

Abstract
Autism spectrum disorders (ASDs) are a group of developmental disabilities that affect social interaction and communication and are characterized by repetitive behaviors. There is now a large body of evidence that suggests a complex role of genetics in ASDs, in which many different loci are involved. Although many current population-scale genomic studies have been demonstrably fruitful, these studies generally focus on analyzing a limited part of the genome or use a limited set of bioinformatics tools. These limitations preclude the analysis of genome-wide perturbations that may contribute to the development and severity of ASD-related phenotypes. To overcome these limitations, we have developed and utilized an integrative clinical and bioinformatics pipeline for generating a more complete and reliable set of genomic variants for downstream analyses. Our study focuses on the analysis of three simplex autism families consisting of one affected child, unaffected parents, and one unaffected sibling. All members were clinically evaluated and widely phenotyped. Genotyping arrays and whole-genome sequencing were performed on each member, and the resulting sequencing data were analyzed using a variety of available bioinformatics tools. We searched for rare variants of putative functional impact that were found to be segregating according to de novo, autosomal recessive, X-linked, mitochondrial, and compound heterozygote transmission models. The resulting candidate variants included three small heterozygous copy-number variations (CNVs), a rare heterozygous de novo nonsense mutation in MYBBP1A located within exon 1, and a novel de novo missense variant in LAMB3. Our work demonstrates how more comprehensive analyses that include rich clinical data and whole-genome sequencing data can generate reliable results for use in downstream investigations.
Keywords: autism
INTRODUCTION
There is a consistent amount of evidence suggesting a complex role of genetics in autism spectrum disorders (ASDs) (Zhao et al. 2007; Betancur 2011; Iossifov et al. 2012; Neale et al. 2012; O'Roak et al. 2012; Zhou and Parada 2012; Bernier et al. 2014; Robinson et al. 2014; Rothwell et al. 2014; Sugathan et al. 2014; Krumm et al. 2015; Lyon and O'Rawe 2015), in which many different loci are involved, but a general understanding of what leads to ASDs on a molecular and physiological level has not yet emerged. There is a large collection of putative disease-contributing variants found in small subpopulations of people with ASDs (Zhou and Parada 2012; Bernier et al. 2014; Iossifov et al. 2014; Ronemus et al. 2014), yet leaving most ASD cases of undetermined etiology. The lack of generality in these findings may be attributed to many factors, including the phenotypic heterogeneity of the disease (Lyon and O'Rawe 2015), the need for larger sample sizes for statistical studies (Iossifov et al. 2012), and the variability in the methodology used to analyze ASD-related data.
Another important factor contributing to the varying results among studies is the diagnosis methodology used for ASDs. Although there are efforts to achieve a clear and consistent method of diagnosis (Schaefer and Mendelsohn 2013), ASD is by definition a spectrum with likely many different contributing factors (Johnson et al. 2011). As a consequence of this, a carefully chosen study cohort is needed for statistical studies to reliably detect shared genetic variants.
Currently, many ASD studies focus on the analysis of microarray and/or exome-sequencing data for understanding the etiological contributions to and mechanisms of ASDs (Levy et al. 2011; Iossifov et al. 2012, 2014; O'Roak et al. 2012). These analyses are generally applied to large cohorts, such as those from the Simons Simplex Collection (Fischbach and Lord 2010; Levy et al. 2011), which consists of families with a single affected child, unaffected parents, and at least one unaffected sibling. These large studies generally use and analyze only one of the high-throughput sequencing technologies, with varying levels of sequence coverage for whole-exome sequencing (WES) or genotyping markers (for genotyping microarrays). Furthermore, these studies use only one or a few analysis tools for detecting sequence variations, which can result in a loss of information in situations where one tool performs poorly. Although these approaches have led to significant genetic discovery (Zhao et al. 2007; Betancur 2011; Levy et al. 2011; Iossifov et al. 2012, 2014; Neale et al. 2012; O'Roak et al. 2012; Zhou and Parada 2012; Bernier et al. 2014; Robinson et al. 2014; Rothwell et al. 2014; Sugathan et al. 2014; Krumm et al. 2015; Lyon and O'Rawe 2015), they are likely to miss-call or simply miss true and disease-relevant genetic variation. Some tools may perform better on just one or a few areas of the genome, and their performance may also differ depending on data set–specific characteristics, which has been studied previously (O'Rawe et al. 2013). To address these problems, we describe an integrative clinical and bioinformatics pipeline that makes use of a variety of analysis tools and orthogonal high-throughput sequencing technologies to obtain a more complete and reliable set of candidate ASD variants for validation and downstream functional analysis, resulting in a collection of different types of variants for a better understanding of the genomic burden of the three autism probands.
RESULTS
This study consisted of the clinical recruitment of three simplex autism families (Fig. 1) for phenotyping and whole-genome studies. Human sequence variation spans a variety of genomic scales, ranging from single nucleotide to megabases and even whole-chromosome differences. Because of the variety of scales and mechanisms that can lead to variation in human sequence between individuals and populations, a variety of algorithms are needed to extract genomic signatures at all scales and that represent a wide variety of variant types. Several variant discovery algorithms and procedures were used during the course of this study, each designed to detect different classes of human sequence variants (Fig. 2).
Figure 1.
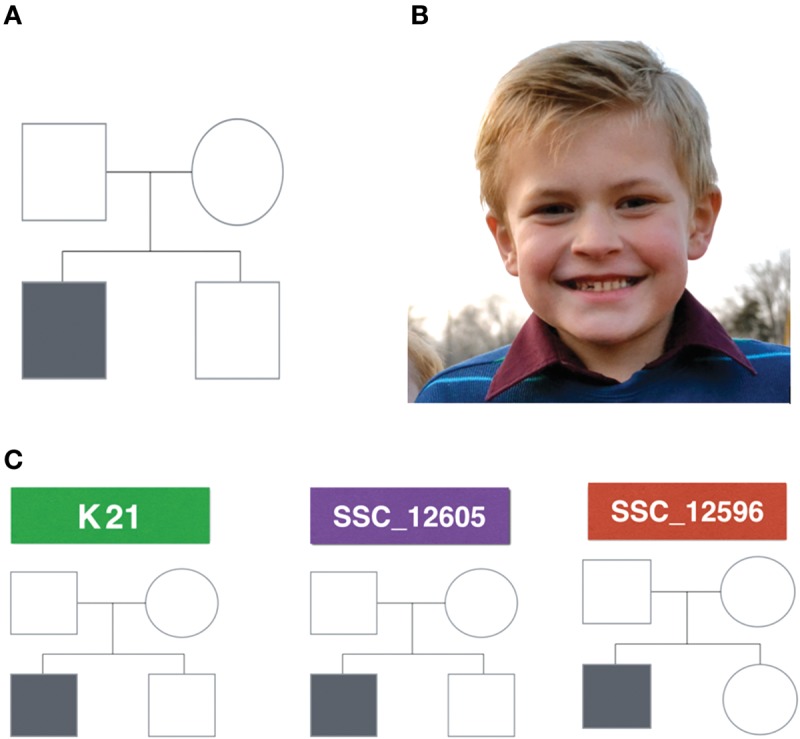
(A) Pedigree structure of a simplex autism family. For a family to be classified as a simplex autism family, it has to be composed of one affected child and at least one unaffected sibling, and both parents should not have obvious autism. Probands and siblings can be either males or females. (B) K21 proband showing no dysmorphology. (C) Analyzed pedigrees. Two of the families have male probands and unaffected male siblings (K21 and SSC_12605), whereas the third family has a male proband and a female unaffected sibling (SSC_12596).
Figure 2.

A conceptual map of human sequence variation. Here, we show approximate sizes, as well as the associated signature, of the various different types of human sequence variation that can be currently detected with whole-genome sequencing (WGS), microarray data, and informatics technologies used in this work. The frequency axis shows the approximate frequency of the various genetic variation types that are currently detectable via germline WGS combined with microarray data. Above the visual signatures of the different types of human sequence variation, the general names of the different informatics software tools for detecting the variation are noted which include, the Genome Analysis Toolkit (GATK), Scalpel, PennCNV, the estimation by read depth with single-nucleotide variants (ERDS) CNV caller, and the FreeBayes caller.
Clinical Presentation
The proband from the Simons Simplex Collection (SSC) pedigree with ID SSC_12596 is an Asian male who was 4 yr and 11 mo old at the time he was evaluated. He was 102 cm tall, weighed 18.1 kg with a body mass index (BMI) of 17.4, and his head circumference was 51.6 cm. He is the second child of the quad, which contains a female unaffected sibling of the same father and mother. No previous miscarriages or medical terminations were reported. According to the Gillberg Optimality Scale, there were no complications during the pregnancy or at birth, and the proband has never presented febrile or nonfebrile seizures. He presented abnormal development at ∼3 mo and a loss of skills relating to communicative intent and social engagement has been reported. The proband is considered nonverbal as he did not formulate phrases at the assessment date and he achieved his first single words at the age of 30 mo, indicating a word delay. His verbal and nonverbal intelligence quotient (IQ) tests resulted in a score of 32 and 89, respectively, and his full-scale IQ test resulted in a score of 61. According to the results of the Autism Diagnostic Interview-Revised, the Autism Diagnostic Observation Schedule, and the clinician's best estimate, the proband was diagnosed with autism with high certainty. Based on a battery of behavioral and cognitive tests applied to the family (Table 1), this proband had a low number of self-injurious behaviors; however, he does have a high-social impairment and inappropriate, stereotyped, compulsive, restrictive, ritualistic, and sameness behaviors. There is no history of autism in the family.
Table 1.
Body measurements and IQ test scores
| Test | SSC_12596 | SSC_2605 |
|---|---|---|
| Diagnostic classification ADI-R | Autism | Autism |
| Diagnostic classification ADOS algorithm | Autism | Autism |
| ADOS module | 1—no words | 4 |
| Certainty of ASD diagnosis | 15 | 15 |
| Verbal IQ | 32 | 136 |
| Nonverbal IQ | 89 | 108 |
| Full-scale IQ | 61 | 120 |
| ABC total score | 53 | 34 |
| Stereotyped behavior | 7 | 1 |
| Self-injurious | 2 | 1 |
| Compulsive behavior | 6 | 13 |
| Ritualistic behavior | 6 | 4 |
| Sameness behavior | 11 | 10 |
| Restricted behavior | 8 | 7 |
| Pregnancy optimality | 0 | 4 |
The proband from the Simons Simplex Collection pedigree ID SSC_12605 is a non-Hispanic-white male adolescent whose assessment was carried out at the age of 16 yr and 4 mo. He was 175 cm tall and weighed 100.9 kg, resulting in a BMI of 32.9 (obese), his head circumference was 60.5 cm. He is the first child of the quad with an unaffected male sibling of the same father and mother. Four miscarriages or medical terminations were reported. Based on the Gillberg Optimality Scale, he presented four nonoptimal events at labor (not specified). He has never presented febrile or nonfebrile seizures. He presented abnormal development at ∼18 mo but no loss of skills relating to communicative intent and social engagement has been reported. He is considered verbal, achieved his first single words at the age of 20 mo and his first phrases at 26 mo with no indication of word or phrase delay. His verbal and nonverbal IQ tests resulted in 136 and 108, respectively, and his full-scale IQ test resulted in a score of 120. According to the results of the Autism Diagnostic Interview-Revised, the Autism Diagnostic Observation Schedule, and the clinician's best estimate, SSC_12596 proband was diagnosed with autism with high certainty. According to a battery of behavioral and cognitive tests applied to the family (Table 1), this proband had a low number of self-injurious or stereotyped behaviors, he presented some inappropriate, ritualistic, sameness, and restricted behaviors as well as compulsive behaviors. There is no history of autism in the family.
The proband with ID K21 is a white male child who was recruited for this study at the age of 8 yr and 11 mo with a previous diagnosis of classical autism with nonsyndromic features; test results and scales used for diagnosis and phenotypical assessment are not available. He reached early milestones in normal time. He rolled over at 6 mo and walked at 12 mo but seemed to undergo regression at ∼15 mo of age. He was 135.5 cm, weighted 31.8 kg and had a head circumference of 53.3 cm. Physical appearance of his eyes, hair, ears, mouth, palate, nose, and in general his appearance was evaluated as normal (Fig. 1B). He has no history of heart murmur or fainting, and there are no concerns for hearing or visual impairment nor for hernia or undescended testis. Ear infections, palate anomalies, breathing problems, unusual scarring or abnormal wound healing, anemia, immunodeficiency, or platelet dysfunction were not detected. There is no concern about limbs, joints, or spine; he has symmetric muscle development, normal strength, and fluent movement. He has a history of constipation and encopresis and has difficulty falling and staying asleep. He is the first child of the quad with an unaffected male sibling of the same father and mother who were 33 and 26 yr old, respectively, at the time the proband was born; there were no concerning exposures to drugs or medications during pregnancy and no acute or chronic illnesses complicated the pregnancy. The proband was born at 41 wk of gestation with an induced labor and C-section for CP disproportion. He was 22 in and weighed 9 pounds, 2 ounces at birth and there were no newborn complications. He has been treated with risperidone and trazodone. He has had a FMR1 test, which was negative for fragile X syndrome. A comparative genomic hybridization (CGH) microarray revealed a duplication on Xq13.1 inherited by the mother and shared by the unaffected sibling. There is no history of autism in close relatives; however, there are other distant cases of autism and pervasive developmental disorder in male individuals in the paternal side of the family (Supplemental Fig. 8).
Concordance between Variant Detection Algorithms
In this section, we explore detection reliability by measuring concordance among algorithm results across all sequenced individuals. Single-nucleotide variants, small insertions or deletions, and the detection of de novo variants of either class were compared across algorithms applied to raw whole-genome sequencing (WGS) data.
Single-Nucleotide Variants (SNVs) and Small Insertions or Deletions (INDELs)
GATK (the Genome Analysis Toolkit) and FreeBayes (FB) are algorithms that detect both SNVs and INDELs across the entire sequenced genome; as such, we report here the concordance between these two algorithms in detecting SNVs and INDELs. The observed mean concordance between GATK and FreeBayes was 79.3% and 56.6% for filtered SNV and INDEL calls, respectively. After filtering for high-quality variants according to each algorithm's recommendation (see Methods), concordance between the algorithms increases by 5.7% and 5.4% for SNVs and INDELs, respectively. Table 2 summarizes the mean per person number of variants called by each algorithm.
Table 2.
Number of variants obtained by each algorithm before and after filtering
| Variants | GATK HC | FreeBayes | Intersection | Unique to GATK % | Unique to FreeBayes % | Intersection % |
|---|---|---|---|---|---|---|
| Raw SNVs | 3,911,804 | 4,216,193 | 3,593,919 | 13.7 | 7 | 79.3 |
| Filtered SNVs | 3,403,728 | 3,714,842 | 3,255,217 | 11.9 | 3.8 | 84.3 |
| Raw INDELs | 814,730 | 790,178 | 580,335 | 20.5 | 22.9 | 56.6 |
| Filtered INDELs | 725,573 | 720,426 | 542,982 | 19.7 | 20.2 | 60.1 |
SNVs, single-nucleotide variants.
De Novo Unique SNVs
The mean number of unfiltered unique de novo SNVs (not shared by siblings) detected by the multinomial analyzer, FreeBayes, and GATK were 65,572, 76,920, and 40,873, respectively. The multinomial analyzer is an algorithm specifically designed to detect de novo SNVs, whereas additional steps were taken to obtain a list of putative de novo variants using FreeBayes and GATK. After filtering variants based on each algorithm's recommendations (refer to the Methods section for details pertaining to the filtering procedures), the mean number of variants detected by each caller dropped to 1692, 24,982, and 31,831 for the multinomial analyzer, FreeBayes, and GATK, respectively. The concordance between the three algorithms was generally low, with FreeBayes and GATK agreeing on 12.4% of their detected variants, and all three agreeing on <1% of the total filtered call set, 0.113%. It is important to note that the low concordance between the multinomial analyzer and the other algorithms is influenced by the fact that its filtering step considers a “de novo score,” which is something that the other algorithms do not use for filtering purposes. Thus, the large difference in overall call rate makes a comparison of the mean overlapping calls somewhat uninformative, as the intersection between all three can only be as large as the smallest set. It is for this reason that the union of the three algorithms was considered during downstream prioritization steps, rather than the intersection.
De Novo Unique INDELs
De novo INDELs from GATK, FreeBayes, and Scalpel were also compared. The mean number of de novo INDELs detected by GATK, FreeBayes, and Scalpel per proband before filtering was 52,631, 55,505, and 128, respectively, and after filtering based on each algorithm's recommendations, this number dropped to 42,425, 37,210, and 70. The concordance between the three algorithms was, again, low. FreeBayes and GATK agreed on 10.7% of the total call set, and all three callers agreed on only one variant and only within the subset of a single family (i.e., there was only one instance in which all three callers found the same variant). One should keep in mind that the filtering criteria and size of call sets are very different across these three callers, so our a priori expectation is that a low number of calls will be within the intersection of all three.
Variant Classification and Prioritization for SNVs and INDELs
After obtaining high-quality call sets from the union of filtered variants from all algorithms and categorizing them according to different disease models, the number of variants was still too large to proceed to more detailed literature searches and putative functional interpretations. Filtering variants by CADD (Combined Annotation-Dependent Depletion) score >20 and MAF (minor allele frequency) <0.01 from the 1000 Genomes Project reduced the number of variants for consideration dramatically (Table 3) and the number of compound heterozygous mutations was reduced to zero. However, variants segregating according to the compound heterozygous model are not necessarily expected to be deleterious on their own, but may be deleterious in combination with other variants in the same gene on the same, or different, chromosome.
Table 3.
Average number of variants by model before and after filtering
| Model | Filtered calls | CADD >20 | 1000 Genomes MAF <0.05 | Databases |
|---|---|---|---|---|
| De novo | 110,794 | 46 | 33 | 2 |
| X-linked | 327,170 | 1017 | 153 | 0 |
| Autosomal recessive | 143,578 | 466 | 14 | 0 |
| Compound heterozygous | 1380 | NA | 0 | 0 |
CADD, combined annotation-dependent depletion; MAF, multiple alignment format.
To narrow the list of possible disease-contributing variants, each call set was annotated and filtered using various criteria and scores described in the Methods section. Out of the resulting prioritized variants, an average of 101 per family were localized to intra- or intergenic regions (Supplemental Table 1) and only three were located within a gene, one of which was found to be common in the SSC controls. Thus, by these filtering criteria, two exonic variants were considered as potentially contributing to the disease (Table 4). The genic variants are described below.
Table 4.
Final set of single-nucleotide variants
| Model | Ref → Alt/effect | Location hg19 | Affected gene | Algorithms that called the variant | Pedigree ID | ExAC allele frequency | CADD score |
|---|---|---|---|---|---|---|---|
| De novo | Sub (C → T) missense | Chr1: 209823359 | LAMB3 | FB, MA, GATK | SSC_12605 | 0 | 22.7 |
| De novo | Sub (G → A) nonsense | Chr17: 4458481 | MYBBP1A | FB, MA, GATK | K21 | 1/74014 = 0.00001351 | 40 |
ExAC, Exome Aggregation Consortium; CADD, combined annotation dependent depletion; FB, FreeBayes; MA, multinomial analyzer; GATK, Genome Analysis Toolkit.
MYBBP1A Stop Gain Variant
A de novo heterozygous nonsense mutation was found on the first exon of MYBBP1A (Chr17: 4,442,191–4,458,926) in pedigree K21 (Fig. 3). This mutation is located at Chr17:4458481, is a G → A substitution, and is annotated as being highly deleterious with a CADD score of 40, which corresponds to being within the top 0.01% of all possible SNVs in terms of its deleteriousness. The variant was not found in dbSNP Human Build 142 (Sherry et al. 2001), the Exome Variant Server (http://evs.gs.washington.edu/EVS/) or in any other person in the Simons Simplex Collection database. One proband from the SSC was found to have a de novo missense G → T substitution in the same gene located at Chr17:4444853 causing an Arg → Ser change. Only one person out of 71,164 unrelated individuals from the Exome Aggregation Consortium (ExAC) (http://exac.broadinstitute.org) is reported to have this exact same mutation, indicating that this is a very rare variant. As the phenotype of this person in the ExAC database with the mutation is unknown, and also given that there are people with neuropsychiatric conditions in ExAC, no conclusions can be made from this alone. Sanger sequencing validated this mutation (Supplemental Fig. 1).
Figure 3.

Genome Browser Screen view for the read depths in the MYBBP1A stop gain (Chr17:4458481) mutation in the K21 family.
LAMB3 Missense Variant
The second de novo mutation detected in the study was found in the SSC_12605 pedigree and was a missense mutation located at Chr1:209823359 on LAMB3 (Fig. 4). Although this mutation was reported in a previous autism exome study in the same proband (Table 6; Iossifov et al. 2012), it was not found in any other person contained within the SSC database and it was not found in any of the other interrogated databases, the Exome Variant Server, or the ExAC database, making it an ultrarare mutation.
Figure 4.

Genome Browser Screen view for the read depths in the LAMB3 missense mutation (Chr1:209823359), showing 34 reads supporting the variant for the proband in SSC_12605 family.
Table 6.
Previous SSC exome study comparison
| Pedigree | Location | Ref → Alt/effect | Gene | Type | HC/Filtered out | FB/Filtered out | MA/Filtered out | CADD score |
|---|---|---|---|---|---|---|---|---|
| SSC_12605 | 10:103908608 | sub (C → T)/missense | PPRC1 | De novo | Yes/no | Yes/yes | Yes/no | 19.5 |
| SSC_12605 | 1:209823359 | sub (C → T)/missense | LAMB3 | De novo | Yes/yes | Yes/yes | Yes/no | 22.7 |
| SSC_12605 | 3:185993461 | sub (C → T)/missense | DGKG | De novo | Yes/no | Yes/yes | Yes/no | 7.5 |
SSC, Simons Simplex Collection; HC, haplotype caller; FB, FreeBayes; MA, multinomial analyzer; CADD, combined annotation dependent depletion.
Variant Classification and Prioritization for Copy-Number Variations
On average, 1500 unfiltered deletions and 450 unfiltered duplications were detected by ERDS applied to the WGS data (see Methods section) for each person in the study. After filtering (see Methods section), 150 deletions and 170 duplications were found on average per person. The number of calls obtained with PennCNV was highly variable, with a mean of 60 unfiltered duplications (SD = 38) and a mean of 80 unfiltered deletions (SD = 29) being detected. After filtering the variants, only 5% and 20% of all duplications and deletions were retained, respectively. After annotation, none of the remaining CNVs were identified as “pathogenic.” However, we detected three CNVs (Figs. 5–7) whose coordinates (Table 5) are embedded within larger CNVs that have been associated with cognitive disease. These CNVs were not found in any other unaffected family member. Two out of the three CNVs were found in pedigree K21; however, only the ERDS algorithm detected them. As described in the Methods section, PennCNV uses the Log R Ratio (LRR) and B Allele Frequencies (BAF) to detect a CNV. Different numbers of copies have different clustering patterns for the LRR and BAF values when plotted. In pedigree K21 (Figs. 5 and 6), both the LRR and BAF are not properly clustered, suggesting, in this case, that these CNVs were not detected by PennCNV but were detected by ERDS as true positives, because of the properties of the microarray data set for this family.
Figure 5.

(A) Genome browser screen view for the read depths in the K21 CNV 3q22.1 region of 16 kb. (B) B allele frequencies (BAF) for Illumina Omni2.5 markers on 3q22.1 region including the markers belonging to the CNV region detected by ERDS in red. (C) Log R ratio (LRR) values for Illumina Omni2.5 markers on 3q22.1 region including the markers belonging to the CNV region detected by ERDS in red.
Figure 7.

Genome Browser Screen view for the read depths in the SSC_12596 CNV 4p16.3 region of 50 kb. The highlighted region is where the four people bear either a homozygous or heterozygous deletion, only the proband has a homozygous deletion including the highlighted area plus the two regions indicated by the red arrows, which could have been generated by inheriting the deleted copy from both parents.
Table 5.
Copy-number variants
| Proband | Location | Genes | Type | ERDS score |
|---|---|---|---|---|
| K21 | Chr16:19945555–19967579 | Intergenic | Homozygous deletion | 2475.9 |
| K21 | Chr3:129763383–129806745 | Intergenic | Homozygous deletion | 3026.58 |
| SSC_596 | Chr4:4108476-4159245 | Intergenic | Homozygous deletion | 1525.08 |
ERDS, estimation by read depth with single-nucleotide variants.
Figure 6.

(A) Genome browser screen view for the read depths in the K21 CNV 16p12.3 region of 22 kb. (B) B allele frequencies (BAF) for Illumina Omni2.5 markers on K21 16p12.3 region including the markers belonging to the CNV region detected by ERDS in red. (C) Log R ratio (LRR) values for Illumina Omni2.5 markers on K21 16p12.3 region including the markers belonging to the CNV region detected by ERDS in red.
CGH microarray and sequence analysis applied to the proband and his mother revealed the presence of a maternally inherited duplication spanning several genes (ChrX:69074860–69512431). The duplication completely overlapped OTUD6A, IGBP1, DGAT2L6, AWAT1, AWAT2, P2RY4, KIF4A, ARR3, GDPD2, RAB41, and PDZD11 and partially overlapped EDA and DLG3. However, WGS-based CNV analyses revealed that the CNV was also present in the healthy sibling (Supplemental Fig. 6A). PennCNV, which was applied to additional Illumina microarray data, did not accurately call the breakpoints for this CNV in any of the three individuals where it was initially detected (mother, proband, sibling), although its presence was clear from manual inspection of the microarray data (see Supplemental Fig. 6B).
FMR1 Test
Fragile X testing resulted in a normal number of CGG trinucleotide repeats for the K21 proband. Analysis of WGS data from all probands did not show any significant difference in CGG-repeat content from the reference genome (Supplemental Figs. 2–5). Traditional clinical fragile X testing does not include sequencing FMR1, thus potentially missing other mutations that can contribute to the development of fragile X syndrome (De Boulle et al. 1993; Lugenbeel et al. 1995; Wang et al. 1997; Collins et al. 2010; Gronskov et al. 2011; Myrick et al. 2014). Although the probands in this study did not present any of the common phenotypic features of fragile X, a profile of all the CGG repeats present in every person was generated using WGS data and these profiles are compared with the reference sequence (Supplemental Figs. 3–5). No point mutations reported in the literature as contributing to fragile X syndrome (FXS) were found in any of the probands (Myrick et al. 2014); therefore, no evidence for FXS was found for any of the probands.
Reproducibility of Previous Exome Studies
As different approaches were taken to obtain the variants for each proband, it was of interest to know whether all of the SSC proband variants detected in the previous exome study, listed in Table 6 (Iossifov et al. 2012), were also detected by the methods used here. In cases where a variant was missed, this type of analysis will enable us to identify which step of the analysis pipeline might be responsible. Out of the three previously reported variants, which belong only to one family (SSC_12605), only one was included in the final list of variants with this pipeline. Two of the three were lost by GATK after the initial filtering step, but they were still included in the downstream analysis because FreeBayes and the multinomial analyzer detected and retained them in their call sets. However, they were ultimately discarded after the CADD score prioritization step, as they were not included in the top 1% most deleterious variants (<20 CADD score). No small variants were found in the SSC_12596 family, and none of the variants reported in Table 6 were found in SSC_12596 or K21.
DISCUSSION
Using a single bioinformatics pipeline for the discovery and analysis of sequence variants stemming from whole-genome sequencing data can result in incomplete data sets. In this study, we used a range of different software tools for detecting and analyzing human sequence variation stemming from WGS data, which has enabled the analysis of a more comprehensive data set composed of SNVs, INDELs, and CNVs. We found that only using the GATK pipeline for detecting SNVs would have resulted in a data set that left validated sequence variants undetected, highlighting the benefits of comprehensive analyses using aggregated data sets stemming from various detection tools. We have shown that the detection of CNVs from WGS data is naturally more sensitive, although the accuracy of WGS-based CNVs was not investigated here, as we did not perform validation experiments for CNVs. It is important to note that detecting CNVs from the same biological sample using microarray data and WGS results in data sets that are rather distinct both in number and in the characteristics of the CNV signal. Indeed, WGS resulted in an average of 1500 unfiltered deletions and 450 unfiltered duplications per sequenced individual whereas microarray resulted in an average of 60 unfiltered deletions and 80 unfiltered duplications per sequenced individual. Despite differences in the number of CNVs detected between microarray and WGS-based methods, having both data sets allowed for quick and relatively simple comparisons between these orthogonal technologies. This allowed for the detection of false-negatives and the evaluation of potential false-positive calls, as CNVs detected by both technologies can, in cases where both have adequate data, be used to cross-validate the calls.
Although we found CNVs and SNVs that fit the filtering and annotation criteria described in the Methods section, there is no obvious connection between any of them and ASDs, so they should be carefully considered only as possible disease-contributing variants that are in need of further functional analysis. In addition, we did not have the statistical power of a larger study to be able to associate our variants as contributing factors in the development of autism, and so our results are restricted to interpretation in the context of the three families studied here.
Aggregated Data Sets Are More Comprehensive
It is known that different algorithms are better at calling particular types of variants, each capable of detecting variants others cannot, and that they all usually agree on a subset of reliably called regions (O'Rawe et al. 2013). For this reason, the results of different algorithms were integrated, and instead of considering only the intersection of variants common to all algorithms, the union of all variant sets was obtained. This enabled the retention of variants that would have otherwise been excluded due to performing intersections with call sets, as only variants agreeing among all callers would have been retained. Indeed, one of the steps in which many variants are lost is during the initial filtering steps applied to each algorithm's raw call set, at which point one can decide how stringent the filter should be. Even recommended filtering parameters resulted in a detectable level of false negatives (i.e., true variants excluded from the final call set), despite these parameters being optimized for both sensitivity and specificity. Because the probands included in our study had already been part of previously reported targeted sequencing experiments, we were able to leverage available validation data to identify which informatics steps would have resulted in false-negative calls. Indeed and as stated above, we found that for the GATK HC (haplotype caller) call set, not all of the previously validated calls (Iossifov et al. 2012) passed the first initial recommended filtering steps.
It is important to note that to measure concordance between the different variant calling algorithms used in this study, we considered variants in agreement if they match in terms of the genomic positions where each algorithm made a call. Because of large differences in INDEL calling and reporting, the same INDEL can sometimes be reported differently (Assmus et al. 2013). For this reason, the reference and alternative fields were not included in the analysis of concordance between the different INDEL callers. Another reason for comparing callers in this way is based on the large differences seen in multiallelic calls reported by GATK HC (∼30,000) and FreeBayes (∼70,000). This nonstandard way of reporting INDELs has made the comparison between algorithms particularly difficult; thus, the comparisons performed in this study are approximate. These issues underscore the importance of carefully integrating sets of variants from different variant callers, as simple intersections can dramatically reduce the number of true positives even if all callers detect them, as their representations may be slightly different between the different callers. New tools that standardize discordant variant reporting into a unified schema have been developed (Tan et al. 2015), and we expect that these tools will aid in the more precise comparison and use of variants stemming from different callers.
Microarray versus WGS Data for Detecting CNVs
Microarray data provide researches with a cheap yet powerful way of detecting CNVs; however, depending on the particular technology used as well as the algorithms used to analyze the generated data, the results can vary widely. Sparse markers in some genomic regions make it difficult to define accurate breakpoints of detected CNVs, something that is less difficult with CNVs detected from WGS data. This is due, in part, to the fact that WGS data are more uniform, having an average coverage of reads which is less variable across the genome. For this study, we had both types of data for one of the quads, making it possible to call variants from both sources and compare the results. We found a large degree of variation in terms of the number of CNVs detected per person and also between the two detection methods used (i.e., WGS-based and DNA microarray–based methods). The genome-wide sensitivity of CNV detection using WGS is higher, because array-based methods do not densely cover the entire human genome with markers. We found that having data from these orthogonal technologies was useful in including or excluding true- or false-positive calls, as each should show some evidence of a CNV, if one does exist. Thus, in regions where both technologies had enough data to detect CNVs, discordant calls could be easily resolved by comparing the data profiles between the two.
Combining Prioritization Methods for More Robust Candidate Lists
Variant prioritization is another potentially delicate and important step in finding candidate disease-contributing variants. One could detect all true variants from WGS analysis yet still discount biologically important variants if the pertinent annotations are not used correctly. When filtering based on annotations that are numerically scaled, filtering threshold values should generally be strict enough to result in a small number of variants in which functional studies are feasible, without letting any biologically important variant go unconsidered. Obtaining this variant set from a single annotation or score is currently not possible, as each individually lacks the power to filter to a small and manageable set, which could otherwise be obtained by using multiple annotations for threshold-based filtering. For these reasons, several tools and annotations were combined to make sure that the results were robust and not due to systematic errors from one prioritization framework. Although two different frameworks were used, they were only slightly different in their results, likely because the VEP-GEMINI toolset has more annotations to determine if a variant is deleterious than does the in-house toolset. Unfortunately, using these two methods, we were unable to find a single candidate SNV or INDEL variant for the SSC_12596 pedigree. One alternative would be to use other prioritizing methods, such as the Variant Annotation, Analysis, and Search Tool (VAAST), which uses an aggregative variant association test that combines both amino acid substitution (AAS) and allele frequencies and incorporates information about phylogenetic conservation (Hu et al. 2013). As human variation not only includes small variations (SNVs and INDELs) and CNVs, but also structural variants and repeats, other software tools have to be used on these WGS data to explore other sources of variation that might contribute to disease.
Diagnostic Yield of ASD Genetic Studies
The diagnostic yield for different studies in the genetics of ASDs have been reported to range from 6% to 15% (Schaefer and Mendelsohn 2013). This variation depends on many factors including the type of technology used to generate the data, the tools used, the type of variants being studied, and the size of the cohort of study. Considering all of these variables, this pipeline tries to improve the strategies of those different studies by integrating them in a single pipeline that in principle would be able to improve the yield of diagnosis, as long as a larger cohort is studied to have statistical power. The diagnostic yield also highly depends on how many cases of the study would fall into the known autism-associated variants versus how many will be novel associated variants, but again, a larger cohort would be necessary to evaluate the improvement of the yield as well as to collect enough evidence to proceed with further functional analysis.
Putative Candidate Variants
After initial filtering, variant prioritization, and segregation analyses, we found two de novo missense variants that were annotated as being highly deleterious (as defined by a CADD score of >20) and rare on the population level (with population allele frequencies <0.01). The first variant was found in the proband of the pedigree K21 and it is a stop gain variant in MYBBP1A, and the second is a missense in LAMB3 found in the proband from the SSC_12605 pedigree.
Stop Gain in MYBBP1A
MYBBP1A codes for a nucleolar transcriptional regulator that was first identified by its ability to bind specifically to the MYB proto-oncogene protein (Favier and Gonda 1994). The encoded protein is thought to play a role in many cellular processes including response to nucleolar stress, tumor suppression and the synthesis of ribosomal DNA, and many cancers have been previously associated with MYBBP1A including brain glioma (Maglott et al. 2005). According to UniProt (Magrane and UniProt Consortium 2011), it may activate or repress transcription via interactions with sequence-specific DNA-binding proteins and repression may be mediated at least in part by histone deacetylase activity. It has been shown that its down-regulation induces apoptosis and mitotic anomalies in mouse embryonic stem cells, embryonic fibroblasts, and human HeLa cells (Mori et al. 2012). The known information about MYBBP1A does not make any obvious connection to ASDs; however, it has not been possible to create a homozygous knockout mouse for MYBBP1A and this is thought to happen as it is essential for early mouse development prior to blastocyst formation (Mori et al. 2012). In this study, the mutation found in this gene is heterozygous and although healthy heterozygous knockout mice have been reported, it is not clear if those mice had any behavioral phenotypes related to autism; further studies are needed before any conclusions about the relevance of this variant in the etiology of ASDs can be made.
LAMB3
LAMB3 codes for a β subunit laminin that belongs to a family of basement membrane proteins. Together with an α and a γ subunit, LAMB3 forms laminin-5. It is known that mutations in this gene can cause autosomal-dominant amelogenesis imperfecta (Kim et al. 2013), epidermolysis bullosa junctional Herlitz type, and generalized atrophic benign epidermolysis bullosa, diseases that are characterized by blistering of the skin (Mellerio et al. 1998; Maglott et al. 2005). According to UniProt (Magrane and UniProt Consortium 2011), its function is to bind to cells via a high affinity receptor, and laminin is thought to mediate the attachment, migration, and organization of cells into tissues during embryonic development by interacting with other extracellular matrix components. Again, the known diseases associated with this gene do not have an obvious link to autism, but its participation during embryonic development makes it an interesting candidate for further functional studies.
CONCLUSIONS
Although a subset of ASD cases are now better understood, with their genetic contributions becoming more clear (Zhao et al. 2007; Betancur 2011; Iossifov et al. 2012, 2014; Bernier et al. 2014; Ronemus et al. 2014; Lyon and O'Rawe 2015), the large degree of phenotypic heterogeneity in ASDs leaves the vast majority of cases still poorly understood. Larger studies have focused on a subset of variant types, but here we have obtained a broader and more complete view of all the different types of genomic variation that could be contributing to the ASD phenotypes observed in this study. By combining different algorithms and variant prioritization methods, we were able to use the strengths of each and compensate for the different weaknesses by integrating their results in one computational framework.
There has been special interest in knowing to what extent de novo mutations contribute to ASD cases (Iossifov et al. 2012). In this study, four different variant detection algorithms and three different prioritization methods were used to detect de novo variants. This allowed us to improve detection sensitivity and to reduce the false-negative rate. We also searched for variants segregating according to other disease transmission models, including autosomal recessive, X-linked, mitochondrial, and compound heterozygote. As expected, the number of variants detected from each model varies widely (Table 3).
As sequencing technologies improve in accuracy and their operational costs decrease, large-sequencing studies including thousands of people at higher sequencing depths are becoming more common. As such, it is useful and perhaps even necessary to design studies that search for and aim to detect all known variant types and to not just focus on a small subset. We suspect that such studies would, in general, obtain more biologically relevant results by doing so. However, study design must also consider the cost/benefit balance of sequencing whole genomes of a large number of people to high-sequence depth, as was done here with the SSC quads (∼75×). The previously reported ideal coverage for accurately detecting SNVs is 40×–45× where the detection saturates (Ajay et al. 2011). It has recently been shown that for accurate INDEL detection in personal genomes, whole-genome sequencing coverage of 60× may be ideal, at least with 100 base pairs (bp) paired-end reads from Illumina (Fang et al. 2014). Given the known complexity and heterogeneity of ASDs (Zhao et al. 2007; O'Roak et al. 2012; Bernier et al. 2014), it is clear that a large study capable of obtaining robust statistical signals is needed; yet a study of this magnitude with 60× coverage is still prohibitively expensive. Our study is useful in terms of contributing a small but rich data set to larger studies, so that the etiology of ASDs can be better understood. While this study was being completed, a study was published using the Complete Genomics (CG) platform to study 85 quartet families with autism (Yuen et al. 2015), although there is a very high false-negative rate associated with this sequencing technology, at least with the CG v2.0 pipeline (O'Rawe et al. 2013), and as a consequence the power of this study to detect recurrent signals may be relatively low. We provide our study as a more comprehensive analysis of three simplex autism quads, with the goal of improving genotype accuracy so that downstream analyses can take advantage of these rich data sets.
METHODS
Sample Collection and Sequencing
A pilot study of two SSC families (SSC_12605 and SSC_12596) and one Utah family (K21) was conducted. The SSC was assembled at 13 clinical centers, with the blood drawn from parents and children (affected and unaffected) sent to the Rutgers University Cell and DNA Repository (RUCDR) for DNA preparation. WGS was performed at Cold Spring Harbor Laboratory (CSHL) on the two SSC families using the Illumina HiSeq 2000 platform at an average coverage of 75×, using paired-end 100-bp reads.
The Utah family had previously undergone fragile X screening and chromosomal microarray (CMA) genotyping for the proband and mother at the University of Utah. K21 blood samples were collected at the Utah Foundation for Biomedical Research, and genomic DNA was extracted and purified. Finally the DNA was quantified using Qubit dsDNA BR Assay Kit (Invitrogen) and 1 µg was sent to the CSHL sequencing facility where WGS was performed on the Illumina HiSeq 2000 platform at an average coverage of 40× using paired-end 100-bp reads, and a parallel DNA sample was genotyped with an Illumina Omni2.5 array at the CHOP core facility.
Clinical and Phenotypic Evaluation for the Simons Simplex Collection Probands
Physical measures such as height, weight, and head circumference were taken for each proband as well as a detailed medical history. To evaluate the pregnancy and birth of each proband the Gillberg Optimality Scale (Gillberg and Gillberg 1983) was used; this scale selects certain events that may happen during pregnancy and labor that are considered “nonoptimal” and get scored as a 1 (if they do not occur, they get scored as a 0).
The Autism Diagnostic Observation Schedule (ADOS) and the Autism Diagnostic Interview Revised (ADI-R) were used by the clinicians to determine whether a proband may fall in the autism, ASD, or nonspectrum category. The ADI-R is a clinical diagnostic instrument for assessing autism; it provides a diagnostic algorithm for autism and focuses on behavior in three main areas: qualities of reciprocal social interaction; communication and language; and restricted and repetitive stereotyped interests and behaviors (Lord et al. 1994). The ADOS is a semistructured assessment of communication, social interaction, and play for individuals suspected of having autism; it consists of four modules, each of which is appropriate for different developmental and language levels, ranging from nonverbal to verbally fluent (Lord et al. 2000). Given the age and language level of the probands, module number 1 for nonverbal individuals was used on proband SSC_12596, and module number 4 was used for proband SSC_12605. Besides ADOS and ADI-R, the diagnosticians were asked to complete a form in which they indicated how certain they were that the proband was on the autism spectrum, compiling a 15-point scale ranging from high certainty that the participant did not have ASD (1) to high certainty that the participant had autism (15). As all probands included in the SSC are required to fall somewhere on the autism spectrum, certainty ratings in this sample may range from 6 (uncertainty whether ASD) to 15 (high certainty of autism). The verbal, nonverbal, and overall cognitive abilities were measured by applying intelligence quotient tests for each ability and are reported in Table 1. The Aberrant Behavior Checklist (Aman et al. 1985; Kaat et al. 2014) is a rating scale that measures the severity of a range of problem behaviors commonly observed in individuals with intellectual and developmental disabilities. Higher scores indicate more inappropriate behavior. The Repetitive Behavior Scale-Revised (RBS-R) is a measure of repetitive behaviors in young children with ASDs (Lam and Aman 2007; Mirenda et al. 2010). Items on the RBS-R are scored from 0 (behavior does not occur) to 3 (behavior occurs and is a severe problem) and items are classified as stereotyped behavior (6 items), self-injurious (8 items), compulsive behavior (8 items), sameness behavior (11 items), and restricted behavior (4 items).
Fragile X Analysis (FMR1 Test)
The pedigree K21 proband was tested for fragile X syndrome, a common inherited form of intellectual disability and autism spectrum disorder with characteristic phenotypic features, in which the majority of patients exhibit a massive CGG-repeat expansion mutation in FMR1 that silences the locus (Myrick et al. 2014). To know if the expansion was present, the fragile X region was amplified by polymerase chain reaction (PCR) using a single chimeric primer set in which one of the primers is fluorescently labeled (FristStepDx [http://www.firststepdx.com/]). The reactions were then separated by capillary electrophoresis on the ABI310xl Genetic Analyzer and analyzed using the GeneMapper software.
Fragile X syndrome can sometimes be misdiagnosed as only autism in the absence of the CGG-repeat expansion. However, there are two missense and other point mutations in the FMR1 gene that have been reported and described as causative of fragile X Syndrome (De Boulle et al. 1993; Lugenbeel et al. 1995; Wang et al. 1997; Collins et al. 2010; Gronskov et al. 2011; Myrick et al. 2014). Because missense mutations cannot be detected using the CGG-repeat test and because WGS data was available for every proband, loci spanning FMR1 were carefully analyzed to see if any of the probands had any possible disease-contributing mutation (e.g., p.Ile304Asn, p.Gly266Glu, IVS10 + 14C → T, and p.Ser27X). A CGG-repeat analysis on the fragile X region (ChrX:146,993,468–147,032,646, http://omim.org/entry/309550) was also performed for all the probands to confirm that the CGG-repeat number was normal compared with the reference genome. This was done by calling variants and generating a gVCF file with the GATK Haplotype Caller software. The gVCF file contains all sites in the FMR1 gene, whether there is a variant present or not. Using the gVCF file, the gene sequences were inferred and each CGG trinucleotide was plotted as it appears within the FMR1 gene region, making evident any subtle difference in the amount or positions of the CGG repeats (Supplemental Fig. 2). This simple method will only work if the CGG-repeat size is covered by the read length of the sequencing technology used to sequence the samples; otherwise it would not align to the reference sequence. However if the reads are not long enough and few or no reads are aligned, we may still infer the presence of an expansion if there is an apparent deletion in the 5' UTR of FMR1.
Chromosomal Microarray Analysis (CMA)
The pedigree K21 proband was genotyped using the Affymetrix Cytogenetics Whole-Genome 2.7 Array, which has a total of 2,141,868 markers across the genome, including 1,742,975 unique nonpolymorphic markers and 398,891 single-nucleotide polymorphism (SNP) markers. After finding a CNV with unknown pathogenicity on chromosome X, the mother was also genotyped using the same array to determine if the CNV was inherited.
SNV and INDEL Variant Calling
Before proceeding to analyze the WGS data, the quality of raw sequencing reads was assessed using FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), which summarizes sequence quality metrics that can indicate whether there was a problem with the sequencing experiment. This quality control procedure is important, because the quality of the raw sequencing data needs to be assessed before performing further downstream analyses. As human genomic variation can range from single-nucleotide changes to whole-chromosome variations, different analyses need to be performed to retrieve most of the true variation present in each person. In this study, several software packages were used in an integrative manner to analyze all the data generated by the different high-throughput technologies. Raw sequence read quality analysis was performed for all samples, followed by aligning them to the reference genome. All analysis prior to the use of variant caller software were applied to the data in a lane by lane fashion; this is done to take account of experimental variation introduced by optical duplicates known to occur in a lane-specific manner (McKenna et al. 2010).
Whole-Genome Sequence Aligning and Precalling Processes
Whole-genome sequence reads from all samples were aligned, lane by lane, to the GRCh37/hg19 human reference sequence using BWA-MEM 0.7.5a-r405 software (Li and Durbin 2009) with default parameters, tagging shorter split hits as secondary for compatibility with Picard tools used downstream from the alignment. Samples from the SSC families were sequenced to a mean coverage of 75×, with six different lanes per sample used to achieve this depth. K21 family samples were sequenced to a mean coverage of 40×, obtained by using 3.5 lanes. The resulting alignments were converted to binary format, and then sorted and indexed using SAMtools version 0.1.19-44428cd (Li et al. 2009). Duplicated reads were marked and read groups were assigned to each lane using Picard tools v1.84 (http://sourceforge.net/projects/picard/). The Qualimap version 2.1 software was used to extract basic statistics on these alignments; Supplemental Document 2 contains a table with the number of mapped reads and coverage by contig aligned to the reference genome. Before any variant calling, all alignments were subjected to the GATK Indel realigner v3.0-0, which was used to correct mapping artifacts that, because of reads aligning to the edges of INDELs, may look like evidence for SNPs. The GATK Base Quality Score Recalibrator was also used to correct systematic errors of sequencing technologies (McKenna et al. 2010; DePristo et al. 2011; Van der Auwera et al. 2013). Finally all lanes were merged by sample with Picard tools to generate a ready-to-use alignment.
Variant Detection for SNV and INDELS
After obtaining ready-to-use alignments, four different variant callers were used to analyze the WGS data for each individual in the three different families. The GATK haplotype caller v2.8-1 and v3.0-0 and FreeBayes variant caller v9.9.2-43- (Garrison and Marth 2012) were both used on the ready-to-use alignments (see Whole-Genome Sequence Aligning and Precalling Processes) to call both SNVs and INDELs. Variants obtained with the GATK Haplotype Caller were filtered for variant quality using the GATK variant quality score recalibration (VQSR) tool; those obtained with the FreeBayes variant caller were also filtered for variant quality, and calls with a QUAL score of <30 or with <10 supporting reads were filtered out. To further support the detection of de novo calls (INDELs and SNVs), two other packages were used. Scalpel, an INDEL variant caller that performs localized micro-assembly to accurately detect mutations (Narzisi et al. 2014), was used in de novo mode considering the four family members in order to make a decision whether an INDEL was de novo. Only de novo variants not shared with the sibling were considered. The multinomial analyzer (MA), a SNV de novo caller (Iossifov et al. 2012), was also used; this implements a multinomial model that also consider all family members of a quad to decide whether a call is a true de novo or not. The filtering thresholds for MA were set to de novo score >60 and χ2 P value >0.0001, as was used in the exome study in which both SSC families were previously analyzed (Iossifov et al. 2012). Variants from the same sample coming from GATK, FreeBayes, Scalpel, and MA were merged into a single VCF file for downstream analysis. See Supplemental Document 1 for a complete list of the parameters or code used for each of the four algorithms.
Variant Classification and Prioritization
The final set of high-quality calls were divided into different models of inheritance, so that the way in which the mutations emerged and how they were possibly contributing to the condition could be interrogated. After obtaining model-specific subsets, the variants were annotated with a combined annotation dependent depletion (CADD) score, a metric that evaluates the deleteriousness of SNV, as well as INDEL, variants in the human genome. CADD scores are generated by integrating multiple annotations, including PolyPhen and SIFT scores, into one metric by contrasting variants that survived natural selection with simulated mutations (Kircher et al. 2014). Those variants with a CADD score of >20 were kept as potentially deleterious, and the number of reads supporting each variant was compared among all family members to decide whether a call was a false positive or not. All variants were further filtered using a MAF <0.01 from the 1000 Genomes Project (October 2014). The final set of variants was annotated with in-house tools as well as the ANNOVAR software (Wang et al. 2010) using the UCSC (Kent et al. 2002) and RefSeq (Raney et al. 2014) gene tables; the SSC (Basu et al. 2009), Exome Variant Server (http://evs.gs.washington.edu/EVS/), and ClinVar databases (Landrum et al. 2014); and the recently released ExAC database (http://exac.broadinstitute.org).
Models
There are several ways in which a disease-contributing genetic variant can be present in an individual. As we were not only interested in the variants, but also in their origin, they were divided into different models before prioritization.
De Novo Model
De novo variants are those that emerge at some stage during the gametogenesis of one of the parents or embryogenesis of the child, so those mutations will be only present in the offspring and not the parents. Only those variants present uniquely in the proband and not in parents or unaffected sibling were kept for downstream annotation and analysis.
X-Linked
Here only variants on chromosome X are considered. As all of the probands in this study are males, the only X chromosome copy they have comes from the mother, who by having two X chromosome copies could be masking the deleteriousness of a mutation, which is then expressed fully in the male offspring. All X chromosome variants present in the proband inherited from the mother but not present in the healthy sibling or father were kept for downstream annotation and analysis.
Autosomal Recessive
In this model, a given variant is required to be present in both probands with one copy inherited by the mother and the second one from the father. The autosomal recessive variants found in the healthy sibling are also excluded.
Compound Heterozygous
Sometimes a gene can bear two different heterozygous mutations; one in each chromosomal copy, affecting both copies of a gene but not with the same exact mutation, as is the case for the autosomal recessive model. For this set of variants, only those combinations of heterozygous mutations on the same gene and present in the proband were considered.
Mitochondrial
In a similar fashion as chromosome X, it is well known that the mitochondrial DNA is passed from mother to offspring; however in this case, if a mutation is contributing to the disease the mother would also be affected so the only mitochondrial mutations considered are under the de novo model described above.
VEP-GEMINI
The VEP (Variant Effect Predictor)-Genome Mining (GEMINI) (Paila et al. 2013) toolset is a framework for annotating and prioritizing genomic variants by different criteria. Built-in analysis tools were used to obtain variants characterized by different classifications: de novo, compound heterozygous, autosomal recessive, and impact severity. The VEP-GEMINI toolset was used to get additional information about each variant, and to compare the results obtained with the model classifications and prioritizations performed with in-house tools. The criteria for keeping variants from each classification scheme were for variants to have a CADD score of >20 or be annotated as having high impact severity for the proband.
Variant Calling for Copy-Number Variants
The estimation by read depth with SNVs (ERDS) software (Zhu et al. 2012) was used with default parameters to call CNVs from WGS data on each individual. It uses WGS data along with previously generated VCF files using the read depth and number of contiguous heterozygous and homozygous SNVs to call CNVs. Only calls with an ERDS score of >300 were kept.
Additionally, CNVs were called with the microarray data from pedigree K21, which was genotyped with an Illumina Omni2.5 array and analyzed with the software package PennCNV (Wang et al. 2007). For kilobase-resolution detection of CNVs, PennCNV uses an algorithm that implements a hidden Markov model, which integrates multiple signal patterns across the genome and uses the distance between neighboring SNPs and the allele frequency of SNPs. The two signal patterns that it uses are the log R ratio (LRR), which is a normalized measure of the total signal intensity for two alleles of the SNP, and the B allele frequency (BAF), a normalized measure of the allelic intensity ratio of two alleles. The combination of both signal patterns is then used to infer copy-number changes in the genome. Microarrays often show variation in hybridization intensity (genomic waves), which is related to the genomic position of the clones, and that correlates to GC content among the genomic features considered. For adjustment of such genomic waves in signal intensities, the cal_gc_snp.pl PennCNV program was used to generate a GC model that considered the GC content surrounding each Illumina Omni2.5 marker within 500 kilobases (kb) on each side (1 Mb total). The joint-calling algorithm designed for parents-offspring trios was used, as it is the most accurate of the algorithms in the package for family-based studies. The hidden Markov model used is contained in the hhall.hmm file provided by the latest PennCNV package, and the custom population frequency of B allele (PFB) file for all the SNPs in the Illumina Omni2.5 array was generated from 600 controls, which consist of 600 unaffected parents from the Simons Simplex Collection (provided by Dr. Stephan Sanders from Yale University). The GC model described above was also used during CNV calling.
Chromosome X CNVs were called separately using the -- test mode with the --Chrx option. Using BEDtools (Quinlan and Hall 2010) and in-house tools, consensus CNV calls were obtained for parents from the two separate trio calling processes that had to be done for each child in the quad. CNVs were quality filtered by considering the length of the CNV event (for both algorithms: ERDS and PennCNV) and for microarray data, the number of SNPs embedded on the CNV region and the number of expected SNPs for that given region (Supplemental Fig. 5), histocompatibility regions, and centromeric and telomeric regions were also filtered out as it is common to find nonpathogenic variants there (both algorithms).
For pedigree K21, ERDS and PennCNV calls were compared and the union of each pipeline's set of variants was annotated with in-house tools and the ANNOVAR software (Wang et al. 2010) using dbVar (Lappalainen et al. 2013), DGV (MacDonald et al. 2014), ClinVar (Landrum et al. 2014), DECIPHER (Firth et al. 2009), ENCODE (Rosenbloom et al. 2013), and the SFARI Gene database (Basu et al. 2009) and those variants in which >90% of their total length overlapped reciprocally with variants found in controls were ruled out. ERDS-filtered output for pedigrees SSC_12596 and SSC_12605 were annotated with the same software and criteria.
Sanger Sequencing
Polymerase chain reaction (PCR) primers for the Chr17:4458481(hg19) variant in MYBBP1A (Supplemental Document 7) were designed to produce a 911-bp amplicon, using Primer3 (http://primer3.sourceforge.net). Primers were obtained from Sigma-Aldrich, and tested for PCR efficiency with an in-house DNA sample using a Phusion Flash High-Fidelity PCR Master Mix (Life Technologies). The optimized PCR reaction was then carried out on patient DNA. PCR products were visually inspected for amplification efficiency using agarose gel electrophoresis and were purified using the QIAquick PCR Purification Kit (QIAGEN). Purified products were then diluted to 5–10 ng/µL in water for use with the ABI 3700 sequencer. The resulting *.ab1 sequence files were loaded into the CodonCode Aligner V5.1.2 for analysis. All sequence traces were manually reviewed to ensure the reliability of the genotype calls.
ADDITIONAL INFORMATION
Ethics Statement
Research was carried out in compliance with the Helsinki Declaration. Two of the families analyzed in this study belong to the SSC (referred as SSC_12596 & SSC_12605), and both families were clinically evaluated and extensively phenotyped as well as whole-exome sequenced for a previous study (Iossifov et al. 2012).
The third family (referred to as K21) was recruited to this study at the Utah Foundation for Biomedical Research (UFBR) where extensive clinical evaluation was performed. Written consent was obtained for phenotyping, use of facial photography, and whole-genome sequencing through Protocol #100 at the Utah Foundation for Biomedical Research, approved by the Independent Investigational Review Board, Inc.
Data Deposition and Access
All of the sequence reads have been deposited in NCBI Sequence Read Archive (SRA; http://www.ncbi.nlm.nih.gov/sra) and are accessible under BioProject PRJNA282537 (BioSamples SAMN03571202, SAMN03571214, SAMN03571217, SAMN03571219). Interpreted variants have been submitted to ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/) under accession numbers SCV000238497, SCV000238498, SCV000239874, SCV000239875, and SCV000239876.
Acknowledgments
Dr. Stephan Sanders provided the PFB file necessary for CNV calling. Dr. Kai Wang assisted in the CNV analysis. The authors would like to thank the Exome Aggregation Consortium and the groups that provided exome variant data for comparison. A full list of contributing groups can be found at http://exac.broadinstitute.org/about. The authors would like to thank Graciela J.B. and the National Autonomous University of Mexico for partly funding this study by granting L.T.J.-B. an undergraduate scholarship for international mobility. L.T.J.-B. thanks Julián Regalado Pérez for all the support.
We thank all the families at the participating SSC sites, as well as the principal investigators (A.L. Beaudet, R. Bernier, J. Constantino, E.H. Cook, Jr., E. Fombonne, D. Geschwind, D.E. Grice, A. Klin, D.H. Ledbetter, C. Lord, C.L. Martin, D.M. Martin, R. Maxim, J. Miles, O. Ousley, B. Peterson, J. Piggot, C. Saulnier, M.W. State, W. Stone, J.S. Sutcliffe, C.A. Walsh, and E. Wijsman) and the coordinators and staff at the SSC sites for the recruitment and comprehensive assessment of simplex families; the SFARI staff for facilitating access to the SSC; and the Rutgers University Cell and DNA Repository (RUCDR) for accessing biomaterials. We would also like to thank the CSHL Woodbury Sequencing Center for generating sequencing data, and W.R. McCombie, E. Antoniou, and E. Ghiban for their assistance in data production at CSHL.
Author Contributions
L.T.J.-B. developed the study design, performed all informatics analyses, and wrote the manuscript. J.A.O. provided in-house tools, contributed to the study design, and participated in bioinformatics analyses and manuscript writing. Y.W. performed the Sanger sequencing validation experiment. M.Y. assisted in general clinical information interpretation. H.F. participated in the study design and bioinformatics analyses. I.I. provided the SSC data and the multinomial analyzer algorithm. G.J.L. participated in the study design, clinical phenotyping, data interpretation, and manuscript writing.
Funding
The laboratory of G.J.L. is supported by funds from the Stanley Institute for Cognitive Genomics at Cold Spring Harbor Laboratory (CSHL). The CSHL Genome Center is supported in part by a Cancer Center Support Grant (CA045508) from the National Cancer Institute. This work was supported in part by SFARI grant (SF362665) to I.I.
Competing Interest Statement
G.J.L. serves on advisory boards for GenePeeks, Inc. and Omicia, Inc.
Supplementary Material
Footnotes
[Supplemental material is available for this article.]
REFERENCES
- Ajay SS, Parker SC, Abaan HO, Fajardo KV, Margulies EH. 2011. Accurate and comprehensive sequencing of personal genomes. Genome Res 21: 1498–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aman MG, Singh NN, Stewart AW, Field CJ. 1985. Psychometric characteristics of the aberrant behavior checklist. Am J Ment Defic 89: 492–502. [PubMed] [Google Scholar]
- Assmus J, Kleffe J, Schmitt AO, Brockmann GA. 2013. Equivalent indels—ambiguous functional classes and redundancy in databases. PLoS One 8: e62803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu SN, Kollu R, Banerjee-Basu S. 2009. AutDB: a gene reference resource for autism research. Nucleic Acids Res 37: D832–D836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernier R, Golzio C, Xiong B, Stessman HA, Coe BP, Penn O, Witherspoon K, Gerdts J, Baker C, Vulto-van Silfhout AT, et al. 2014. Disruptive CHD8 mutations define a subtype of autism early in development. Cell 158: 263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur C. 2011. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res 1380: 42–77. [DOI] [PubMed] [Google Scholar]
- Collins SC, Bray SM, Suhl JA, Cutler DJ, Coffee B, Zwick ME, Warren ST. 2010. Identification of novel FMR1 variants by massively parallel sequencing in developmentally delayed males. Am J Med Genet A 152A: 2512–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Boulle K, Verkerk AJ, Reyniers E, Vits L, Hendrickx J, Van Roy B, Van den Bos F, de Graaff E, Oostra BA, Willems PJ. 1993. A point mutation in the FMR-1 gene associated with fragile X mental retardation. Nat Genet 3: 31–35. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. 2011. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43: 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Wu Y, Narzisi G, O'Rawe JA, Barron LT, Rosenbaum J, Ronemus M, Iossifov I, Schatz MC, Lyon GJ. 2014. Reducing INDEL calling errors in whole genome and exome sequencing data. Genome Med 6: 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favier D, Gonda TJ. 1994. Detection of proteins that bind to the leucine zipper motif of c-Myb. Oncogene 9: 305–311. [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, Carter NP. 2009. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84: 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach GD, Lord C. 2010. The Simons Simplex Collection: a resource for identification of autism genetic risk factors. Neuron 68: 192–195. [DOI] [PubMed] [Google Scholar]
- Garrison E, Marth G. 2012. Haplotype-based variant detection from short-read sequencing. arXiv preprint arXiv:12073907.
- Gillberg C, Gillberg IC. 1983. Infantile autism: a total population study of reduced optimality in the pre-, peri-, and neonatal period. J Autism Dev Disord 13: 153–166. [DOI] [PubMed] [Google Scholar]
- Gronskov K, Brondum-Nielsen K, Dedic A, Hjalgrim H. 2011. A nonsense mutation in FMR1 causing fragile X syndrome. Eur J Hum Genet 19: 489–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Huff CD, Moore B, Flygare S, Reese MG, Yandell M. 2013. VAAST 2.0: improved variant classification and disease-gene identification using a conservation-controlled amino acid substitution matrix. Genet Epidemiol 37: 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, Yamrom B, Lee YH, Narzisi G, Leotta A, et al. 2012. De novo gene disruptions in children on the autistic spectrum. Neuron 74: 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. 2014. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515: 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson HM, Gaitanis J, Morrow EM. 2011. Genetics in autism diagnosis: adding molecular subtypes to neurobehavioral diagnoses. Med Health R I 94: 124–126. [PMC free article] [PubMed] [Google Scholar]
- Kaat AJ, Lecavalier L, Aman MG. 2014. Validity of the aberrant behavior checklist in children with autism spectrum disorder. J Autism Dev Disord 44: 1103–1116. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. 2002. The human genome browser at UCSC. Genome Res 12: 996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lee KE, Ko J, Yildirim M, Tuna EB, Gencay K, Shin TJ, Kyun HK, Simmer JP, et al. 2013. LAMB3 mutations causing autosomal-dominant amelogenesis imperfecta. J Dent Res 92: 899–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. 2014. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 46: 310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumm N, Turner TN, Baker C, Vives L, Mohajeri K, Witherspoon K, Raja A, Coe BP, Stessman HA, He ZX, et al. 2015. Excess of rare, inherited truncating mutations in autism. Nat Genet 47: 582–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam KS, Aman MG. 2007. The Repetitive Behavior Scale-Revised: independent validation in individuals with autism spectrum disorders. J Autism Dev Disord 37: 855–866. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Riley GR, Jang W, Rubinstein WS, Church DM, Maglott DR. 2014. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 42: D980–D985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen I, Lopez J, Skipper L, Hefferon T, Spalding JD, Garner J, Chen C, Maguire M, Corbett M, Zhou G, et al. 2013. DbVar and DGVa: public archives for genomic structural variation. Nucleic Acids Res 41: D936–D941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy D, Ronemus M, Yamrom B, Lee YH, Leotta A, Kendall J, Marks S, Lakshmi B, Pai D, Ye K, et al. 2011. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 70: 886–897. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R; Genome Project Data Processing S. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord C, Rutter M, Le Couteur A. 1994. Autism Diagnostic Interview-Revised: a revised version of a diagnostic interview for caregivers of individuals with possible pervasive developmental disorders. J Autism Dev Disord 24: 659–685. [DOI] [PubMed] [Google Scholar]
- Lord C, Risi S, Lambrecht L, Cook EH Jr, Leventhal BL, DiLavore PC, Pickles A, Rutter M. 2000. The autism diagnostic observation schedule-generic: a standard measure of social and communication deficits associated with the spectrum of autism. J Autism Dev Disord 30: 205–223. [PubMed] [Google Scholar]
- Lugenbeel KA, Peier AM, Carson NL, Chudley AE, Nelson DL. 1995. Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nat Genet 10: 483–485. [DOI] [PubMed] [Google Scholar]
- Lyon GJ, O'Rawe J. 2015. Human genetics and clinical aspects of neurodevelopmental disorders. In The genetics of neurodevelopmental disorders (ed. Mitchell K). Wiley, Hoboken, NJ. [Google Scholar]
- MacDonald JR, Ziman R, Yuen RK, Feuk L, Scherer SW. 2014. The Database of Genomic Variants: a curated collection of structural variation in the human genome. Nucleic Acids Res 42: D986–D992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maglott D, Ostell J, Pruitt KD, Tatusova T. 2005. Entrez Gene: gene-centered information at NCBI. Nucleic Acids Res 33: D54–D58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrane M, UniProt Consortium. 2011. UniProt Knowledgebase: a hub of integrated protein data. Database (Oxford) 2011: bar009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellerio JE, Eady RA, Atherton DJ, Lake BD, McGrath JA. 1998. E210K mutation in the gene encoding the β3 chain of laminin-5 (LAMB3) is predictive of a phenotype of generalized atrophic benign epidermolysis bullosa. Br J Dermatol 139: 325–331. [DOI] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirenda P, Smith IM, Vaillancourt T, Georgiades S, Duku E, Szatmari P, Bryson S, Fombonne E, Roberts W, Volden J, et al. 2010. Validating the Repetitive Behavior Scale-revised in young children with autism spectrum disorder. J Autism Dev Disord 40: 1521–1530. [DOI] [PubMed] [Google Scholar]
- Mori S, Bernardi R, Laurent A, Resnati M, Crippa A, Gabrieli A, Keough R, Gonda TJ, Blasi F. 2012. Myb-binding protein 1A (MYBBP1A) is essential for early embryonic development, controls cell cycle and mitosis, and acts as a tumor suppressor. PLoS One 7: e39723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myrick LK, Nakamoto-Kinoshita M, Lindor NM, Kirmani S, Cheng X, Warren ST. 2014. Fragile X syndrome due to a missense mutation. Eur J Hum Genet 22: 1185–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narzisi G, O'Rawe JA, Iossifov I, Fang H, Lee YH, Wang Z, Wu Y, Lyon GJ, Wigler M, Schatz MC. 2014. Accurate de novo and transmitted indel detection in exome-capture data using microassembly. Nat Methods 11: 1033–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L, Ma'ayan A, Samocha KE, Sabo A, Lin CF, Stevens C, Wang LS, Makarov V, et al. 2012. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature 485: 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, Bodily P, Tian L, Hakonarson H, Johnson WE, et al. 2013. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med 5: 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Roak BJ, Vives L, Fu W, Egertson JD, Stanaway IB, Phelps IG, Carvill G, Kumar A, Lee C, Ankenman K, et al. 2012. Multiplex targeted sequencing identifies recurrently mutated genes in autism spectrum disorders. Science 338: 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paila U, Chapman BA, Kirchner R, Quinlan AR. 2013. GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput Biol 9: e1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raney BJ, Dreszer TR, Barber GP, Clawson H, Fujita PA, Wang T, Nguyen N, Paten B, Zweig AS, Karolchik D, et al. 2014. Track data hubs enable visualization of user-defined genome-wide annotations on the UCSC Genome Browser. Bioinformatics 30: 1003–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson EB, Samocha KE, Kosmicki JA, McGrath L, Neale BM, Perlis RH, Daly MJ. 2014. Autism spectrum disorder severity reflects the average contribution of de novo and familial influences. Proc Natl Acad Sci 111: 15161–15165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronemus M, Iossifov I, Levy D, Wigler M. 2014. The role of de novo mutations in the genetics of autism spectrum disorders. Nat Rev Genet 15: 133–141. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Sloan CA, Malladi VS, Dreszer TR, Learned K, Kirkup VM, Wong MC, Maddren M, Fang R, Heitner SG, et al. 2013. ENCODE data in the UCSC Genome Browser: year 5 update. Nucleic Acids Res 41: D56–D63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Sudhof TC. 2014. Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158: 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer GB, Mendelsohn NJ. 2013. Clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet Med 15: 399–407. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, Sirotkin K. 2001. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugathan A, Biagioli M, Golzio C, Erdin S, Blumenthal I, Manavalan P, Ragavendran A, Brand H, Lucente D, Miles J, et al. 2014. CHD8 regulates neurodevelopmental pathways associated with autism spectrum disorder in neural progenitors. Proc Natl Acad Sci 111: E4468–E4477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan A, Abecasis GR, Kang HM. 2015. Unified representation of genetic variants. Bioinformatics 31: 2202–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 11: 11.10.1–11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Lin ML, Lin SJ, Li YC, Li SY. 1997. Novel point mutation within intron 10 of FMR-1 gene causing fragile X syndrome. Hum Mutat 10: 393–399. [DOI] [PubMed] [Google Scholar]
- Wang K, Li M, Hadley D, Liu R, Glessner J, Grant SF, Hakonarson H, Bucan M. 2007. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 17: 1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38: e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen RK, Thiruvahindrapuram B, Merico D, Walker S, Tammimies K, Hoang N, Chrysler C, Nalpathamkalam T, Pellecchia G, Liu Y, et al. 2015. Whole-genome sequencing of quartet families with autism spectrum disorder. Nat Med 21: 185–191. [DOI] [PubMed] [Google Scholar]
- Zhao X, Leotta A, Kustanovich V, Lajonchere C, Geschwind DH, Law K, Law P, Qiu S, Lord C, Sebat J, et al. 2007. A unified genetic theory for sporadic and inherited autism. Proc Natl Acad Sci 104: 12831–12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Parada LF. 2012. PTEN signaling in autism spectrum disorders. Curr Opin Neurobiol 22: 873–879. [DOI] [PubMed] [Google Scholar]
- Zhu M, Need AC, Han Y, Ge D, Maia JM, Zhu Q, Heinzen EL, Cirulli ET, Pelak K, He M, et al. 2012. Using ERDS to infer copy-number variants in high-coverage genomes. Am J Hum Genet 91: 408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.