

Summary
ABPN is a retinoic acid derivative that selectively inhibits pancreatic cancer cell growth with low toxicity towards normal cells. ABPN downregulates ErbB3 and Met while inducing Nrdp1 expression. ABPN significantly reduces tumor size in vivo with a higher potency than erlotinib.
Abstract
Combination chemotherapy for the treatment of pancreatic cancer commonly employs gemcitabine with an EGFR inhibitor such as erlotinib. Here, we show that the retinoic acid derivative, ABPN, exhibits more potent anticancer effects than erlotinib, while exhibiting less toxicity toward noncancerous human control cells. Low micromolar concentrations of ABPN induced apoptosis in BxPC3 and HPAC pancreatic cancer cell lines, concomitant with a reduction in phosphorylated EGFR as well as decreased ErbB3, Met and BRUCE protein levels. The degradation of ErbB3 is a result of proteasomal degradation, possibly due to the ABPN-dependent upregulation of Nrdp1. Administration of ABPN showed significant reductions in tumor size when tested using a mouse xenograft model, with higher potency than erlotinib at the same concentration. Analysis of the tumors demonstrated that ABPN treatment suppressed ErbB3 and Met and induced Nrdp1 in vivo. The data suggest that ABPN may be more suitable in combination chemotherapy with gemcitabine than the more widely used EGFR inhibitor, erlotinib.
Introduction
Pancreatic cancer remains the fourth most common cause of cancer-related deaths worldwide (1). The majority of neoplasms are thought to arise from precancerous lesions developing in the exocrine components of the organ. Detection of the disease is typically accompanied by a poor prognosis, due in part to an absence of symptoms in its early stages. Locally advanced and metastatic disease accounts for over 80% of diagnosed individuals, with median survival rates at ~10 and 6 months, respectively (2). Treatment options are limited, with only a minority of cases suitable for surgical tumor removal and adjuvant radiation therapy. For these and other reasons, pancreatic cancer holds the dubious distinction of having the lowest survival rate amongst all cancers (3).
Deregulation of the epidermal growth factor receptor (i.e. EGFR; ErbB1; HER1) has been implicated in the progression of multiple cancer types, including pancreatic cancer (4). The receptor is a membrane-bound tyrosine kinase that forms heterodimers with other members of the same family, including ErbB3. Overexpression of EGFR and ErbB3 is associated with decreased survival and downstream activation of cell survival mediators including the Akt, MAPK and JNKs pathways (5–7). Nrdp1 is an important negative regulator of ErbB3 acting as an ubiquitin ligase responsible for ErbB3 proteasomal degradation (8).
The majority of current treatment strategies for pancreatic cancer include chemotherapeutic agents, which became prevalent after the discovery of the thymidylate synthase inhibitory activity of 5-fluorouracil (5-FU). 5-FU is a pyrimidine analog that disrupts the synthesis of DNA, leading to cell cycle arrest and apoptosis (9). The compound is a member of the antimetabolite family of drugs, which includes the more commonly prescribed gemcitabine, a nucleoside analog. Gemcitabine is frequently used for palliative purposes in advanced pancreatic cancers and was the first FDA-approved drug for a clinical nonsurvival endpoint (9,10). Recently, the drug has been used in combination with EGFR inhibitors, such as erlotinib, for more effective therapeutic outcomes (11).
The wide cellular influence of EGFR has made it a prominent target of interest for monoclonal antibody design (i.e. cetuximab, panitumumab and matuzumab) and small molecule kinase inhibitors (gefitinib, erlotinib and lapatinib) (12). EGFR-positive patients often have good initial treatment outcomes; however, the development of resistance remains a serious obstacle. This is partly because EGFR is susceptible to mutations that amplify its activity or render it insensitive to inhibition. Two major EGFR-related resistance mechanisms include the T790M mutation and amplification of the Met oncogene (13–15). Due to the higher likelihood of late stage cancers developing such resistance, novel strategies that focus on downregulation or degradation of EGFR/ErbB3 could represent more effective solutions.
Retinoic acid is a metabolite of vitamin A that plays critical roles in chordate embryogenesis, as well as diverse roles in development, differentiation and homeostasis (16). Transcriptional responses occur after it binds with the retinoic acid receptor and subsequent activation of Hox family genes (17). Despite its use in therapy for a number of cancer types, including breast cancer and leukemia, undesirable side-effects include teratogenicity (18). This has led to interest in synthesizing novel derivatives of retinoic acid, also known as retinoids, presenting opportunities for reduced side effects and enhanced antitumor activity.
Following the synthesis of various retinoic acid derivatives, we previously identified one compound, designated ABPN (A4-amino-2-(butyrylamino)phenyl (2E,4E,6E,8E)-3,7-dimethyl-9- (2,6,6-trimethyl-1-cyclohexenyl)-2,4,6,8-nonatetraenoate), with potent antitumor activity against several cancer cell lines (19). In a previous report, ABPN showed markedly enhanced anticancer potency compared to all-trans retinoic acid (ATRA), but activated retinoic acid receptor isotypes to an extent similar to ATRA, suggesting that the improved anticancer effect of ABPN relies on a retinoic acid receptor-independent mechanism (19). Here we report that its mode of action is achieved through significant downregulation of ErbB3 expression, which led us to hypothesize that the observed effects of ABPN may involve upregulation of Nrdp1. Our results indicate that ABPN downregulates multiple components of the EGFR/ErbB3 signaling pathway through Nrdp1 activation. This causes induction of apoptosis in specific pancreatic cancer cell lines that normally show a minimal response to both retinoic acid and erlotinib treatment, an effect confirmed in vivo.
Materials and methods
Materials
ATRA, 5-FU, fetal bovine serum (FBS) and the antibody against β-actin were purchased from Sigma-Aldrich (St Louis, MO). Gemcitabine and erlotinib were purchased from LC Laboratories (Woburn, MA). Paclitaxel was purchased from BioTang Inc. (Lexington, MA). Antibodies to detect phosphorylated ErbB3, ErbB2, EGFR, Met and p70S6K and PARP, caspase-3, ErbB3, Met and Akt were purchased from Cell Signaling Technology (Beverly, MA). Antibodies to detect ErbB2, EGFR, Cdk4, and cyclin D1 were purchased from Santa Cruz (Santa Cruz, CA) and the BRUCE antibody was from BD Biosciences (San Jose, CA). The antibody against Nrdp1/RNF41 was obtained from Bethyl Laboratories (Montgomery, TX) and the antibody to detect phosphorylated Akt was purchased from GenScript (Piscataway, NJ). The chemiluminescence detection kit was purchased from Amersham Pharmacia Biotech (Piscataway, NJ) and the protein assay kit was obtained from Bio-Rad Laboratories (Hercules, CA). The MTS reagent powder was purchased from Promega (Madison, WI).
Cell lines
BxPC3, HPAC, Panc-1 and Mia Paca-2 human pancreatic cancer cell lines were grown in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS and penicillin-streptomycin. Normal CCD-112CoN human colon fibroblasts were obtained from the American Type Culture Collection and grown in Eagle’s Minimum Essential Medium supplemented with 10% FBS and penicillin–streptomycin. Primary cultured human dermal fibroblasts were a kind gift from Dr. Sam W. Lee (Massachusetts, General Hospital) and were cultured in Dulbecco’s Modified Eagle Medium supplemented with 10% FBS and penicillin-streptomycin. All cells were maintained as monolayer cultures at 37°C in an incubator with an atmosphere of 5% CO2.
Cell viability assay
Cells were seeded in 96-well plates (1000–4000 cells per well depending on the cell type) and incubated overnight before treatment. Cell viability was measured using the CellTiter 96® AQueous MTS Reagent (Promega). Cell viability was also determined using the Sulforhodamine B Based In Vitro Toxicology Assay Kit (Sigma-Aldrich). Cells were plated in 60-mm dishes, and were treated the following day with compounds at the indicated concentrations.
Cell cycle analysis
Cells (1.5×105 cells per well) were seeded overnight in 60-mm dishes with culture medium followed by treatment for the indicated times with compounds in Dulbecco’s Modified Eagle Medium containing 10% FBS. The cells were trypsinized and then washed twice with cold phosphate-buffered saline and fixed with ice-cold 70% ethanol at −20°C overnight. Cells were then washed twice with phosphate-buffered saline, incubated with 20mg/ml RNase A and 200mg/ml propidium iodide (PI) in phosphate-buffered saline at room temperature for 30min in the dark and subjected to flow cytometry analysis using the FACS Calibur flow cytometer. Data were analyzed using the ModFit LT (Verity Software House, Topsham, ME) software program.
Immunoblotting
Cells (5×105) were seeded in 10-cm dishes overnight and treated with ABPN and harvested at the designated time points. The harvested cells were disrupted with cell lysis buffer (Pierce, Rockford, IL) and the proteins were collected. The protein concentration was determined using a dye-binding protein assay kit (Bio-Rad, Hercules, CA) as described in the manufacturer’s manual. Protein lysates (20–80 μg) were subjected to SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to polyvinylidene difluoride membranes. After blotting, the membranes were incubated with a specific primary antibody at 4°C overnight. Protein bands were visualized on film using a chemiluminescence detection kit after hybridization with an alkaline phosphatase-linked secondary antibody. Protein bands were also visualized by LAS 4000 imaging system (GE Healthcare Biosciences, Pittsburgh, PA). All blots presented in the manuscript are from a film scan. Blots were quantified using the Image J (NIH) software program.
Real-time PCR
Total RNA was extracted from cultured cells using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA) following the manufacturer’s instructions. The reverse transcription reaction was performed with the amfiRivert cDNA Synthesis Platinum Master Mix (GenDepot, Barker, TX). Expression of the indicated genes was assessed with a 7500 Real Time PCR system (Applied Biosystems, Carlsbad, CA) using the Power SYBR Green Master Mix (Life Technology, Grand Island, NY). Reaction plates were incubated in a 96-well thermal cycling plate at 95°C for 10min and then underwent 40 cycles of 15 s at 95°C and 1min at 59°C. All reactions were performed in triplicate. Relative quantitation (RQ) was calculated using the 2−ΔCt method, where ΔCt symbolizes the change in Ct between the sample and reference mRNA. The following primers were used to detect expression–GAPDH: 5′-AGCCACATCGCTCAGACAC-3′ (forward), 5′-GCCCAATACGACCAAATCC-3′ (reverse); Met: 5′-TTGGATAGGCTTGTAAGTGCCC-3′ (forward), 5′-TACTGCA CTTGTCGGCATGAA-3′ (reverse); Egfr: 5′-AGGACCAAGCAACATGGTCA-3′ (forward), 5′-CCTTGCAGCTGTTTTCACCT-3′ (reverse); ErbB3: 5′-CCCTGC CATGAGAACTGCAC-3′ (forward), 5′-TCACTGTCAAAGCCATTGTCAGAT-3′ (reverse); Nrdp1: 5′-GAGGAGGGATGGTGGTAGAGA-3′ (forward), 5′-TTCCCAG TGACAAGCTCCAT-3′ (reverse); BRUCE: 5′-CTTGGTCTGAACACGAAAGACA-3′ (forward), 5′-TCCATCCGTACAAGGAAACTGT-3′ (reverse).
RNA interference
BxPC3 cells were grown in 60-mm dishes and transfected with an Nrdp1-specific small interfering RNA oligonucleotide (si-RNF41/Nrdp1; Cat no: #1130460; Bioneer, Seoul, Korea) or scrambled oligonucleotides (si-scrambled; Cat no: #SN-1001 Bioneer), using Lipofectamine™ 2000 (Invitrogen, Grand Island, NY) according to the manufacturer’s instructions. To confirm knockdown, cells transfected with si-RNF41/Nrdp1 or scrambled oligonucleotide were harvested for protein extraction and immunoblotting.
Animals
All animal procedures were conducted in accordance with animal care guidelines provided by Seoul National University (Seoul, Korea). Male athymic nude mice (6-week-old) were purchased from the Institute of Laboratory Animal Resources at Seoul National University. Animals were acclimated for 1 week prior to the study and had free access to food and water. The animals were housed in climate-controlled quarters with a 12-h light/dark cycle.
Xenograft model
BxPC3 pancreatic cancer cells (100 μl) were mixed with 100 μl BD Matrigel (BD Biosciences, San Jose, CA) and cells (5×106) were implanted s.c. into a hind flank of each mouse. Eight mice were used per experimental group. Mice were treated when their tumor volume reached 50–100mm3 as measured using calipers and the volume was estimated using the equation V = π/6 (l × h × w). ABPN (0.2 or 1mg/kg) or erlotinib (1 or 50mg/kg) was administered intraperitoneally 5 days per week. Tumor volume was measured every 7 days, and tumor weight was measured after excision on the final day of the experiment. After all mice were killed, a portion of the tumor tissue was fixed in formalin and embedded in paraffin for slide production with the remainder frozen in liquid nitrogen for disruption and protein analysis.
Immunoblotting of tumor tissues
After pulverizing, tumor tissues were homogenized (Bullet Blender, Next Advance) and protein was extracted using the T-PER tissue protein extraction reagent (Thermo Scientific, Waltham, MA). Protein concentration was quantified and loaded on gels for analysis as described above.
Immunohistochemistry
Tumor tissues were prepared for immunohistochemical analysis of phosphorylated ErbB3. Tissue sections (5 μm thick) from 10% neutral formalin solution-fixed paraffin-embedded tissues were cut on silane-coated glass slides and then deparaffinized with xylene and dehydrated through a graded alcohol bath. The deparaffinized sections were boiled in citric acid buffer (pH 6.0) for antigen retrieval. Each section was treated with hydrogen peroxide solution. The VECTASTAIN Elite ABC Kit (Vector Labs, Southfield, MI) was used for further detection and slides were incubated with primary antibodies overnight at 4°C. ImmPACT DAB (Vector Labs) was used for staining and Mayer’s hematoxylin (Sigma-Aldrich) was applied as a counterstain.
Synergy assessment
Combination index (CI) was used to quantify synergism or antagonism for two drugs (20) using the following formula:
In this case, CI < 1, = 1 or > 1 indicate synergism, independence or antagonism, respectively. In the denominators, (Dx)1 represents D1 ‘alone’ that inhibits a system x%, and (Dx)2 is for D2 ‘alone’ that inhibits a system x%. In the numerators, (D)1 and (D)2 ‘in combination’ also inhibit x%. CI was calculated for every dose of two drug pairs. Fraction affected (F a) is fractional inhibition of a phenotype by compound treatment(s). Fa of a group was calculated as F a = percent inhibition of cell viability/100. Fraction affected-combination index plot was drawn with every group treated with more than one compound.
Statistical analysis
Bar graphs indicate the mean values of all replicates performed independently with error ranges indicated. Experiments using cells are shown as representative, and have been repeated a minimum of three times. The software used for statistical analysis was GraphPad Prism v.6 and MATLAB. One-way ANOVA was used to compare two groups. ANOVA with Dunnett’s test was used for multiple comparisons within experiments. As indicated in each Figure legend, data are presented as mean values ± standard deviation (SD). P values are indicated with and asterisk (*) and/or pound sign (#); (*P < 0.05; **P < 0.01 and ***P < 0.001).
Results
ABPN selectively targets cancer cells and enhances gemcitabine-induced cell death
We examined the effect of ABPN on BxPC3 pancreatic cancer cells compared to all-transretinoic acid (ATRA), 5-FU, gemcitabine (Gem) and erlotinib (Er), which are common therapeutics for pancreatic cancer treatment (9). ABPN exhibited significant inhibitory effects on BxPC3 cell growth and was more potent than ATRA, 5-FU or erlotinib (Figure 1A and Supplementary Figure1, available at Carcinogenesis Online). We determined the effect of ABPN (1 μM) against multiple human pancreatic cancer cell lines, including BxPC3, HPAC, Panc-1 and Mia Paca-2. BxPC3 and HPAC cells were sensitive to ABPN, whereas Panc-1 and Mia Paca-2 cells were relatively insensitive (Figure 1B). The effect of ABPN on normal cells, human CCD-112CoN colon fibroblasts and human dermal fibroblasts was relatively low (Figure 1C), suggesting that ABPN selectively targets certain types of cancer cells. We also measured apoptosis in normal cells treated with ABPN. Whereas paclitaxel induced significant apoptosis, ABPN had much less effect on human dermal fibroblast cells, further confirming that ABPN shows minimal toxicity toward normal cells (Supplementary Figure 2A, available at Carcinogenesis Online). Interestingly, a previous study reported that erlotinib treatment had a similar sensitivity profile against the pancreatic cancer cell lines BxPC3, HPAC, Panc-1 and Mia Paca-2 (21). Thus, we further analyzed the effect of ABPN compared to erlotinib in these four pancreatic cancer cell lines. ABPN showed a marked dose-dependent inhibitory effect against BxPC3 and HPAC cells. However, Panc-1 and Mia Paca-2 pancreatic cancer cells were relatively resistant (Figure 1D). Even though the sensitivity profile of ABPN showed a similar pattern to erlotinib, ABPN was significantly more potent (Figure 1D). Erlotinib is clinically prescribed with gemcitabine to enhance gemcitabine’s chemotherapeutic effect (9,11). We treated cells with ABPN combined with gemcitabine to compare with erlotinib and gemcitabine cotreatment. Results show that combining gemcitabine (30 or 100nM) with ABPN (0.5 or 1 μM) inhibited growth substantially better than the combination of gemcitabine and erlotinib (Figure 1E). Also, calculation of the combination index based on viability demonstrated that ABPN and gemcitabine cotreatment generates synergistic inhibitory effects (Figure 1E and Supplementary Figure 2B, available at Carcinogenesis Online). These results suggest that ABPN selectively targets cancer cells with low toxicity toward normal cells and that ABPN might enhance the chemotherapeutic effects of gemcitabine in pancreatic cancer.
Figure 1.
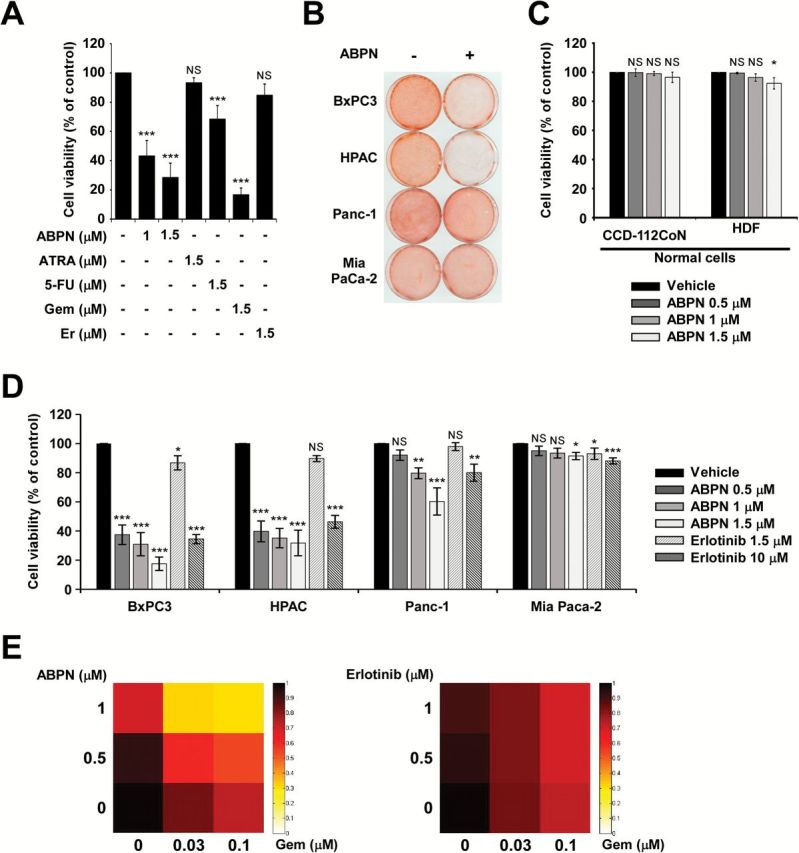
Effect of ABPN, ATRA, 5FU, gemcitabine or erlotinib on pancreatic cancer cell viability. (A) BxPC3 pancreatic cancer cells were treated with ABPN, ATRA, 5-FU, gemcitabine (Gem) or erlotinib (Er) for 72h. Data shown are from three independent experiments. (B) ABPN differentially affects pancreatic cancer cell growth. BxPC3, HPAC, Panc-1 and Mia Paca-2 pancreatic cancer lines were treated with ABPN (1 μM) for 72h and then stained with sulforhodamine B-based solution to detect viable cells. (C) Effect of ABPN on normal cells. CCD-112CoN (normal colon) or HDF (human dermal fibroblasts) were treated with ABPN at the indicated concentrations for 72h before measuring cell viability. All data are presented as mean values ± SD. Data shown are from three independent experiments. (D) Viability of BxPC3, HPAC, Panc-1 and Mia Paca-2 pancreatic cancer cells was measured after treatment with ABPN or erlotinib for 72h. All data are presented as mean values ± SD and data shown are from three independent experiments. One-way ANOVA and Dunnett’s test. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, significant differences between DMSO-treated control and drug-treated groups. All data are presented as mean values ± SD (in triplicate). (E) Cotreatment of ABPN with gemcitabine enhances growth inhibition. BxPC3 cells were treated as indicated for 48h and then viability was measured in triplicate. Heat maps were generated using the MATLAB software program and the color intensity represents viability.
ABPN induces apoptosis of BxPC3 and HPAC pancreatic cancer cells
To further study the potential anticancer effects of ABPN, we performed cell cycle analysis. ABPN induced G1 cell cycle arrest in BxPC3 cells (Table 1) with a concomitant increase in apoptosis, which was more pronounced than that induced by ATRA or erlotinib treatment (Figure 2A). Examination of cell cycle progression after synchronization at the G2/M phase further confirmed that ABPN strongly prevents the G1 to S phase transition (Supplementary Figure 3A, available at Carcinogenesis Online). In addition, the expression of cyclin D1, a cell cycle regulator required for progression through the G1 phase, was downregulated by ABPN (Supplementary Figure 3B, available at Carcinogenesis Online). Western blotting showed that ABPN treatment led to activation of caspase-3 and PARP in BxPC3 and HPAC cells (Figure 2B and Supplementary Figure 4A, available at Carcinogenesis Online). However, the ABPN-resistant Panc-1 and Mia Paca-2 cells did not show significant cleavage of PARP compared to the sensitive cell lines (Figure 2C and Supplementary Figure 4B, available at Carcinogenesis Online). Because the sensitivity of pancreatic cancer cells to ABPN was similar to that of erlotinib, and erlotinib sensitivity is known to depend on ErbB3 levels (21), we analyzed the expression levels of ErbB3 and related proteins in pancreatic cancer cells. The results indicated that higher levels of ErbB3 and Met were associated with sensitivity towards ABPN, suggesting that ABPN-induced cytotoxicity could be linked to ErbB3 and Met (Figure 2D and Supplementary Figure 4C, available at Carcinogenesis Online).
Table 1.
Effect of ABPN on cell cycle distribution
| Group | G1 (%) | S (%) | G2/M (%) |
|---|---|---|---|
| Control (48 h) | 38.85±1.38 | 44.10±0.91 | 17.38±0.48 |
| ABPN 1.5 μM (48 h) | 59.90±2.56*** | 24.48±3.04*** | 15.61±0.60* |
| ATRA 1.5 μM (48 h) | 37.43±1.14 | 43.58±1.07 | 18.99±0.85* |
| Er 1.5 μM (48 h) | 48.70±1.64** | 35.80±1.06 *** | 15.49±1.58 |
| Control (72 h) | 40.16±0.55 | 42.04±0.47 | 17.81±0.30 |
| ABPN 1.5 μM (72 h) | 61.41±0.77*** | 29.73±1.32 *** | 8.86±0.56*** |
| ATRA 1.5 μM (72 h) | 41.93±1.29 | 40.76±1.26 | 17.31±1.23 |
| Er 1.5 μM (72 h) | 47.67±1.73** | 40.02±0.58** | 12.30±1.19** |
BxPC3 cells were seeded on 60-mm dishes and treated with ABPN for 48 or 72h. Cell cycle was analyzed using flow cytometry as described in Materials and methods (*P < 0.01; **P < 0.01; ***P < 0.001, significant differences between DMSO-treated control and drug-treated group). Data were generated using three different plates per group measured at the same time.
Figure 2.

ABPN triggers apoptosis. (A) Treatment of cells with ABPN increases the subG1 fraction. BxPC3 cells were treated with ABPN, ATRA or erlotinib as indicated and cell cycle was analyzed using flow cytometry. All data are presented as mean values ± SD (in triplicate). (B) ABPN induces cleavage of caspase-3 and PARP in BxPC3 and HPAC cells. (C) Differential effects of ABPN on PARP cleavage. (D) Expression of ErbB3, Met, EGFR, ErbB2 in BxPC3, HPAC, Panc-1 and Mia Paca-2 cancer cells. β-Actin served as a loading control and immunoblots are representative images of three independent experiments (one-way ANOVA and Dunnett’s test. NS, not significant; *P < 0.05, **P < 0.01, ***P < 0.001, significant differences between DMSO-treated control and drug-treated groups). All data are presented as mean values ± SD (in triplicate).
ABPN decreases ErbB3 and Met protein levels
Because cells expressing high levels of ErbB3 and Met were more sensitive to ABPN treatment, we investigated whether ErbB3 and Met themselves were a target of inhibition. Treatment with ABPN decreased the expression of ErbB3 and Met and also reduced phosphorylation of Akt and p70S6K in BxPC3 cells (Figure 3A and Supplementary Figure 5A, available at Carcinogenesis Online). Treatment with ABPN did not alter the mRNA levels of ErbB3 and Met, implying the possible involvement of a post-translational control mechanism in ABPN-induced ErbB3 downregulation (Figure 3B and Supplementary Figure 6A, available at Carcinogenesis Online). Previous studies suggest that Nrdp1 is an E3 ligase responsible for the degradation of ErbB3 and BRUCE (8,22). Because ABPN caused a decrease in the expression of ErbB3 with no change in the mRNA level, we sought to determine whether a change was occurring in the Nrdp1 protein level. Treatment with ABPN reduced phosphorylation of ErbB3, Met and EGFR and suppressed the expression of ErbB3 and Met in a dose-dependent manner (Figure 3C and Supplementary Figure 5B, available at Carcinogenesis Online). More importantly, Nrdp1 expression was significantly induced when ErbB3 was downregulated, and BRUCE, another substrate of Nrdp1, was also dose-dependently attenuated by ABPN (Figure 3C and Supplementary Figure 5B, available at Carcinogenesis Online). Treatment of HPAC pancreatic cancer cells with ABPN also caused a significant suppression of ErbB3 and Met levels accompanied by Nrdp1 induction (Supplementary Figure 6B, available at Carcinogenesis Online). The mRNA levels of Bruce, Nrdp1 and EGFR did not show any significant change (Figure 3B and Supplementary Figure 6A, available at Carcinogenesis Online). Whereas ABPN downregulated the protein levels of ErbB3 and BRUCE, their mRNA levels remained stable, implying the possibility of ABPN affecting the stability of these proteins.
Figure 3.
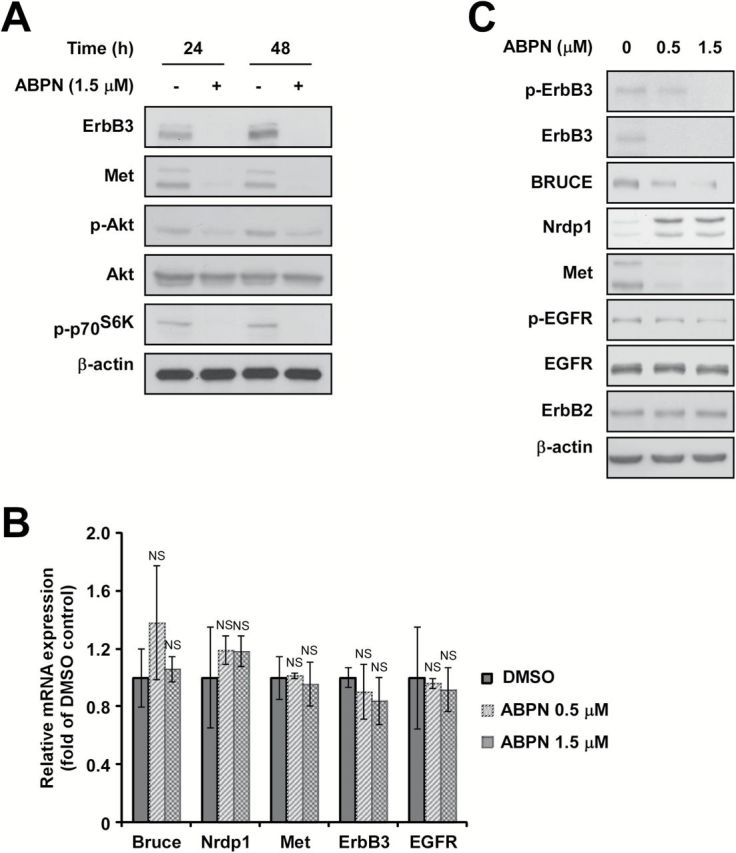
ABPN decreases ErbB3 and Met protein levels and induces Nrdp1 expression in BxPC3 cells. (A) Effect of ABPN on ErbB3 and Met protein expression. (B) Effect of ABPN on ErbB3, Met, Bruce, EGFR and Nrdp1 mRNA levels. (C) Effect of ABPN on ErbB3, Met, BRUCE and Nrdp1 expression. Cells were treated with ABPN at the indicated concentration for 12h followed by disruption for immunoblot analysis. β-actin served as a loading control and immunoblots are representative images of three independent experiments (one-way ANOVA and Dunnett’s test. NS, not significant).
ABPN induces downregulation of ErbB3 through the proteasome degradation pathway
In order to further assess the mechanism of downregulation, we used the proteasome inhibitor, MG132. Cotreatment of cells with MG132 reversed the downregulation of ErbB3 and BRUCE levels induced by ABPN in BxPC3 and HPAC cells (Figure 4A and Supplementary Figure 7A, available at Carcinogenesis Online). We also found that cotreatment with the protein synthesis inhibitor cycloheximide (CHX) exacerbated the decline in ErbB3 and BRUCE expression caused by ABPN in BxPC3 and HPAC cells (Figure 4B and Supplementary Figure 7B, available at Carcinogenesis Online). To determine whether Nrdp1 was responsible for proteasome-dependent downregulation of ErbB3, we induced Nrdp1 knockdown with siRNA, which increased ErbB3 levels and protected the ABPN-induced reduction of ErbB3 (Figure 4C and Supplementary Figure 7C, available at Carcinogenesis Online).
Figure 4.
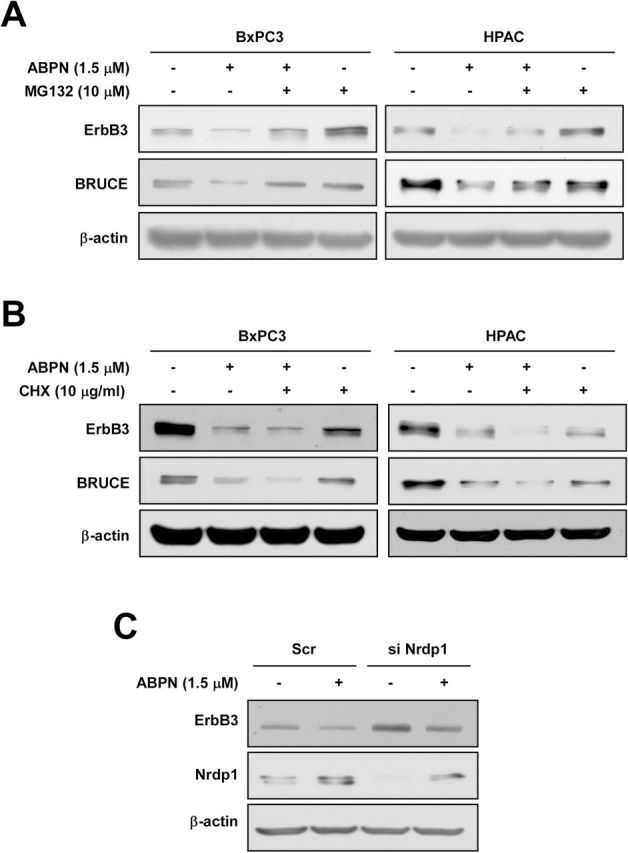
ABPN induces proteasomal degradation of ErbB3 and BRUCE. (A) MG132 recovers ABPN-induced downregulation of ErbB3 and BRUCE. BxPC3 and HPAC cells were treated with MG132 for 3h with or without ABPN. (B) CHX enhances ABPN induction of downregulation of ErbB3 and BRUCE. BxPC3 and HPAC cells were treated with CHX for 3h with or without ABPN. (C) Nrdp1 knockdown suppresses ABPN-induced ErbB3 degradation. BxPC3 cells were transfected with either scrambled siRNA as a control or siNrdp1 for 24h before treatment with ABPN. β-Actin served as a loading control and immunoblots are representative images of three independent experiments.
ABPN suppresses tumor growth in vivo
ABPN’s antitumor activity was evaluated using BxPC3 xenografts in nude mice. ABPN treatment (0.2 or 1mg/kg) showed a dramatic in vivo therapeutic effect by reducing overall tumor size (Figure 5A and B). After killing, we analyzed tumor weight and found that ABPN-treated mice displayed a significant reduction in tumor weight compared to controls or erlotinib-treated animals at the same dosage (Figure 5C). Analysis of tumor tissues obtained from mice showed marked inhibition of phosphorylation and expression of ErbB3 and Met and increased Nrdp1 protein levels in the ABPN-treated group (Figure 6A and Supplementary Figure 8, available at Carcinogenesis Online). Immunohistochemical analysis further demonstrated attenuation of phosphorylated ErbB3 in the ABPN-treated group (Figure 6B).
Figure 5.
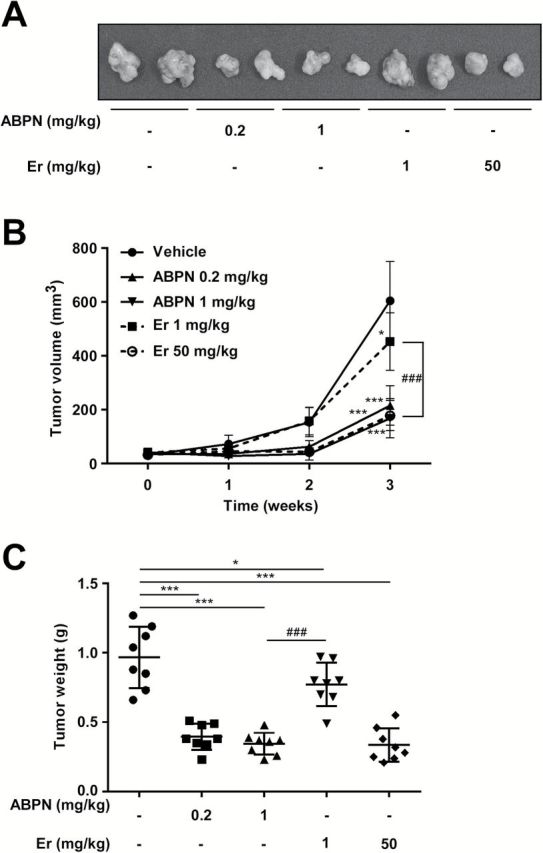
ABPN inhibits BxPC3 tumor growth in vivo. (A) Representative photographs of BxPC3 xenograft tumors. Tumors were excised on the final day of the experiment. Eight mice were used per experimental group. (B) Effect of ABPN or erlotinib on tumor volume. Tumor volumes were measured every 7 days with a caliper. (C) Effect of ABPN and erlotinib (Er) on tumor weight. Tumors were weighed after killing the mice on the final day (one-way ANOVA and Dunnett’s test after log-transformation of data; *P < 0.05, ***P < 0.001, significant differences between vehicle-only control and ABPN- or erlotinib-treated groups. ###P < 0.001, significant difference between ABPN 1mg/kg and erlotinib 1mg/kg-treated groups).
Figure 6.

ABPN reduces ErbB3 and Met expression in tumor tissues. (A) Effect of ABPN on protein expression in tumor tissues. BxPC3 tumor tissues were homogenized and proteins were extracted for immunoblotting. Tumor lysates from three randomly selected mice per group were used. (B) Effect of ABPN on phosphorylation of ErbB3 in tumor tissues. Tissues were processed as described in the Materials and Methods and stained with the corresponding antibody.
Discussion
Our results demonstrate the potential for chemotherapeutic application of the retinoid, ABPN, against pancreatic cancer. We previously identified ABPN as having potent anticancer activity, although the mechanism of action was not clearly understood (19). We now report that concentrations of ABPN in the low micromolar range are sufficient to substantially downregulate phosphorylation of EGFR, as well as phosphorylation and total protein content of the oncoproteins ErbB3 and Met in specific pancreatic cancer cell lines. This is accompanied by decreased cell viability and induction of apoptotic factors, an outcome reflected in reduced tumor size in mouse xenografts. Unexpectedly, ABPN treatment also resulted in downregulation of BRUCE, a large 530kDa membrane-associated inhibitor of apoptosis protein (IAP). BRUCE antagonizes both precursor and mature forms of Smac and caspase 9, and its degradation can directly lead to apoptosis (22,23).
At identical concentrations, we observed that ABPN was more effective at inhibiting cancer growth than erlotinib or all-trans retinoic acid. More importantly, in combination with gemcitabine, the effect of ABPN cotreatment was greater than that achieved with the erlotinib/gemcitabine combination. We hypothesize that this is due to erlotinib’s mechanism of action being limited only to inhibition of EGFR/ErbB3 phosphorylation, whereas ABPN affects the expression of a wider range of oncogenic targets. Importantly, ABPN did not induce deleterious effects in normal cells, including CCD-112CoN human colon fibroblasts and human dermal fibroblasts. Such selectivity is likely due to the fact that EGFR/ErbB3 and Met are normally present at relatively lower levels in normal cells whereas uncontrolled proliferation of cancer cells can sometimes be dependent upon EGFR/ErbB3 overexpression (24). This is supported by the observation that ABPN treatment elicited stronger effects in BxPC3 and HPAC pancreatic cancer cell lines with relatively higher expression of ErbB3 and Met, while having less pronounced effects in the Panc-1 and Mia Paca-2 cell lines, which have lower relative endogenous levels of these proteins. Previous reports have shown that EGFR is highly expressed in pancreatic cancer and its expression correlates with worsened outcomes (9,25,26). Gemcitabine can induce phosphorylation of EGFR at Tyr845 and Tyr1173 and combined treatment with cetuximab or erlotinib leads to better growth inhibition (27).
Patients with advanced pancreatic cancers have limited treatment options, and some of the current strategies focus on combinations of gemcitabine with complementary drugs including erlotinib (26). Gemcitabine exerts its antitumor effects in the nucleus, whereas erlotinib is involved in suppression of oncogenic EGFR-mediated signaling pathways. However, membrane receptors including EGFR are known to have a high frequency of mutation in cancer, which directly impacts overall survival when treatment methods involve an EGFR inhibitor (28). The development of resistance to cytotoxic agents such as gemcitabine and other common chemotherapeutics with EGFR inhibitors has been reported (29).
In contrast to direct inhibition of receptor tyrosine kinase activity, we determined that ABPN treatment results in degradation of ErbB3 and Met, thereby addressing some potential issues arising from receptor mutation. The advantages of oncoprotein degradation (as opposed to activity inhibition) has spawned recent interest in regulators of protein turnover, including the role of deubiquitinating enzymes (29–31) that influence proteasome-mediated degradation. ABPN-induced Nrdp1-dependent downregulation of ErbB3 is proteasome-dependent and does not affect mRNA levels. We suggest that the anticancer activity exhibited by ABPN is a result of previously reported Nrdp1-dependent ubiquitination of ErbB3 and BRUCE (8,22) in addition to downregulation of Met by an unknown mechanism. The binding of EGFR and ErbB3 is known to activate several survival mediators relevant to pancreatic cancer, including the PI3-K/Akt pathway (32), while the Met oncogene has been identified as a mediator of drug resistance (33). Our results support previous findings that Nrdp1 functions as a potentially important tumor suppressor (34), although the full nature of its function in pancreatic cancer proliferation needs further investigation. Exactly how ABPN induces upregulation of Nrdp1 also remains to be elucidated. The possibility exists that ABPN directly inhibits an unidentified negative regulator of Nrdp1, such as a transcriptional repressor or ubiquitinating protein of which Nrdp1 is a substrate. Alternatively, it may play a role in attenuating Nrdp1 autoubiquitination.
The mechanism responsible for Nrdp1-dependent degradation of Met is also an intriguing question. Previous reports indicate that crosstalk exists between RTK signaling pathways, with some receptors able to regulate the activity of others (35). Similarly, we observed that knockdown of ErbB3 by siRNA led to reduction in Met levels (data not shown), implying that ErbB3 might be involved in Met stability. Such a mechanism, if it exists, would explain why Met levels decrease in response to ABPN treatment. However, our finding that Met mRNA levels remain stable during treatment suggests that ErbB3 may block Met degradation at the protein level.
The rationale for undertaking combination chemotherapy is to enhance cancer cell death by suppressing multiple signaling pathways and to reduce the likelihood of cancer cells developing resistance through mutation of alternate survival pathways. The suppression of multiple targets by a single compound may represent a more effective chemotherapeutic strategy for a number of reasons. It simplifies dosing regimens and reduces the extent of clinical study needed to determine pharmacokinetic parameters and interactions with other drugs, as well as lowering the risk of unforeseen toxicity emerging in a wider population. It also helps to address the propensity of cancer cells to hijack signaling pathways to compensate for chemotherapeutic challenge. The Met oncogene is known to play a causative role in gefitinib-resistance in lung cancer (13). This has led to a suggestion for new strategies to incorporate simultaneous blocking of Met and EGFR, thereby reducing the probability of emerging resistance. Drugs that influence multiple signaling factors are also more likely to be effective against a broader range of malignancies because cancers arising from different tissues may be dependent upon alternative signaling pathways. For example, gene copy number analysis suggests that the PI3-K/Akt pathway is the most frequently altered pathway in ovarian cancers (36), whereas Ras overexpression is commonly associated with invasive breast cancers and pancreatic cancers (2). ABPN could be effective in diverse cancer types that are commonly dependent or addicted to membrane receptors including ErbB3, Met or BRUCE. In our study, we have found that ABPN treatment led to strong induction of cell death as well as G1 cell cycle arrest (Figure 2A and Supplementary Figure 3A, available at Carcinogenesis Online and Table 1). Because ErbB3, EGFR and Nrdp1 have been implicated in the regulation of survival and cell cycle progression (37–40), the effect of ABPN on these proteins likely induced a significant impact on cell cycle transition and survival. Further studies will be required to determine the precise cellular mechanisms affected by ABPN treatment.
In conclusion, we have identified multiple oncogenic factors that are downregulated by a single compound, ABPN. In combination with gemcitabine, ABPN was superior to erlotinib at suppressing pancreatic cancer cell growth while exhibiting no detectable toxicity toward noncancerous human cells. This selectivity may be due to cancer cells relying heavily on the suppression by BRUCE of normal apoptosis pathways, while becoming addicted to EGFR, ErbB3 and Met signaling for proliferation. For cancers overexpressing such factors, ABPN may represent a less toxic and more potent alternative to erlotinib treatment.
Supplementary material
Supplementary Figures 1–8 can be found at http://carcin.oxfordjournals.org/
Funding
National Leap Research Program (2010-0029233) of the National Research Foundation of Korea funded by the Ministry of Science, ICT and Future Planning (MSIP) of Korea, Republic of Korea, and The Hormel Foundation and National Institutes of Health (CA172457, CA166011 and R37 CA081064).
Conflict of Interest Statement: None declared.
Supplementary Material
Glossary
Abbreviations
- ABPN
A4-amino-2-(butyrylamino)phenyl (2E,4E,6E,8E)- 3,7-dimethyl-9-(2,6,6-trimethyl-1-cyclohexenyl)-2,4,6,8-nonatetraenoate
- ATRA
all-trans retinoic acid
- FBS
fetal bovine serum
- 5-FU
5-fluorouracil
References
- 1. Hariharan D., et al. et al. (2008) Analysis of mortality rates for pancreatic cancer across the world. HPB (Oxford), 10, 58–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bardeesy N., et al. et al. (2002) Pancreatic cancer biology and genetics. Nat. Rev. Cancer, 2, 897–909. [DOI] [PubMed] [Google Scholar]
- 3. Jemal A., et al. et al. (2007) Cancer statistics, 2007. CA. Cancer J. Clin., 57, 43–66. [DOI] [PubMed] [Google Scholar]
- 4. Liles J.S., et al. et al. (2010) ErbB3 expression promotes tumorigenesis in pancreatic adenocarcinoma. Cancer Biol. Ther., 10, 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dent P., et al. et al. (2003) Stress and radiation-induced activation of multiple intracellular signaling pathways. Radiat. Res., 159, 283–300. [DOI] [PubMed] [Google Scholar]
- 6. Witta S.E., et al. et al. (2009) ErbB-3 expression is associated with E-cadherin and their coexpression restores response to gefitinib in non-small-cell lung cancer (NSCLC). Ann. Oncol., 20, 689–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yamanaka Y., et al. et al. (1993) Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumor aggressiveness. Anticancer Res., 13, 565–569. [PubMed] [Google Scholar]
- 8. Cao Z., et al. et al. (2007) Neuregulin-induced ErbB3 downregulation is mediated by a protein stability cascade involving the E3 ubiquitin ligase Nrdp1. Mol. Cell. Biol., 27, 2180–2188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sutton J.M., et al. et al. (2014) Neoadjuvant therapy for pancreas cancer: past lessons and future therapies. World J. Gastroenterol., 20, 15564–15579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mahadevan D., et al. et al. (2007) Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol. Cancer Ther., 6, 1186–1197. [DOI] [PubMed] [Google Scholar]
- 11. Bareschino M.A., et al. et al. (2007) Erlotinib in cancer treatment. Ann. Oncol., 18 Suppl 6, vi35–vi41. [DOI] [PubMed] [Google Scholar]
- 12. Thomas S.M., et al. et al. (2004) Pharmacokinetic and pharmacodynamic properties of EGFR inhibitors under clinical investigation. Cancer Treat. Rev., 30, 255–268. [DOI] [PubMed] [Google Scholar]
- 13. Engelman J.A., et al. et al. (2007) MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science, 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- 14. Jun H.J., et al. et al. (2012) Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme. Oncogene, 31, 3039–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yun C.H., et al. et al. (2008) The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc. Natl. Acad. Sci. USA, 105, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chambon P. (1996) A decade of molecular biology of retinoic acid receptors. FASEB J., 10, 940–954. [PubMed] [Google Scholar]
- 17. Duester G. (2008) Retinoic acid synthesis and signaling during early organogenesis. Cell, 134, 921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hendrickx A.G., et al. et al. (1992) Teratogenicity of all-trans retinoic acid during early embryonic development in the cynomolgus monkey (Macaca fascicularis). Teratology, 45, 65–74. [DOI] [PubMed] [Google Scholar]
- 19. Um S.J., et al. et al. (2003) Novel retinoic acid derivative ABPN has potent inhibitory activity on cell growth and apoptosis in cancer cells. Int. J. Cancer, 107, 1038–1046. [DOI] [PubMed] [Google Scholar]
- 20. Chou T.C. (2006) Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol. Rev., 58, 621–681. [DOI] [PubMed] [Google Scholar]
- 21. Buck E., et al. et al. (2006) Inactivation of Akt by the epidermal growth factor receptor inhibitor erlotinib is mediated by HER-3 in pancreatic and colorectal tumor cell lines and contributes to erlotinib sensitivity. Mol. Cancer Ther., 5, 2051–2059. [DOI] [PubMed] [Google Scholar]
- 22. Qiu X.B., et al. et al. (2004) Nrdp1-mediated degradation of the gigantic IAP, BRUCE, is a novel pathway for triggering apoptosis. EMBO J., 23, 800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qiu X.B., et al. et al. (2005) The membrane-associated inhibitor of apoptosis protein, BRUCE/Apollon, antagonizes both the precursor and mature forms of Smac and caspase-9. J. Biol. Chem., 280, 174–182. [DOI] [PubMed] [Google Scholar]
- 24. Troiani T., et al. et al. (2012) Targeting EGFR in pancreatic cancer treatment. Curr. Drug Targets, 13, 802–810. [DOI] [PubMed] [Google Scholar]
- 25. Fjällskog M.L., et al. et al. (2003) Expression of molecular targets for tyrosine kinase receptor antagonists in malignant endocrine pancreatic tumors. Clin. Cancer Res., 9, 1469–1473. [PubMed] [Google Scholar]
- 26. Ueda S., et al. et al. (2004) The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas, 29, e1–e8. [DOI] [PubMed] [Google Scholar]
- 27. Morgan M.A., et al. et al. (2008) The combination of epidermal growth factor receptor inhibitors with gemcitabine and radiation in pancreatic cancer. Clin. Cancer Res., 14, 5142–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maughan T., S., et al. et al. (2011) Addition of cetuximab to oxaliplatin-based first-line combination chemotherapy for treatment of advanced colorectal cancer: results of the randomised phase 3 MRC COIN trial. Lancet, 377, 2103–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Byun S., et al. et al. (2013) USP8 is a novel target for overcoming gefitinib resistance in lung cancer. Clin. Cancer Res., 19, 3894–3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pal A., et al. et al. (2014) Emerging potential of therapeutic targeting of ubiquitin-specific proteases in the treatment of cancer. Cancer Res., 74, 4955–4966. [DOI] [PubMed] [Google Scholar]
- 31. Yang J.M. (2007) Emerging roles of deubiquitinating enzymes in human cancer. Acta Pharmacol. Sin., 28, 1325–1330. [DOI] [PubMed] [Google Scholar]
- 32. Frolov A., et al. et al. (2007) ErbB3 expression and dimerization with EGFR influence pancreatic cancer cell sensitivity to erlotinib. Cancer Biol. Ther., 6, 548–554. [DOI] [PubMed] [Google Scholar]
- 33. Krumbach R., et al. et al. (2011) Primary resistance to cetuximab in a panel of patient-derived tumour xenograft models: activation of MET as one mechanism for drug resistance. Eur. J. Cancer, 47, 1231–1243. [DOI] [PubMed] [Google Scholar]
- 34. Yen L., et al. et al. (2006) Loss of Nrdp1 enhances ErbB2/ErbB3-dependent breast tumor cell growth. Cancer Res., 66, 11279–11286. [DOI] [PubMed] [Google Scholar]
- 35. Jo M., et al. et al. (2000) Cross-talk between epidermal growth factor receptor and c-Met signal pathways in transformed cells. J. Biol. Chem., 275, 8806–8811. [DOI] [PubMed] [Google Scholar]
- 36. Huang J., et al. et al. (2011) Frequent genetic abnormalities of the PI3K/AKT pathway in primary ovarian cancer predict patient outcome. Genes Chromosomes Cancer, 50, 606–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Factor V.M., et al. et al. (2010) Loss of c-Met disrupts gene expression program required for G2/M progression during liver regeneration in mice. PLoS One, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holbro T., et al. et al. (2003) The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA, 100, 8933–8938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu H., et al. et al. (2014) Nrdp1 inhibits growth of colorectal cancer cells by nuclear retention of p27. Tumour Biol., 35, 8639–8643. [DOI] [PubMed] [Google Scholar]
- 40. Zhou X., et al. (2009) Gefitinib inhibits the proliferation of pancreatic cancer cells via cell cycle arrest. Anat. Rec. (Hoboken)., 292, 1122–1127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.