

Abstract
Granular cell tumors are an uncommon soft tissue neoplasm. Malignant granular cell tumors comprise <2% of all granular cell tumors, are associated with aggressive behavior and poor clinical outcome, and are poorly understood in terms of tumor etiology and systematic treatment. Because of its rarity, the genetic basis of malignant granular cell tumor remains unknown. We performed whole-genome sequencing of one malignant granular cell tumor with metabolic response to pazopanib. This tumor exhibited a very low mutation rate and an overall stable genome with local complex rearrangements. The mutation signature was dominated by C>T transitions, particularly when immediately preceded by a 5′ G. A loss-of-function mutation was detected in a newly recognized tumor suppressor candidate, BRD7. No mutations were found in known targets of pazopanib. However, we identified a receptor tyrosine kinase pathway mutation in GFRA2 that warrants further evaluation. To the best of our knowledge, this is only the second reported case of a malignant granular cell tumor exhibiting a response to pazopanib, and the first whole-genome sequencing of this uncommon tumor type. The findings provide insight into the genetic basis of malignant granular cell tumors and identify potential targets for further investigation.
Keywords: pharyngeal neoplasm
INTRODUCTION
Granular cell tumors are soft tissue neoplasms believed to be of neural origin (Lage et al. 2013). Most of these tumors are benign and can be treated with surgical resection, without need for adjuvant radiation and/or chemotherapy. However, malignant granular cell tumors, comprising <2% of all granular cell tumors, are extremely rare and aggressive. They are associated with poor prognosis, with 39% of patients succumbing to this disease within 3 yr (Fanburg-Smith et al. 1998). The genetic basis of malignant granular cell tumor is currently not known, and there is little knowledge with respect to its tumor biology. The current criteria for distinguishing benign, atypical, and malignant granular cell tumors are based on histological evidence. Although surgical resection can be curative for patients with localized disease, there is no effective treatment for unresectable or metastatic malignant granular cell tumors.
Recently, Conley et al. (2014) reported the first documented response of a malignant granular cell tumor to pazopanib. A gene expression profiling analysis was conducted on this patient's tumor, and dasatinib, an inhibitor of Src family kinases, was initiated primarily because SRC was the most significantly overexpressed gene. After the patient failed to respond to dasatinib, pazopanib was selected because of the overexpression of VEGFA. Pazopanib is a potent receptor tyrosine kinase inhibitor selectively targeting EGFR types 1, 2, and 3, PDGFR α and β, C-Kit, FGFR 1 and 3, ITK, LCK, and C-FMS (encoded by CSF1R) (van der Graaf et al. 2012). The patient responded to treatment with pazopanib, with an improvement in pulmonary and soft tissue metastases that was ongoing after 4 mo of therapy. Although the exact mode of action responsible for tumor response to pazopanib in this case remains unknown, this study suggests the utility of genome and expression profiling in treatment selection.
In the present study, we performed whole-genome sequencing (WGS) of a case of malignant granular cell tumor from a patient who had a metabolic response to pazopanib. This provides the first characterization of the genomic landscape of this rare disease. Our results provide insight about the genetic basis of this poorly understood and aggressive disease.
RESULTS
Clinical Presentation
A 26-yr old female with no significant past medical history sought medical attention for dysphagia, multiple subcutaneous nodules, and unintentional weight loss. A laryngoscopy revealed a mass involving the posterior hypopharyngeal wall and surrounding areas. Computed tomography (CT) imaging detected a 7.2-cm heterogeneously enhancing mass centered in the posterior pharyngeal space with bilateral enlarged level II cervical lymph nodes. 18-fluorodeoxy-glucose positron emission tomography (FDG-PET) revealed FDG-avid lesions in bilateral breasts, scalp, pelvis, subcutaneous tissue of buttocks and in the left proximal humerus. Pathology review of the resection of the hypopharyngeal mass and biopsies of a subcutaneous nodule and breast lesion revealed a neoplasm that consisted of large polygonal cells with intracytoplasmic eosinophilic globules. These cells had large nuclei with mild-to-moderate atypia and occasional macronucleoli. Necrosis or spindling was not identified. The neoplastic cells were positive for S100, CD68 (Fig. 1), and CD57 (not shown) immunohistochemical stains. The morphologic and immunohistochemical features were consistent with “granular cell tumor of uncertain malignant potential.” However the clinical features were suggestive of a malignant phenotype with the hypopharyngeal mass presumed to be the primary site.
Figure 1.

Hematoxylin and eosin (H&E)-stained images of a granular cell tumor. (A) High magnification of pleomorphic polygonal cells with granular cytoplasm. (B) Granular cell neoplasm infiltrating skeletal muscle. (C) Positive immunohistochemical staining for S100. (D) Positive immunohistochemical staining for CD68.
Treatment was initiated with gemcitabine 675 mg/m2 on days 1 and 8, plus docetaxel 100 mg/m2 on day 8 with growth factor support in a 21-d cycle. Because there was no objective response after three cycles, the patient was switched to pazopanib at 800 mg/day. Restaging FDG-PET after 3 mo of therapy showed significant interval decrease in FDG avidity of multiple lesions, including soft tissue and skeletal metastases consistent with metabolic response to therapy. FDG uptake within a large left pelvic mass resolved completely from prepazopanib maximized standard uptake value (SUVmax) of 5.0 (Fig. 2). After 6 mo on pazopanib, restaging revealed minimal increase in FDG avidity and size in some of the metastatic lesions, but with new uptake identified in the left ventricular myocardium and pericardial areas. CT angiography confirmed numerous nodules (<1.5 cm) scattered throughout the pericardium consistent with disease progression. The patient was then changed to daily sunitinib (37.5 mg/day). Restaging after 2 mo of therapy revealed a mixed response with decreased FDG avidity within the pericardial and subcutaneous lesions, but increased uptake within the right vocal cord and in bilateral retro-orbital areas along with a new hepatic metastasis. Following two additional months on sunitinib, the patient developed further disease progression. Subsequent interventions included surgical fixation of a femoral lytic lesion, palliative resection of symptomatic pharyngeal recurrence, placement of a gastrostomy feeding tube, and radiotherapy for painful sacral and femoral bony metastases. She received fourth-line systemic treatment with doxorubicin for two cycles without response and eventually expired 28 mo from her original diagnosis of granular cell tumor.
Figure 2.

Representative fused FDG-PET images prior to (A,B) and 3 mo after (C,D) starting pazopanib. SUVmax at the left pelvic metastatic lesion was 5.0 prepazopanib (A) and not hypermetabolic at 3 mo (C). SUVmax in the right breast metastatic nodules was 5.8 prepazopanib (B) and 2.7 after treatment (D).
Genomic Analyses
Overall Mutation Landscape
The current granular cell tumor sample demonstrated a low mutation burden (Fig. 3). Using a previous definition (Zhang et al. 2012b), the current WGS analysis identified 17 tier-1 mutations in gene coding regions, splice sites or noncoding RNA; 81 tier-2 mutations in regulatory or evolutionarily conserved regions of the genome; and 541 tier-3 mutations in other nonrepetitive regions of the genome. All 17 tier-1 mutations were confirmed by Sanger sequencing. The background mutation rate (BMR) was estimated to be 0.18 mutations per million covered base pairs (Mbp), which is low compared with most other common adult cancers (Kandoth et al. 2013). The mutation signature was dominated by C>T transitions, mostly when adjacent to a 5′ G (Fig. 4). No small insertion/deletions (indels) were detected in gene coding regions in this tumor. In addition to point mutations, we identified and validated 12 structural variations (SVs), and 22 copy-number variations (CNVs).
Figure 3.
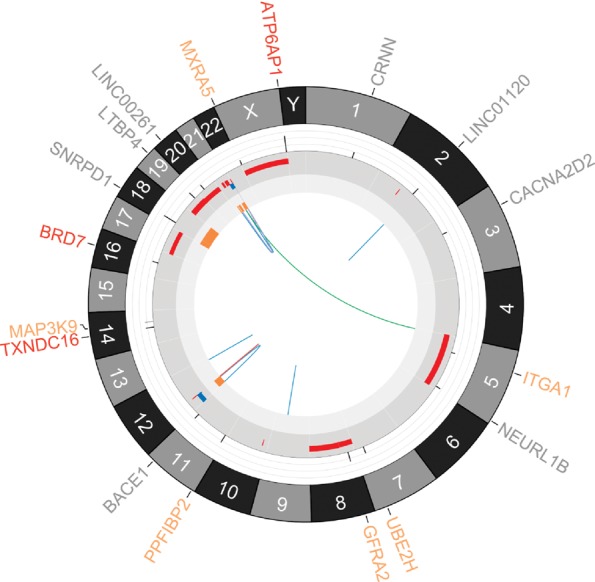
Circos plot (Krzywinski et al. 2009) depicting genetic alterations identified by WGS analyses in granular cell tumor. From outer to inner components: amino-acid-changing point mutations in genes (red, truncating mutations including nonsense and splice-site single-nucleotide variants (SNVs); orange, alternating mutations including missense SNVs; gray, nonamino-acid-changing mutations, including silent or noncoding SNVs), chromosomes, variant allele frequency (VAF) bars for the corresponding point mutation (range = [0:1], the color of the VAF bar indicates coverage: light gray = 0–9×; gray = 10–29×; black = ≥30×), copy-number variations (red, gain; blue, loss), loss of heterozygosity, structural variations (red, insertion; blue, deletion; purple, intrachromosomal translocation; green, interchromosomal translocation).
Figure 4.

The mutational signature of the granular cell tumor. Each single-nucleotide substitution is categorized into the 96 distinct trinucleotide mutation types, depending on the substitution class (C>A, C>G, C>T, T>A, T>C, or T>G) and the neighboring bases (A, C, G, or T) immediately 3′ and 5′ to the mutated base. The number of mutations in each category is divided by the abundance of its trinucleotide in the reference genome to give a fractional count. The fractional count is then normalized and displayed as the percent attribution to a specific mutation type in the plot.
Genes Affected by Point Mutations
The 17 validated tier-1 mutations in genes included six missense single-nucleotide variants (SNVs), three nonsense SNVs, and eight other SNVs not predicted to cause amino acid changes (Fig. 3). For the nine amino-acid-changing SNVs, their variant allele frequencies (VAFs) ranged from 14% to 60% (Table 1). No mutations were found in Cancer Gene Census genes (Futreal et al. 2004). However, one nonsense SNV was identified in BRD7 (Fig. 5), a newly recognized tumor suppressor candidate that is required for TP53 function (Drost et al. 2010). Additionally, one missense SNV was found in a neurotrophic factor receptor GFRA2, which, upon binding of neurturin, activates RET tyrosine kinase (Airaksinen and Saarma 2002).
Table 1.
Point mutations predicted to introduce amino acid changes
| GeneName | Chr | GRCh37 position | Ref>Alt | GenBank accession | Predicted effect | Class | WGS reads (mut/total) | VAF in tumor % | |
|---|---|---|---|---|---|---|---|---|---|
| Tumor | Normal | ||||||||
| ATP6AP1 | chrX | 153664066 | C>A | NM_001183 | Y414* | Nonsense | 18/30 | 0/16 | 60 |
| GFRA2 | chr8 | 21608169 | G>A | NM_001495 | S242F | Missense | 18/49 | 0/27 | 37 |
| TXNDC16 | chr14 | 52937263 | A>C | NM_020784 | L483* | Nonsense | 12/40 | 0/44 | 30 |
| BRD7 | chr16 | 50353864 | G>A | NM_001173984 | R613* | Nonsense | 7/34 | 0/27 | 21 |
| MAP3K9 | chr14 | 71199588 | G>A | NM_033141 | T847M | Missense | 8/26 | 0/18 | 31 |
| PPFIBP2 | chr11 | 7670871 | C>T | NM_003621 | R703W | Missense | 11/35 | 0/29 | 31 |
| UBE2H | chr7 | 129497389 | C>T | NM_003344 | C87Y | Missense | 12/47 | 0/47 | 26 |
| MXRA5 | chrX | 3240233 | G>A | NM_015419 | R1165W | Missense | 6/32 | 0/22 | 19 |
| ITGA1 | chr5 | 52225500 | G>A | NM_181501 | E914K | Missense | 9/65 | 0/33 | 14 |
WGS, whole-genome sequencing; VAF, variant allele frequency.
Figure 5.

The BRD7 nonsense mutation identified by WGS. (A) The protein domain view of BRD7 with the R613* nonsense mutation. (B) Sanger sequencing chromatograms confirming the somatic status of the mutation. The mutant peak is off-center and only slightly above baseline, presumably because of the allelic burden of this variant being barely above 20%. We repeated the experiment twice and the pattern was consistent in both runs.
Somatic Structural Variations
We identified 12 somatic SVs, including six deletions (DELs), four intrachromosomal translocations (ITXs), one insertion (INS), and one interchromosomal translocation (CTX) (Table 2). The majority of the intrachromosomal rearrangements occurred within Chromosome 12 (two DELs, two ITXs, one INS) and Chromosome 21 (two ITXs, two DELs). Additionally, Chromosomes 2 and 9 were found to each harbor one DEL. The only CTX occurred between Chromosomes 5 and 21. The complex rearrangements on Chromosome 21 contained five SVs, where several breakpoints were located within the same region (Fig. 6). All identified somatic SVs were experimentally validated by polymerase chain reaction and Sanger sequencing.
Table 2.
Structural variations identified by whole-genome sequencing
| ChrA | GRCh37 PosA | Orient A | Num Reads A | ChrB | GRCh37 PosB | Orient B | Num Reads B | Type | Genea |
|---|---|---|---|---|---|---|---|---|---|
| 12 | 512012 | + | 2 | 12 | 28683009 | + | 3 | DEL | CCDC77_CCDC91 |
| 12 | 31929644 | − | 1 | 12 | 31221648 | + | 2 | ITX | DDX11-AS1 |
| 12 | 31835817 | + | 3 | 12 | 30028522 | − | 0 | ITX | AMN1 |
| 21 | 15950538 | + | 4 | 21 | 48047093 | + | 2 | DEL | SAMSN1 |
| 21 | 36981223 | + | 4 | 21 | 48033053 | + | 2 | DEL | |
| 21 | 48048095 | − | 6 | 21 | 36989776 | + | 3 | ITX | |
| 21 | 37092441 | + | 1 | 5 | 2243357 | + | 6 | CTX | |
| 9 | 92290776 | + | 3 | 9 | 92290847 | + | 0 | DEL | |
| 12 | 120004464 | + | 3 | 12 | 120025731 | + | 0 | DEL | |
| 12 | 32142064 | + | 4 | 12 | 31797004 | + | 2 | INS | KIAA1551 |
| 21 | 20846107 | + | 16 | 21 | 48047298 | − | 0 | ITX | |
| 2 | 133400741 | + | 2 | 2 | 133400857 | + | 6 | DEL |
DEL, deletion; INS, insertion; ITX, intrachromosomal translocation; CTX, interchromosomal translocation.
aIncludes genes overlapping with the breakpoints according to Ref-seq.
Figure 6.
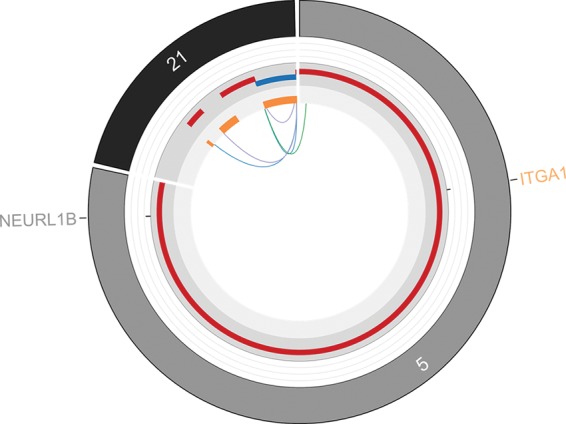
Complex rearrangement of Chromosome 21. A total of five structural variations were identified on Chromosome 21. The lines connecting different genomic locations, as well as their orientations, were identified by the CREST program. All structural variations were experimentally validated. The sizes of segments and their distances are not in proportion to the actual genomic distance.
None of the identified SVs were predicted to cause an overexpressed gene fusion. One DEL on Chromosome 12 joined two genes—CCDC77 and CCDC99—but was not in the same reading frame. Predicted truncating SVs were identified in four genes: DDX11-AS1, AMN1, SAMSN1, and KIAA1551.
Somatic Copy-Number Variations and Loss of Heterozygosity
Analyzing WGS data identified 22 regions with CNVs. Several whole-chromosome gains were detected for Chromosomes 5, 8, 17, 19, 20, and X. The gain of Chromosomes 8 was composed of several segments with different copy-number amplitudes. Chromosome 21 contained several gains as well as losses coupled with loss of heterozygosity, which was consistent with the identified SVs. Chromosome 12 had a one-copy loss of the p-arm with loss of heterozygosity (LOH). Copy-neutral LOH was observed across the entire Chromosome 18. Focal CNVs was one mechanism of introducing tumorigenic alterations (Zack et al. 2013). We identified four focal CNV segments containing <10 genes (Table 3). However, none of the contained genes have been implicated in cancer.
Table 3.
Copy-number variations identified by whole-genome sequencing
| Chr | Start | End | num.marka | seg.meana | Focal CNVb genes | Overexpressed in Conley et al. (2014) |
|---|---|---|---|---|---|---|
| 2 | 89830501 | 89880100 | 469 | 1.362 | ||
| 5 | 11701 | 180785400 | 1,646,623 | 0.416 | ||
| 8 | 33501 | 43820800 | 400,545 | 0.377 | GNRH1 | |
| 8 | 46847001 | 47000100 | 1141 | 0.374 | ||
| 8 | 47064701 | 47086300 | 216 | 0.425 | ||
| 8 | 47175401 | 47237700 | 598 | 0.308 | ||
| 8 | 47255701 | 47284200 | 268 | 0.395 | ||
| 8 | 47359501 | 146303800 | 934,229 | 0.413 | LYN | |
| 10 | 18841301 | 18862700 | 200 | 0.521 | NSUN6 | |
| 12 | 60701 | 28683000 | 265,733 | –0.201 | ||
| 12 | 30028501 | 31221500 | 11,065 | –0.252 | IPO8, CAPRIN2, TSPAN11 | |
| 12 | 31798301 | 32143600 | 3,267 | 0.393 | C12orf72, AMN1, H3F3C, C12orf35 | |
| 17 | 1 | 81195100 | 704,846 | 0.273 | BRCA1, SSTR2, TOP2A | |
| 19 | 89201 | 59118800 | 510,602 | 0.346 | ||
| 20 | 60001 | 62965000 | 566,176 | 0.339 | OGFR, SRC | |
| 21 | 15810801 | 20846100 | 47,794 | 0.461 | ||
| 21 | 27262601 | 36981200 | 92,904 | 0.401 | ||
| 21 | 36981201 | 36989800 | 86 | –0.212 | ||
| 21 | 36989801 | 37092400 | 1014 | 0.36 | ||
| 21 | 37092401 | 48033100 | 104,439 | –0.203 | ||
| 21 | 48033101 | 48119800 | 647 | 0.332 | PRMT2 | |
| X | 2699501 | 154930300 | 1,313,803 | 0.401 | POLA1 |
num.mark, number of fixed-size windows (100 bp) in the segment; seg.mean, the estimated GC-corrected signal difference between tumor and normal. Definitions according to CONSERTING (Chen et al. 2015).
a“Focal CNV” defined as copy-number variations containing less than 10 genes.
In the previous gene expression study of one malignant granular cell tumor, Conley et al. (2014) identified 16 overexpressed genes. In the current WGS data, eight of these 16 genes were located in CNV regions including SRC, TOP2A, GNRH1, BRCA1, OGFR, POLA1, SSTR2, and LYN. All of the involved CNVs were gains (Table 3).
DISCUSSION
Malignant granular cell tumor, often associated with unresectable disease or metastatic presentation, is exceedingly rare with no effective treatment available. In addition, there is a paucity of knowledge of the genomic profile of this neoplasm. A recent study by Conley et al. (2014) reported the first documented response of a malignant granular cell tumor to a systemic agent, pazopanib. We report the first whole-genome sequencing of a malignant granular cell tumor in a patient who had a striking metabolic response to pazopanib.
We found a loss-of-function mutation in BRD7 (bromodomain-containing 7), a subunit of PBAF-specific SWI/SNF chromatin remodeling complexes. As a recently recognized tumor-suppressor candidate, BRD7 is required for TP53 function by affecting histone acetylation during p53 activation, and is also a regulator of BRCA1 (Harte et al. 2010). In breast cancers, it has been reported that BRD7 expression is lost in as many as 20% of the cases that contained wild-type TP53 (Drost et al. 2010; Harte et al. 2010). The locus 16q12 containing the BRD7 gene is a known target in cancer, which has shown to be affected by focal deletions in 2.68% of all cancers (Burrows et al. 2010). In addition to deletions, there have been efforts to locate BRD7 point mutations in cancer. One such study failed to identify any truncating mutation of BRD7 in triple negative breast cancers (Pern et al. 2012). The same BRD7 mutation as the current one (R613*) has been found in four previously published cancer samples according to COSMIC (annotated as R612* using a different isoform) (Forbes et al. 2008). Notably, the BRD7 nonsense mutation reported here was predicted to remove the last 40 amino acids near the carboxy terminus (Fig. 5). Further experiments are warranted to investigate the functional role of BRD7 in malignant granular cell tumors.
Pazopanib is a potent tyrosine kinase inhibitor. The known targets of pazopanib include VEGFR types 1, 2, and 3, PDGFR α and β, C-Kit, FGFR 1 and 3, ITK, LCK, and C-FMS (encoded by CSF1R) (Conley et al. 2014). However, in our case study, no somatic mutations were identified in any of these genes. The only identified mutation in receptor tyrosine kinase pathways was GFRA2 (GDNF family receptor alpha 2), encoding for the primary receptor for neurturin. Neurturin is a neurotrophic factor, which is responsible for the growth, survival, and maintenance of neurons (Deister and Schmidt 2006). Neurturin belongs to the glial cell line–derived neurotrophic factor family of ligands and has been considered a candidate for treating Parkinson disease (Hong et al. 2008). Upon activation by neurturin, GFRA2 mediates tyrosine autophosphorylation and activates the RET tyrosine kinase receptor (Airaksinen and Saarma 2002). If GFRA2 is altered by a gain-of-function mutation, GFRA2 and the RET signaling pathway could potentially be activated. The GFRA2 mutation in the current case is predicted to have changed amino acid 242, a conservative residue across many species (data not shown), from serine to phenylalanine. Further functional studies using in vivo or in vitro systems to determine whether increased phosphorylation of the RET or other receptor tyrosine kinase targets of pazopanib is observed with the GFRA2 mutant gene will be essential to fully characterize the identified GFRA2 mutation.
The current whole-genome sequencing only generated a limited 38.6× average coverage in tumor. Given the fact that in the current case tumor cellularity was high (estimated at 90%), which was also supported by the VAFs of observed coding mutations, the current analysis should be sensitive in capturing major clone mutations. In the future, targeted deep sequencing on a larger set of variants may provide higher sensitivity in finding subclonal mutations. Moreover, additional RNA-seq analyses may provide important insight into the gene expression profile, potential fusion transcripts, and splicing variants.
In summary, we present the first, to our knowledge, whole-genome characterization of a malignant granular cell tumor. The tumor exhibited low mutation burden and an overall stable genome with local complex rearrangements. A loss-of-function mutation was detected in a newly identified tumor suppressor candidate, BRD7. None of the known targets of pazopanib were mutated in this patient who demonstrated metabolic response to pazopanib, but our mutation list contains a receptor tyrosine kinase pathway mutation in GFRA2, which may help to elucidate the mechanism of pazopanib response. While this current study includes only one malignant granular cell tumor, it provides the first reference for future research and clinical efforts aiming to advance our understanding of this rare cancer type, which could ultimately lead to the development of focused genetic testing and personalized treatment.
METHODS
Whole-Genome Sequencing (WGS)
The current tumor was estimated to have 90% cellularity. DNA libraries for the tumor and matching germline blood DNA were prepared using the TruSeq DNA Sample v2 preparation kit (Illumina) according to the manufacturer's instructions. Briefly, 1 µg of each DNA sample was fragmented using Covaris shearing to a size range of 300–400 bp (Covaris). Following end repair and 3′ adenylation, indexing adapters were ligated to the fragment ends. Following purification, the fragmented DNA was PCR amplified for five cycles, purified, and validated for appropriate size on a 2100 Bioanalyzer High Sensitivity DNA chip (Agilent Technologies). Each DNA library was quantitated using quantitative PCR (KAPA Biosystems) prior to normalization. Libraries were loaded and clustered to individual lanes of a HiSeq Flow Cell using an Illumina cBot (TruSeq PE Cluster Kit v3), followed by 2 × 100 cycle paired-end WGS on a HiSeq2000 sequencer according to the manufacturer's recommended protocol (Illumina). Image analysis and base calling were carried out by Illumina Software CASAVA v 1.8.2 with default parameters.
WGS Data Analyses
Using a high-throughput paired-end sequencing approach, 2.6 billion 100-bp reads were generated from the tumor sample; 87% of the reads were successfully mapped to the human reference genome. Sequencing the normal sample yielded 1.3 billion reads with a 91% mapping rate. The average genome coverage was 38.6× and 30.5× in the tumor and normal samples, respectively, where 89.9% and 86.4% of the genome was covered with at least 20× coverage (Supplemental Table S1).
The bioinformatics analysis began by utilizing high quality paired-end reads passing Illumina RTA filter aligned to the NCBI human reference genome (hg19) using BWA (Li and Durbin 2009). PCR duplicated reads were marked using Picard (http://picard.sourceforge.net/). Putative SNVs and indels were identified by running variation detection module of Bambino (Edmonson et al. 2011). After initial variant calling, the predicted mutations were further filtered to remove potential false calls: (1) the alternative alleles were present in both tumor and the matched normal samples and the Fisher's exact test P value equal to or larger than 0.05; (2) the mutant alleles were only present in one of the two stands and the Fisher's exact test P < 0.05; (3) the putative mutation occurred at a site with systematically dropped base quality scores, defined by >70% of the mutant reads with Phred quality score reduced by at least 10 at the mutant site compared with the 5′ or 3′ side neighboring bases. Putative indels were evaluated by a realignment process to filter out false calls introduced by germline events, mapping artifacts, and homopolymer. Prior to validation, the predicted mutations were manually inspected using Bambino viewer (Edmonson et al. 2011) to remove false calls introduced by mapping or alignment artifacts. The putative mutations passing manual review were segregated into high and low confidence groups according to their supporting information, such as the number of supporting reads, the variant allele fraction, and the coverage in the matched normal sample. The identified somatic mutations were compared with the public human germline databases, including dbSNP (Sherry et al. 2001), 1000 Genomes Project (The 1000 Genomes Project Consortium 2012), National Heart, Lung, and Blood Institute's Exome Sequencing Project (NHLBI 2013). The mutations were annotated by ANNOVAR (Wang et al. 2010) using RefSeq sequence database downloaded from NCBI on 22 March 2015. Mutations were classified into four tiers following previously described definitions (Zhang et al. 2012a,b, 2013), including tier-1 mutations occurring in gene coding regions, splice sites, or noncoding RNA according to RefSeq; tier-2 mutations in regulatory regions annotated by UCSC annotation (regulatory annotations included are targetScanS, ORegAnno, tfbsConsSites, vistaEnhancers, eponine, firstEF, L1 TAF1 Valid, Poly(A), switchDbTss, encodeUViennaRnaz, laminB1, cpgIslandExt) or evolutionarily conserved genomic regions defined by a conservation score equal to or >500 based on either the phastConsElements28way table or the phastConsElements17way table from the UCSC Genome Browser (http://genome.ucsc.edu/); tier-3 mutations in any other nonrepetitive regions; and tier-4 all other mutations. Structural variations were initially detected by using CREST (Wang et al. 2011) and then manually reviewed using Bambino viewer (Edmonson et al. 2011). Potential fusion products between genetic regions were also predicted by considering the orientation and break points of the SVs. CNVs were detected using CONSERTING, a copy-number analysis method for WGS data (Chen et al. 2015). Only CNVs with “seg.mean” and “log.ratio” absolute value no less than 0.2 were considered for downstream analyses.
Sanger Sequencing—Variant Confirmation
PCR primers (IDT) were designed to flank the variant positions using Primer3 (Koressaar and Remm 2007; Untergrasser et al. 2012). All primers are included in Supplemental Table S2. Amplification products for each sample were generated by 35 cycles of PCR using 20-ng template DNA. The products were purified and thermal cycled for 35 cycles of 96°C (20 sec), 50°C (5 sec), and 60°C (3 min) with an appropriately diluted BigDye Terminator v.3.1 Cycle Sequencing Kit (Life Technologies). Following incubation at 12°C, the products were purified over hydrated Sephadex G-50 (Sigma-Aldrich) in 45 micron Multiscreen HV filter plates (Merck Millipore). The eluted fluorescently tagged products were then detected on an ABI Prism 3130xl Genetic Analyzer (Life Technologies) using POP-7 Polymer on a 50-cm Array according to manufacturer's specifications. The data were then analyzed for variants using Mutation Surveyor DNA Variant Analysis Software (Softgenetics). In the current study, 9 out of 11 high confidence SNVs and 8 out of 19 low confidence SNVs were validated as somatic mutations. For validation of SVs, two sets of primers were designed spanning junction. Amplification of the novel amplicon with these primers sets was visualized on an ethidium bromide agarose gel and then sequenced to confirm breakpoints. We validated 12 out of the 13 putative SVs.
ADDITIONAL INFORMATION
Ethics Statement
The Roswell Park Cancer Institute (RPCI) Institutional Review Board (IRB) gave approval for this study. The patient was consented for next-generation sequencing, remnant tissue procurement, and collection of blood through RPCI IRB-approved protocols.
Data Deposition and Access
The sequencing data from the tumor and normal blood samples has been submitted to the NCBI Database of Genotypes and Phenotypes (dbGaP) under study accession phs000978.v1.p1. The interpreted variants have been submitted to ClinVar under accession numbers SVC000243887 and SVC000243888.
Acknowledgments
The RPCI Bioinformatics Shared Resource, Clinical Data Network, Data Bank and BioRepository, Genomics Shared Resource and Pathology Resource Network are CCSG Shared Resources supported by National Institutes of Health grant P30 CA016056.
Author Contributions
L.W., S.L., J.C., J.W., A.P.-S., and C.D.M. designed the study. L.W., S.L., J.C., J.W., A.P.-S., S.T.G., M.M., L.L., Q.H., J.C., K.M.M., D.E.N., B.L., M.Q., W.B., A.R.O., K.H., M.B., B.B., C.D., J.K., M.M., R.C., A.F., K.S., C.T., N.I.K., D.L.T., C.S.J., and C.D.M. performed experiments and analyzed data. L.W., S.L., J.C., J.W., A.P.-S., N.I.K., and C.D.M wrote the manuscript, K.M.M., D.E.N., A.R.O., M.M., R.C., K.S., and D.L.T. assisted in preparing the manuscript.
Funding
This work was supported by the Roswell Park Alliance Foundation.
Competing Interest Statement
The authors have declared no competing interest.
Referees
Obi L. Griffith
Anonymous
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Airaksinen MS, Saarma M. 2002. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 3: 383–394. [DOI] [PubMed] [Google Scholar]
- Burrows AE, Smogorzewska A, Elledge SJ. 2010. Polybromo-associated BRG1-associated factor components BRD7 and BAF180 are critical regulators of p53 required for induction of replicative senescence. Proc Natl Acad Sci 107: 14280–14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Gupta P, Wang J, Nakitandwe J, Roberts K, Dalton JD, Parker M, Patel S, Holmfeldt L, Payne D, et al. 2015. CONSERTING: integrating copy-number analysis with structural-variation detection. Nat Methods 12: 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conley AP, Koplin S, Caracciollo JT, Reed DR, Webber NP, Attia S. 2014. Dramatic response to pazopanib in a patient with metastatic malignant granular cell tumor. J Clin Oncol 32: e107–e110. [DOI] [PubMed] [Google Scholar]
- Deister C, Schmidt CE. 2006. Optimizing neurotrophic factor combinations for neurite outgrowth. J Neural Eng 3: 172–179. [DOI] [PubMed] [Google Scholar]
- Drost J, Mantovani F, Tocco F, Elkon R, Comel A, Holstege H, Kerkhoven R, Jonkers J, Voorhoeve PM, Agami R, et al. 2010. BRD7 is a candidate tumour suppressor gene required for p53 function. Nat Cell Biol 12: 380–389. [DOI] [PubMed] [Google Scholar]
- Edmonson MN, Zhang J, Yan C, Finney RP, Meerzaman DM, Buetow KH. 2011. Bambino: a variant detector and alignment viewer for next-generation sequencing data in the SAM/BAM format. Bioinformatics 27: 865–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanburg-Smith JC, Meis-Kindblom JM, Fante R, Kindblom LG. 1998. Malignant granular cell tumor of soft tissue: diagnostic criteria and clinicopathologic correlation. Am J Surg Pathol 22: 779–794. [DOI] [PubMed] [Google Scholar]
- Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, Menzies A, Teague P, Futreal A, Stratton MR. 2008. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet Chapter 10: Unit 10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. 2004. A census of human cancer genes. Nat Rev Cancer 4: 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harte MT, O'Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT, Mullan PB, Harkin DP. 2010. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res 70: 2538–2547. [DOI] [PubMed] [Google Scholar]
- Hong M, Mukhida K, Mendez I. 2008. GDNF therapy for Parkinson's disease. Expert Rev Neurother 8: 1125–1139. [DOI] [PubMed] [Google Scholar]
- Kandoth C, McLellan MD, Vandin F, Ye K, Niu B, Lu C, Xie M, Zhang Q, McMichael JF, Wyczalkowski MA, et al. 2013. Mutational landscape and significance across 12 major cancer types. Nature 502: 333–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koressaar T, Remm M. 2007. Enhancements and modifications of primer design program Primer3. Bioinformatics 23: 1289–1291. [DOI] [PubMed] [Google Scholar]
- Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. 2009. Circos: an information aesthetic for comparative genomics. Genome Res 19: 1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lage TL, Miranda MF, Bittencourt Mde J, Dias CM, Parijos AM, Raiol TK. 2013. Case for diagnosis. Granular cell tumor. An Bras Dermatol 88: 1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NHLBI. 2013. Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP). Seattle, WA: http://evs.gs.washington.edu/EVS/ [4 June 2013 accessed]. [Google Scholar]
- Pern F, Bogdanova N, Schurmann P, Lin M, Ay A, Langer F, Hillemanns P, Christiansen H, Park-Simon TW, Dork T. 2012. Mutation analysis of BRCA1, BRCA2, PALB2 and BRD7 in a hospital-based series of German patients with triple-negative breast cancer. PLoS One 7: e47993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491: 56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Untergrasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. 2012. Primer3—new capabilities and interfaces. Nucleic Acids Res 40: e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schoffski P, Aglietta M, Staddon AP, Beppu Y, et al. 2012. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379: 1879–1886. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, et al. 2011. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nat Methods 8: 652–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack TI, Schumacher SE, Carter SL, Cherniack AD, Saksena G, Tabak B, Lawrence MS, Zhang CZ, Wala J, Mermel CH, et al. 2013. Pan-cancer patterns of somatic copy number alteration. Nat Genet 45: 1134–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, et al. 2012a. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481: 329–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, Easton J, Chen X, Wang J, Rusch M, et al. 2012b. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature 481: 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, Orisme W, Punchihewa C, Parker M, Qaddoumi I, et al. 2013. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 45: 602–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.