

Abstract
Objective
Chronic kidney disease (CKD) amplifies atherosclerosis, which involves renin-angiotensin system (RAS) regulation of macrophages. RAS influences peroxisome proliferator-activated receptor-γ (PPARγ), a modulator of atherogenic functions of macrophages, however, little is known about its effects in CKD. We examined the impact of combined therapy with a PPARγ agonist and angiotensin receptor blocker on atherogenesis in a murine uninephrectomy model.
Methods
Apolipoprotein E knockout mice underwent uninephrectomy (UNx) and treatment with pioglitazone (UNx + Pio), losartan (UNx + Los), or both (UNx + Pio/Los) for 10 weeks. Extent and characteristics of atherosclerotic lesions and macrophage phenotypes were assessed; RAW264.7 and primary peritoneal mouse cells were used to examine pioglitazone and losartan effects on macrophage phenotype and inflammatory response.
Results
UNx significantly increased atherosclerosis. Pioglitazone and losartan each significantly reduced the atherosclerotic burden by 29.6% and 33.5%, respectively; although the benefit was dramatically augmented by combination treatment which lessened atherosclerosis by 55.7%. Assessment of plaques revealed significantly greater macrophage area in UNx + Pio/Los (80.7 ± 11.4% vs. 50.3 ± 4.2% in UNx + Pio and 57.2 ± 6.5% in UNx + Los) with more apoptotic cells. The expanded macrophage-rich lesions of UNx + Pio/Los had more alternatively activated, Ym-1 and arginine 1-positive M2 phenotypes (Ym-1: 33.6 ± 8.2%, p < 0.05 vs. 12.0 ± 1.1% in UNx; arginase 1: 27.8 ± 0.9%, p < 0.05 vs. 11.8 ± 1.3% in UNx). In vitro, pioglitazone alone and together with losartan was more effective than losartan alone in dampening lipopolysaccharide-induced cytokine production, suppressing M1 phenotypic change while enhancing M2 phenotypic change.
Conclusion
Combination of pioglitazone and losartan is more effective in reducing renal injury-induced atherosclerosis than either treatment alone. This benefit reflects mitigation in macrophage cytokine production, enhanced apoptosis, and a shift toward an anti-inflammatory phenotype.
Keywords: Pioglitazone, Losartan, PPARγ, Chronic kidney disease, Atherosclerosis, Macrophage phenotype
1. Introduction
Chronic kidney disease (CKD) increases the risk of cardiovascular disease (CVD) [1–3]. Individuals with early CKD are more likely to develop fatal CVD than to progress to end-stage kidney disease [4]. These observations are significant because CKD now affects some 10–16% of the population world-wide, a figure that is projected to rise [5,6]. Cardiovascular mortality in CKD is due to many causes; however, compared with the general population, atherosclerotic coronary artery disease is persistently overrepresented at every stage of CKD [7]. Further, although lipid-lowering treatment with HMG-CoA reductase inhibitors (statins) benefit patients with mild to moderate CKD, a large (50–80%) residual cardiovascular risk remains [8–10]. This residual risk reaches >90% in individuals with advanced CKD, and statins do not reduce CAD risk [8–10]. These considerations underscore the urgent need for additional therapeutic options to reduce CVD in the CKD population.
Previously, we and others have shown that dysregulation of the renin-angiotensin system (RAS) contributes to the acceleration of atherosclerosis caused by renal injury, and that the angiotensin II receptor blocker (ARB), losartan, reduces this effect [11]. Using bone marrow transplantation from angiotensin II receptor 1a (AT1a) deficient animals, we also showed that deletion of macrophage AT1a lessens uninephrectomy (UNx)-induced acceleration of atherosclerosis and alters macrophage phenotype distribution patterns in the lesions [12].
Peroxisome proliferator-activated receptor-γ (PPARγ) is a member of the ligand-activated nuclear receptor family with critical functions in energy balance and prominent atheroprotective effects [13,14]. Interestingly, PPARγ has been shown to have a reciprocal relationship with RAS [15,16]. Pioglitazone, a synthetic selective PPARγ agonist used in the treatment of type 2 diabetes mellitus is reported to reduce CVD [17–20]. The beneficial effects have been linked to improvement in glucose, lipid and insulin homeostasis, anti-inflammatory effects [17,18,20] and phenotype modulation of vascular macrophages [21]. Because CKD-accelerated atherosclerosis includes dramatic changes in plaque composition and inflammatory macrophage phenotype [12], the current study examined if the PPARγ agonist, pioglitazone, can modulate renal injury-aggravated atherosclerosis and mediate a synergistic anti-atherogenic interaction with the angiotensin II receptor blocker, losartan.
2. Materials and methods
2.1. Experimental groups
All experiments were done using female apolipoprotein E knockout (apoE−/−) mice on a C57BL/6 background (Jackson Laboratories, Bar Harbor, ME) maintained on normal mouse chow (RP5015; PMI Feeds, St. Louis, MO) containing 18.9% protein, 11.0% fat, 0.8% calcium, 0.5% phosphate, and 3.3 IU/gm vitamin D3. UNx (n = 40) or sham operation (Sham, n = 10) were performed at 8 weeks of age [11,22]. UNx mice were further divided into four groups: no treatment, pioglitazone [Takeda Chemical Industries, Osaka, Japan, 0.016% (w/w) in food, n = 10; UNx + Pio], losartan (Sigma–Aldrich, St. Louis, MO, 100 mg/L in drinking water, n = 10; UNx + Los), and pioglitazone together with losartan (n = 10; UNx + Pio/Los) until sacrifice at 20 weeks of age. Care and experimental procedures were in accordance with National Institutes of Health and Vanderbilt University Institutional Animal Care and Usage guidelines.
2.2. Systemic parameters
Blood pressure (BP) was measured biweekly in conscious trained mice using the Bp-2000 blood pressure analysis system (Visitech Systems Inc, Apex, NC) automated tail cuff as described [23]. The animals were acclimated to the procedure, and the mean values were based on an average of ten stable readings. Body weight was assessed biweekly. Serum level of creatinine was measured by HPLC [22]. Serum total cholesterol and triglycerides levels were determined at the end of the experiment [24].
2.3. Assessment of atherosclerotic lesions
Mice were sacrificed under phenobarbital anesthesia and perfused with PBS through the left ventricle. The heart, together with proximal aorta, was embedded in OCT and snap-frozen in liquid nitrogen. Ten μm thick cryosections were cut from the proximal aorta beginning at the end of the aortic sinus and stained with Oil-Red-O to assess lipid deposition [24]. Quantitative analysis of lesions was performed using Imaging System KS300 (Release 2.0; Kontron Elektronik GmbH, Poway, CA). To assess necrotic areas in atherosclerotic lesions, cryosections were stained with Harris H&E (Sigma–Aldrich) as previously described [22,25,26]. Cellular-stained and acellular areas in intimal lesions were quantified as total atherosclerotic lesion area. The necrotic core was defined as acellular areas that were also clear as evidenced by complete lack of staining with H&E in atherosclerotic lesion. As reported by others [22,25,26], we did not include very small clear areas as these are unlikely to represent substantial areas of necrosis. The quantification of those areas was obtained by image analysis software, Imaging System KS300 (Release 2.0; Kontron Elektronik GmbH, Poway, CA). Percentage of necrotic core areas in the lesions was calculated from the ratio of necrotic core to total atherosclerotic lesion area. To assess collagen, 5-μm thick sections of proximal aortas were stained with aniline blue, and the collagen-positive area was expressed as a ratio of aniline-to-total atherosclerotic area [27]. Calcium deposition was assessed on sections stained by von Kossa as previously described [28]. Immunohistochemistry for MCP-1 was assessed (1:200, Novus, Littleton, CO) on 5-μm cryosections. Images were acquired by Nikon ECLIPSE E400 system. The percentage of positive area was analyzed by AxioVision software (Carl Zeiss, Germany). Apoptosis of macrophages was detected by TUNEL method (Roche). All images of atherosclerotic area were acquired at 40× magnification, and fraction of CD68-positive cells assessed.
2.4. Assessment of macrophage content and phenotype in atherosclerotic lesions
Serial, five μm thick cryosections of proximal aorta were fixed in acetone and incubated with monoclonal rat antibody to mouse macrophages (MOMA-2, Serotec, Raleigh, NC) to measure macrophage-positive area within atherosclerotic lesions [11,22]. Rat anti-mouse CD68 (AbD Serotec, UK) and nuclear DAPI were used to stain macrophages. Rabbit anti-mouse CCR7 and iNOS (BD bioscience, San Jose, CA) were used to stain for M1 macrophage phenotype [29–32], while rabbit anti-mouse Ym-1 (Stemcell Technologies, Canada) or arginase1 (BD bioscience) were used to stain for M2 macrophage phenotype [33,34]. M1 phenotype staining was assessed on images acquired by Nikon ECLIPSE E400 system and M2 phenotype staining was assessed on images acquired by co-focal microscopy (Zeiss LSM 510 META Inverted Confocal microscope). The percentage of M1 or M2 subtypes was determined as the ratio of positive cells for each phenotype marker to total CD68 positive cells [12].
2.5. Macrophage inflammatory reaction in vitro
RAW264.7 cells (American Type Culture Collection, Manassas, VA) or C57BL6 mouse peritoneal macrophages harvested by peritoneal lavage 3 days after peritoneal injection of 3% thioglycollate were plated at 20×106/35-mm well in RPMI1640 containing 10% FBS. After overnight incubation, macrophages were exposed to lipopolysaccharide (LPS) (50 ng/ml, Sigma–Aldrich, MO), pioglitazone (10 μM, Sigma–Aldrich) and/or losartan (10 μM) for 24 h. Total RNA was extracted by Genelute mammalian total RNA miniprep Kit (Sigma–Aldrich). Quantitative real-time PCR was performed using One Step SYBR Plus RT PCR Kit on a Thermal Cycler Dice Real-time System (TP900, Takara, Shiga, Japan). Primers for mouse iNOS (MA102694, Takara), CCR7 (MA078789, Takara), TNF-α (Fw: CATGAGCACAGAAAGCATGATCCG, Rv: AAGCAGGAATGAGAAGAGGCTGAG) [35], MCP-1 (Fw: CTTCTGGGCCTGCTGTTCA, Rv: CCAGCCTACTCATTGGGATCA) [36], arginase 1 (MA116062, Takara) and GAPDH (MA050371, Takara) were used to quantitate mRNA expression. GAPDH was used as an internal control.
2.6. Statistical analysis
Results are expressed as means ± SEM. Differences were determined by one-way ANOVA (Bonferroni posttest), and p < 0.05 was considered to be significant.
3. Results
3.1. Systemic parameters
Table 1 shows the systemic parameters. There were no differences in body weight or blood glucose among the groups. In agreement with previous reports [11,37], UNx caused a modest but significant increase in serum creatinine and this was not modified by pioglitazone or losartan. BP decreased in mice treated with losartan alone and in combination with pioglitazone. Pioglitazone treatment alone did not affect BP, however total cholesterol and triglycerides levels increased both in mice treated with pioglitazone alone and in combination with losartan.
Table 1.
Systemic parameters.
| Sham | UNx | UNx + Pio | UNx + Los | UNx + Pio/Los | |
|---|---|---|---|---|---|
| Body weight (g) | 24.1 ± 0.4 | 22.7 ± 0.6 | 23.3 ± 0.6 | 23.5 ± 0.4 | 22.4 ± 0.5 |
| Systolic blood pressure (mmHg) | 102 ± 3 | 108 ± 4 | 100 ± 2 | 88 ± 3* | 85 ± 2* |
| Blood glucose (mg/dl) | 81.9 ± 3.9 | 82.9 ± 7.2 | 94.8 ± 4.7 | 93.6 ± 2.4 | 83.8 ± 5.0 |
| Serum cholesterol (mg/dl) | 348 ± 24 | 338 ± 24 | 496 ± 24* | 330 ± 18 | 495 ± 29* |
| Serum triglycerides (mg/dl) | 69 ± 12 | 89 ± 12 | 230 ± 10* | 139 ± 9 | 240 ± 16* |
| Serum creatinine (mg/dl) | 0.098 ± 0.006 | 0.131 ± 0.008† | 0.132 ± 0.007† | 0.134 ± 0.006† | 0.133 ± 0.004† |
Sham, sham-operated apolipoprotein E knockout mice; UNx, uninephrectomised apolipoprotein E knockout mice; UNx + Pio, UNx treated with pioglitazone; UNx + Los, UNx mice treated with losartan; UNx + Pio/Los, UNx treated with both pioglitazone and losartan;
p < 0.05 versus UNx,
p < 0.05 versus Sham.
3.2. Atherosclerotic lesions and necrotic area
UNx significantly increased atherosclerotic lesion area as assessed by Oil-Red-O staining of aortic cross-sections by 67.7% compared to sham (331,385 ± 25,020 μm2 in UNx vs. 197,670 ± 19,131 μm2 in Sham, p < 0.05). These results are in agreement with previous findings in this model [11,37] (Fig. 1). Pioglitazone and losartan each significantly reduced UNx-dependent atherosclerosis by 29.6% and 33.5%, respectively (233,408 ± 17,116 μm2 in UNx + Pio and 220,335 ± 24,382 μm2 in UNx + Los, both p < 0.05 vs. UNx). The combination of pioglitazone and losartan provided greater benefit than pioglitazone or losartan treatment alone with a 55.7% reduction (146,979 ± 17046 μm2, p < 0.05 vs. UNx + Pio or UNx + Los).
Fig. 1.
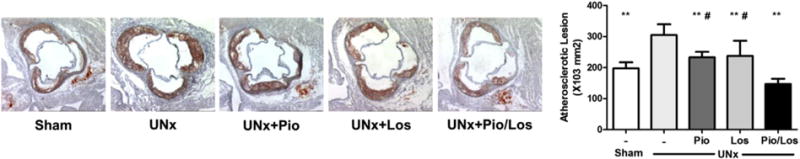
Synergistic effects of pioglitazone and losartan, on renal injury-induced acceleration in atherosclerosis. Apolipoprotein E knockout mice underwent sham operation (Sham, n = 10) or uninephrectomy (UNx, n = 40) and were divided into four groups: no treatment (UNx, n = 10), pioglitazone (UNx + Pio, n = 10), losartan (UNx + Los, n = 10), and pioglitazone together with losartan (UNx + Pio/Los, n = 10). Atherosclerosis was assessed on cryosections from the proximal aorta and stained with Oil-Red-O. **p < 0.05 vs. UNx, and #p < 0.05 vs. UNx + Pio/Los.
Compared with sham-operated mice with intact kidneys, the necrotic lesions of UNx mice had more than a 3-fold increase in area (9.15 ± 1.38% in UNx vs. 2.69 ± 0.35% in Sham, p < 0.05). Compared to untreated UNx, all treatment regimens decreased the necrotic area, with the Pio/Los combination causing the greatest reduction. (4.67 ± 1.00% in UNx + Pio, 5.03 ± 0.97% in UNx + Los, and 2.98 ± 0.89% in UNx + Pio/Los, p < 0.05 vs. UNx, Fig. 2A). Aortic expression of MCP-1, assessed by immunostaining, was decreased by all treatment regimens (7.31 ± 0.72% in UNx + Pio, 6.65 ± 1.71% in UNx + Los, and 6.64 ± 1.04% in UNx + Pio/Los, p < 0.05 vs. 12.76 ± 0.18% UNx, Supplemental Fig. 1A). Collagen deposition, assessed by aniline blue staining and compared to total atherosclerotic area of individual animals was not significantly affected (21.6 ± 3.8% in Sham, 23.1 ± 1.4% in UNx, 18.0 ± 1.7% in UNx + Pio, 21.1 ± 1.9% in UNx + Los, 18.6 ± 0.8% in UNx + Pio/Los, NS, Supplemental Fig. 1B). Also calcium deposition, assessed by von Kossa staining, showed sporadic staining of the aortic valve and only occasional deposition in the vascular intima, however, the extent was very slight and not significantly affected by treatment with pioglitazone, losartan, or the combination (Supplemental Fig. 1C).
Fig. 2.
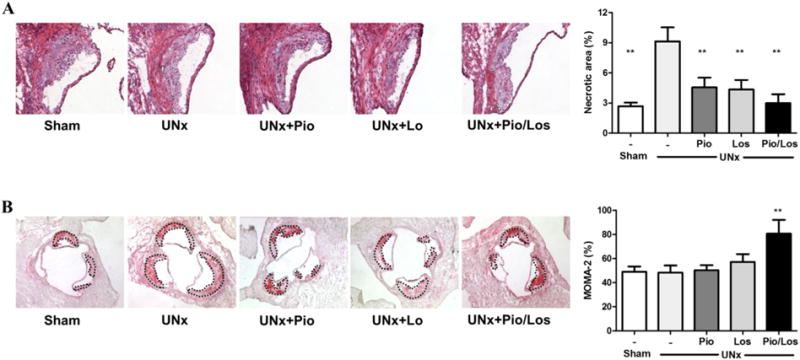
Pioglitazone and losartan combination lessens necrotic areas in proximal atherosclerotic lesions and increases macrophage content. Apolipoprotein E knockout mice underwent sham operation (Sham, n = 10) or uninephrectomy (UNx, n = 40) and were divided into four groups: no treatment (UNx, n = 10), pioglitazone (UNx + Pio, n = 10), losartan (UNx + Los, n = 10), and pioglitazone together with losartan (UNx + Pio/Los, n = 10). Necrotic area assessed by acellular area in Harris H&E sections (A) and macrophages content assessed by MOMA-2 staining (B), calculated as ratio with atherosclerotic lesion, respectively. **p < 0.05 vs. UNx.
3.3. Macrophage phenotype in atherosclerotic lesions
Macrophage-positive area assessed by MOMA-2 immunostaining was dramatically increased in UNx + Pio/Los mice compared to the other groups (80.7 ± 11.4% in UNx + Pio/Los vs. 40.9 ± 4.5% in Sham, 48.3 ± 5.9% in UNx, 50.3 ± 4.2% in UNx + Pio, 57.2 ± 6.5% in UNx + Los, p < 0.05 for each comparison, Fig. 2B). The macrophage phenotype within the atherosclerotic lesions was also affected by treatment. UNx significantly increased the subtype of macrophages expressing markers of the M1 phenotype, including CCR7 (75.2 ± 4.8% vs. 48.5 ± 5.5% in Sham, p < 0.05) and iNOS (61.9 ± 4.8% vs. 36.1 ± 4.1% in Sham, p < 0.05) (Fig. 3A and B). The lesions of UNx mice also had fewer cells with markers of the M2 phenotype, including Ym-1 (12.0 ± 1.1% vs. 31.6 ± 1.9% in Sham, p < 0.05) and arginase 1 (11.8 ± 1.3% vs. 26.9 ± 1.1% in Sham, p < 0.05) (Fig. 3C and D). In contrast, pioglitazone and losartan treatment reduced M1 phenotype prevalence (CCR7: 40.3 ± 4.3% in UNx + Pio and 29.1 ± 6.0% in UNx + Los, p < 0.05 vs. UNx; iNOS: 29.5 ± 2.5% in UNx + Pio and 38.6 ± 3.3% in UNx + Los, p < 0.05 vs. UNx). Although each intervention numerically increased the M2 phenotype (Ym-1: 26.0 ± 1.6% in UNx + Pio and 25.4 ± 3.6% in UNx + Los, NS vs. 12.0 ± 1.1% in UNx; arginase 1: 21.3 ± 1.8% in UNx + Pio and 13.6 ± 5.7% in UNx + Los, NS vs. 11.8 ± 1.3% in UNx) (Fig.3A–D), a significant increase in abundance of the M2 was seen in lesions of animals treated by the combination of pioglitazone and losartan (Ym-1: 33.6 ± 8.2%, p < 0.05 vs. UNx; arginase 1: 27.8 ± 0.9%, p < 0.05 vs. UNx) (Fig. 3C and D). The percent apoptotic macrophages, assessed by TUNEL staining, was significantly increased in UNx + Pio/Los mice compared to untreated UNx, UNx + Los, and UNx + Pio (23.50 ± 1.32% vs 3.82 ± 1.63%, 9.62 ± 0.92%, 9.91 ± 1.89%, Fig. 4)
Fig. 3.
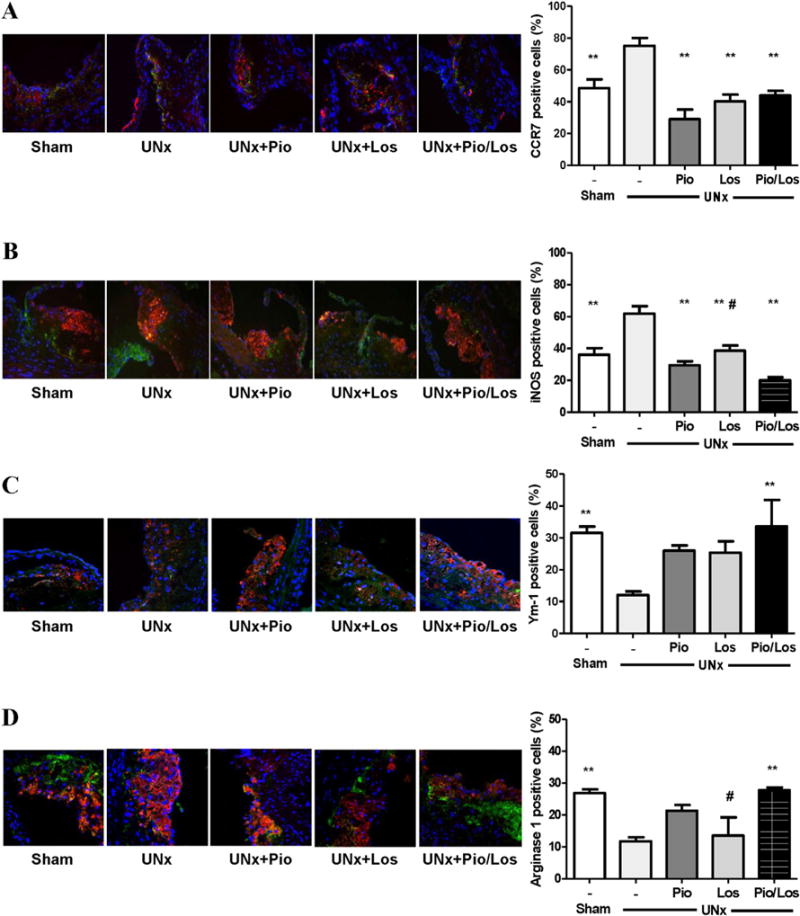
Pioglitazone and losartan modulate renal damage-induced macrophage phenotype. Immunofluorescent staining for CCR7 (A), iNOS (B), Ym-1 (C) and arginase 1 (D) assessed as fractions of total macrophages stained with CD68 in atherosclerotic lesions of mice with sham-operation or uninephrectomy with no treatment (UNx), pioglitazone (UNx + Pio), losartan (UNx + Los), and pioglitazone together with losartan (UNx + Pio/Los). **p < 0.05 vs. UNx, and #p < 0.05 vs. UNx + Pio/Los.
Fig. 4.
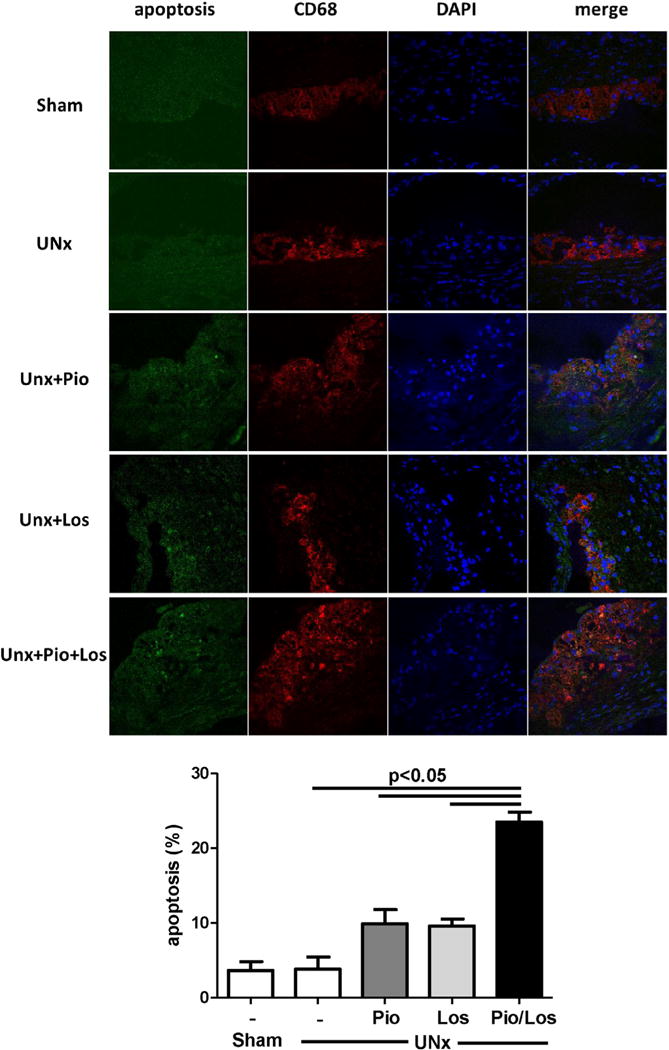
Combination treatment with pioglitazone and losartan increased apoptotic macrophages in proximal atherosclerotic lesions. Apoptoric macrophage in the atherosclerotic lesion assessed by staining with TUNEL, CD68 and DAPI in atherosclerotic lesions of mice with sham-operation or uninephrectomy with no treatment (UNx), pioglitazone (UNx + Pio), losartan (UNx + Los), and pioglitazone together with losartan (UNx + Pio/Los).
3.4. Macrophage inflammation and phenotype modulation in vitro
Pioglitazone alone and together with losartan modulated the LPS-induced response of iNOS, CCR7, TNF-α and MCP-1 expression in RAW264.7 macrophages (Fig. 5A–D) and thioglycollate-elicited peritoneal macrophages from C57BL/6 mice (Supplemental Fig. 2A–D). Losartan alone had a smaller effect on cytokine stimulation, iNOS production (Fig. 5A and Supplemental Fig. 2A), and expression of other inflammatory cytokines in both cell types (Fig. 5B–D and Supplemental Fig. 2B–D). By contrast, pioglitazone, alone or with losartan, increased macrophage arginase1 mRNA expression in both cell types (Fig. 5E and Supplemental Fig. 2E).
Fig. 5.
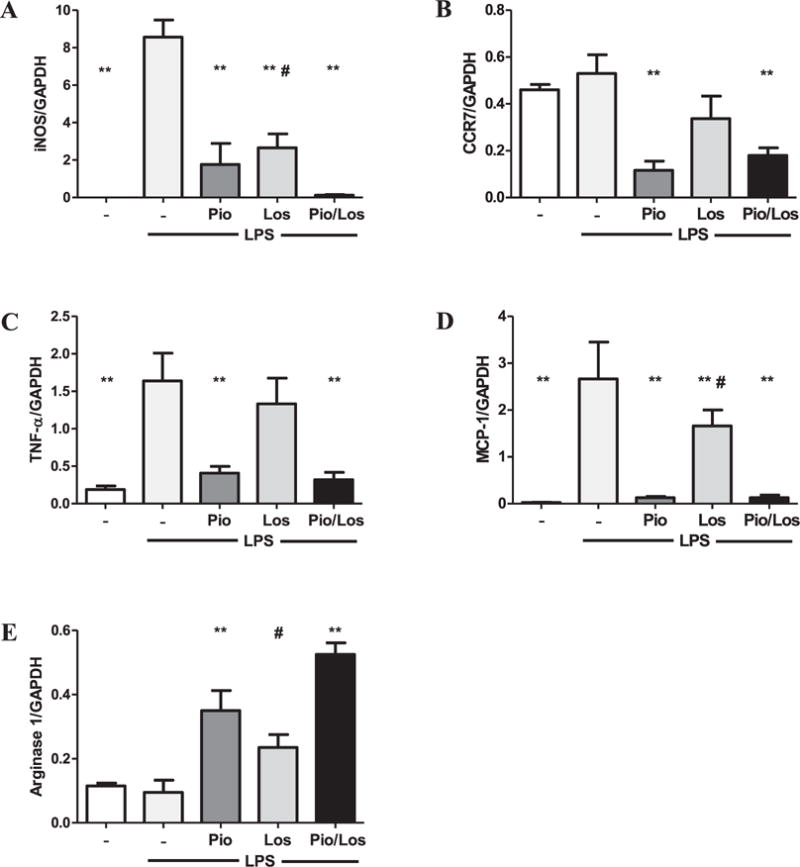
Pioglitazone and losartan modulate LPS-induced macrophage M1 phenotypic change and inflammatory reaction. RAW264.7 macrophages were reacted with LPS (50 ng/ml, n = 4), LPS and pioglitazone (10 μM) (n = 4), LPS and losartan (10 μM) (n = 4), and LPS and both pioglitazone and losartan (n = 4) for 24 h. Quantitative real-time PCR for iNOS (A), CCR7 (B), TNF-α (C), MCP-1 (D), and arginase-1 (E) was performed. GAPDH was used as an internal control. **p < 0.05 vs. UNx, and #p < 0.05 vs. UNx + Pio/Los.
4. Discussion
We report the novel observation that the PPARγ agonist pioglitazone combined with the ARB losartan inhibit renal injury-induced acceleration of atherosclerosis. The underlying mechanisms include changes in plaque morphology with enhanced apoptosis and fewer pro-inflammatory M1 versus more anti-inflammatory M2 macrophages in the atherosclerotic lesion.
Therapeutic strategies for CKD-induced atherosclerosis targeting traditional risk factors such as dyslipidemia and hypertension are inadequate [38]. The current study indicates that pioglitazone inhibits renal injury-induced acceleration of atherosclerosis independent of blood pressure and glucose levels. These results complement experimental findings that pioglitazone inhibits acceleration of atherosclerosis in low-density lipoprotein receptor knockout mice [39,40], and clinical reports that pioglitazone reduces cardiovascular events in patients with type 2 diabetes and lessens carotid intima media thickness in patients with impaired glucose tolerance [18,19]. Although those findings suggest therapeutic effects of pioglitazone on atherosclerosis, the current study is the first to show significant inhibition of atherosclerosis by pioglitazone in a CKD setting.
The beneficial effect occurred even in the face of higher plasma cholesterol levels, which rather associate with exacerbation of atherosclerosis. A divergence between systemic lipid levels and vascular pathology is similar to our results in mice treated with losartan, which lessened atherosclerosis but did not affect the plasma lipid profile. Together these findings reiterate the concept that unique, non-traditional risks and local vascular mechanisms drive atherosclerotic disease in the CKD population. While uninephrectomy increased the extent of atherosclerosis, there was little change in abundance of collagen or calcium deposition which were not affected by the therapeutic interventions (Supplemental Fig. 1). The sporadic calcium deposition echoes our previous observations in this model but differs from vascular calcification observed in other studies and may reflect differences in the method of renal ablation (step-wise removal of renal parenchyma that involved uninephrectomy followed by removal of two-thirds of the remaining kidney at a later point in the previous studies) and/or a difference in diet composition (lower levels of vitamin D in the previous studies) that can affect calcium deposition [41,42]. Further, although we did not observe a change in the collagen content in the current study, it is possible that as the atherosclerotic process progresses, therapeutic interventions will impact the plaque composition. Indeed, we have previously shown that longer treatment with losartan (until 36 weeks as compared to 20 weeks in the current study) increases collagen content in aortic lesions of animals with intact kidneys [27], raising the possibility that longer treatment may be beneficial in the renal injury setting. The current study suggests that at this stage of atherogenesis, macrophage infiltration and changes in their phenotype are the major drivers of atherosclerosis and that losartan, pioglitazone but especially the combination treatment modulates the process. In this regard, our previous studies indicate that UNx potentiates atherogenic processes such as macrophage foam cell formation by decreasing cholesterol efflux via repression of cellular ABCA1 transporter expression [43]. Notably, incubation of human macrophages with the ARB telmisartan caused an upregulation of ABCA1 and increase in cholesterol efflux via a PPARγ-dependent pathway [40]. Moreover, macrophage-specific knockout of PPARγ increased atherosclerosis and inflammation in low-density lipoprotein receptor knockout mice [21]. Rőszer T et al. reported that peritoneal macrophages of PPARγ knockout mice have impaired apoptotic cell uptake, i.e., efferocytosis, a function ascribed to the M2 macrophages [44]. On the other hand, pioglitazone was found to increase macrophage cholesterol efflux by upregulation of ATP cassette binding transporters [45,46]. While there is abundant evidence that PPARγ can affect atherosclerosis through its modulation of macrophage function, there is very little information on the accelerated atherosclerosis in the setting of kidney disease. In the current study, we show that one of the beneficial effects of pioglitazone on UNx-induced acceleration of atherosclerosis occurs through modulation of macrophage phenotype. Pioglitazone decreased iNOS and CCR7-positive M1 macrophages in atherosclerotic lesion which was accompanied by reduced necrotic area and MCP-1 expression in aorta (Fig. 2A and Supplemental Fig. 1A). These data complement our previous study showing that decrease of M1 macrophages is associated with a decrease in necrotic area in the atherosclerotic lesion [12]. These findings suggest activation of PPARγ in macrophages may be an attractive strategy for prevention or treatment of CKD-induced atherosclerosis.
Losartan also inhibited UNx-induced acceleration of atherosclerosis and, similar to pioglitazone, modulated macrophage phenotype within the lesions (Fig. 3). These findings echo our previous study showing that macrophage AT1a knockout modulates UNx-induced acceleration of atherosclerosis increasing the ratio of M1–M2 phenotype [12]. In addition to the direct impact on macrophages in the lesion, losartan may slow atherosclerosis by reducing systemic blood pressure. Also, previous reports show it can modulate other macrophage functions, including chemotaxis, oxidative stress, and cellular lipid handling that can bring about atheroprotective effects [11,43].
The current study reveals that the losartan driven reduction of UNx-induced atherosclerosis is potentiated by pioglitazone (Fig. 1). The synergistic effect is due, at least in part, to a shift in macrophage phenotype with fewer M1 and more M2 macrophages, an effect that is more pronounced than with either pioglitazone or losartan alone (Fig. 3). Thus, monotherapy by RAS inhibition with losartan, PPARγ agonism with pioglitazone, as well as the combination similarly reduces macrophages bearing the M1 markers in lesions compared to atherosclerosis in untreated mice, e.g., reduction in macrophages bearing M1 markers, CCR7 and iNOS as well as aortic expression of MCP-1. Critically, compared to untreated UNx, only the combination treatment and not losartan alone or pioglitazone alone were effective in increasing the abundance of macrophages with M2 markers, arginase-1 and Ym-1. These results underscore that the combination treatment is linked to increased anti-inflammatory M2 macrophages and also raises potential contributions of other, non-M1/M2 mechanisms modulating atherosclerosis. Indeed, although all interventions increase plaque apoptosis, the combination therapy increase was statistically greater than UNx, or UNx + losartan or UNx + pioglitazone. The observations are in agreement with the recognition that apoptosis is an integral component in vascular remodeling that accompanied resorption of atherosclerotic plaque. Arai et al. showed that knockout of AIM (apoptosis inhibitor expressed by macrophage, also called Spα or Api6) increase apoptosis and lessen atherosclerosis in low density lipoprotein receptor knockout (LDLR−/−) mice while knockout of Bax and p53 decreased apoptosis that was accompanied by increased atherosclerosis in LDLR−/− and apoE−/− mice, respectively [47–49]. Further, our previous studies in LDLR−/− mice reconstituted with fetal liver cells deficient in prostaglandin 4 showed suppressed development of atherosclerosis with increased apoptosis through mechanisms involving inhibition in the PI3K/Akt and NF-kappaB pathways [50].
A recent report indicates that angiotensin II decreases PPARγ expression via secretion of transforming growth factor-β1 and phosphorylation of p38 mitogen-activated protein kinase and his-tone deacetylase 3 in aortic smooth muscle cells [15]. Taken together, these results suggest that combining PPARγ activation with inhibition of the angiotensin II receptor induces a synergic effect that modulates macrophage function in atherosclerotic lesions in the CKD setting. In this current study, both pioglitazone and losartan treatments reduced the extent of necrosis within the atherosclerotic lesions, while the combination treatment had the largest effect. Indeed, the necrotic area in mice on combination treatment was not different than shams having intact kidneys (Fig. 2A). It is possible that this beneficial effect of combination treatment on plaque necrosis becomes more important in more advanced stages of atherosclerosis.
In summary, combining the PPARγ agonist, pioglitazone, together with the ARB, losartan, reduces renal injury-induced amplification of atherosclerosis by increasing apoptosis and the ratio of M2–M1 phenotype macrophages, which leads to a reduction of necrotic plaque.
Supplementary Material
Acknowledgments
The authors acknowledge the expert technical assistance of Cathy Xu and Youmin Zhang.
Funding sources
This work was supported in part by grants NIH HL087061, DK44757, HL65709 and HL57986 and the Lipid, Lipoprotein and Atherosclerosis Core of the Vanderbilt Mouse Metabolic Phenotyping Center (NIH DK59637-01).
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.atherosclerosis.2015.06.055.
Footnotes
Disclosures
None.
References
- 1.Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med. 2004;351:1296–1305. doi: 10.1056/NEJMoa041031. [DOI] [PubMed] [Google Scholar]
- 2.Klausen KP, Scharling H, Jensen JS. Very low level of microalbuminuria is associated with increased risk of death in subjects with cardiovascular or cerebrovascular diseases. J Intern Med. 2006;260:231–237. doi: 10.1111/j.1365-2796.2006.01679.x. [DOI] [PubMed] [Google Scholar]
- 3.Schmieder RE, Mann JF, Schumacher H, Gao P, Mancia G, Weber MA, McQueen M, Koon T, Yusuf S. Changes in albuminuria predict mortality and morbidity in patients with vascular disease. J Am Soc Nephrol. 2011;22:1353–1364. doi: 10.1681/ASN.2010091001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keith DS, Nichols GA, Gullion CM, Brown JB, Smith DH. Longitudinal follow-up and outcomes among a population with chronic kidney disease in a large managed care organization. Arch Intern Med. 2004;164:659–663. doi: 10.1001/archinte.164.6.659. [DOI] [PubMed] [Google Scholar]
- 5.Levey AS, de Jong PE, Coresh J, Nahas MEl, Astor BC, Matsushita K, Gansevoort RT, Kasiske BL, Eckardt KU. The definition, classification, and prognosis of chronic kidney disease: a KDIGO controversies conference report. Kidney Int. 2011;80:17–28. doi: 10.1038/ki.2010.483. [DOI] [PubMed] [Google Scholar]
- 6.Mahmoodi BK, Fox CS, Astor BC, Nelson RG, Matsushita K, Coresh J. Association of chronic kidney disease with adverse outcomes – authors’ reply. Lancet. 2013;381:532–533. doi: 10.1016/S0140-6736(13)60273-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu H, Yan L, Ma GS, Zhang LP, Gao M, Wang YL, Wang SP, Liu BC. Association of chronic kidney disease and coronary artery disease in 1,010 consecutive patients undergoing coronary angiography. J Nephrol. 2012;25:219–224. doi: 10.5301/JN.2011.8478. [DOI] [PubMed] [Google Scholar]
- 8.Fellstrom BC, Jardine AG, Schmieder RE, Holdaas H, Bannister K, Beutler J, Chae DW, Chevaile A, Cobbe SM, Gronhagen-Riska C, De Lima JJ, Lins R, Mayer G, McMahon AW, Parving HH, Remuzzi G, Samuelsson O, Sonkodi S, Sci D, Suleymanlar G, Tsakiris D, Tesar V, Todorov V, Wiecek A, Wuthrich RP, Gottlow M, Johnsson E, Zannad F. Rosuvastatin and cardiovascular events in patients undergoing hemodialysis. N Engl J Med. 2009;360:1395–1407. doi: 10.1056/NEJMoa0810177. [DOI] [PubMed] [Google Scholar]
- 9.Wanner C, Krane V, Marz W, Olschewski M, Mann JF, Ruf G, Ritz E. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med. 2005;353:238–248. doi: 10.1056/NEJMoa043545. [DOI] [PubMed] [Google Scholar]
- 10.Baigent C, Landray MJ, Reith C, Emberson J, Wheeler DC, Tomson C, Wanner C, Krane V, Cass A, Craig J, Neal B, Jiang L, Hooi LS, Levin A, Agodoa L, Gaziano M, Kasiske B, Walker R, Massy ZA, Feldt-Rasmussen B, Krairittichai U, Ophascharoensuk V, Fellstrom B, Holdaas H, Tesar V, Wiecek A, Grobbee D, de Zeeuw D, Gronhagen-Riska C, Dasgupta T, Lewis D, Herrington W, Mafham M, Majoni W, Wallendszus K, Grimm R, Pedersen T, Tobert J, Armitage J, Baxter A, Bray C, Chen Y, Chen Z, Hill M, Knott C, Parish S, Simpson D, Sleight P, Young A, Collins R. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (study of heart and renal protection): a randomised placebo-controlled trial. Lancet. 2011;377:2181–2192. doi: 10.1016/S0140-6736(11)60739-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Suganuma E, Zuo Y, Ayabe N, Ma J, Babaev VR, Linton MF, Fazio S, Ichikawa I, Fogo AB, Kon V. Antiatherogenic effects of angiotensin receptor antagonism in mild renal dysfunction. J Am Soc Nephrol. 2006;17:433–441. doi: 10.1681/ASN.2005080883. [DOI] [PubMed] [Google Scholar]
- 12.Yamamoto S, Yancey PG, Zuo Y, Ma LJ, Kaseda R, Fogo AB, Ichikawa I, Linton MF, Fazio S, Kon V. Macrophage polarization by angiotensin II-type 1 receptor aggravates renal injury-acceleration of atherosclerosis. Arterioscler Thromb Vasc Biol. 2011;31:2856–2864. doi: 10.1161/ATVBAHA.111.237198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DeFronzo RA, Tripathy D, Schwenke DC, Banerji M, Bray GA, Buchanan TA, Clement SC, Henry RR, Hodis HN, Kitabchi AE, Mack WJ, Mudaliar S, Ratner RE, Williams K, Stentz FB, Musi N, Reaven PD. Pioglitazone for diabetes prevention in impaired glucose tolerance. N Engl J Med. 2011;364:1104–1115. doi: 10.1056/NEJMoa1010949. [DOI] [PubMed] [Google Scholar]
- 14.Miyazaki Y, DeFronzo RA. Rosiglitazone and pioglitazone similarly improve insulin sensitivity and secretion, glucose tolerance and adipocytokines in type 2 diabetic patients. Diabetes Obes Metab. 2008;10:1204–1211. doi: 10.1111/j.1463-1326.2008.00880.x. [DOI] [PubMed] [Google Scholar]
- 15.Subramanian V, Golledge J, Heywood EB, Bruemmer D, Daugherty A. Regulation of peroxisome proliferator-activated receptor-gamma by angiotensin II via transforming growth factor-beta1-activated p38 mitogen-activated protein kinase in aortic smooth muscle cells. Arterioscler Thromb Vasc Biol. 2012;32:397–405. doi: 10.1161/ATVBAHA.111.239897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alexis JD, Wang N, Che W, Lerner-Marmarosh N, Sahni A, Korshunov VA, Zou Y, Ding B, Yan C, Berk BC, Abe J. Bcr kinase activation by angiotensin II inhibits peroxisome-proliferator-activated receptor gamma transcriptional activity in vascular smooth muscle cells. Circ Res. 2009;104:69–78. doi: 10.1161/CIRCRESAHA.108.188409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nicholls SJ, Tuzcu EM, Wolski K, Bayturan O, Lavoie A, Uno K, Kupfer S, Perez A, Nesto R, Nissen SE. Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: insights from the PERISCOPE (pioglitazone effect on regression of intravascular sonographic coronary obstruction prospective evaluation) study. J Am Coll Cardiol. 2011;57:153–159. doi: 10.1016/j.jacc.2010.06.055. [DOI] [PubMed] [Google Scholar]
- 18.Saremi A, Schwenke DC, Buchanan TA, Hodis HN, Mack WJ, Banerji M, Bray GA, Clement SC, Henry RR, Kitabchi AE, Mudaliar S, Ratner RE, Stentz FB, Musi N, Tripathy D, DeFronzo RA, Reaven PD. Pioglitazone slows progression of atherosclerosis in prediabetes independent of changes in cardiovascular risk factors. Arterioscler Thromb Vasc Biol. 2013;33:393–399. doi: 10.1161/ATVBAHA.112.300346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilcox R, Kupfer S, Erdmann E. Effects of pioglitazone on major adverse cardiovascular events in high-risk patients with type 2 diabetes: results from PROspective pioglitAzone clinical trial in macro vascular events (PROactive 10) Am Heart J. 2008;155:712–717. doi: 10.1016/j.ahj.2007.11.029. [DOI] [PubMed] [Google Scholar]
- 20.Davidson M, Meyer PM, Haffner S, Feinstein S, D’Agostino R, Sr, Kondos GT, Perez A, Chen Z, Mazzone T. Increased high-density lipoprotein cholesterol predicts the pioglitazone-mediated reduction of carotid intima-media thickness progression in patients with type 2 diabetes mellitus. Circulation. 2008;117:2123–2130. doi: 10.1161/CIRCULATIONAHA.107.746610. [DOI] [PubMed] [Google Scholar]
- 21.Babaev VR, Yancey PG, Ryzhov SV, Kon V, Breyer MD, Magnuson MA, Fazio S, Linton MF. Conditional knockout of macrophage PPARgamma increases atherosclerosis in C57BL/6 and low-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:1647–1653. doi: 10.1161/01.ATV.0000173413.31789.1a. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto S, Zuo Y, Ma J, Yancey PG, Hunley TE, Motojima M, Fogo AB, Linton MF, Fazio S, Ichikawa I, Kon V. Oral activated charcoal adsorbent (AST-120) ameliorates extent and instability of atherosclerosis accelerated by kidney disease in apolipoprotein E-deficient mice. Nephrol Dial Transpl. 2011;26:2491–2497. doi: 10.1093/ndt/gfq759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma J, Weisberg A, Griffin JP, Vaughan DE, Fogo AB, Brown NJ. Plasminogen activator inhibitor-1 deficiency protects against aldosterone-induced glomerular injury. Kidney Int. 2006;69:1064–1072. doi: 10.1038/sj.ki.5000201. [DOI] [PubMed] [Google Scholar]
- 24.Fazio S, Major AS, Swift LL, Gleaves LA, Accad M, Linton MF, Farese RV., Jr Increased atherosclerosis in LDL receptor-null mice lacking ACAT1 in macrophages. J Clin Investig. 2001;107:163–171. doi: 10.1172/JCI10310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Investig. 2009;119:886–898. doi: 10.1172/JCI37262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yancey PG, Blakemore J, Ding L, Fan D, Overton CD, Zhang Y, Linton MF, Fazio S. Macrophage LRP-1 controls plaque cellularity by regulating efferocytosis and Akt activation. Arterioscler Thromb Vasc Biol. 2010;30:787–795. doi: 10.1161/ATVBAHA.109.202051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Suganuma E, Babaev VR, Motojima M, Zuo Y, Ayabe N, Fogo AB, Ichikawa I, Linton MF, Fazio S, Kon V. Angiotensin inhibition decreases progression of advanced atherosclerosis and stabilizes established atherosclerotic plaques. J Am Soc Nephrol. 2007;18:2311–2319. doi: 10.1681/ASN.2006090967. [DOI] [PubMed] [Google Scholar]
- 28.Westenfeld R, Schafer C, Smeets R, Brandenburg VM, Floege J, Ketteler M, Jahnen-Dechent W. Fetuin-A (AHSG) prevents extraosseous calcification induced by uraemia and phosphate challenge in mice. Nephrol Dial Transpl. 2007;22:1537–1546. doi: 10.1093/ndt/gfm094. [DOI] [PubMed] [Google Scholar]
- 29.Mungrue IN, Gros R, You X, Pirani A, Azad A, Csont T, Schulz R, Butany J, Stewart DJ, Husain M. Cardiomyocyte overexpression of iNOS in mice results in peroxynitrite generation, heart block, and sudden death. J Clin Investig. 2002;109:735–743. doi: 10.1172/JCI13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie QW, Cho HJ, Calaycay J, Mumford RA, Swiderek KM, Lee TD, Ding A, Troso T, Nathan C. Cloning and characterization of inducible nitric oxide synthase from mouse macrophages. Science. 1992;256:225–228. doi: 10.1126/science.1373522. [DOI] [PubMed] [Google Scholar]
- 31.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 108:7166–7171. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Feig JE, Rong JX, Shamir R, Sanson M, Vengrenyuk Y, Liu J, Rayner K, Moore K, Garabedian M, Fisher EA. HDL promotes rapid atherosclerosis regression in mice and alters inflammatory properties of plaque monocyte-derived cells. Proc Natl Acad Sci U S A. 2011;108:7166–7171. doi: 10.1073/pnas.1016086108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Guo L, Johnson RS, Schuh JC. Biochemical characterization of endogenously formed eosinophilic crystals in the lungs of mice. J Biol Chem. 2000;275:8032–8037. doi: 10.1074/jbc.275.11.8032. [DOI] [PubMed] [Google Scholar]
- 34.Teupser D, Burkhardt R, Wilfert W, Haffner I, Nebendahl K, Thiery J. Identification of macrophage arginase I as a new candidate gene of atherosclerosis resistance. Arterioscler Thromb Vasc Biol. 2006;26:365–371. doi: 10.1161/01.ATV.0000195791.83380.4c. [DOI] [PubMed] [Google Scholar]
- 35.Wong BL, Zhu SL, Huang XR, Ma J, Xia HH, Bucala R, Wong BC, Lan HY. Essential role for macrophage migration inhibitory factor in gastritis induced by Helicobacter pylori. Am J Pathol. 2009;174:1319–1328. doi: 10.2353/ajpath.2009.080708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang XR, Chung AC, Zhou L, Wang XJ, Lan HY. Latent TGF-beta1 protects against crescentic glomerulonephritis. J Am Soc Nephrol. 2008;19:233–242. doi: 10.1681/ASN.2007040484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bro S, Bentzon JF, Falk E, Andersen CB, Olgaard K, Nielsen LB. Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice. J Am Soc Nephrol. 2003;14:2466–2474. doi: 10.1097/01.asn.0000088024.72216.2e. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto S, Kon V. Mechanisms for increased cardiovascular disease in chronic kidney dysfunction. Curr Opin Nephrol Hypertens. 2009;18:181–188. doi: 10.1097/MNH.0b013e328327b360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Subramanian V, Golledge J, Ijaz T, Bruemmer D, Daugherty A. Pioglitazone-induced reductions in atherosclerosis occur via smooth muscle cell-specific interaction with PPAR{gamma} Circ Res. 2010;107:953–958. doi: 10.1161/CIRCRESAHA.110.219089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakaya H, Summers BD, Nicholson AC, Gotto AM, Jr, Hajjar DP, Han J. Atherosclerosis in LDLR-knockout mice is inhibited, but not reversed, by the PPARgamma ligand pioglitazone. Am J Pathol. 2009;174:2007–2014. doi: 10.2353/ajpath.2009.080611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Westenfeld R, Schafer C, Kruger T, Haarmann C, Schurgers LJ, Reutelingsperger C, Ivanovski O, Drueke T, Massy ZA, Ketteler M, Floege J, Jahnen-Dechent W. Fetuin-A protects against atherosclerotic calcification in CKD. J Am Soc Nephrol. 2009;20:1264–1274. doi: 10.1681/ASN.2008060572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aikawa E, Aikawa M, Libby P, Figueiredo JL, Rusanescu G, Iwamoto Y, Fukuda D, Kohler RH, Shi GP, Jaffer FA, Weissleder R. Arterial and aortic valve calcification abolished by elastolytic cathepsin S deficiency in chronic renal disease. Circulation. 2009;119:1785–1794. doi: 10.1161/CIRCULATIONAHA.108.827972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zuo Y, Yancey P, Castro I, Khan WN, Motojima M, Ichikawa I, Fogo AB, Linton MF, Fazio S, Kon V. Renal dysfunction potentiates foam cell formation by repressing ABCA1. Arterioscler Thromb Vasc Biol. 2009;29:1277–1282. doi: 10.1161/ATVBAHA.109.188995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roszer T, Menendez-Gutierrez MP, Lefterova MI, Alameda D, Nunez V, Lazar MA, Fischer T, Ricote M. Autoimmune kidney disease and impaired engulfment of apoptotic cells in mice with macrophage peroxisome proliferator-activated receptor gamma or retinoid X receptor alpha deficiency. J Immunol. 2011;186:621–631. doi: 10.4049/jimmunol.1002230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khera AV, Cuchel M, de la Llera-Moya M, Rodrigues A, Burke MF, Jafri K, French BC, Phillips JA, Mucksavage ML, Wilensky RL, Mohler ER, Rothblat GH, Rader DJ. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N Engl J Med. 2011;364:127–135. doi: 10.1056/NEJMoa1001689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ozasa H, Ayaori M, Iizuka M, Terao Y, Uto-Kondo H, Yakushiji E, Takiguchi S, Nakaya K, Hisada T, Uehara Y, Ogura M, Sasaki M, Komatsu T, Horii S, Mochizuki S, Yoshimura M, Ikewaki K. Pioglitazone enhances cholesterol efflux from macrophages by increasing ABCA1/ABCG1 expressions via PPARgamma/LXRalpha pathway: findings from in vitro and ex vivo studies. Atherosclerosis. 2011;219:141–150. doi: 10.1016/j.atherosclerosis.2011.07.113. [DOI] [PubMed] [Google Scholar]
- 47.Arai S, Shelton JM, Chen M, Bradley MN, Castrillo A, Bookout AL, Mak PA, Edwards PA, Mangelsdorf DJ, Tontonoz P, Miyazaki T. A role for the apoptosis inhibitory factor AIM/Spalpha/Api6 in atherosclerosis development. Cell Metab. 2005;1:201–213. doi: 10.1016/j.cmet.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Thewke DP, Su YR, Linton MF, Fazio S, Sinensky MS. Reduced macrophage apoptosis is associated with accelerated atherosclerosis in low-density lipoprotein receptor-null mice. Arterioscler Thromb Vasc Biol. 2005;25:174–179. doi: 10.1161/01.ATV.0000148548.47755.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boesten LS, Zadelaar AS, van Nieuwkoop A, Hu L, Teunisse AF, Jochemsen AG, Evers B, van de Water B, Gijbels MJ, van Vlijmen BJ, Havekes LM, de Winther MP. Macrophage p53 controls macrophage death in atherosclerotic lesions of apolipoprotein E deficient mice. Atherosclerosis. 2009;207:399–404. doi: 10.1016/j.atherosclerosis.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 50.Babaev VR, Chew JD, Ding L, Davis S, Bryer MD, Bryer RM, Oates JA, Fazio S, Linton MF. Macrophage EP4 deficiency increases apoptosis and supresses early atherosclerosis. Cell Metab. 2008;8:492–501. doi: 10.1016/j.cmet.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.