

Abstract
Background
Matrix-metalloproteinases (MMP) and cancer cell invasion are crucial for solid tumour metastasis. Important signalling events triggered by inflammatory cytokines, such as tumour necrosis factor α (TNFα), include Src-kinase-dependent activation of Akt and focal adhesion kinase (FAK) and phosphorylation of caveolin-1. Based on previous studies where we demonstrated amide-type local anaesthetics block TNFα-induced Src activation in malignant cells, we hypothesized that local anaesthetics might also inhibit the activation and/or phosphorylation of Akt, FAK and caveolin-1, thus attenuating MMP release and invasion of malignant cells.
Methods
NCI-H838 lung adenocarcinoma cells were incubated with ropivacaine or lidocaine (1 nM-100 µM) in absence/presence of TNFα (20 ng ml−1) for 20 min or 4 h, respectively. Activation/phosphorylation of Akt, FAK and caveolin-1 were evaluated by Western blot, and MMP-9 secretion was determined by enzyme-linked immunosorbent assay. Tumour cell migration (electrical wound-healing assay) and invasion were also assessed.
Results
Ropivacaine (1 nM–100 μM) and lidocaine (1–100 µM) significantly reduced TNFα-induced activation/phosphorylation of Akt, FAK and caveolin-1 in NCI-H838 cells. MMP-9 secretion triggered by TNFα was significantly attenuated by both lidocaine and ropivacaine (half-maximal inhibitory concentration [IC50]=3.29×10−6 M for lidocaine; IC50=1.52×10−10 M for ropivacaine). The TNFα-induced increase in invasion was completely blocked by both lidocaine (10 µM) and ropivacaine (1 µM).
Conclusions
At clinically relevant concentrations both ropivacaine and lidocaine blocked tumour cell invasion and MMP-9 secretion by attenuating Src-dependent inflammatory signalling events. Although determined entirely in vitro, these findings provide significant insight into the potential mechanism by which local anaesthetics might diminish metastasis.
Keywords: anesthetics, local; inflammation; neoplasm metastasis
Editor's key points.
Anaesthetic techniques may have an impact on outcome from cancer surgery.
Local anaesthetics have anti-inflammatory effects which may impact upon cancer cell invasion.
Lung adenocarcinoma cells were exposed to ropivacaine or lidocaine at clinically relevant concentrations.
Both local anaesthetics blocked tumour cell invasion and metalloproteinase expression via effects on Src signalling.
This provides some insight into the mechanisms behind the effects of local anaesthetics on cancer cell invasion.
There is conflicting evidence regarding the effect of anaesthesia technique and patient outcome after cancer surgery.1 Retrospective analyses indicate the perioperative use of regional anaesthesia using amide-linked local anaesthetics (LAs) may decrease cancer recurrence and metastasis in these patients.2–4 Anti-inflammatory effects of LAs in various epithelial and endothelial cell types are well known.5,6 The fact that LAs have anti-inflammatory properties in malignant cells lead to our recent observations that amide-type LAs inhibit the activation of inflammatory Src tyrosine protein kinase (Src), a critical mediator of endothelial hyperpermeability and also cancer cell extravasation and metastasis.7,8
Matrix-metalloproteinases (MMPs) are important in the pathogenesis of new metastatic sites from solid tumours as these enzymes degrade the extracellular matrix and basal lamina, thus allowing malignant cells to enter or escape the primary tumour, enter the circulation, and extravasate to form satellite lesions.9,10 De novo synthesis of MMPs [e.g. due to stimulation with tumour necrosis factor α (TNFα)], largely depends on Src-dependent activation of Akt and focal adhesion kinase (FAK) and phosphorylation of caveolin-1 (Cav-1) by Src,11 signalling pathways which are also required for cancer cell migration.12–14
In the present study, we tested the hypothesis that amide-type local anaesthetics such as lidocaine and ropivacaine inhibit TNFα-induced Akt and FAK activation and Src-dependent Cav-1 phosphorylation, thus attenuating MMP-9 secretion, migration, and invasion of lung adenocarcinoma cells (Fig. 1).
Fig 1.
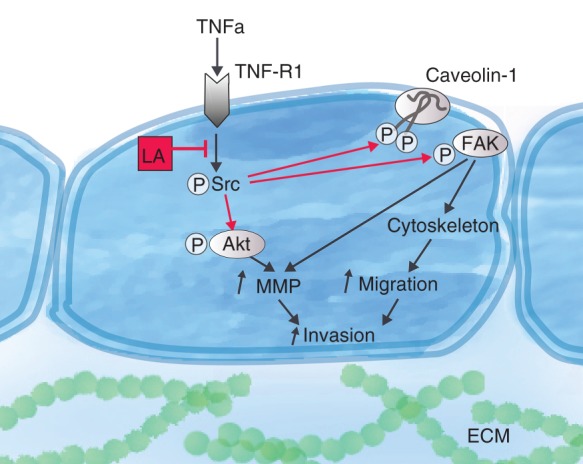
Summary of proposed mechanism by which amide-linked local anaesthetics inhibit cancer cell invasion and metastasis. As demonstrated previously, local anaesthetics (LA) block tumour necrosis factor α (TNFα)-induced activation of Src tyrosine protein kinase (Src) by inhibiting signal propogation downstream of TNF receptor 1 (TNF-R1).7,8 Through this mechanism, amide-linked LAs indirectly prevent activation of Akt kinase (Akt), phosphorylation of caveolin-1, and activation of focal adhesion kinase (FAK). These signalling steps are crucial during metastasis, as they promote cancer cell migration by regulating the cytoskeleton and are necessary for the secretion of matrix-metalloproteinases (MMP), thus enabling the cells to break up the basal lamina and the extracellular matrix (ECM) and further invade into the surrounding tissue.
Methods
Cell culture
Human NCI-H838 lung adenocarcinoma cells (CRL-5844, ATCC, Rockville, MD) were cultured as previously described.7 All cells were maintained in 5% CO2 and 95% room air in a water-jacketed 37°C incubator.
Experimental procedure
NCI-H838 cell monolayers were incubated with TNFα (Gibco Invitrogen, Carlsbad, CA, final concentration 20 ng ml−1) in the absence or presence of lidocaine (lidocaine-HCl 2%, APP Pharmaceuticals, Schaumburg, IL) or ropivacaine (Naropin® 0.5%, APP Pharmaceuticals) at concentrations ranging from 1 pM–100 µM, depending on the experiment. Incubation times varied between 20 min for Western blot analysis (Akt, FAK and Cav-1) and 4 h for determination of MMP-9 secretion. For some experiments, cells were pre-treated with wortmannin (100 nM, Sigma Aldrich, St. Louis, MO), an inhibitor of phosphoinositide-3 kinase (PI3K), which is required for the activation of Akt, or the specific FAK inhibitor FI 14 (5 µM, Tocris Bioscience, Minneapolis, MN) (Fig. 1).
Cell harvest and lysis
After indicated incubation times, cells were harvested and lysed as described previously.15 Total protein concentration was determined using the DC Protein Assay Kit (BioRad, Hercules, CA) in accordance with the manufacturer's instructions. Immunoglobulin G was used for the generation of a standard curve.
Western blot analysis
Whole cell lysates were subjected to Western blot analysis following a published protocol.16 Antibodies against FAK, phosphorylated at tyrosine 397 (pY397), total FAK, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and total Akt were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), against phospho-tyrosine 14 (pY14) Cav-1, total Cav-1, and β-actin from BD Biosciences (Franklin Lakes, NJ), and for Akt, phosphorylated at threonine 308 (pT308), from Cell Signaling Technology (Danvers, MA).
Enzyme-linked immunosorbent assay (ELISA)
The concentration of MMP-9 in cell culture supernatant was determined using the human MMP-9 DuoSet from R&D Systems (Minneapolis, MN) in accordance with the manufacturer's instructions. The total amount of MMP-9 secreted by the cells in each well was calculated from the ELISA values (pg ml−1) and normalized to the amount of protein (pg mg−1 protein) found in whole cell lysates.
Electrical wound-healing assay
NCI-H838 lung cancer cells were grown to confluence on gold-plated microelectrodes (ECIS cultureware; Applied Biophysics, Troy, NY) coated with 0.2% gelatin (Sigma-Aldrich, St. Louis, MO). Electrical impedance measured over time across the monolayer at 1 V, 4000 Hz with the Electric Cell-Substrate Impedance Sensing (ECIS) system (Applied Biophysics), was used to calculate transepithelial resistance. After an equilibration period of 30 min, TNFα (final concentration 20 ng ml−1) was added in absence or presence of lidocaine (10 µM) or ropivacaine (1 µM). After that, cells growing in the middle of the gold electrode (i.e. on the small active electrode) were injured via the application of 2.5 V at 40 000 Hz for 10 s, leading to an immediate decrease in transepithelial resistance.17 Recovery of transepithelial resistance after electrical injury was used as a surrogate for cell migration.17 For some experiments, cells were pre-treated for 30 min before the addition of TNFα with either wortmannin (100 nM) or FAK inhibitor (FI) 14 (5 µM).
In vitro invasion assay
Transwell inserts with a polycarbonate membrane with 8 μm pore size (Falcon® Permeable Support, BD Biosciences) were coated with 20 µl of Matrigel® (Growth Factor Reduced, from Corning, Tewksbury MA). NCI-H838 cells were suspended in RPMI-1640 medium with 0% FBS and treated with either lidocaine (10 µM) or ropivacaine (1 µM) in the absence or presence of TNFα (20 ng ml−1). The cell suspension was inoculated in the upper chamber of the Transwell, which was then inserted into a 24-well cell culture plate (TPP Techno Plastic Products, Trasadingen, Switzerland). The bottom chamber of the system was pre-filled with RPMI-1640 medium supplemented with 10% FBS plus TNFα and LA as in each corresponding upper chamber. Cells were allowed to invade into the Matrigel-coated membrane for 6 h. Non-invading cells were removed, invading cells were fixed with 2% paraformaldehyde solution (Kantonsapotheke Zurich, Zurich, Switzerland) and stained with 4′,6-diamidino-2-phenylindole (DAPI, Life Technologies, Zug, Switzerland). Membranes were mounted on glass slides for microscopy and quantitative image analysis. Images were acquired at 20× magnification using a Leica DM6000B microscope equipped with a DFC350FX camera and Leica Application Suite Advanced Fluorescence software, Version 3.x for Windows (all from Leica Microsystems, Heerbrugg, Switzerland). Semi-automated counting of cell nuclei stained with DAPI was carried out using the ImageJ software, Version 1.47v for Mac (http://imagej.nih.gov/ij). Eight fields of view (0.2 mm2 each) were quantified per slide and averaged for further analysis.
Statistical analysis
Normal distribution was assessed using Shapiro–Wilk testing. Normally distributed data of transepithelial resistance were analysed by one-way ANOVA. Non-normally distributed data (invasion) were analysed with non-parametric testing (Kruskal-Wallis and subsequent Mann-Whitney U-tests) with post hoc corrections using the Simes-Hochberg method in order to keep the family-wise error rate below 0.05. All other data were evaluated with two-way ANOVA with the local anaesthetic (or other treatments) and the absence or presence of TNFα as factors to be tested, whereas we report the F ratio with the degrees of freedom for each of these factors and for the interaction between them. Bonferroni post hoc testing was conducted to detect differences between groups. The exact number of experiments is indicated in the respective figure legend. All tests were performed two-sided and non-blinded using GraphPad Prism software for Mac, Version 6 (GraphPad Software). A p-value of <0.05 was considered statistically significant.
The half-maximal inhibitory concentration (IC50)18 was assessed for lidocaine and ropivacaine on TNFα-induced MMP-9 secretion by NCI-H838 lung cancer cells by plotting the MMP-9 concentration against the (logarithmic) concentration of the local anaesthetic. Curve fitting (Hill slope fixed at −1.0) was conducted using non-linear regression analysis with the following equation:
Results reported in Table 1 are values with 95% confidence interval (CI, for IC50, top, and bottom).
Table 1.
Inhibitory properties of lidocaine and ropivacaine on TNFα-induced MMP-9 secretion by NCI-H838 lung adenocarcinoma cells. IC50, half maximal inhibitory concentration; CI, confidence interval
| Drug | IC50 [M] (95% CI) | Bottom (95% CI) | Top (95% CI) |
|---|---|---|---|
| Lidocaine | 3.291×10−6 (1.358×10−6–7.978×10−6) | 37.62 (25.5–49.74) | 114.0 (103.3–124.7) |
| Ropivacaine | 1.521×10−10 (4.155×10−11–5.57×10−10) | 70.15 (57.72–82.58) | 126.9 (115.3–138.5) |
Results
Cytotoxic effects of local anaesthetics at the concentrations used were excluded in a previous study.7
Representative Western blots of the activated form of Akt, phosphorylated at threonine 308 (pT308 Akt), total Akt, and GAPDH are shown in Figure 2A and B. Quantitative densitometry analysis revealed a 2.6-fold increase in Akt activation after treatment with TNFα alone compared with untreated cells (2.71 [sd 0.92] Figure 2Ci and 2.35 [0.45], Figure 2 Cii, respectively). In the experiments involving lidocaine, analysis with two-way ANOVA showed a significant effect of TNFα (F(1,90)=17.55, P<0.001). Post hoc analysis revealed a significant reduction in TNFα-induced Akt activation in presence of lidocaine at a concentration of 1 µM (p=0.03), Figure 2Ci. As shown in Figure 2Cii, TNFα-induced Akt activation was also significantly blocked by ropivacaine at 1 nM (p=0.03).
Fig 2.

(a) Representative Western blots of Akt phosphorylated at threonine 308 (pT308 Akt, row 1), total Akt (row 2), and GAPDH, (row 3) after treatment with either (i) lidocaine or (ii) ropivacaine (1 nM–100 µM) for 20 min in absence (a) or presence (b) of tumour necrosis factor α (TNFα) (c) Quantitative analysis of densitometry of Western blots showing the ratio of pT308 Akt over total Akt in NCI-H838 cells treated with (i) lidocaine or (ii) ropivacaine compared with untreated cells (set as 1.0, dashed line) in absence (green bars) or presence (blue bars) of TNFα (20 ng ml−1). Data shown are mean (sd) (n=10 for lidocaine; n=7 for ropivacaine; *P<0.05 compared with TNFα alone. (d) Representative Western blots of FAK phosphorylated at tyrosine 397 (pY397 FAK, row 1), total FAK (row 2), and GAPDH (row 3) after treatment with either (i) lidocaine or (ii) ropivacaine (1 nM–100 µM) alone for 20 min, or (e) after treatment with TNFα in absence or presence of (i) ropivacaine or (ii.) lidocaine (1 nM–100 µM) for 20 min. (f) Quantitative analysis of densitometry of Western blots showing the ratio of pY397 FAK over total FAK in NCI-H838 cells treated with (i.) lidocaine or (ii.) ropivacaine compared with untreated cells (set as 1.0, dashed line) in absence (blue bars) or presence (green bars) of TNFα (20 ng ml−1). Data shown are mean (sd), n=8 for lidocaine, n=7 for ropivacaine; *P<0.05 compared with TNFα alone, #P<0.05 vs. control.
The same samples were also evaluated for FAK phosphorylation at tyrosine 397 (pY397 FAK) as compared to the total amount of FAK protein (Fig. 2D and E). We again observed a significant effect of TNFα on FAK phosphorylation in both sets of experiments (F(1,69)=15.81 P<0.001 for lidocaine experiments), Figure 2Fi; (F(1,60)=23.14 P<0.001 for ropivacaine experiments, Fig. 2Fii). The effect of the two different LAs reached statistical significance by two-way ANOVA (F(4,69)=4.07 P=0.005) for lidocaine experiments, Figure 2Fi. Post hoc analysis was comparable with those observed with Akt: 10 µM lidocaine attenuated TNFα-induced FAK activation by 51% [25%] (P=0.04) and ropivacaine completely abolished the TNFα-induced increase in pY397 FAK at a concentration of 1 nM (P=0.01). At higher concentrations (beginning at 10 µM) and in absence of TNFα, ropivacaine significantly decreased FAK phosphorylation below baseline values (P=0.01, Fig. 2Fii).
We also assessed the effect of lidocaine and ropivacaine on TNFα-induced Cav-1 phosphorylation at tyrosine 14 via Western blot (pY14 Cav-1, Fig 3A and B). TNFα significantly increased pY14 Cav-1 by 75% [51%], (F(1,60)=4.22, P=0.04, Fig. 3Ci) and 49% [36%] (F(1,65)=5.42, P=0.02, Fig. 3Cii), respectively. It also became evident that both LAs completely blocked TNFα-induced Cav-1 phosphorylation at concentrations of 1 µM (P=0.002 for lidocaine, P=0.004 for ropivacaine).
Fig 3.

Representative Western blots of caveolin-1 (Cav-1) phosphorylated at tyrosine 14 (pY14 Cav-1, row 1), total Cav-1 (row 2), and either (i.) β-actin or (ii.) (GAPDH, row 3 respectively) after treatment with either (i.) lidocaine or (ii.) ropivacaine (1 nM–100 µM) alone for 20 min (a), or following (b) treatment with TNFα in absence or presence of (i.) ropivacaine or (ii.) lidocaine (1 nM–100 µM) for 20 min. (c) Quantitative analysis of densitometry of Western blots showing the ratio of pY14 Cav-1 over total Cav-1 in NCI-H838 cells treated with (i.) lidocaine or (ii.) ropivacaine compared with untreated cells (set as 1.0, dashed line) in absence (green bars) or presence (blue bars) of TNFα (20 ng ml−1). Data shown are mean (sd), n=7 for lidocaine; n=8 for ropivacaine; *P<0.05 compared with TNFα alone.
Non-linear curve fitting was used to determine IC50 values of lidocaine and ropivacaine on TNFα-induced MMP-9 secretion as shown in Table 1: the IC50 for lidocaine was 3.29 µM (95% CI, 1.36–7.98 µM) with a maximum effect of 62% (95% CI, 51–75%) whereas ropivacaine had a lower IC50 (152 pM, 95% CI, 42–557 pM) and a lower maximum effect of 30% (95% CI, 17–42%, all Table 1). NCI-H838 cells pre-treated with either wortmannin (100 nM) or FI 14 (5 µM) had significantly lower TNFα-induced MMP-9 secretion of 37% [7%] and 52% [5%], (P<0.001 compared with TNFα alone for both, all Fig. 4A).
Fig 4.
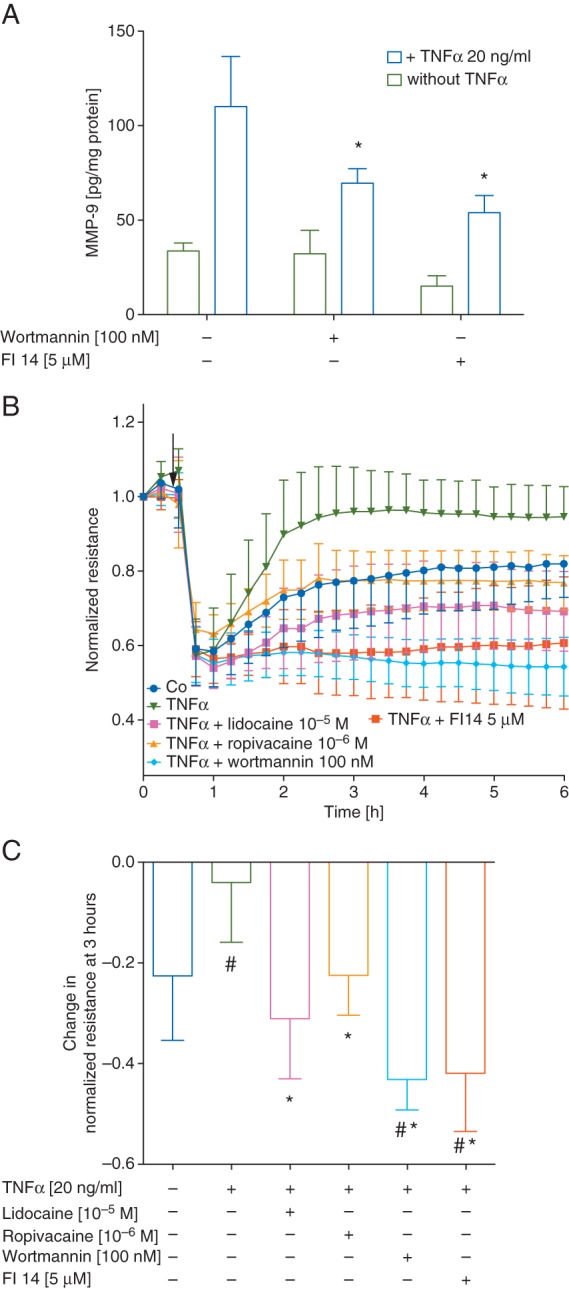
(a) Amount of MMP-9 (in pg normalized to mg of cell protein content) in cell culture supernatants of NCI-H838 cells treated with 100 nM Wortmannin (an inhibitor of phosphoinositide-3 kinase) or 5 µM FI 14 in absence (green bars) or presence (blue bars) of TNFα (20 ng ml−1) for 4 h. Data shown are mean (sd), n=6; *P<0.05 compared with TNFα alone. (b) Normalized resistance of NCI-H838 lung cancer cell monolayers over time after electrical injury with 2.5 V at 40 000 Hz for 10s (indicated by arrow). Treatment with either TNFα (20 ng ml−1, green), TNFα+lidocaine (10 µM, pink), TNFα+ropivacaine (1 µM, gold), TNFα+wortmannin (100 nM, turquoise, pre-treatment for 30 min before addition of TNFα), TNFα+FI 14 (5 µM, orange, pre-treatment for 30 min before addition of TNFα), or vehicle (control, blue). Data shown are mean (sd) of normalized resistance. (c) Quantification of normalized resistance at 3 h. Data shown are mean (sd); untreated cells (blue bar), TNFα alone (green bar), lidocaine at 10 µM (pink bar), TNFα+ropivacaine at 1 µM (gold bar), TNFα+wortmannin (turquoise bar) and TNFα+FI 14 (orange bar); n=10 for control, TNFα, TNFα+ropivacaine, TNFα+lidocaine; n=8 for TNFα+wortmannin, TNFα+FI 14; #P<0.05 vs untreated cells, *P<0.05 compared with TNFα alone.
Resistance values measured over NCI-H838 monolayers were normalized to baseline values and plotted against time as shown in Fig. 4b. Treatment with TNFα resulted in complete restoration of monolayer integrity (96% [12%] of pre-injured baseline resistance) compared with untreated cells in which the resistance was still 23 [13%] below basal at the 3 h time point (Fig. 4c; one-way ANOVA, P=0.006). Lidocaine and ropivacaine both completely abolished TNFα-induced migration and recovery of barrier properties in the presence of lidocaine, (P<0.001); or ropivacaine, (P=0.01) compared with TNFα alone. Pre-treatment with either wortmannin or FI 14 also decreased the restoration of barrier properties compared with TNFα alone (both P<0.001 Fig. 4c).
Supplementary data, Figure 1 shows representative microscopic images of Matrigel®-coated membranes after the invasion assay. Counting of the number of cells per field of view reveals a significant increase in invasion when cells were stimulated with TNFα (median 125 [minimum–maximum 88–160] cells/field vs control, 64 [53–71] cells/field, P<0.001, Fig. 5). Incubation of the cells with ropivacaine or lidocaine alone did not alter the number of invading cells compared with unstimulated cells (data not shown). However, both lidocaine (10 µM) and ropivacaine (1 µM) completely blocked TNFα-induced invasion of tumour cells, similar to that observed in presence of wortmannin or FI 14 (P<0.01 vs TNFα alone for all comparisons, Fig. 5).
Fig 5.
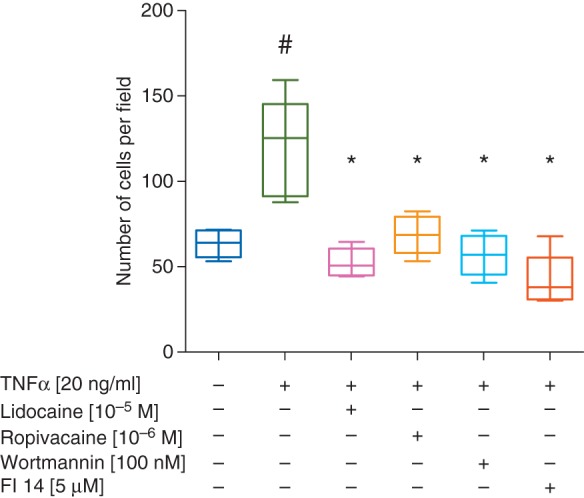
Quantitative analysis of the number of invading cells found per field of view in DAPI stained images of Matrigel coated membranes. Cells were treated with and without TNF plus lidocaine, ropivacaine, wortmannin or FI 14. Data presented as boxplots showing the median, with interquartile and full range. N=5, whereas the values for each slide were calculated as the mean of 8 fields of view. #P<0.05 vs untreated cells, *P<0.05 compared with TNFα alone.
Discussion
In this study, we evaluated the effect of lidocaine and ropivacaine on TNFα-induced, Src-dependent inflammatory signalling events in malignant cells, namely activation of Akt and FAK and phosphorylation of Cav-1.
As hypothesized, these signalling events were blocked in presence of the amide-linked LAs. We then also linked inhibition of this inflammatory signalling downstream of TNFα and Src to function, assessing the effect of LAs on MMP-9 production, migration, and invasion of H838 cells, further elucidating a potential mechanism by which local anaesthetics might be able to attenuate metastasis of solid epithelial tumours.
Mechanisms similar to inflammatory processes may play an important role in the development, growth, and metastasis of solid tumours.19–21 We have previously shown that amide-linked local anaesthetics, such as lidocaine and ropivacaine, inhibit TNFα-induced activation of Src by blocking the early propagation of signalling at the level of TNFα receptor-1 (TNF-R1).7,8 Activated Src functions to increase vascular permeability,22 cancer and inflammatory cell adhesion to the endothelium, and cancer cell migration. Additionally, Src is involved in signalling epithelial-to-mesenchymal transformation and extravasation of cancer cells, processes that are necessary for solid tumour metastasis.19,23
The release of MMPs by neutrophils and malignant cells alike is known to contribute to tumour growth and metastasis by degrading the extracellular matrix and breaking up the basal lamina, thereby enabling cancer cells to enter and then exit the circulation at a remote location.9,10 TNFα-induced expression of MMPs by malignant cells is Src-dependent and involves the PI3 K/Akt pathway.11 Activation of Akt by PI3K is Src-dependent and involved in numerous downstream signalling events that contribute to cancer cell growth, proliferation and survival and has therefore become a very interesting protein for targeted therapies.24 The results of our study clearly show that both lidocaine and ropivacaine significantly inhibit Akt activation in NCI-H838 non-small cell lung cancer (NSCLC) cells, thus attenuating MMP-9 production.
Cav-1 is a primary Src substrate22 and Src-dependent Cav-1 Y14 phosphorylation is important for migration of (malignant) cells via the regulation of focal adhesion dynamics and assembly,12,14 which also involves the PI3K/Akt pathway.25 pY14-Cav-1 promotes tumour growth whereas loss of Cav-1 expression in either the tumour microenvironment or cancer cells per se leads to a significant increase in invasiveness and overall worse prognosis.26–28 As we have previously shown that tyrosine phosphorylation of Cav-1 promotes its degradation,16 reduction of pY14 Cav-1 as seen in our study may be a mechanism by which the LAs reduce the metastatic potential of malignant cells.
FAK is the most important kinase in focal adhesion signalling and its activation is not only PI3K- and Src-dependent,29,30 but also crucial for cell orientation, actin cytoskeletal assembly/re-organization, and ultimately cancer cell migration.12,13,31 The results from our study confirm these previous findings. We showed that enhanced cancer cell migration induced by TNFα was impaired by the PI3K inhibitor wortmannin, the specific FAK inhibitor FI 14, and importantly, lidocaine and ropivacaine had a similar effect.
In order to combine findings on the effect of LAs on TNFα-signalling, migration, and MMP secretion, we assessed the impact of lidocaine and ropivacaine on tumour cell invasion into a collagen-rich environment in vitro. We demonstrated that the amide-linked local anaesthetics lidocaine and ropivacaine were able to completely mitigate TNFα-induced enhancement in invasion and showed that inhibition of Akt activation and FAK had the same effect. These findings are in accordance with results from experiments conducted by Baptista and colleagues32, where ropivacaine was also demonstrated to inhibit invasion of metastatic colon cancer cells (SW620), as a result of an inhibition of the Nav1.5 subset of voltage-gated sodium channels (VGSC). However, we previously showed that lidocaine and ropivacaine (amide-linked LAs), but not chloroprocaine (ester-type LA), inhibit TNFα-induced Src activation and downstream signalling in NCI-H838 lung cancer cells independently of VGSC blockade,7 further supporting our hypothesis that LAs exhibit anti-inflammatory and anti-metastatic effects, by blocking TNFα signalling at the level of its receptor, TNF-R1.8
Taken together with results of PI3K inhibitor wortmannin and FAK inhibitor FI 14, the observed attenuation of cancer cell invasion by lidocaine and ropivacaine is likely to be as a result of the blockade of Src-dependent Akt and FAK activation and of Cav-1 phosphorylation.
In summary, cancer dissemination is a multi-step process where many cellular and molecular regulatory mechanisms may be targets for therapeutic interventions. This study clearly demonstrates the importance of the blockade of the Src pathway by clinically relevant concentrations of amide LAs and—most importantly—explores this pathway further downstream, linking the interference with this particular pathway to the invasion of malignant cells.
Although determined entirely in vitro, the current study provides new and significant insight into a potential mechanism by which amide-LAs might attenuate metastasis.
Authors' contributions
Study design/planning: T.P., D.E.S., A.B., R.D.M., B.B.S.
Study conduct: T.P., M.S.
Data analysis: T.P., R.O.D., R.D.M., B.B.S.
Writing paper: T.P., M.S., R.O.D., D.E.S., A.B., R.D.M., B.B.S.
Revising paper: all authors
Supplementary material
Supplementary material is available at British Journal of Anaesthesia online.
Declaration of interest
None declared.
Funding
This work was supported by the Young Investigator Start-Up Grant 2014 by the European Society of Anaesthesiology as well as by the European Society of Regional Anaesthesia & Pain Medicine Research Grant (both to T.P.).
Supplementary Material
Acknowledgements
The authors would like to thank Irene Odermatt (Art Designer, Institute of Anesthesiology, University Hospital Zurich, Switzerland) for assistance with the illustration of Fig. 1 of this manuscript.
References
- 1.Heaney A, Buggy DJ. Can anaesthetic and analgesic techniques affect cancer recurrence or metastasis? Brit J Anaesth 2012; 109(Suppl 1): i17–28 [DOI] [PubMed] [Google Scholar]
- 2.Christopherson R, James KE, Tableman M, Marshall P, Johnson FE. Long-term survival after colon cancer surgery: a variation associated with choice of anesthesia. Anesth Analg 2008; 107: 325–32 [DOI] [PubMed] [Google Scholar]
- 3.Biki B, Mascha E, Moriarty DC, Fitzpatrick JM, Sessler DI, Buggy DJ. Anesthetic technique for radical prostatectomy surgery affects cancer recurrence: a retrospective analysis. Anesthesiology 2008; 109: 180–7 [DOI] [PubMed] [Google Scholar]
- 4.Exadaktylos AK, Buggy DJ, Moriarty DC, Mascha E, Sessler DI. Can anesthetic technique for primary breast cancer surgery affect recurrence or metastasis? Anesthesiology 2006; 105: 660–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blumenthal S, Borgeat A, Pasch T, et al. Ropivacaine decreases inflammation in experimental endotoxin-induced lung injury. Anesthesiology 2006; 104: 961–9 [DOI] [PubMed] [Google Scholar]
- 6.Piegeler T, Dull RO, Hu G, et al. Ropivacaine attenuates endotoxin plus hyperinflation-mediated acute lung injury via inhibition of early-onset Src-dependent signaling. BMC Anesthesiol 2014; 14: 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Piegeler T, Votta-Velis EG, Liu G, et al. Antimetastatic Potential of Amide-linked Local Anesthetics: Inhibition of Lung Adenocarcinoma Cell Migration and Inflammatory Src Signaling Independent of Sodium Channel Blockade. Anesthesiology 2012; 117: 548–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Piegeler T, Votta-Velis EG, Bakhshi FR, et al. Endothelial Barrier Protection by Local Anesthetics: Ropivacaine and Lidocaine Block Tumor Necrosis Factor-alpha-induced Endothelial Cell Src Activation. Anesthesiology 2014; 120: 1414–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nicoud IB, Jones CM, Pierce JM, et al. Warm hepatic ischemia-reperfusion promotes growth of colorectal carcinoma micrometastases in mouse liver via matrix metalloproteinase-9 induction. Cancer Res 2007; 67: 2720–8 [DOI] [PubMed] [Google Scholar]
- 10.Muller-Edenborn B, Roth-Z'graggen B, Bartnicka K, et al. Volatile Anesthetics Reduce Invasion of Colorectal Cancer Cells through Down-regulation of Matrix Metalloproteinase-9. Anesthesiology 2012; 117: 293–301 [DOI] [PubMed] [Google Scholar]
- 11.Lee CW, Lin CC, Lin WN, et al. TNF-alpha induces MMP-9 expression via activation of Src/EGFR, PDGFR/PI3 K/Akt cascade and promotion of NF-kappaB/p300 binding in human tracheal smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2007; 292: L799–812 [DOI] [PubMed] [Google Scholar]
- 12.Goetz JG, Joshi B, Lajoie P, et al. Concerted regulation of focal adhesion dynamics by galectin-3 and tyrosine-phosphorylated caveolin-1. J Cell Biol 2008; 180: 1261–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joshi B, Bastiani M, Strugnell SS, Boscher C, Parton RG, Nabi IR. Phosphocaveolin-1 is a mechanotransducer that induces caveola biogenesis via Egr1 transcriptional regulation. J Cell Biol 2012; 199: 425–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi B, Strugnell SS, Goetz JG, et al. Phosphorylated caveolin-1 regulates Rho/ROCK-dependent focal adhesion dynamics and tumor cell migration and invasion. Cancer Res 2008; 68: 8210–20 [DOI] [PubMed] [Google Scholar]
- 15.Place AT, Chen Z, Bakhshi FR, Liu G, O'Bryan JP, Minshall RD. Cooperative Role of Caveolin-1 and C-Terminal Src Kinase Binding Protein in C-Terminal Src Kinase-Mediated Negative Regulation of c-Src. Mol Pharmacol 2011; 80: 665–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bakhshi FR, Mao M, Shajahan AN, et al. Nitrosation-dependent caveolin 1 phosphorylation, ubiquitination, and degradation and its association with idiopathic pulmonary arterial hypertension. Pulm Circ 2013; 3: 816–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keese CR, Wegener J, Walker SR, Giaever I. Electrical wound-healing assay for cells in vitro. Proc Natl Acad Sci U S A 2004; 101: 1554–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sebaugh JL. Guidelines for accurate EC50/IC50 estimation. Pharm Stat 2011; 10: 128–34 [DOI] [PubMed] [Google Scholar]
- 19.Guarino M. Src signaling in cancer invasion. J Cell Physiol 2010; 223: 14–26 [DOI] [PubMed] [Google Scholar]
- 20.Looney M, Doran P, Buggy DJ. Effect of anesthetic technique on serum vascular endothelial growth factor C and transforming growth factor beta in women undergoing anesthesia and surgery for breast cancer. Anesthesiology 2010; 113: 1118–25 [DOI] [PubMed] [Google Scholar]
- 21.Weis S, Cui J, Barnes L, Cheresh D. Endothelial barrier disruption by VEGF-mediated Src activity potentiates tumor cell extravasation and metastasis. J Cell Biol 2004; 167: 223–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu G, Minshall RD. Regulation of transendothelial permeability by Src kinase. Microvasc Res 2009; 77: 21–5 [DOI] [PubMed] [Google Scholar]
- 23.Kim MP, Park SI, Kopetz S, Gallick GE. Src family kinases as mediators of endothelial permeability: effects on inflammation and metastasis. Cell Tissue Res 2009; 335: 249–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis 2004; 9: 667–76 [DOI] [PubMed] [Google Scholar]
- 25.Park JH, Han HJ. Caveolin-1 plays important role in EGF-induced migration and proliferation of mouse embryonic stem cells: involvement of PI3 K/Akt and ERK. Am J Physiol Cell Physiol 2009; 297: C935–44 [DOI] [PubMed] [Google Scholar]
- 26.Chanvorachote P, Chunhacha P. Caveolin-1 regulates endothelial adhesion of lung cancer cells via reactive oxygen species-dependent mechanism. PLoS One 2013; 8: e57466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shan T, Lu H, Ji H, et al. Loss of stromal caveolin-1 expression: a novel tumor microenvironment biomarker that can predict poor clinical outcomes for pancreatic cancer. PLoS One 2014; 9: e97239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faggi F, Mitola S, Sorci G, et al. Phosphocaveolin-1 enforces tumor growth and chemoresistance in rhabdomyosarcoma. PLoS One 2014; 9: e84618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson C, Nicholes K, Bustos D, et al. Overcoming EMT-associated resistance to anti-cancer drugs via Src/FAK pathway inhibition. Oncotarget 2014; 5: 7328–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Basuroy S, Dunagan M, Sheth P, Seth A, Rao RK. Hydrogen peroxide activates focal adhesion kinase and c-Src by a phosphatidylinositol 3 kinase-dependent mechanism and promotes cell migration in Caco-2 cell monolayers. Am J Physiol Gastroint Liver Physiol 2010; 299: G186–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gupton SL, Waterman-Storer CM. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration. Cell 2006; 125: 1361–74 [DOI] [PubMed] [Google Scholar]
- 32.Baptista-Hon DT, Robertson FM, Robertson GB, et al. Potent inhibition by ropivacaine of metastatic colon cancer SW620 cell invasion and NaV1.5 channel function. Br J Anaesth 2014; 113(Suppl 1): i39–48 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}