

Abstract
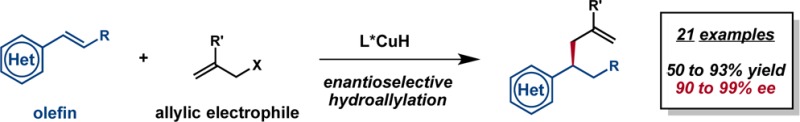
The enantioselective, intermolecular hydroallylation of vinylarenes employing allylic phosphate electrophiles has been achieved through a copper hydride catalyzed process. The protocol described herein can be applied to a diverse set of vinylarene substrates and allows for the installation of the parent allyl group as well as a range of 2-substituted allylic fragments.
The creation of configurationally well-defined stereogenic centers during the course of carbon–carbon bond formation is of great importance for the synthesis of complex organic molecules. Due to the synthetic versatility of the olefin functional group, the enantioselective installation of an allylic fragment has long been regarded as a particularly valuable subset of stereoselective C–C bond-forming transformations.1−3 Among the numerous transition-metal-catalyzed methods for enantioselective allylation, the copper-catalyzed addition of organometallic reagents to allylic electrophiles is distinguished by its applicability to non-heteroatom-stabilized carbon nucleophiles.4 As a consequence, these methods can be readily applied to the construction of C–C bonds remote from polar functional groups in an enantioselective manner, a process for which few other catalytic methods are available. In typical copper-catalyzed allylic substitution reactions, the organocopper intermediate undergoes addition to a prochiral allylic electrophile, resulting in the formation of a stereocenter α to a double bond. In contrast, the addition of an α-chiral organocopper species to an allylic electrophile to furnish a β-stereocenter has seldom been reported and would represent a significant advance (Figure 1A).5
Figure 1.
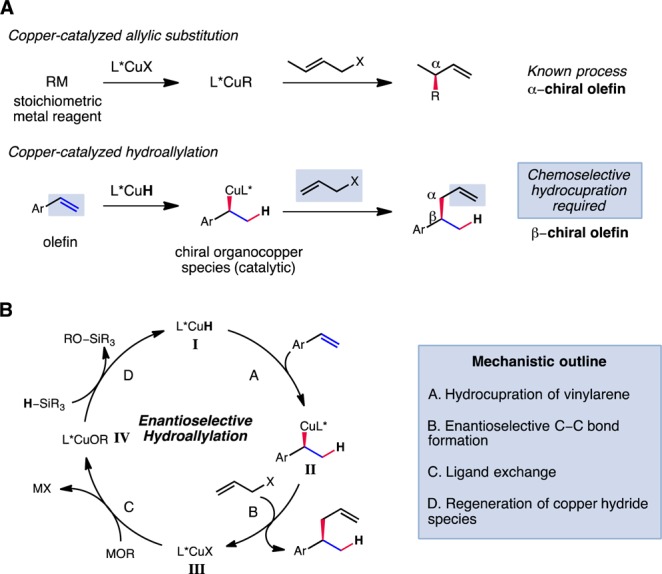
Comparison between copper-catalyzed allylic substitution and hydroallylation (A) and postulated catalytic cycle (B).
Recently, our group has described the reactions of copper hydride (CuH) catalysts with olefins as a means of accessing putative chiral organocopper intermediates. This approach avoids the preparation of a stoichiometric organometallic reagent and allows for the use of mild, functional group tolerant conditions. Using this strategy, we developed several hydroamination reactions,6 an asymmetric indoline synthesis,7 as well as an intramolecular hydroalkylation protocol for the synthesis of four- to six-membered carbo- and heterocycles.8
We anticipated that further advances in CuH chemistry would enable the enantioselective formation of C–C bonds in an intermolecular sense. In particular, we reasoned that the use of allylic electrophiles would permit the development of a hydroallylation process in which simple styrenes and allylic electrophiles could be brought together to form enantioenriched β-chiral olefins. Previous methods to access these products have generally required the use of either stoichiometric chiral reagents or multistep synthetic sequences.9
A postulated catalytic cycle for the proposed formal hydroallylation process is shown in Figure 1B. In analogy to previously reported CuH-catalyzed reactions, insertion of the olefin into the ligated copper hydride species L*CuH (I) would afford enantioenriched benzylcopper species II. This could then be intercepted by the allylic electrophile to furnish the organic product and copper (pseudo)halide species III. Ligand exchange of III with a metal alkoxide and σ-bond metathesis of the resultant copper alkoxide IV with a hydrosilane would regenerate I and complete the catalytic cycle. We recognized that the undesired reactions of I with the allylic electrophile or the β-chiral olefin product were also possible and represented potential complications in efforts to implement the proposed hydroallylation process. In this manuscript, we report that the requisite selectivity could indeed be achieved, permitting the development of a highly enantioselective hydroallylation reaction which affords a wide range of functionalized and unfunctionalized β-chiral olefins with good to excellent yields and outstanding levels of enantioselectivity.
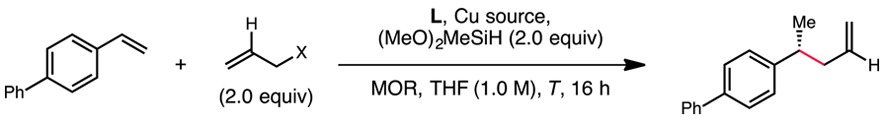
We initiated our investigation by examining the reactivity of 4-phenylstyrene in the presence of common allyl electrophiles under conditions similar to those previously developed for the intramolecular hydroalkylation reaction. When Ph-BPE was employed as the supporting ligand, allyl chloride was found to provide the desired product in moderate yield and with excellent enantioselectivity, while allyl benzoate was unreactive (Table 1, entries 1 and 2). Although reactivity improved upon substitution of allyl chloride with allyl diethylphosphate, the enantioselectivity decreased significantly (entry 3). An evaluation of different bisphosphine ligands at this point revealed that the catalyst derived from Ph-BPE exhibited the best reactivity, although that based on DTBM-SEGPHOS provided product with higher enantiomeric excess (entries 4–6). The enhanced enantioselectivity obtained using allyl chloride as the substrate led us to postulate a beneficial effect from the presence of a chloride anion in the reaction mixture. Indeed, the use of a 1:1 complex of copper(I) chloride and Ph-BPE10 provided product in high yield and with excellent enantioselectivity. The reactivity, however, was somewhat attenuated, and a higher reaction temperature was required (entry 7).11 Switching from LiOMe to LiOt-Bu as the metal alkoxide dramatically improved reactivity while maintaining a high level of enantioselectivity (entry 8). Lastly, the use of diphenylphosphate as the leaving group allowed the desired product to be obtained in high yield and high enantioselectivity at room temperature with a reduced catalyst loading of 2 mol % (entry 12).
Table 1. Optimization of Reaction Conditions.
| entry | X | precatalyst (mol %) | MOR (equiv) | T (°C) | % yielda (% ee)b |
|---|---|---|---|---|---|
| 1 | Cl | Cu(OAc)2/L1c | LiOMe (4.0) | 40 | 47 (96) |
| 2 | OBz | Cu(OAc)2/L1c | LiOMe (4.0) | 40 | <5 |
| 3 | OP(O)(OEt)2 | Cu(OAc)2/L1c | LiOMe (4.0) | 40 | 76 (49) |
| 4 | OP(O)(OEt)2 | Cu(OAc)2/L2c | LiOMe (4.0) | 40 | 50 (95) |
| 5 | OP(O)(OEt)2 | Cu(OAc)2/L3c | LiOMe (4.0) | 40 | 21 (57) |
| 6 | OP(O)(OEt)2 | Cu(OAc)2/L4c | LiOMe (4.0) | 40 | 32 (36) |
| 7 | OP(O)(OEt)2 | L1CuCl (5) | LiOMe (4.0) | 60 | 83 (98) |
| 8 | OP(O)(OEt)2 | L1CuCl (5) | LiOt-Bu (2.0) | rt | 95 (98) |
| 9 | OP(O)(OEt)2 | L1CuCl (5) | NaOt-Bu (2.0) | rt | <5 |
| 10 | OP(O)(OEt)2 | L1CuCl (5) | KOt-Bu (2.0) | rt | <5 |
| 11 | OP(O)(OEt)2 | L1CuCl (2) | LiOt-Bu (2.0) | rt | 74 (98) |
| 12 | OP(O)(OPh)2 | L1CuCl (2) | LiOt-Bu (2.0) | rt | 93 (99) |
Yields determined by NMR with 1,3,5-(MeO)3C6H3 as internal standard, 0.25 mmol scale.
ee determined by HPLC.
LCuH solution was
first prepared
from Cu(OAc)2 (5 mol %), ligand (5.5 mol %), THF, and hydrosilane
(see Supporting Information for experimental
details).
Having found reaction conditions that resulted in an excellent outcome, we set out to investigate the scope of the electrophilic component of this hydroallylation protocol (Table 2). In addition to the parent allyl group, a variety of 2-substituted electrophiles could be handled. For instance, substrates with alkyl groups of varying steric demand were efficiently transformed (3b–3d). A chloro group was also tolerated (3e), providing the desired coupling product in high yield without reduction of the vinyl chloride functional group. Moreover, electron-rich (3f) and electron-poor (3g) aryl substituents could be incorporated. Heteroaryl substituents were similarly feasible (3h and 3i). Regardless of the nature of the 2-substituent, products were isolated in good to high yield, and the enantioselectivity was found to be uniformly high (≥97% ee) in all cases.12
Table 2. Scope of the Allylic Electrophilea.
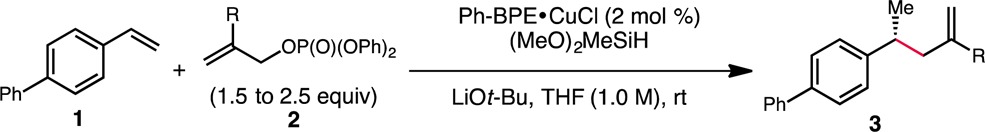
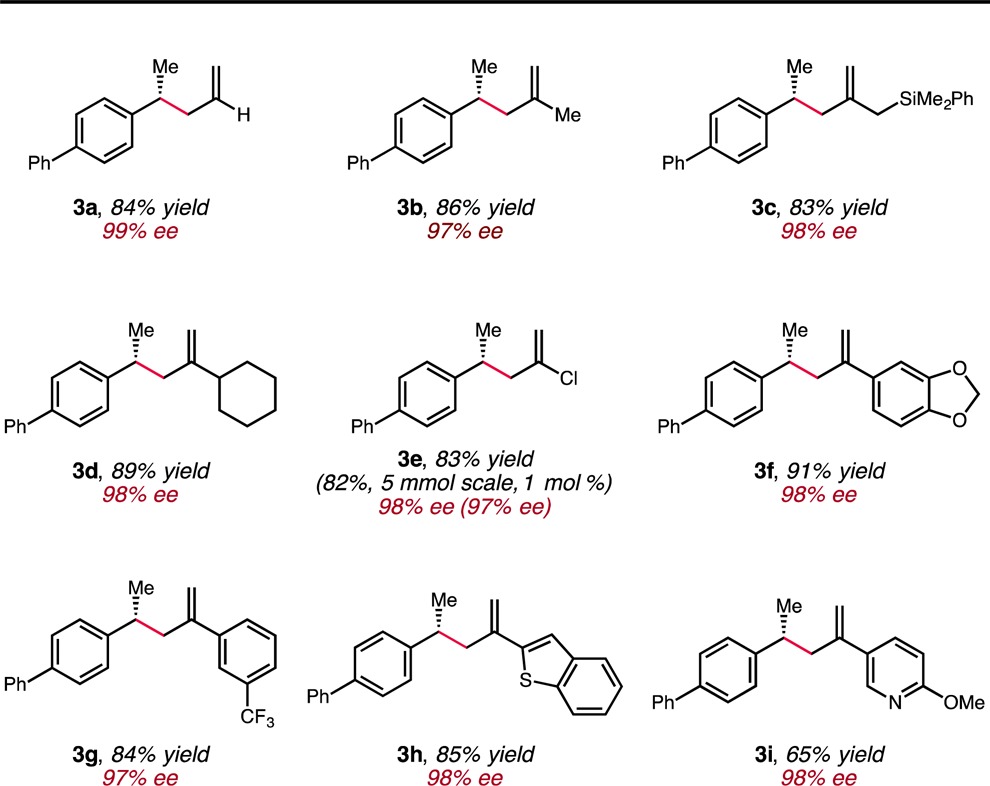
Reactions were conducted on 0.5 mmol scale. The yields reported are the average of two runs.
To evaluate the scalability of the current procedure, the synthesis of 3e was conducted on a 5 mmol scale with the catalyst loading reduced to 1 mol %. Under these conditions, the desired product was isolated in essentially unchanged yield and enantioselectivity (1.05 g of 3e isolated, 82% yield, 97% ee). Finally, the connectivity and absolute configuration of 3h was ascertained by X-ray crystallography. The observed sense of stereoinduction was consistent with that of the previously reported CuH-catalyzed indoline synthesis, which also utilized Ph-BPE as the supporting ligand.7
Subsequently, the scope of the olefin coupling partner was surveyed (Table 3). A range of olefin substrates could be coupled to give the desired β-chiral olefin in moderate to excellent yield and high enantioselectivity (≥90% ee). Styrenes with a range of electronic properties could be employed in this transformation to provide the respective products with good synthetic efficiency (3j–l). Chlorinated styrenes (ortho and para) could also be used to deliver products which are potentially amenable to further functionalization through metal-catalyzed cross-coupling reactions (3j and 3m). Moreover, a tert-butyl ester (3o) and tertiary amide (3n) were readily accommodated. Highlighting the mildness and functional group tolerance of this protocol, the presence of a conjugated diene in 3o and a vinyl bromide in 3n did not lead to the formation of undesired overreduction products. Vinylheteroarenes were also explored as substrates. Vinyl-substituted 2-aminopyrimidine (3p), quinoline (3q), and carbazole (3r) substrates all proved to be suitable coupling partners. A styrene with β-substitution was also found to react efficiently (3s). Remarkably, the hydroallylation reaction could be applied to vinylferrocene as well as a vinylsilane derivative, providing rapid access to highly enantioenriched ferrocene (3t) and alkylsilane (3u) derivatives. In the case of the latter, the product was isolated as a 10:1 mixture of regioisomers, with the minor regioisomer formed from competitive allylation of the terminal position of the vinylsilane.
Table 3. Scope of the Olefin Coupling Partnera.
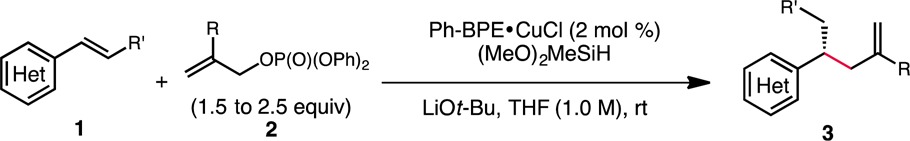
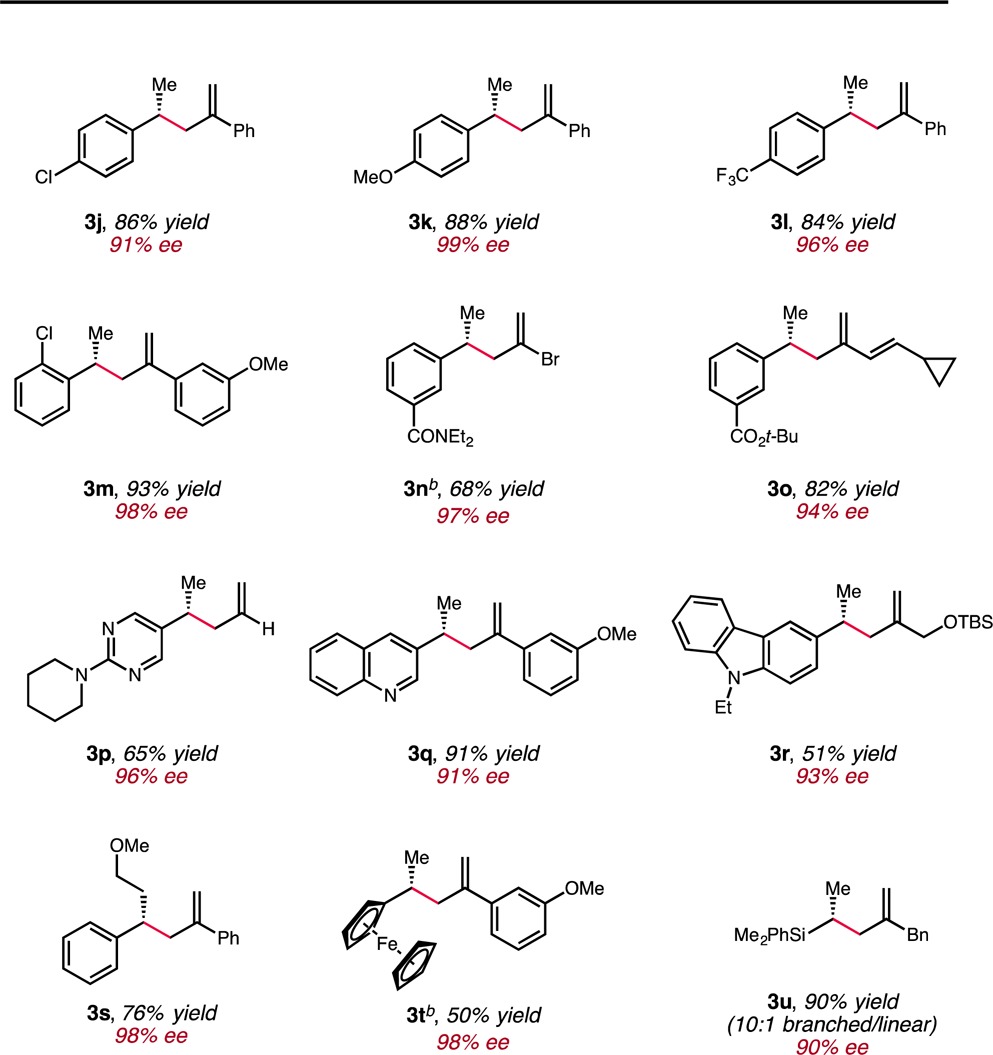
Reactions were conducted on 0.5 mmol scale. The yields reported are the average of two runs.
Conducted with 4 mol % catalyst at 35 °C.
To gain an understanding of the C–C bond forming process in this hydroallylation reaction, a 3,3-dideuterated allylic phosphate was prepared to distinguish between substitution at the 1- and 3-positions of the electrophile. Upon subjecting a mixture of 4-(trifluoromethyl)styene and the deuterated substrate to CuH-catalyzed hydroallylation conditions, 3l-d2 was obtained in high yield, with full deuterium incorporation observed at the internal allylic positions, rather than the terminal olefinic positions (eq 1). This result suggests that substitution occurs through an SN2′-like process via attack of the postulated organocopper species at the 3-position of the allylic phosphate.
 |
1 |
In summary, an intermolecular, enantioselective hydroallylation of vinylarenes has been developed. This process was suitable for a wide range of vinyl(hetero)arenes, allowing for the installation of unsubstituted and 2-substituted allylic fragments in a highly enantioselective manner. Furthermore, the reaction proceeds with a relatively low catalyst loading, and the mild conditions were suitable for substrates containing a variety of functional groups. Efforts to develop other broadly applicable hydroalkylation protocols are currently underway.
Acknowledgments
Research reported in this publication was supported by the National Institutes of Health under award No. GM46059. Y.-M.W. thanks the National Institutes of Health for a postdoctoral fellowship (GM112218). We thank Dr. Peter Mueller for X-ray crystallographic data and Drs. Michael Pirnot and Aaron Sather for their advice on this manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.6b02527.
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews on enantioselective formation of C—C bonds using electrophilic π-allyl species (Tsuji–Trost-type allylation), see:; a Trost B. M. Tetrahedron 2015, 71, 5708. 10.1016/j.tet.2015.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhuo C.-X.; Zheng C.; You S.-L. Acc. Chem. Res. 2014, 47, 2558. 10.1021/ar500167f. [DOI] [PubMed] [Google Scholar]; c Hong A. Y.; Stoltz B. M. Eur. J. Org. Chem. 2013, 2013, 2745. 10.1002/ejoc.201201761. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tosatti P.; Nelson A.; Marsden S. P. Org. Biomol. Chem. 2012, 10, 3147. 10.1039/c2ob07086c. [DOI] [PubMed] [Google Scholar]; e Hartwig J. F.; Stanley L. M. Acc. Chem. Res. 2010, 43, 1461. 10.1021/ar100047x. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Lu Z.; Ma S. Angew. Chem., Int. Ed. 2008, 47, 258. 10.1002/anie.200605113. [DOI] [PubMed] [Google Scholar]; g Helmchen G.; Dahnz A.; Dübon P.; Schelwies M.; Weihofen R. Chem. Commun. 2007, 675. 10.1039/B614169B. [DOI] [PubMed] [Google Scholar]; h Mohr J. T.; Stoltz B. M. Chem. - Asian J. 2007, 2, 1476. 10.1002/asia.200700183. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Trost B. M.; Crawley M. L. Chem. Rev. 2003, 103, 2921. 10.1021/cr020027w. [DOI] [PubMed] [Google Scholar]
- For selected reviews on enantioselective Cu-catalyzed SN2′-like electrophilic allylation reactions, see:; a Langlois J.-B.; Alexakis A. Top. Organomet. Chem. 2011, 38, 235. 10.1007/3418_2011_12. [DOI] [Google Scholar]; b Falciola C. A.; Alexakis A. Eur. J. Org. Chem. 2008, 2008, 3765. 10.1002/ejoc.200800025. [DOI] [Google Scholar]; c Alexakis A.; Bäckvall J.-E.; Krause N.; Pàmies O.; Diéguez M. Chem. Rev. 2008, 108, 2796. 10.1021/cr0683515. [DOI] [PubMed] [Google Scholar]; d Harutyunyan S. R.; den Hartog T.; Geurts K.; Minnaard A. J.; Feringa B. L. Chem. Rev. 2008, 108, 2824. 10.1021/cr068424k. [DOI] [PubMed] [Google Scholar]
- For selected reviews on the enantioselective nucleophilic addition of allylmetal species to C=X bonds, see:; a Yus M.; González-Gómez J. C.; Foubelo F. Chem. Rev. 2011, 111, 7774. 10.1021/cr1004474. [DOI] [PubMed] [Google Scholar]; b Yamada K.-i.; Tomioka K. Chem. Rev. 2008, 108, 2874. 10.1021/cr078370u. [DOI] [PubMed] [Google Scholar]; c Chemler S. R.; Roush W. R. In Modern Carbonyl Chemistry; Otera J., Ed.; Wiley-VCH: Weinheim, Germany, 2000; pp 403–490. For a transfer hydrogenation strategy for the in situ generation of carbon nucleophiles, including allylmetal species, see: [Google Scholar]; d Ketcham J. M.; Shin I.; Montgomery T. P.; Krische M. J. Angew. Chem., Int. Ed. 2014, 53, 9142. 10.1002/anie.201403873. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Patman R. L.; Bower J. F.; Kim I. S.; Krische M. J. Aldrichimica Acta 2008, 41, 95. [PMC free article] [PubMed] [Google Scholar]; For mechanistically distinct hydrovinylation processes leading to similar products, see:; f RajanBabu T. V. Chem. Rev. 2003, 103, 2845. 10.1021/cr020040g. [DOI] [PubMed] [Google Scholar]
- For seminal reports on Cu-catalyzed allylic alkylation, see:; a van Klaveren M.; Persson E. S. M.; del Villar A.; Grove D. M.; Bäckvall J.-E.; van Koten G. Tetrahedron Lett. 1995, 36, 3059. 10.1016/0040-4039(95)00426-D. [DOI] [Google Scholar]; b Dübner F.; Knochel P. Angew. Chem., Int. Ed. 1999, 38, 379.. [DOI] [PubMed] [Google Scholar]; c Luchaco-Cullis C. A.; Mizutani H.; Murphy K. E.; Hoveyda A. H. Angew. Chem., Int. Ed. 2001, 40, 1456.. [DOI] [PubMed] [Google Scholar]; d Alexakis A.; Malan C.; Lea L.; Benhaim C.; Fournioux X. Synlett 2001, 2001, 927. 10.1055/s-2001-14626. [DOI] [Google Scholar]; e Van Veldhuizen J. J.; Campbell J. E.; Giudici R. E.; Hoveyda A. H. J. Am. Chem. Soc. 2005, 127, 6877. 10.1021/ja050179j. [DOI] [PubMed] [Google Scholar]; f Lee Y.; Akiyama K.; Gillingham D. G.; Brown M. K.; Hoveyda A. H. J. Am. Chem. Soc. 2008, 130, 446. 10.1021/ja0782192. [DOI] [PubMed] [Google Scholar]; g Shintani R.; Takatsu K.; Takeda M.; Hayashi T. Angew. Chem., Int. Ed. 2011, 50, 8656. 10.1002/anie.201103581. [DOI] [PubMed] [Google Scholar]; h Shido Y.; Yoshida M.; Tanabe M.; Ohmiya H.; Sawamura M. J. Am. Chem. Soc. 2012, 134, 18573. 10.1021/ja3093955. [DOI] [PubMed] [Google Scholar]; For examples of enantioselective allylation using nonstabilized organometallic nucleophiles catalyzed by other transition metals, see:; i Menard F.; Chapman T. M.; Dockendorff C.; Lautens M. Org. Lett. 2006, 8, 4569. 10.1021/ol061777l. [DOI] [PubMed] [Google Scholar]; j Alexakis A.; El Hajjaji S.; Polet D.; Rathgeb X. Org. Lett. 2007, 9, 3393. 10.1021/ol0713842. [DOI] [PubMed] [Google Scholar]; k Son S.; Fu G. C. J. Am. Chem. Soc. 2008, 130, 2756. 10.1021/ja800103z. [DOI] [PubMed] [Google Scholar]; l Zhang P.; Brozek L. A.; Morken J. P. J. Am. Chem. Soc. 2010, 132, 10686. 10.1021/ja105161f. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Hamilton J. Y.; Sarlah D.; Carreira E. M. J. Am. Chem. Soc. 2013, 135, 994. 10.1021/ja311422z. [DOI] [PubMed] [Google Scholar]; For transition metal-free N-heterocyclic carbene catalyzed allylation, see:; n Lee Y.; Hoveyda A. H. J. Am. Chem. Soc. 2006, 128, 15604. 10.1021/ja067456m. [DOI] [PubMed] [Google Scholar]; o Lee Y.; Li B.; Hoveyda A. H. J. Am. Chem. Soc. 2009, 131, 11625. 10.1021/ja904654j. [DOI] [PMC free article] [PubMed] [Google Scholar]; p Jackowski O.; Alexakis A. Angew. Chem., Int. Ed. 2010, 49, 3346. 10.1002/anie.201000577. [DOI] [PubMed] [Google Scholar]
- A Pd/Cu cooperative catalytic system proceeding through borylated α-chiral organocopper species has recently been reported for enantioselective allylboration:; a Jia T.; Cao P.; Wang B.; Lou Y.; Yin X.; Wang M.; Liao J. J. Am. Chem. Soc. 2015, 137, 13760. 10.1021/jacs.5b09146. [DOI] [PubMed] [Google Scholar]; For the stoichiometric addition of sulfoxide-stabilized α-chiral organocopper species to propargyl electrophiles, see:; b García Ruano J. L.; Marcos V.; Alemán J. Angew. Chem., Int. Ed. 2008, 47, 6836. 10.1002/anie.200802158. [DOI] [PubMed] [Google Scholar]
- a Zhu S.; Niljianskul N.; Buchwald S. L. J. Am. Chem. Soc. 2013, 135, 15746. 10.1021/ja4092819. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhu S.; Buchwald S. L. J. Am. Chem. Soc. 2014, 136, 15913. 10.1021/ja509786v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Niljianskul N.; Zhu S.; Buchwald S. L. Angew. Chem., Int. Ed. 2015, 54, 1638. 10.1002/anie.201410326. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Shi S.; Buchwald S. L. Nat. Chem. 2014, 7, 38. 10.1038/nchem.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Yang Y.; Shi S.-L.; Niu D.; Liu P.; Buchwald S. L. Science 2015, 349, 62. 10.1126/science.aab3753. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Bandar J. S.; Pirnot M. T.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 14812. 10.1021/jacs.5b10219. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Zhu S.; Niljianskul N.; Buchwald S. L. Nat. Chem. 2016, 8, 144. 10.1038/nchem.2418. [DOI] [PMC free article] [PubMed] [Google Scholar]; h Shi S.-L.; Wong Z. L.; Buchwald S. L.. Nature 2016, doi: 10.1038/nature17191.For the independent, contemporaneous development of a related system, see: [DOI] [Google Scholar]; i Miki Y.; Hirano K.; Satoh T.; Miura M. Angew. Chem., Int. Ed. 2013, 52, 10830. 10.1002/anie.201304365. [DOI] [PubMed] [Google Scholar]; j Miki Y.; Hirano K.; Satoh T.; Miura M. Org. Lett. 2014, 16, 1498. 10.1021/ol5003219. [DOI] [PubMed] [Google Scholar]; k Nishikawa D.; Hirano K.; Miura M. J. Am. Chem. Soc. 2015, 137, 15620. 10.1021/jacs.5b09773. [DOI] [PubMed] [Google Scholar]; For a recent report of directed hydroalkylation of internal alkenes, see:; l Xi Y.; Butcher T. W.; Zhang J.; Hartwig J. F. Angew. Chem., Int. Ed. 2016, 55, 776. 10.1002/anie.201509235. [DOI] [PubMed] [Google Scholar]
- Ascic E.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 4666. 10.1021/jacs.5b02316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wang Y.-M.; Bruno N. C.; Placeres Á. L.; Zhu S.; Buchwald S. L. J. Am. Chem. Soc. 2015, 137, 10524. 10.1021/jacs.5b07061. [DOI] [PMC free article] [PubMed] [Google Scholar]; For the CuH-catalyzed hydroalkylation of alkynes, see:; b Uehling M. R.; Suess A. M.; Lalic G. J. Am. Chem. Soc. 2015, 137, 1424. 10.1021/ja5124368. [DOI] [PubMed] [Google Scholar]; c Suess A. M.; Uehling M. R.; Kaminsky W.; Lalic G. J. Am. Chem. Soc. 2015, 137, 7747. 10.1021/jacs.5b03086. [DOI] [PubMed] [Google Scholar]; For a related Cu-catalyzed hydroalkylation of dienes, see:; d Iwasaki T.; Shimizu R.; Imanishi R.; Kuniyasu H.; Kambe N. Angew. Chem., Int. Ed. 2015, 54, 9347. 10.1002/anie.201503288. [DOI] [PubMed] [Google Scholar]
- For an approach using stoichiometric boron reagents, see:; a Nave S.; Sonawane R. P.; Elford T. G.; Aggarwal V. K. J. Am. Chem. Soc. 2010, 132, 17096. 10.1021/ja1084207. [DOI] [PubMed] [Google Scholar]; For a multistep approach using asymmetric methylzirconation, see:; b Novak T.; Tan Z.; Liang B.; Negishi E.-i. J. Am. Chem. Soc. 2005, 127, 2838. 10.1021/ja043534z. [DOI] [PubMed] [Google Scholar]; Limited examples of a dynamic kinetic allylation of benzylic ethers using a chiral Ti complex has been reported:; c Braun M.; Kotter W. Angew. Chem., Int. Ed. 2004, 43, 514. 10.1002/anie.200352128. [DOI] [PubMed] [Google Scholar]
- Initial preparation of the Ph-BPE·CuCl complex:Yazaki R.; Kumagai N.; Shibasaki M. J. Am. Chem. Soc. 2010, 132, 5522. 10.1021/ja101687p. [DOI] [PubMed] [Google Scholar]
- The addition of LiCl to the catalyst prepared from Cu(OAc)2 also led to a significant increase in enantioselectivity. The nature of this chloride anion effect is the subject of ongoing investigations.
- In preliminary investigations, 3-substituted allylic phosphate substrates were also found to undergo the desired transformation. However, under the present reaction conditions, poor diastereoselectivities were obtained. For example, the reaction of cinnamyl diphenylphosphate and 4-phenylstyrene afforded the hydroallylation product as a 1:1.1 mixture of diastereomers in 70% combined yield (NMR). Attempts to prepare a 1,1-dimethyl substituted allylic phosphate resulted in decomposition.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.