°C for an additional 48h. Then 20 µL of CellTiter 96® AQueous reagent solution was pipetted into each well, and the plates were incubated for 4 hours at 37 °C in a humidified 5% CO2 atmosphere. The absorbance at 490 nm was recorded using a 96-well Elisa plate reader.
2.10.3. Statistical Analysis
Treatment groups were compared for differences in mean spheroid, supernatant doxorubicin concentration, and cell cytotoxicity using analysis of variance (ANOVA) followed by Tukeys multiple comparison post-hoc test. All analyses were performed using GraphPad Prism 5.0 (GraphPad Software Inc.). All p-values were two-sided, and a p-value less than 0.05 indicated statistical significance. Values are reported as mean±SEM unless otherwise indicated.
Results
3.1 Validation of 1,3-PD encapsulation in E-LTSLs using GC-MS
1,3-PD was readily detected in the E-LTSL after 2 cycles of column purification. As shown in Fig. 1a–b, derivatized 1,3-PD (MW: 162) was readily detected when referenced to known ethylene glycol and PD external standards.
3.2 TEM analysis of emulsion formation in liposomes
TEM studies showed that E-LTSLs effectively emulsified PFP within the liposome core. Like LTSLs, E-LTSLs were spherical at 37 °C (Fig. 2a–b), whereas they became deformed at ~42 °C (Fig. 2c). E-LTSLs retained PFP-1,3-PD emulsions at both 37 °C and 42°C (Fig. 2b–c).
Figure 2.
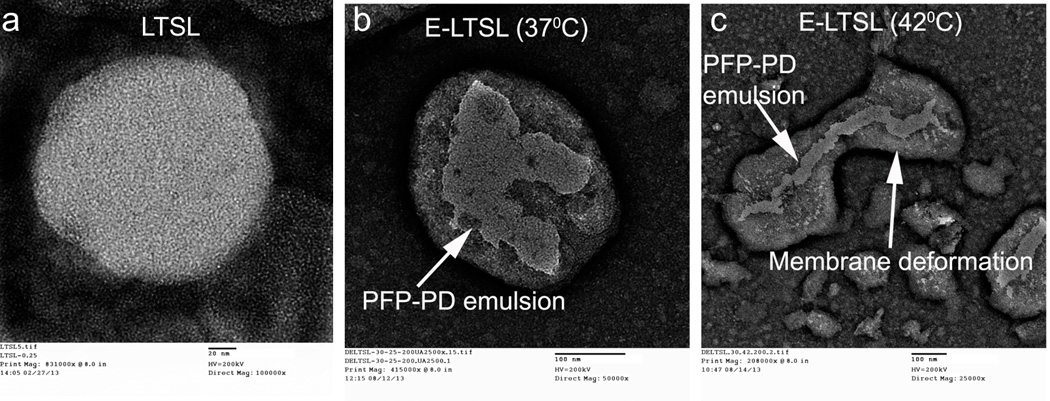
TEM images. (A) LTSL, (B) spherical E-LTSL encapsulating PFP-1,3-PD emulsion at 37 °C, (C) E-LTSL encapsulating PFP-PD emulsion with membrane deformation at 42 °C caused by hyperthermic relaxation of liposome membrane.
3.3 Echogenic behavior of E-LTSLs
In the tissue mimicking phantom, unpurified PBS mixtures containing PFP were used as a positive control, and they were found to be echogenic as expected (Fig. 3a). The column-purified PBS mixtures containing PFP alone or 1,3-PD plus PFP, which were used as additional controls, indicated effective removal of free PFP (20µL/100 mg lipid) external to the LTSLs (Fig. 3a). At optimized ratios of PFP and PD (PFP (20µL):PD (40µL), V/V per mL of LTSL (100 mg total lipid)), the PD-10 column effectively removed > 95% of free PFP. Furthermore, column-purified E-LTSL samples usually exhibited small amounts of echogenicity at ~30 °C (image not shown), and the intensity increased substantially at 37–42 °C (Fig. 3b). In general, as the E-LTSL samples were heated from 25 to 42 °C, US signal intensity progressively increased (4–5 fold from 25 to 39.8 °C, and at ~40 °C, a further 2–3 fold increase occurred),
Figure 3.
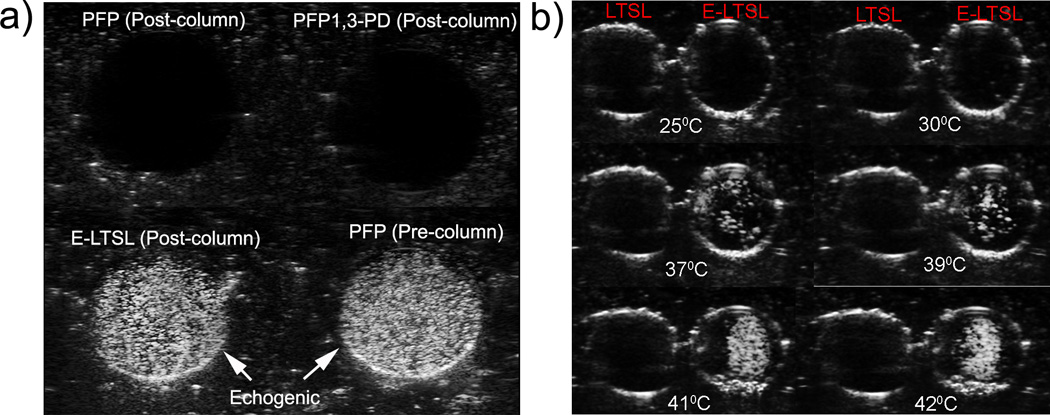
Ultrasound images of LTSL and E-LTSL from the phantom study. (A) Column purified PFP mixture containing PFP or 1,3-PD plus PFP showing no echogenicity (dark round black circle, top panel), whereas column purified E-LTSL sample showing similar echogenicity like unpurified PBS mixture containing PFP (white circular regions, bottom panel), (B) US intensity of LTSL and E-LTSL from 25–42 °C. A substantial increase in US intensity is noted at higher temperatures.
3.4 Confirmation of in vivo imageability of E-LTSL
Continuous high-resolution US images obtained in two-dimensional B-mode from time zero until ~15–20 min showed significant contrast enhancement in the tumor blood vessels (Fig. 4 a–d). In general, the E-LTSL showed maximal contrast for 15–20 min. but in some cases contrast enhancement could be seen post injection till 30–40 min (data not shown).
Figure 4.
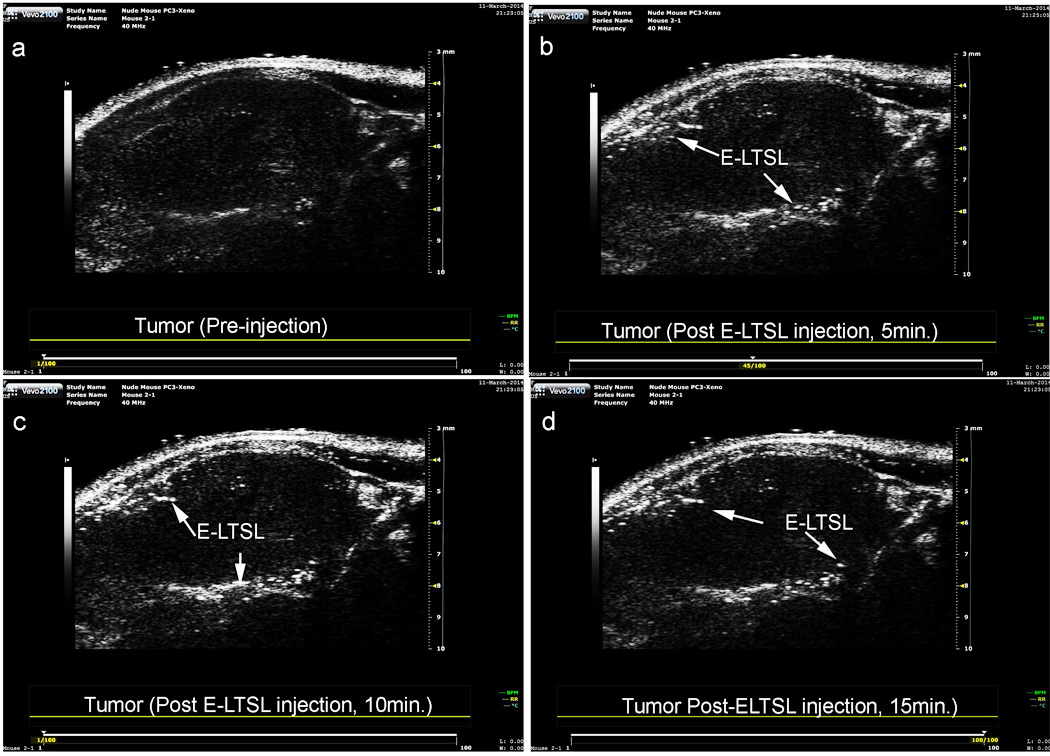
Tumour vascular contrast enhancement following intravenous injection of E-LTSL in a mouse model. A sustained increase in contrast compared to preinjection control was noted at (A) 0 min, (B) 5 min, (C) 10 min, (D) 15 min.
3.5 Size to temperature analysis of LTSL and E-LTSL for thermal stability
The hydrodynamic diameter of LTSLs and E-LTSLs at room temperature (25 °C) was 141 ± 1.5 and 144 ± 1.0 nm, and the polydispersity index values were 0.086 ±0.02 and 0.130 ±0.71, respectively. Also, the sizes of LTSL and E-LTSLs remained stable throughout the 25–37 °C range, with a hydrodynamic size of ~140–150 nm (Fig. 5a). Similar to LTSL, the synthesized E-LTSLs were also stable in an aqueous environment and exhibited no visual evidence of particle accumulation after 48 h of storage at 4 °C (Fig. 5b).
Figure 5.
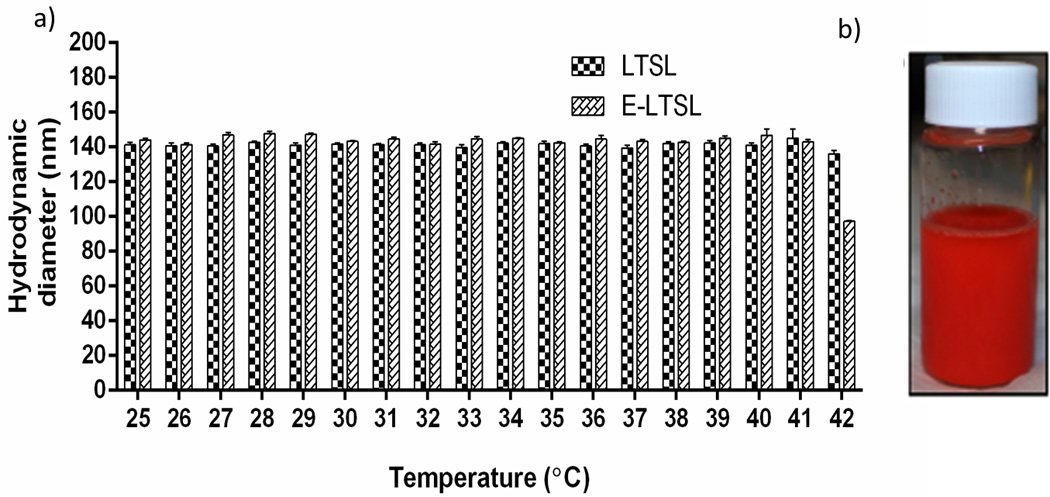
(A) Size to temperature analysis of LTSL and E-LTSL demonstrating stable hydrodynamic diameter (~150 nm) throughout the 25–42 °C range, (B) E-LTSL vial following 48 h storage at 4 °C with no visual evidence of particle accumulation.
3.6 Dox loading and release in physiological buffer
Active loading of Dox by transmembrane pH gradient yielded an encapsulation efficiency of > 95% for LTSLs. Compared to LTSLs; E-LTSL encapsulated ~70–80% of the added drug upon sonication.
3.6.1 Thermoscan release assay
Percent Dox release from E-LTSL and LTSL alike in PBS by its fluorescence dequenching was minimal (<5%) at 25–39°C (Fig. 6a–b); was followed by a more gradual release at 40°C (~20%), and was rapid and complete (>95%) near the temperature giving maximum release rate (~41–42°C). In 10% FBS, Dox release from E-LTSL and LTSL was ~20% at 37°C, and ~ 90% at 40°C. Greater than 95% release was attained at 41–42°C.
Figure 6.
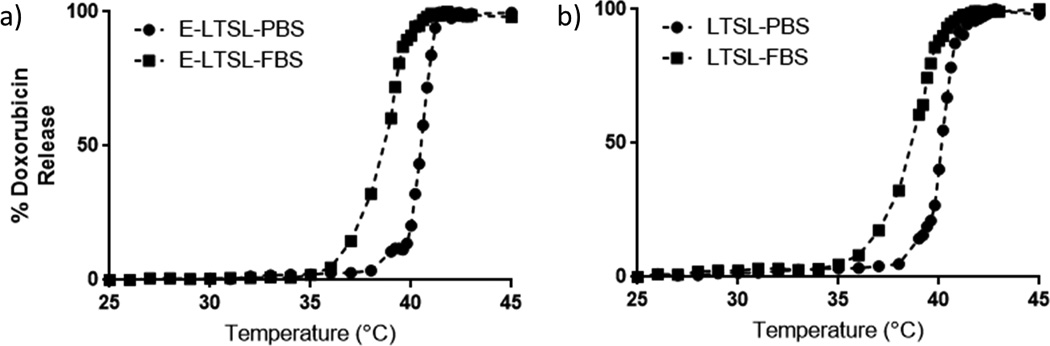
Thermoscan assay in physiological buffer between 25–42 °C. (A) Release of Dox from E-LTSL was relatively greater in serum than in PBS. (B) Release profile of LTSL demonstrates a similar profile to E-LTSL (A).
3.6.2 Dox release kinetics
When incubated for 30min. each at various temperature points (25–42°C) in PBS, both E-LTSL and LTSL exhibited similar kinetics (Fig.7c–d). In general, between 25–38°C release was <10%, and ~10–15% release was seen at 39°C. At 40°C within 10 minutes, there was a gradual increase in Dox release to 50–60%, and >95% release was achieved by 42°C. Compared to PBS, release was more prominent in FBS with 20–30% of the encapsulated drug releasing at 37°C (Fig. 7a–b), and by 40°C, about 90% release was observed. However, >95% release was achieved between 41–42°C.
Figure 7.
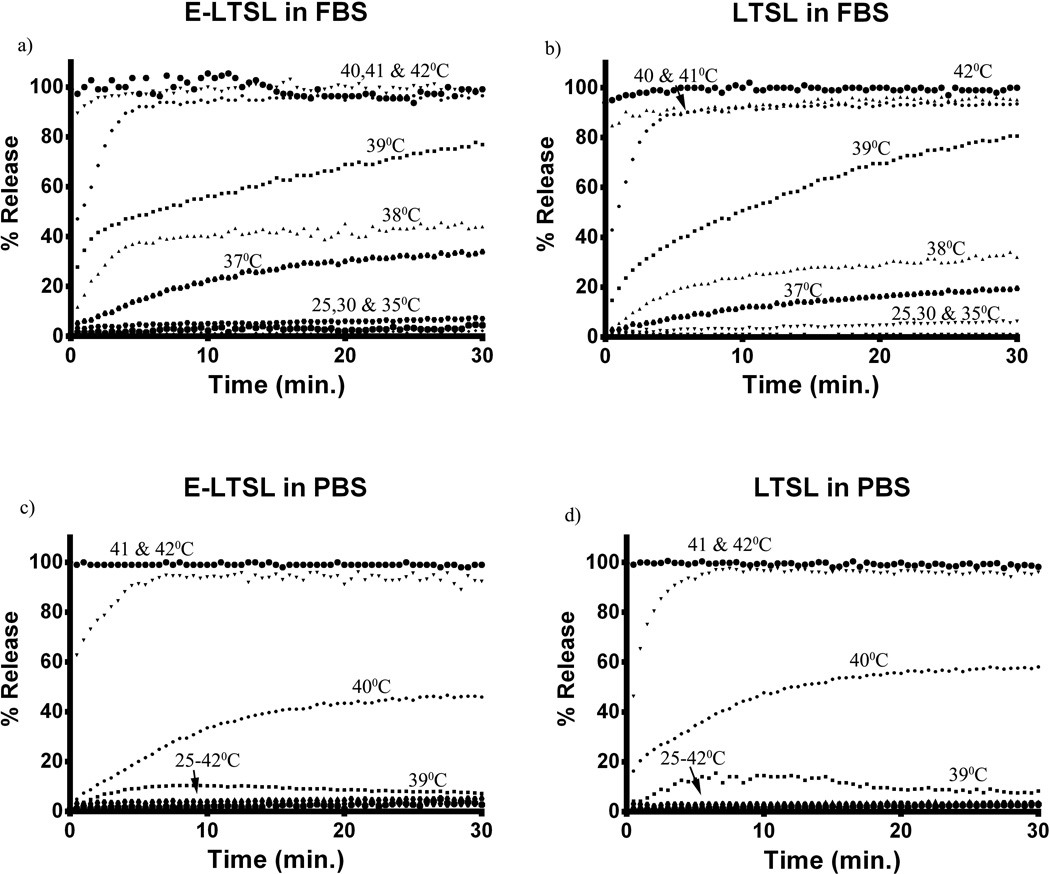
Dox release kinetics. (A, B) Release from E-LTSL and LTSL in serum as a function of time (30 min) at constant temperature. (C, D) Release from E-LTSL and LTSL in PBS as a function of time (30 min) at constant temperature.
3.6.3 Dox release as a function of sonication time and US exposure
Release of Dox from E-LTSLs was proportional to sonication time. Dox release was ~10% for 30 sec–1 min, 20–40% for 1–2 min, and almost 70% upon 5min. sonication (Fig. 8a). Release of Dox upon sonication was similar for LTSLs and E-LTSLs, and it was independent of PFP loading. When E-LTSLs were suspended at 37 °C in the presence of continuous-wave US (13–24 MHz) for greater than 20 min. in a tissue-mimicking agarose-phantom model, no significant time-related change in the fluorescence readings was detected (Fig. 8b)
Figure 8.
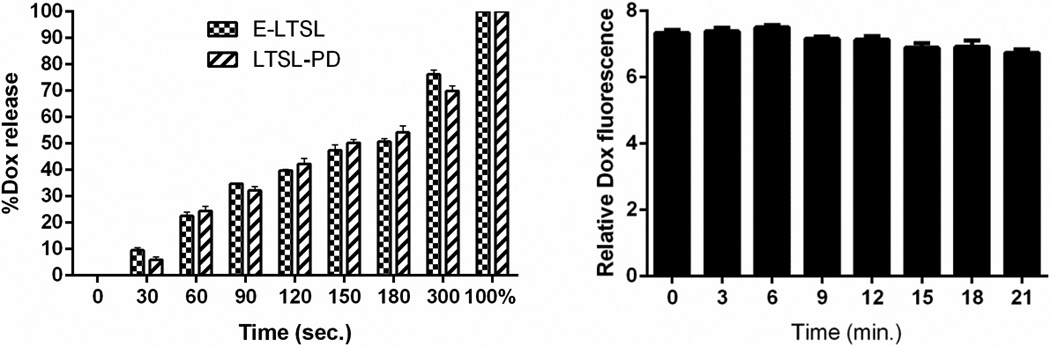
(A) Dox release as a function of sonication time (0–5 min). (B) Dox release in the presence of continuous-wave focused US (3–12 MHz).
3.7 Cellular uptake of Dox in combination with HIFU
The mild hyperthermia heating parameters resulted in an accurate (41±1 °C, target=40–42 °C) and homogeneous (SD=1.0 °C) temperature within the targeted region of interest (ROI). It took approximately 15s to achieve the target temperature range of 40–41 °C, after which the temperature was maintained consistently for 5min. by manually switching off or turning on the sonication. The optimized method resulted in complete release of encapsulated drug in the tumor spheroid supernatant was 13.52±0.1, 12.52±0.08, 35.02±0.18, 34.15±0.2 µM Dox for LTSL, E-LTSL, LTSL+ HIFU, and E-LTSL + HIFU respectively (Fig. 9a). Also, the LTSL+HIFU & ELTSL+HIFU demonstrated significantly greater cellular uptake compared to LTSL and E-LTSL alone in the tumor spheroids (p<0.05, Tukeys). Spheroid doxorubicin concentrations were 2.82±0.03, 2.97±0.01, 7.76±0.15, 9.85±0.1 µM Dox for LTSL, E-LTSL, LTSL+ HIFU, and E-LTSL + HIFU respectively (Fig. 9b).
Figure 9.
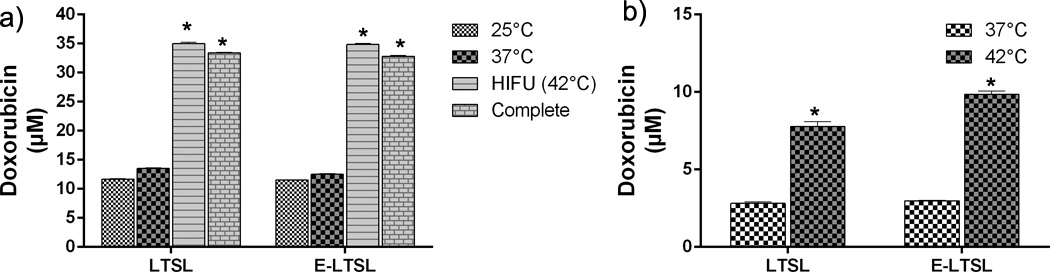
Mild hyperthermia-induced drug release. (A) Significantly greater release in heated sample (LTSL & ELTSL, 42°C) relative to unheated control at 37°C was noted in cell supernatant. (B) Greater intracellular accumulation of the released drug was noted in 3D tumour spheroid upon HIFU plus E-LTSL and LTSL mild hyperthermia treatment compared to untreated samples (*p<0.05).
3.8. Cytotoxicity assay
Co-incubation of C26 and A549cells with LTSL and E-LTSL at body temperature did not result in significant differences in toxicity compared to untreated control. In contrast, cells treated with pre-HIFUed LTSL and E-LTSL showed significant toxicity in the dose range of 10µM Dox compared to untreated and body temperature samples in both the cell lines (p<0.05, Fig. 10a– b). Additionally, the toxicity at the indicated dose range was greater in C26 than A549 presumably due to higher sensitivity to Dox for C26 cell lines.
Figure 10.
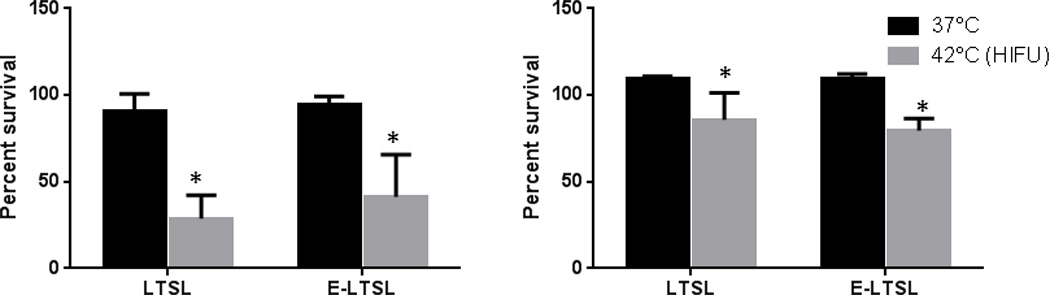
Cytotoxicity of LTSL and E-LTSL at body temperature and HIFU-induced mild hyperthermia in (A) C-26 cells, (B) A549 cells. All data were normalised to untreated control. Significant toxicity of released drug was noted at 42 °C compared to body temperature (*p<0.05).
4. Discussion
The objective of this study was to formulate and characterize a heat-activated US-imageable liposome-based drug delivery system to improve tumor targeted drug delivery. Successful translation of this liposome can help guide and localize treatment, improve clinical practice and therapeutic outcomes, even with currently available chemotherapeutic agents such as Dox.
4.1 Choice of liposome, PFP and imaging modality
To permit delivery of nonselective, fairly toxic anticancer drugs selectively to tumors, an encapsulated-drug delivery system that allows externally triggered, localized release of drug payloads is needed. In this study we used Dox-encapsulated LTSLs that are sensitive to mild, non-destructive temperature elevations above normal body temperature. Additionally, to provide imaging and precision-warming capabilities, we co-encapsulated LTSLs with PFP, an echogenic contrast agent (Fig. 2a–b). This can permit in vivo tracking of liposome distribution using an US device and improve/fine-tune real-time control of drug delivery. The imageability of E-LTSL was verified in in vitro tissue-mimicking phantoms and mouse prostate tumors where E-LTSLs provided significantly enhanced contrast compare to relevant controls (Fig.3a–b, 4a–d). As a contrast agent, PFP has many benefits25,27. PFP transitions from a liquid (~29°C) to an echogenic state at body temperature, which can allow us to track LTSL redistribution during locally-applied hyperthermia to induce Dox release from the liposomes. Also, when stabilized by a lipid shell, the Laplace pressure (i.e. the pressure difference between the inside and the outside of PFP) substantially increases its boiling temperature. This is caused by the surface tension at the interface between PFP and bulk liquids34. As shown in Fig.3b, as the samples were heated from 25 to 42 °C, US signal intensity progressively increased (4–5 fold from 25 to 42°C). This predictable property of PFP boiling point changes can be hypothetically applied for nanothermometry and nanomonitoring of drug delivery. Studies are currently underway in to understand this phenomenon in an in vivo model.
One major advantage of our E-LTSL technology is that the co-encapsulation of PFP in LTSL is very stable. As shown previously by Ibsen et al and our study, the addition of a water soluble surfactant such as 1,3-PD into the aqueous core of the liposome is important in reducing the interfacial tension against water29,35 and induce PFP emulsion formation. (Fig.2b). 1,3-PD is also nontoxic, even with repeated inhalation exposures in rats, and thus may have an easier translation path36. Because 1,3-PD is a polar but uncharged molecule it is conceivable that during storage at 4°C it may escape the liposomal membrane in its native state, but the process of PFP emulsification and resulting increase in emulsion size likely reduces its permeability through the liposomal membrane. Further, 1,3-PD contains two OH groups that enhance the strength of hydrogen bonding, thereby preventing its rapid exit from the liposomal core and escape through the lipid bilayer. The theoretical basis of our rationale was supported by Gas Chromatography-Mass Spectrophotometry (GC-MS) analysis, which showed 1,3-PD was readily detected in the E-LTSL sample after 2 cycles of column purification (Fig 1a–b).
Our in vitro data suggests that the emulsion-loaded E-LTSL has an excellent thermal stability. Size-vs.-temperature analysis revealed that E-LTSLs remained stable throughout the 25–37°C range, with a hydrodynamic size of ~100–150 nm (Fig. 5a), close to that of TSL currently used in phase II and III studies (http://celsion.com/). The synthesized E-LTSLs were also stable in aqueous environment, with no visual evidence of particle accumulation after 48 hr storage at 4 °C (Fig. 5b). These promising results indicate that our synthesis procedure confers PFP with the crucial needed properties of water miscibility, lack of major hyperthermia-induced changes in liposomal size, and proof that PFP is retained in emulsion form within liposomes under the relevant conditions and temperatures, rather simply forming an admixture with the LTSL.
4.2 Key challenges of MB-assisted delivery and proposed E-LTSL based solutions
As drug delivery system, MBs have two major shortcomings: 1) MBs are large and have relatively low stability and circulation half-life (<~5min) compared with LTSLs; and 2) MBs cannot encapsulate a large quantity of drug within their thin shells (~ 3 nm). In contrast, LTSLs have a much higher drug capacity due to incorporation of the drug in their aqueous core. The results of our study indicate that encapsulation of PFP in a size-controlled manner within LTSLs can theoretically increase their half-life and confer echogenicity (~15–20 min.), which may make their use more clinically feasible than that of MBs. Since the resolution of US transducer is in the micrometer range, it is not clear if the E-LTSLs at higher temperature (~40°C) remains nanosized dispersion or complexes with other E-LTSLs to give a microbubble-like appearance. Balancing drug release, imageability and therapeutic efficacy, while maintaining systemic stability, is the major consideration for optimization of any drug-plus-contrast agent formulation. Regardless, our data indicate that co-encapsulation of PFP in E-LTSL had no effect on Dox release as a function of temperature or with incubation at 37°C for >20 min in the presence of continuous-wave focused US (13–24 MHz), vs. its release from conventional LTSL (Fig. 8a–b). Thus, Dox release from E-LTSL and LTSL alike was minimal (<5%) at 25–39°C (Fig. 8a); was followed by a more gradual release at 40°C (~20%), and was rapid and complete (>95%) near the temperature giving maximum release rate (~41–42°C). Similarly, when E-LTSLs were suspended at 37°C in the presence of continuous-wave focused US (13–24 MHz) in a tissue-mimicking agarose-psyllium fiber phantom model used to mimic in vivo tissue warming/imaging conditions, and sampled at various times (0,3,6,9,12,15,18,21 min), no time-related change in the fluorescence readings was seen (Fig. 8b). It is noteworthy however that the Dox release in serum rich medium was relatively greater than PBS. This is in agreement with previous studies37,38 and needs more fine-tuning to improve in vivo stability.
Nonetheless, our data suggests that a continuous MHz-frequency US exposure, or bubble formation inside E-LTSL at 37°C, does not alter liposome integrity or induce premature Dox release. This was further evidenced by TEM images showing that E-LTSL liposomes were spherical at 37°C (Fig. 2a–b), whereas they became deformed at ~42°C due to hyperthermic relaxation of the liposomal membrane yet nonetheless retained emulsions (Fig. 2c), indicating that warming did not cause liposomes to burst, but that instead the melting of lipid components created pores on the membrane that triggered drug release. This is in line with previous works by Ickenstein et al. who reported appearance of open liposomes and bilayer discs when LTSLs were cycled through the gel to liquid-crystalline state (~40–42°C)39. At melting temperatures, DSPE-PEG and lysolipids adopts a thermodynamically favored micelle-like conformation within the bilayer to stabilize the rim of open liposome structures. Depending on the degree of lateral lipid domain formation, this process may lead to the formation of membrane pores, open liposomes, or bilayer discs. Thus, E-LTSLs can be encapsulated with anticancer agents and plausibly mediate intravascular release of Dox and cellular uptake in tumors at higher temperatures. This is important since unlike non-thermosensitive liposomes, E-LTSL formulation does not rely on EPR-mediated drug delivery, which requires a long carrier half-life. Furthermore, unlike micro/nanobubbles that generally have a bimodal size range (100 nm to ~1–2 µm) that can adversely affect in vivo half-life and efficiency, intravascular drug release from size-controlled E-LTSLs (100–150 nm) can be triggered immediately following injection by localized hyperthermia of the tumor region even with a slower residence time (~20–30 min.). As a proof of principle to the proposed approach, we tested the hypothesis that Dox release and uptake can be improved in combination with mild-hyperthermia mediated by HIFU. Dox estimation in spheroid and monolayer cell model suggests that drug delivery and cytotoxicity with LTSL+HIFU and E-LTSL+HIFU hyperthermia was significantly greater compared to unheated samples (Fig. 9a–b, 10a–b). Studies are currently underway in a mouse tumor model to further demonstrate the utility of this approach. However, our initial in vitro studies suggest that E-LTSL may be potentially combined with HIFU for targeted drug delivery.. Thus, this therapeutic method needs further investigation for possible clinical translation.
5. Conclusion
Building on prior success with MR-imageable liposomes and LTSL-mediated drug delivery12,13,17,21, we have developed an US imageable formulation. Our data suggest that E-LTSLs incorporate Dox and release it in a temperature-controlled manner upon US-directed site-specific warming and that PFP forms stable emulsions within the liposomal core in the presence of water-soluble surfactant. E-LTSLs provide significantly enhanced US contrast compared to that seen with relevant controls. Thus, E-LTSLs have the potential to permit IGDD in tumors.
Acknowledgments
Research reported in this publication was supported by the Center for Veterinary Health Sciences Seed Support, National Cancer Institute of the National Institutes of Health under Award Number R15CA179369, and Oklahoma Center for Advancement in Science and Technology (OCAST). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or OCAST.
References
- 1.Bae YH, Park K. Targeted drug delivery to tumors: myths, reality and possibility. Journal of controlled release : official journal of the Controlled Release Society. 2011;153:198–205. doi: 10.1016/j.jconrel.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- 3.O'Brien ME, et al. Reduced cardiotoxicity and comparable efficacy in a phase III trial of pegylated liposomal doxorubicin HCl (CAELYX/Doxil) versus conventional doxorubicin for first-line treatment of metastatic breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2004;15:440–449. doi: 10.1093/annonc/mdh097. [DOI] [PubMed] [Google Scholar]
- 4.Lyseng-Williamson KA, Duggan ST, Keating GM. Pegylated liposomal doxorubicin: a guide to its use in various malignancies. BioDrugs : clinical immunotherapeutics, biopharmaceuticals and gene therapy. 2013;27:533–540. doi: 10.1007/s40259-013-0070-1. [DOI] [PubMed] [Google Scholar]
- 5.Park K. Questions on the role of the EPR effect in tumor targeting. Journal of controlled release : official journal of the Controlled Release Society. 2013;172:391. doi: 10.1016/j.jconrel.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Ranjan A, et al. Image-guided drug delivery with magnetic resonance guided high intensity focused ultrasound and temperature sensitive liposomes in a rabbit Vx2 tumor model. Journal of controlled release : official journal of the Controlled Release Society. 2012;158:487–494. doi: 10.1016/j.jconrel.2011.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Needham D, Anyarambhatla G, Kong G, Dewhirst MW. A new temperature-sensitive liposome for use with mild hyperthermia: characterization and testing in a human tumor xenograft model. Cancer research. 2000;60:1197–1201. [PubMed] [Google Scholar]
- 8.Kong G, et al. Efficacy of liposomes and hyperthermia in a human tumor xenograft model: importance of triggered drug release. Cancer research. 2000;60:6950–6957. [PubMed] [Google Scholar]
- 9.Tashjian JA, Dewhirst MW, Needham D, Viglianti BL. Rationale for and measurement of liposomal drug delivery with hyperthermia using non-invasive imaging techniques. Int J Hyperther. 2008;24:79–90. doi: 10.1080/02656730701840147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kircher MF, Willmann JK. Molecular body imaging: MR imaging, CT, and US. part I. principles. Radiology. 2012;263:633–643. doi: 10.1148/radiol.12102394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Smet M, et al. Magnetic resonance guided high-intensity focused ultrasound mediated hyperthermia improves the intratumoral distribution of temperature-sensitive liposomal doxorubicin. Investigative radiology. 2013;48:395–405. doi: 10.1097/RLI.0b013e3182806940. [DOI] [PubMed] [Google Scholar]
- 12.Viglianti BL, et al. Chemodosimetry of in vivo tumor liposomal drug concentration using MRI. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2006;56:1011–1018. doi: 10.1002/mrm.21032. [DOI] [PubMed] [Google Scholar]
- 13.Ponce AM, et al. Magnetic resonance imaging of temperature-sensitive liposome release: drug dose painting and antitumor effects. Journal of the National Cancer Institute. 2007;99:53–63. doi: 10.1093/jnci/djk005. [DOI] [PubMed] [Google Scholar]
- 14.Viglianti BL, et al. In vivo monitoring of tissue pharmacokinetics of liposome/drug using MRI: illustration of targeted delivery. Magnetic resonance in medicine : official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2004;51:1153–1162. doi: 10.1002/mrm.20074. [DOI] [PubMed] [Google Scholar]
- 15.Tashjian JA, Dewhirst MW, Needham D, Viglianti BL. Rationale for and measurement of liposomal drug delivery with hyperthermia using non-invasive imaging techniques. Int J Hyperthermia. 2008;24:79–90. doi: 10.1080/02656730701840147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kircher MF, Willmann JK. Molecular body imaging: MR imaging, CT, and US. Part II. Applications. Radiology. 2012;264:349–368. doi: 10.1148/radiol.12111703. [DOI] [PubMed] [Google Scholar]
- 17.Negussie AH, et al. Formulation and characterisation of magnetic resonance imageable thermally sensitive liposomes for use with magnetic resonance-guided high intensity focused ultrasound. Int J Hyperthermia. 2010;27:140–155. doi: 10.3109/02656736.2010.528140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.de Smet M, Heijman E, Langereis S, Hijnen NM, Grull H. Magnetic resonance imaging of high intensity focused ultrasound mediated drug delivery from temperature-sensitive liposomes: an in vivo proof-of-concept study. Journal of controlled release : official journal of the Controlled Release Society. 2010;150:102–110. doi: 10.1016/j.jconrel.2010.10.036. [DOI] [PubMed] [Google Scholar]
- 19.Deckers R, et al. Absolute MR thermometry using nanocarriers. Contrast Media Mol I. 2014;9:283–290. doi: 10.1002/cmmi.1572. [DOI] [PubMed] [Google Scholar]
- 20.Hijnen NM, et al. The magnetic susceptibility effect of gadolinium-based contrast agents on PRFS-based MR thermometry during thermal interventions. Journal of therapeutic ultrasound. 2013;1:8. doi: 10.1186/2050-5736-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Davis RM, et al. A method to convert MRI images of temperature change into images of absolute temperature in solid tumours. Int J Hyperther. 2013;29:569–581. doi: 10.3109/02656736.2013.790091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hernot S, Klibanov AL. Microbubbles in ultrasound-triggered drug and gene delivery. Advanced drug delivery reviews. 2008;60:1153–1166. doi: 10.1016/j.addr.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Javadi M, Pitt WG, Belanp DM, Tsosie NH, Hartley JM. Encapsulating Nanoemulsions Inside eLiposomes for Ultrasonic Drug Delivery. Langmuir : the ACS journal of surfaces and colloids. 2012 doi: 10.1021/la303464v. [DOI] [PubMed] [Google Scholar]
- 24.Fokong S, et al. Image-guided, targeted and triggered drug delivery to tumors using polymer-based microbubbles. Journal of controlled release : official journal of the Controlled Release Society. 2012;163:75–81. doi: 10.1016/j.jconrel.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 25.Rapoport N, et al. Ultrasound-mediated tumor imaging and nanotherapy using drug loaded, block copolymer stabilized perfluorocarbon nanoemulsions. Journal of controlled release : official journal of the Controlled Release Society. 2011;153:4–15. doi: 10.1016/j.jconrel.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kandadai MA, et al. Comparison of surfactants used to prepare aqueous perfluoropentane emulsions for pharmaceutical applications. Langmuir : the ACS journal of surfaces and colloids. 2010;26:4655–4660. doi: 10.1021/la100307r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rapoport N, Gao ZG, Kennedy A. Multifunctional nanoparticles for combining ultrasonic tumor imaging and targeted chemotherapy. Journal of the National Cancer Institute. 2007;99:1095–1106. doi: 10.1093/jnci/djm043. [DOI] [PubMed] [Google Scholar]
- 28.Javadi M, Pitt WG, Belnap DM, Tsosie NH, Hartley JM. Encapsulating Nanoemulsions Inside eLiposomes for Ultrasonic Drug Delivery. Langmuir : the ACS journal of surfaces and colloids. 2012;28:14720–14729. doi: 10.1021/la303464v. [DOI] [PubMed] [Google Scholar]
- 29.Ibsen S, et al. A novel nested liposome drug delivery vehicle capable of ultrasound triggered release of its payload. Journal of controlled release : official journal of the Controlled Release Society. 2011;155:358–366. doi: 10.1016/j.jconrel.2011.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen CC, Borden MA. The role of poly(ethylene glycol) brush architecture in complement activation on targeted microbubble surfaces. Biomaterials. 2011;32:6579–6587. doi: 10.1016/j.biomaterials.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Senavirathna LK, et al. Tumor Spheroids as an In Vitro Model for Determining the Therapeutic Response to Proton Beam Radiotherapy and Thermally Sensitive Nanocarriers. Theranostics. 2013;3:687–691. doi: 10.7150/thno.6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El Hilali N, Rubio N, Blanco J. Noninvasive in vivo whole body luminescent analysis of luciferase labelled orthotopic prostate tumours. Eur J Cancer. 2004;40:2851–2858. doi: 10.1016/j.ejca.2004.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Hu Z, et al. Release of endogenous danger signals from HIFU-treated tumor cells and their stimulatory effects on APCs. Biochemical and biophysical research communications. 2005;335:124–131. doi: 10.1016/j.bbrc.2005.07.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rapoport N. Phase-shift, stimuli-responsive perfluorocarbon nanodroplets for drug delivery to cancer. Wiley interdisciplinary reviews. Nanomedicine and nanobiotechnology. 2012;4:492–510. doi: 10.1002/wnan.1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kandadai MA, et al. Comparison of Surfactants Used to Prepare Aqueous Perfluoropentane Emulsions for Pharmaceutical Applications. Langmuir : the ACS journal of surfaces and colloids. 2010;26:4655–4660. doi: 10.1021/la100307r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Scott RS, Frame SR, Ross PE, Loveless SE, Kennedy GL. Inhalation toxicity of 1,3-propanediol in the rat. Inhalation toxicology. 2005;17:487–493. doi: 10.1080/08958370590964485. [DOI] [PubMed] [Google Scholar]
- 37.Hossann M, et al. In vitro stability and content release properties of phosphatidylglyceroglycerol containing thermosensitive liposomes. Biochimica et biophysica acta. 2007;1768:2491–2499. doi: 10.1016/j.bbamem.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 38.Hossann M, et al. Proteins and cholesterol lipid vesicles are mediators of drug release from thermosensitive liposomes. Journal of controlled release : official journal of the Controlled Release Society. 2012;162:400–406. doi: 10.1016/j.jconrel.2012.06.032. [DOI] [PubMed] [Google Scholar]
- 39.Ickenstein LM, Arfvidsson MC, Needham D, Mayer LD, Edwards K. Disc formation in cholesterol-free liposomes during phase transition. Biochimica et biophysica acta. 2003;1614:135–138. doi: 10.1016/s0005-2736(03)00196-2. [DOI] [PubMed] [Google Scholar]