

Abstract
The most abundant mRNA post-transcriptional modification is N6-methyladenosine (m6A) that has broad roles in RNA biology1-5. In mammalian cells, the asymmetric distribution of m6A along mRNAs leaves relatively less methylation in the 5′ untranslated region (5′UTR) compared to other regions6,7. However, whether and how 5′UTR methylation is regulated is poorly understood. Despite the crucial role of the 5′UTR in translation initiation, very little is known whether m6A modification influences mRNA translation. Here we show that in response to heat shock stress, m6A is preferentially deposited to the 5′UTR of newly transcribed mRNAs. We found that the dynamic 5′UTR methylation is a result of stress-induced nuclear localization of YTHDF2, a well characterized m6A “reader”. Upon heat shock stress, the nuclear YTHDF2 preserves 5′UTR methylation of stress-induced transcripts by limiting the m6A “eraser” FTO from demethylation. Remarkably, the increased 5′UTR methylation in the form of m6A promotes cap-independent translation initiation, providing a mechanism for selective mRNA translation under heat shock stress. Using Hsp70 mRNA as an example, we demonstrate that a single site m6A modification in the 5′UTR enables translation initiation independent of the 5′ end m7G cap. The elucidation of the dynamic feature of 5′UTR methylation and its critical role in cap-independent translation not only expands the breadth of physiological roles of m6A, but also uncovers a previously unappreciated translational control mechanism in heat shock response.
Given the reversible nature of m6A mRNA methylation8,9, we sought to assess the potential impact of heat shock stress on m6A modification of eukaryotic mRNAs. Using immunofluorescence staining, we first examined the subcellular localization of the entire m6A machinery in a mouse embryonic fibroblast (MEF) cell line before and after heat shock stress. It is believed that m6A modification occurs primarily at nuclear speckles, whereas its functionality takes place in the cytosol (Fig. 1a). Consistent with this notion, both the m6A “writers” (METTL3, METTL14, WTAP) and the “eraser” FTO were predominantly present in the nucleus, whereas the majority of the “reader” YTHDF2 resided in the cytosol (Fig. 1b and Extended Data Fig. 1). In response to heat shock stress, neither the “writers” nor the “eraser” changed their nuclear localization (Extended Data Fig. 1). Surprisingly, nearly all of the YTHDF2 molecules were relocated into the nucleus from the cytosol upon heat shock stress (Fig. 1b). The same phenomenon holds true in HeLa cells. Intriguingly, the protein level of YTHDF2 was also markedly increased after heat shock stress in a manner similar to Hsp70 induction (Fig. 1c). In contrast, neither the m6A “writers” nor the “eraser” showed any differences in protein levels upon stress. Supporting the stress-induced transcriptional up-regulation of YTHDF2, real-time PCR revealed a nearly 4-fold increase of mRNA abundance after heat shock stress (Fig. 1d). The increased YTHDF2 abundance was not due to altered mRNA degradation since heat shock stress had negligible effects on mRNA stability (Extended Data Fig. 2a). Notably, YTHDF2 exhibited a relatively short half-life (t1/2 <1 h) in cells, supporting the importance of stress-induced transcriptional up-regulation. Genes encoding other YTH domain family proteins like YTHDF1 and YTHDF3 also showed up-regulation, although to a lesser extent (Extended Data Fig. 2b). Using a mouse fibroblast cell line lacking the heat shock transcription factor 1 (HSF1)10, we confirmed that YTHDF2 is subject to regulation by HSF1 (Extended Data Fig. 2c). The unexpected stress inducible feature of YTHDF2 suggests a potential role of m6A modification in heat shock response.
Figure 1. YTHDF2 changes cellular localization and expression levels in response to heat shock stress.
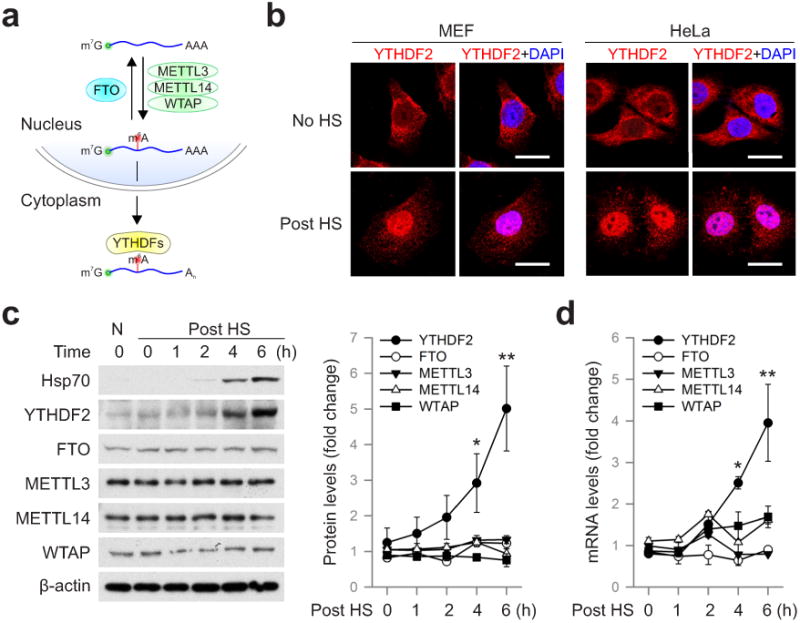
a, Schematic of m6A modification machinery in mammalian cells. b, Subcellular localization of YTHDF2 in MEF and HeLa cells before or 2 h after heat shock (42°C, 1 h). Bar, 10 μm. Representative of at least 50 cells. c, Immunoblotting of MEF cells after heat shock stress (42°C, 1 h). N: no heat shock. The right panel shows the relative protein levels quantified by densitometry and normalized to β-actin. Representative of three biological replicates. d, Same samples in c were used for RNA extraction and real-time PCR. Relative levels of indicated transcripts are normalized to β-actin. Error bars, mean ± s.e.m.; *p < 0.05, **p < 0.01, unpaired two-tailed t-test; n=3, biological replicates (c and d).
Although YTHDF2 primarily serves as the “reader” of m6A, recent proteomic data revealed that YTHDF2 has an extensive physical interaction with the components of m6A “writers”11. Given their co-localization upon heat shock stress, we postulated that the nuclear presence of YTHDF2 could influence the m6A modification and alter the landscape of mRNA methylomes. Using an optimized MeRIP-seq procedure6,12, we sequenced the entire methylated RNA species purified from MEF cells with or without heat shock stress. From a total of 15,454 putative methylation sites, we confirmed the m6A consensus sequence motif as GGAC (Extended Data Fig. 3 and Table 1). Consistent with previous reports6,7, the majority of m6A sites are enriched in the vicinity of the stop codon and in the 3′UTR (Fig. 2a). Unexpectedly, heat shock stress led to an elevated m6A peak in the 5′UTR but not other regions. Reasoning that only a handful of genes undergo up-regulation as a result of heat shock response13,14, we compared the levels of m6A modification between stress-inducible and non-inducible transcripts defined by RNA-seq. It is clear that the up-regulated transcripts showed greater m6A modification in the 5′UTR than the transcripts down-regulated upon stress (Fig. 2b). We next stratified transcripts based on differential changes of m6A modification in the 5′UTR. While transcripts with elevated 5′UTR methylation are mostly up-regulated in response to stress, transcripts exhibiting decreased 5′UTR methylation are largely down-regulated (Fig. 2c, p < 0.001, Mann-Whitney Test). One particular example of stress-induced transcripts is the Hsp70 gene HSPA1A, which not only showed a 90-fold increase of mRNA levels after heat shock, but also displayed a prominent m6A peak in the 5′UTR (Fig. 2d). By contrast, the constitutively expressed Hsc70 gene HSPA8 showed only minor increase in both the mRNA level and the m6A modification in response to heat shock stress (Extended Data Fig. 4). These results suggest that the increased 5′UTR methylation selectively occurs on the stress-inducible mRNAs.
Figure 2. Altered m6A profiles in MEF cells in response to heat shock stress.
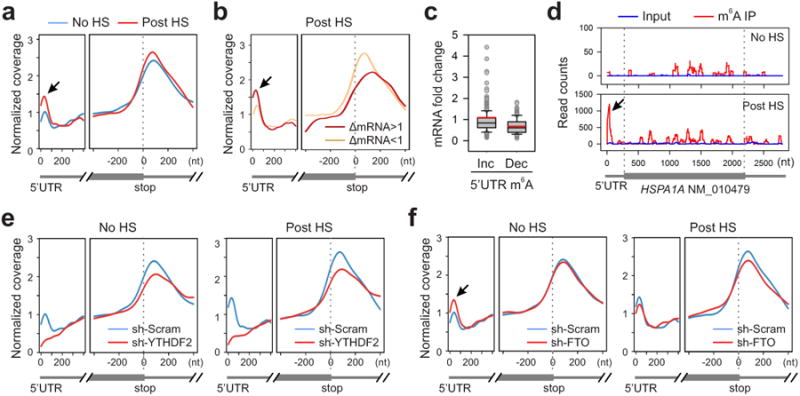
a, Metagene profiles of m6A distribution across the transcriptome of cells before or 2 h after heat shock (42°C, 1 h). Black arrow indicates the m6A peak in the 5′UTR region. b, Transcripts are stratified by different expression levels after heat shock stress, followed by metagene profiles of m6A distribution. c, A box plot depicting fold changes of mRNA levels after heat shock for transcripts showing increased or decreased m6A modification in the 5′UTR. Red line, mean value. d, An example of stress-induced transcript HSPA1A harboring m6A peaks. e, Metagene profiles of m6A distribution across the transcriptome of cells with or without YTHDF2 knockdown, before or after heat shock stress. f, Metagene profiles of m6A distribution across the transcriptome of cells with or without FTO knockdown, before or after heat shock stress.
To examine whether the elevated 5′UTR methylation upon heat shock stress is a result of nuclear localization of YTHDF2, we silenced YTHDF2 in MEF cells using shRNA-expressing lentiviruses. Remarkably, MEF cells lacking YTHDF2 demonstrated a substantial loss of m6A modification in the 5′UTR (Fig. 2e). Upon heat shock stress, these cells no longer showed the elevated 5′UTR methylation as seen in control cells. The abolished 5′UTR methylation in the absence of YTHDF2 was clearly exemplified in HSPA1A that exhibited only background m6A modification in the 5′UTR (Extended Data Fig. 5). This result indicates a novel function of YTHDF2 in heat shock response by promoting 5′UTR methylation on mRNAs transcribed during stress.
YTHDF2 is not a methytransferase per se, and does not bind to mRNAs without prior m6A modification3,7. How does the nuclear presence of YTHDF2 promote selective methylation in the 5′UTR? One possibility is that YTHDF2 protects the pre-existing m6A from FTO-mediated demethylation. Upon heat shock stress, the nuclear localization of YTHDF2 likely limits the accessibility of FTO to newly minted m6A sites, thereby tilting the equilibrium towards methylation. Indeed, an in vitro m6A binding and demethylation assay confirmed direct competition between FTO and YTHDF2 (Extended Data Fig. 6). To demonstrate whether FTO preferentially removes m6A modification from the 5′UTR, we knocked down FTO from MEF cells and examined the m6A distribution across the entire transcriptome. Interestingly, only the 5′UTR region showed an increase of m6A density in cells lacking FTO (Fig. 2f). Additionally, the 5′UTR methylation showed no further increase upon heat shock stress in the absence of FTO.
The 5′UTR is crucial in mediating translation initiation of eukaryotic mRNAs15,16. Under stress conditions, the cap-dependent translation is generally suppressed. However, subsets of transcripts are selectively translated via a poorly understood cap-independent mechanism17-19. To investigate whether differential methylation of 5′UTR influences the translational status of these mRNAs, we conducted ribosome profiling of MEF cells with or without heat shock stress. Among the genes undergoing stress-induced transcriptional up-regulation, many of them not only showed elevated m6A modification in the 5′UTR, but also demonstrated increased ribosome occupancy in the coding region (Fig. 3a). Several prominent examples are genes encoding heat shock proteins, in particular Hsp70 (Extended Data Table 2). Therefore, the coordinated up-regulation of transcription and 5′UTR methylation is coupled with robust translation in response to heat shock stress.
Figure 3. m6A modification promotes selective translation under heat shock stress.
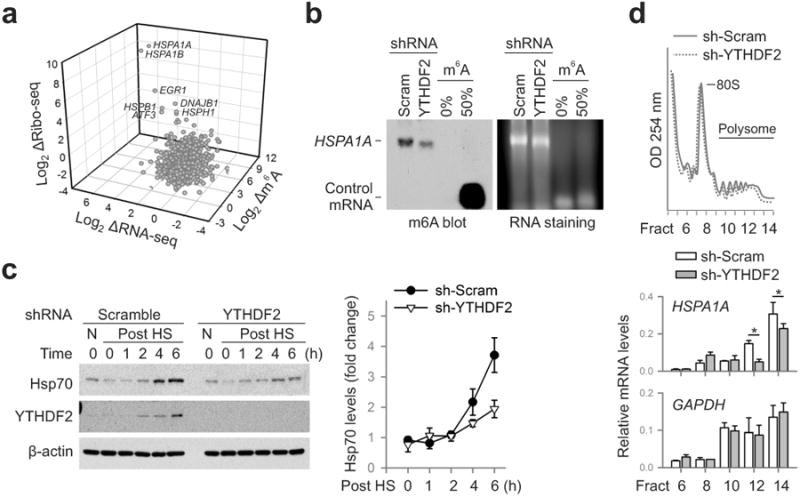
a, A 3-D plot depicting fold changes (log2) of mRNA abundance, CDS ribosome occupancy, and 5′UTR m6A levels in MEF cells after heat shock stress. b, m6A blotting of HSPA1A purified from MEF with or without YTHDF2 knockdown. mRNAs synthesized by in vitro transcription in the absence or presence of m6A were used as control. Representative of two biological replicates. c, Immunoblotting of MEF cells with or without YTHDF2 knockdown after heat shock stress (42°C, 1 h). N: no heat shock. The right panel shows the relative protein levels quantified by densitometry and normalized to β-actin. Representative of three biological replicates. d, MEF cells with or without YTHDF2 knockdown were subject to heat shock stress followed by sucrose gradient sedimentation. Specific mRNA levels in polysome fractions were measured by qPCR. The values are first normalized to the spike in control then to the total. Error bars, mean ± s.e.m.; * p < 0.05, unpaired two-tailed t-test; n=3, biological replicates (c and d).
To validate the causal relationship between stress-induced 5′UTR methylation and selective translation, we examined Hsp70 synthesis in cells with differential m6A modification. Knocking down YTHDF2 leads to depleted 5′UTR methylation as revealed by MeRIP-seq (Fig. 2e). Indeed, direct m6A blotting of HSPA1A purified from heat shock stressed MEFs confirmed the marked reduction of methylation in cells lacking YTHDF2 (Fig. 3b). Remarkably, the heat shock-induced Hsp70 synthesis was substantially dampened in the absence of YTHDF2 (Fig. 3c). The comparable Hsp70 mRNA levels in cells with or without YTHDF2 knockdown indicate that the reduced Hsp70 synthesis is a result of translational deficiency (Extended Data Fig. 7). Further supporting this notion, the Hsp70 transcript but not GAPDH showed much less enrichment in the polysomes of MEF cells lacking YTHDF2 (Fig. 3d).
Reasoning that YTHDF2 competes with FTO in preserving 5′UTR m6A modification, we speculated that FTO knockdown would increase the 5′UTR methylation as well as the translation efficiency of Hsp70 mRNA. This was indeed the case. Direct m6A blotting of HSPA1A purified from stressed MEFs lacking FTO revealed a clear increase of methylation when compared to the scramble control (Extended Data Fig. 8). Importantly, FTO knockdown potentiated the synthesis of Hsp70 after heat shock stress. Collectively, these results established the functional connection between dynamic 5′UTR methylation and selective mRNA translation during stress.
It is commonly believed that the 5′UTR of Hsp70 mRNA recruits the translational machinery via an internal ribosome entry site (IRES)20-23. However, conflicting results exist and the exact cap-independent translation-promoting determinants remain elusive23,24. Given the fact that the normal 5′ end cap structure is a methylated purine (N7-methylguanosine, m7G), we hypothesize that the stress-induced m6A in the 5′UTR enables selective translation by acting as a functional cap substitute. To test this hypothesis, we performed a firefly luciferase (Fluc) reporter assay in MEF cells by transfecting mRNAs synthesized in the absence or presence of m6A (Fig. 4a). For the messenger without 5′UTR, random incorporation of m6A slightly reduced the Fluc activity after mRNA transfection. In the presence of 5′UTR from Hsp70, but not tubulin, the incorporation of m6A markedly increased the Fluc activity in transfected MEF cells. Notably, m6A incorporation does not affect the stability of the synthesized mRNAs in transfected cells (Extended Data Fig. 9a). We next replaced the 5′ end m7G cap with a non-functional cap analog ApppG. As expected, the resultant mRNA did not support translation in the absence of 5′UTR or in the presence of tubulin 5′UTR (Fig. 4a and Extended Data Fig. 9b). Only when the Hsp70 5′UTR was present, was the translating-promoting feature clearly manifested after m6A incorporation, in particular under stress conditions (Fig. 4a). This effect is specific to m6A modification but not m6Am because ribose methylation in the form of 2′-O-MeA suppressed translation of the Fluc reporter bearing Hsp70 5′UTR (Fig. 4a). Therefore, methylation of Hsp70 5′UTR in the form of m6A promotes cap-independent translation.
Figure 4. Selective 5′UTR m6A modification mediates cap-independent translation.
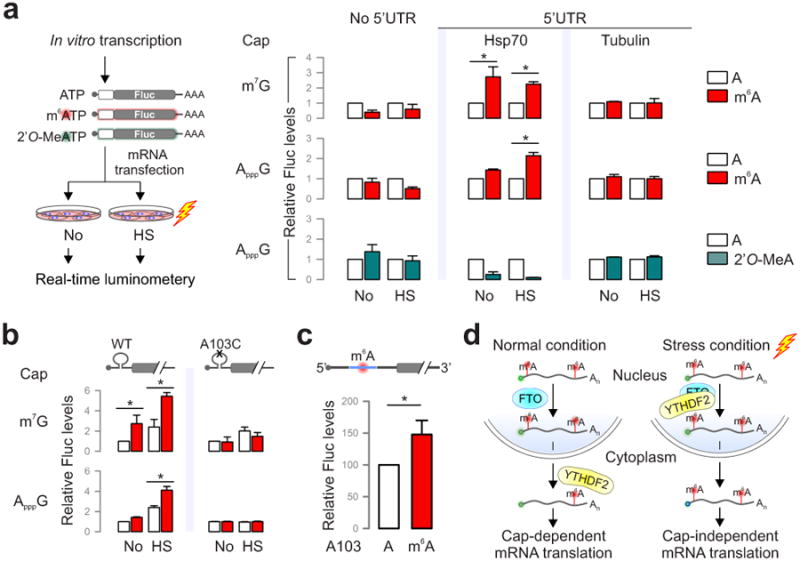
a, MEF cells transfected with Fluc mRNA reporters were subject to heat shock treatment and the Fluc activity was measured by real-time luminometry. Fluc activities were quantified and normalized to the one containing normal As. b, Constructs expressing Fluc reporter with Hsp70 5′UTR or the one with A103C mutation are depicted on the top. Fluc activities in transfected MEF cells were quantified and normalized to the control containing normal A without stress. c, Fluc mRNAs bearing Hsp70 5′UTR with a single m6A site were constructed using sequential splint ligation. After in vitro translation in rabbit reticulate lysates, Fluc activities were quantified and normalized to the control lacking m6A. Error bars, mean ± s.e.m.; * p < 0.05, unpaired two-tailed t-test; n=3, biological replicates (a, b and c). d, A proposed model for dynamic m6A 5′UTR methylation in response to stress and its role in cap-independent translation. Under the normal growth condition, nuclear FTO demethylates the 5′UTR m6A from nascent transcripts and the matured transcripts are translated via a cap-dependent mechanism. Under stress conditions, nuclear localization of YTHDF2 protects the 5′UTR of stress-induced transcripts from demethylation. With enhanced 5′UTR methylation, these transcripts are selectively translated via a cap-independent mechanism.
To further demonstrate the 5′UTR specificity in m6A-facilitated cap-independent translation, we examined 5′UTRs from a constitutively expressed chaperone Hsc70 (HSPA8) and another stress-inducible chaperone Hsp105 (HSPH1) (Fig. 3a). Only the 5′UTR of Hsp105 enhanced translation of the non-capped message after m6A incorporation (Extended Data Fig. 9c). This result is consistent with the selective 5′UTR methylation of stress-inducible transcripts upon heat shock stress.
5′UTR contains multiple As although not all of them are methylated. Based on the predicted m6A sequence motif, the A residue at the 103 position of Hsp70 mRNA is likely to be methylated. Using a single nucleotide m6A detection method25, we confirmed the methylation event at this position upon heat shock stress (Extended Data Fig. 10a). To demonstrate the significance of methylation at this single site, we introduced an A103C mutation into the Hsp70 5′UTR. Remarkably, m6A incorporation no longer promoted translation of the Fluc reporter in transfected cells (Fig. 4b). To directly demonstrate the importance of single site m6A without changing nucleotide, we employed a sequential RNA splint ligation strategy to construct a Fluc reporter bearing Hsp70 5′UTR with or without A103 methylation (Fig. 4c)26,27. Using an in vitro translation system, the Fluc reporter containing the single m6A at the 103 position showed about 50% increase in translation efficiency in comparison to the one with normal A (Fig. 4c). Notably, both messages showed comparable turnover during the entire course of in vitro translation (Extended Data Fig. 10c). Collectively, these results firmly established a crucial role of 5′UTR m6A modification in non-canonical translation initiation.
Much of our current understanding of cap-independent translation is limited to IRES28,29. However, beyond a few examples, many cellular genes capable of cap-independent translation do not seem to contain any IRES elements. The results presented here demonstrate a surprising role of m6A in mediating mRNA translation initiation independent of the normal m7G cap. How exactly the methylated adenosine recruits the translation machinery merits further investigation. m6A modification has been shown to alter RNA secondary structures4. It is possible that distinct translation initiation factors are recruited to the methylatd 5′UTR, thereby facilitating cap-independent translation.
In contrast to the wide belief that m6A modification is static on mRNAs, we found that 5′UTR methylation in the form of m6A is dynamic. Methylation often serves as a mark to distinguish self and foreign DNAs or parental and daughter DNA strands30. The stress-inducible mRNA 5′UTR methylation permits ribosomes to distinguish nascent transcripts from pre-existing messages, thereby achieving selective mRNA translation (Fig. 4d). The unexpected stress-inducible feature of YTHDF2 offers an elegant mechanism for temporal control of m6A modification on subsets of mRNAs. The mechanistic connection between 5′UTR methylation and cap-independent translation solves the central puzzle how selective translation is achieved when global translation is suppressed in responding to stress.
Methods
Cell lines and reagents
HeLa (cervical cancer) was originally purchased from ATCC and MEF cells were a gift from D.J. Kwiatkowski (Harvard Medical School). Cells were not authenticated recently but tested negative for mycoplasma contamination. Both cells were maintained in Dulbecco's Modified Eagle's Medium (DMEM) with 10% fetal bovine serum (FBS). Antibodies used in the experiments are listed below: anti-YTHDF2 (Proteintech 24744-1-AP, 1:1000 WB, 1:600 IF); anti-Hsp70 (Stressgen SPA-810, 1:1000 WB); anti-FTO (Phosphosolutions 597-Fto, 1:1000 WB, 1:600 IF); anti-METTL3 (Abnova H00056339-B01P, 1:1000 WB, 1:600 IF); anti-METTL14 (sigma HPA038002, 1:1000 WB, 1:600 IF); anti-WTAP (Santa Cruz sc-374280, 1:1000 WB, 1:600 IF); anti-m6A (Millipore ABE572, 1:1000 m6A immunoblotting); Alexa Fluor 488 donkey anti-mouse secondary antibody (Invitrogen A21202. 1:600 IF); Alexa Fluor 546 donkey anti-rabbit secondary antibody (Invitrogen A10040, 1:600 IF).
Construction of 5′UTR reporters
The Fluc reporter with Hsp70 5′UTR has been reported previously23. For Fluc reporters bearing other 5′UTRs, the following primers were used for 5′UTR cloning:
| Hsc70 (HSPA8) | |
| Fwd: | 5′-CCCAAGCTTGGTCTCATTGAACGCGG-3′; |
| Rev: | 5′-CGGGATCCCCTTAGACATGGTTGCTT-3′. |
| Tubulin (TUBG2) | |
| Fwd: | 5′-GGCAAGCTTTGCGCCTGTGCTGAATTCCAGCTGC-3′; |
| Rev: | 5′-GGCGGATCCGCATCGCCGATCAGACCTAG-3′. |
| Hsp105 (HSPH1) | |
| Fwd: | 5′-CCCAAGCTTGTAAAATGCTGCAGATTC-3′; |
| Rev: | 5′-CGGGATCCCCACCGACATGGCTGGCCCG-3′. |
Lentiviral shRNAs
All shRNA targeting sequences were cloned into DECIPHER™ pRSI9-U6-(sh)-UbiC-TagRFP-2A-Puro (Cellecta, CA). shRNA targeting sequences listed below were based on RNAi consortium at Broad Institute (http://www.broad.mit.edu/rnai/trc). YTHDF2 (mouse): 5′-GCTCCAGGCATGAATACTATA-3′; FTO (mouse): 5′-GCTGAGGCAGTTCTGGTTTCA 3′; Scramble control sequence: 5′-AACAGTCGCGTTTGCGACTGG-3′. Lentiviral particles were packaged using Lenti-X 293T cells (Clontech). Virus-containing supernatants were collected at 48-h after transfection and filtered to eliminate cells. MEF cells were infected by the lentivirus for 48 hr before selection by 1 μg ml-1 puromycin.
Recombinant protein expression
YTHDF2 and FTO were cloned into vector pGEX-6P-1 using the following primers:
| YTHDF2 | |
| Fwd: | 5′-ATGAATTCCCATCGGCCAGCAGCCTCTTG-3′; |
| Rev: | 5′-CCGCTCGAGTTCTATTTCCCACGACCTTGA-3′. |
| FTO | |
| Fwd: | 5′-ATGAATTCAGCATGAAGCGCGTCCAGACC-3′; |
| Rev: | 5′-CCGCTCGAGCCTCTAGGATCTTGC-3′. |
The resulting clones were transfected into the E coli strain BL21 and expression was induced at 22 °C with 1 mM IPTG for 16-18 h. The pellet collected from 1 litres of bacteria culture was then lysed in 15 ml PBS (50 mM NaH2PO4, 150 mM NaCl, pH 7.2, 1 mM PMSF, 1 mM DTT, 1 mM EDTA, 0.1% (v/v) Triton X-100) and sonicated for 10 min. After removing cell debris by centrifuge at 12,000 rpm for 30 min, the protein extract was mixed with 2 ml equilibrated Pierce® glutathione agarose and mixed on an end-over-end rotator for 2 h at 4°C. The resin was washed three times with 10 resin-bed volumes of Equilibration/Wash Buffer (50mM Tris, 150mM NaCl, pH 8.0). YTHDF2 and FTO protein was cleaved from the glutathione agarose using PreScission Protease (Genscript) in Cleavage Buffer (50 mM Tris-HCl, pH 7.0, 150 mM NaCl, 1 mM EDTA, 1 mM DTT) at 4 °C overnight.
Immunoblotting
Cells were lysed on ice in TBS buffer (50 mM Tris, pH7.5, 150 mM NaCl, 1 mM EDTA) containing protease inhibitor cocktail tablet, 1% Triton X-100, and 2 U ml-1 DNase. After incubating on ice for 30 min, the lysates were heated for 10 min in SDS/PAGE sample buffer [50 mM Tris (pH6.8), 100 mM dithiothreitol, 2% SDS, 0.1% bromophenol blue, 10% glycerol). Proteins were separated on SDS-PAGE and transferred to Immobilon-P membranes (Millipore). Membranes were blocked for 1-h in TBS containing 5% non-fat milk and 0.1 % Tween-20, followed by incubation with primary antibodies overnight at 4°C. After incubation with horseradish peroxidase-coupled secondary antibodies at room temperature for 1 h, immunoblots were visualized using enhanced chemiluminescence (ECLPlus, GE Healthcare).
Immunofluorescence staining
Cells grown on glass coverslips were fixed in 4% paraformaldehyde for 10 min at 4°C. After permeabilization in 0.2% Triton X-100 for 5 min at room temperature, the cover slips were blocked with 1% BSA for 1 h. Cells were stained with indicated primary antibody overnight at 4°C, followed by incubation with Alexa Fluor 488 donkey anti-mouse secondary antibody or Alexa Fluor 546 donkey anti-rabbit secondary antibody for 1 hr at room temperature. The nuclei were counter-stained with DAPI (1:1000 dilution) for 10 min. Cover slips were mounted onto slides and visualized using a Zeiss LSM710 confocal microscope.
mRNA stability measurement
Cells were treated with actinomycin D (5 μg/ml) for 4 h, 2 h, and 0 h before trypsinization collection. RNA spike-in control was added proportional to the total cell numbers and total RNA was isolated by Trizol kit (Life Technologies). After reverse transcription, the mRNA level of interested transcripts were detected by real-time quantitative PCR.
Real-time quantitative PCR
Total RNA was isolated by TRIzol reagent (Invitrogen) and reverse transcription was performed using High Capacity cDNA Reverse Transcription Kit (Invitrogen). Real-time PCR analysis was conducted using Power SYBR Green PCR Master Mix (Applied Biosystems) and carried on a LightCycler 480 Real-Time PCR System (Roche Applied Science). Primers for amplifying each target are listed below:
| YTHDF2 | |||
| Fwd: | 5′-CAGTTTGCCTCCAGCTACTATT-3′; | Rev: | 5′-GCAATGCCATTCTTGGTCTTC-3′. |
| FTO | |||
| Fwd: | 5′-TCAGCAGTGGCAGCTGAAAT-3′; | Rev: | 5′-CTTGGATCCTCACCACGTCC-3′. |
| Hsp70 | |||
| Fwd: | 5′-TGGTGCAGTCCGACATGAAG-3′; | Rev: | 5′-GCTGAGAGTCGTTGAAGTAGGC-3′. |
| METTL3 | |||
| Fwd: | 5′-ATCCAGGCCCATAAGAAACAG-3′; | Rev: | 5′-CTATCACTACGGAAGGTTGGG-3′. |
| METTL14 | |||
| Fwd: | 5′-CAGGCAGAGCATGGGATATT-3′; | Rev: | 5′-TCCGACCTGGAGACATACAT-3′. |
| ALKBH5 | |||
| Fwd: | 5′-AGTTCCAGTTCAAGCCCATC-3′; | Rev: | 5′-GGCGTTCCTTAATGTCCTGAG-3′. |
| WTAP | |||
| Fwd: | 5′-CTGGCAGAGGAGGTAGTAGTTA-3′; | Rev: | 5′-ACTGGAGTCTGTGTCATTTGAG-3′. |
| β-actin | |||
| Fwd: | 5′-TTGCTGACAGGATGCAGAAG-3′; | Rev: | 5′-ACTCCTGCTTGCTGATCCACAT-3′. |
| GAPDH | |||
| Fwd: | 5′-CAAGGAGTAAGAAACCCTGGAC-3′; | Rev: | 5′-GGATGGAAATTGTGAGGGAGAT-3′. |
| Fluc | |||
| Fwd: | 5′-ATCCGGAAGCGACCAACGCC-3′; | Rev: | 5′-GTCGGGAAGACCTGCCACGC-3′. |
In vitro transcription
Plasmids containing the corresponding 5′-untranslated region sequences of mouse HSPA1A and full length firefly luciferase were used as templates. Transcripts with normal m7G cap were generated using the mMessage mMachine T7 Ultra kit (Ambion) and transcripts with non-functional cap analog GpppA were synthesized using MEGAscript® T7 Transcription Kit (Ambion). To obtain mRNAs with the adenosine replaced with m6A, in vitro transcription was conducted in a reaction in which 5% of the adenosine was replaced with N6-methyladenosine. All mRNA products were purified using the MEGAclear kit (Ambion) according to the manufacturer's instruction.
In vitro translation
In vitro translation was performed using the Rabbit Reticulocyte Lysate System (Promega) according to the manufacturer's instructions. Luciferase activity was measured using a luciferase reporter assay system (Promega) on a SynergyTM HT Multi-detection Microplate Reader (BioTek Instruments).
Real-time luciferase assay
Cells grown in 35 mm dishes were transfected with in vitro synthesized mRNA containing the luciferase gene. Luciferase substrate D-luciferin (1 mM, Regis Tech) was added into the culture medium immediately after transfection. Luciferase activity was monitored and recorded using Kronos Dio Luminometer (Atto).
Site-specific m6A detection
For site-specific m6A detection, DNA primers were firstly 5′ labeled with 32P using T4 polynucleotide kinase (Invitrogen) and purified by ethanol precipitation. The primer 5′- AGGGATGCTCTGGGGAAGGCTGG-3′ was used to detect potential m6A site and the primer 5′-CGCCGCTCGCTCTGCTTCTCTTGTCTTCGCT-3′ was used to detect the non-methylated site. Synthesized mRNA 5′- CGATCCTCGGCCAGG(m6A)CCAGCCTTCCCCAG -3′ and 5′- CGATCCTCGGCCAGGACCAGCCTTCCCCAG -3′ were served as positive and negative control templates. To set up the reaction, a 2× annealing solution was prepared in a total volume of 8 μL with 1× Tth buffer (Promega) or AMV buffer (Invitrogen), 1 μL of each radiolabeled primer and 10 μg mRNA from MEF cells that had been heat shock treated. The mixture was heated at 95°C for 10 min and cooled slowly to room temperature. 3 μL of annealing solution were combined with 2 μL of enzyme and heated at 37°C (AMV Reverse Transcriptase) or 55°C (Tth DNA Polymerase) for two minutes. After adding the dTTP solution (final dTTP concentration: 100 μM), the reactions were heated for 5 minutes at 37°C (AMV) or 10 minutes at 55°C (Tth). Reaction products were resolved on a 20% denaturing polyacrylamide gel and exposed overnight.
RNA splint ligation
The ligation method was optimized from previous reports26,27,31. The RNA oligonucleotide covering the 82-117 nt region of HSPA1A was synthesized by Thermo Scientific, whereas RNA fragments corresponding to other regions were generated by in vitro transcription. For sequential splint ligation, two DNA bridging oligos were designed:
DNA Bridge 1: 5′-GGTCCTGGCCGAGGATCGGGAACGCGCCGCTCGCTC-3′
DNA Bridge 2: 5′-CTCCGCGGCAGGGATGCTCTGGGGAAGGCTGGTCCT-3′
For 3′ RNA oligo (donor) phosphorylation, mix 1 μl of 20 μM donor oligo with 1 μl of 10 × PNK buffer, 6 μl of ATP (10 mM), 0.5 μl of RNasin (20 units) and 1 μl of T4 PNK (5 units). The reaction mixture was incubated at 37°C for 30 min followed by inactivation of T4 PNK at 65 °C for 20 min. Next, the DNA bridge oligo was hybridized with the 3′ RNA oligo and the 5′ RNA oligo (acceptor) at a 1: 1.5: 2 ratio (5′RNA: bridge: 3′RNA). Oligos were annealed (95°C for 1 min followed by 65°C for 2 min and 37°C for 10 min) in the presence of 1 × T4 DNA dilution buffer. To ligate the 5′ and the 3′ RNA together, T4 DNA ligase and the T4 DNA ligation buffer were added and the reaction mixture was incubated at 37°C for 1 hr. The ligation was stopped by adding 1 μl of 0.5 M EDTA followed by phenol-chloroform extraction and ethanol precipitation. Ligation products were analyzed by 10% TBE-Urea gels or formaldehyde gels. The expected RNA ligation products in TBE-Urea gels were eluted in RNA gel elution buffer (300 mM NaOAc pH 5.5, 1 mM EDTA and 0.1 U/μl SUPERase_In) followed by ethanol precipitation. The final products in formaldehyde gels were isolated by Zymoclean™ Gel RNA Recovery Kit (Zymo Research).
Hsp70 mRNA pulldown and m6A immunoblotting
To isolate endogenous Hsp70 mRNA, 400 pmol of biotin-labeled probe (5′-TTCATAACATATCTCTGTCTCTT-3′) was incubated with of 2 mg M-280 Streptavidin Dynabeads (Life Technologies) in 1 ml 1× B & W buffer (5 mM Tris-HCL pH 7.5, 0.5 mM EDTA and 1 M NaCl) at 4°C for 1 hr. 2 mg total RNA was denatured at 75°C for 2 min and added to the pre-coated Dynabeads for an additional incubation of 2 h at 4°C. Captured RNA was eluted by heating beads for 2 min at 90°C in 10mM EDTA with 95% formamide followed by Trizol LS isolation. Isolated RNA was quantified using NanoDrop ND-1000 UV-Vis Spectrophotometer and equal amounts of RNAs were mixed with 2× RNA Loading Dye (Thermo Scientific) and denatured for 3 min at 70°C. In vitro transcribed mRNA containing 50% N6-methyladenosine or 100% adenosine was used as positive and negative controls, respectively. Samples were separated on a formaldehyde denaturing agarose gel and transferred to a positively charged nylon membrane by siphonage in transfer buffer (10 mM NaOH, 3 M NaCl) for overnight at room temperature. After transfer, the membrane was washed 5 min in 2× SSC buffer and RNA was UV crosslinked to the membrane. Membrane was blocked for 1 hr in PBST containing 5% non-fat milk and 0.1 % Tween-20, followed by incubation with anti-m6A antibody (1:1000 dilution) for overnight at 4°C. After extensive washing with 0.1 % PBST for 3 times, the membrane was incubated with HRP-conjugated anti-rabbit IgG (1:5000 dilution) for 1 hr. Membrane was visualized by using enhanced chemiluminescence (ECLPlus, GE Healthcare).
YTHDF2 and FTO in vitro pull down
Synthesize mRNA (100 pmol) with a single m6A at A103 was label by biotin using the Pierce RNA 3′ End Desthiobiotinylation Kit. Binding of the labeled RNA to streptavidin magnetic beads was performed in RNA capture buffer (20mM Tris, pH 7.5, 1M NaCl, 1mM EDTA) for 30 min at room temperature with rotation. The RNA-protein binding reaction was conducted in protein-RNA binding buffer (20mM Tris (pH 7.5), 50M NaCl, 2mM MgCl2, 0.1% Tween™-20 Detergent) at 4 °C for 60 min with rotation. After washing three times with the wash buffer (20mM Tris pH 7.5, 10mM NaCl, 0.1% Tween-20 Detergent), protein was eluted by Biotin Elution Buffer (Pierce) and detected by western blot.
YTHDF2 and FTO in vitro competition assay
The YTHDF2 and FTO in vitro competition assay was performed in 100 μl of reaction mixture containing 5 μM RNA incorporated with 50% m6A, 283 μM of (NH4)2Fe(SO4)2·6H2O, 300 μM of α-KG, 2 mM of L-ascorbic acid, 50 μg/ml of BSA, and 50 mM of HEPES buffer (pH 7.0). The reaction was incubated for 3 h at room temperature, and quenched by adding 5 mM EDTA followed by heating for 5 min at 95 °C. RNA was isolated by Trizol LS and quantified using NanoDrop ND-1000 UV-Vis Spectrophotometer. Equal amount of RNA was used for dot blot and methylene blue staining was used to show the amount of RNA on hybridization membranes.
Polysome profiling analysis
Sucrose solutions were prepared in polysome buffer (10 mM HEPES, pH 7.4, 100 mM KCl, 5 mM MgCl2, 100 μg ml-1 cycloheximide and 2% Triton X-100). A 15%- 45% (w/v) Sucrose density gradients were freshly prepared in SW41 ultracentrifuge tubes (Backman) using a Gradient Master (BioComp Instruments). Cells were pre-treated with 100 μg ml-1 cycloheximide for 3 min at 37°C followed by washing using ice-cold PBS containing 100 μg ml-1 cycloheximide. Cells were then lysed in polysome lysis buffer. Cell debris were removed by centrifugation at14,000 rpm for 10 min at 4°C. 500 μl of supernatant was loaded onto sucrose gradients followed by centrifugation for 2 h 28 min at 38,000 rpm 4°C in a SW41 rotor. Separated samples were fractionated at 0.75 ml/min through an automated fractionation system (Isco) that continually monitores OD254 values. An aliquot of ribosome fraction were used to extract total RNA using Trizol LS reagent (Invitrogen) for real-time PCR analysis.
RNA-seq and m6A–seq
For m6A immunoprecipitation, total RNA was first isolated using Trizol reagent followed by fragmentation using freshly prepared RNA fragmentation buffer (10 mM Tris-HCl, pH 7.0, 10 mM ZnCl2). 5 μg fragmented RNA was saved as input control for RNA-seq. 1 mg fragmented RNA was incubated with 15 μg anti-m6A antibody (Millipore ABE572) in 1× IP buffer (10 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% Igepal CA-630) for 2 hr at 4°C. The m6A-IP mixture was then incubated with Protein A beads for additional 2 hr at 4°C on a rotating wheel. After washing 3 times with IP buffer, bound RNA was eluted using 100 μl elution buffer (6.7 mM N6-Methyladenosine 5′-monophosphate sodium salt in 1× IP buffer), followed by ethanol precipitation. Precipitated RNA was used for cDNA library construction and high-throughput sequencing described below.
Ribo-seq
Ribosome fractions separated by sucrose gradient sedimentation were pooled and digested with E. coli RNase I (Ambion, 750 U per 100 A260 units) by incubation at 4 °C for 1 h. SUPERase inhibitor (50 U per 100 U RNase I) was then added into the reaction mixture to stop digestion. Total RNA was extracted using TRIzol reagent. Purified RNA was used for cDNA library construction and high-throughput sequencing described below.
cDNA library construction
Fragmented RNA input and m6A-IP elutes were dephosphorylated for 1 hr at 37°C in 15 μl reaction (1× T4 polynucleotide kinase buffer, 10 U SUPERase_In and 20 U T4 polynucleotide kinase). The products were separated on a 15 % polyacrylamide TBE-urea gel (Invitrogen) and visualized using SYBR Gold (Invitrogen). Selected regions of the gel corresponding to 40-60 nt (for RNA-seq and m6A-seq) or 25-35 nt (for Ribo-seq) were excised. The gel slices were disrupted by using centrifugation through the holes at the bottom of the tube. RNA fragments were dissolved by soaking overnight in 400 μl gel elution buffer (300 mM NaOAc, pH 5.5, 1 mM EDTA, 0.1 U/ml SUPERase_In). The gel debris was removed using a Spin-X column (Corning), followed by ethanol precipitation. Purified RNA fragments were re-suspended in nuclease-free water. Poly-(A) tailing reaction was carried out for 45 min at 37°C (1 × poly-(A) polymerase buffer, 1 mM ATP, 0.75 U/μl SUPERase_ In and 3 U E. coli poly-(A) polymerase).
For reverse transcription, the following oligos containing barcodes were used:
MCA02: 5′-pCAGATCGTCGGACTGTAGAACTCTCAAGCAGAAGACGGCATACGATTT TTTTTTTTTTTTTTTTTVN-3′;
LGT03: 5′-pGTGATCGTCGGACTGTAGAACTCTCAAGCAGAAGACGGCATACGATT TTTTTTTTTTTTTTTTTTVN-3′;
YAG04: 5′-pAGGATCGTCGGACTGTAGAACTCTCAAGCAGAAGACGGCATACGATT TTTTTTTTTTTTTTTTTTVN-3′;
HTC05: 5′-pTCGATCGTCGGACTGTAGAACTCTCAAGCAGAAGACGGCATACGATT TTTTTTTTTTTTTTTTTTVN-3′.
In brief, the tailed-RNA sample was mixed with 0.5 mM dNTP and 2.5 mM synthesized primer and incubated at 65 °C for 5 min, followed by incubation on ice for 5 min. The reaction mix was then added with 20 mM Tris (pH 8.4), 50 mM KCl, 5 mM MgCl2, 10 mM DTT, 40 U RNaseOUT and 200 U SuperScript III. Reverse transcription reaction was performed according to the manufacturer's instruction. Reverse transcription products were separated on a 10% polyacrylamide TBE-urea gel as described earlier. The extended first-strand product band was expected to be approximately 100 nt, and the corresponding region was excised. The cDNA was recovered by using DNA gel elution buffer (300 mM NaCl, 1 mM EDTA). First-strand cDNA was circularized in 20 μl of reaction containing 1× CircLigase buffer, 2.5 mM MnCl2, 1M Betaine, and 100 U CircLigase II (Epicentre). Circularization was performed at 60 °C for 1 h, and the reaction was heat inactivated at 80 °C for 10 min. Circular single-strand DNA was re-linearized with 20 mM Tris-acetate, 50 mM potassium acetate, 10 mM magnesium acetate, 1 mM DTT, and 7.5 U APE 1 (NEB). The reaction was carried out at 37 °C for 1 h. The linearized single-strand DNA was separated on a Novex 10% polyacrylamide TBE-urea gel (Invitrogen) as described earlier. The expected 100-nt product bands were excised and recovered as described earlier.
Deep sequencing
Single-stranded template was amplified by PCR by using the Phusion High-Fidelity enzyme (NEB) according to the manufacturer's instructions. The oligonucleotide primers qNTI200 (5′-CAAGCAGAAGACGGCATA- 3′) and qNTI201 (5′-AATGATACGGCGACCACCG ACAGGTTCAGAGTTCTACAGTCCGACG- 3′) were used to create DNA suitable for sequencing, i.e., DNA with Illumina cluster generation sequences on each end and a sequencing primer binding site. The PCR contains 1× HF buffer, 0.2 mM dNTP, 0.5 μM oligonucleotide primers, and 0.5 U Phusion polymerase. PCR was carried out with an initial 30 s denaturation at 98 °C, followed by 12 cycles of 10 s denaturation at 98 °C, 20 s annealing at 60 °C, and 10 s extension at 72 °C. PCR products were separated on a nondenaturing 8% polyacrylamide TBE gel as described earlier. Expected DNA at 120 bp (for Ribo-seq), or 140 bp (for RNA-seq and m6A-seq) was excised and recovered as described earlier. After quantification by Agilent BioAnalyzer DNA 1000 assay, equal amount of barcoded samples were pooled into one sample. Approximately 3–5 pM mixed DNA samples were used for cluster generation followed by deep sequencing by using sequencing primer 5′-CGACAGGTTCAGAGTTCTAC AGTCCGACGATC-3′ (Illumina HiSeq).
Preprocessing of sequencing reads
For Ribo-seq, the sequencing reads were first trimmed by 8 nt from the 3′ end and trimmed reads were further processed by removing adenosine (A) stretch from the 3′ end (one mismatch is allowed). The processed reads between 25nt and 35nt were first mapped by Tophat using parameters (−−bowtie1 –p 10 –no-novel-juncs) to mouse transcriptome (UCSC Genes)32. The unmapped reads were then mapped to corresponding mouse genome (mm10). Non-uniquely mapped reads were disregarded for further analysis due to ambiguity. The same mapping procedure was applied to RNA-seq and m6A-seq. For Ribo-seq, the 13th position (12nt offset from the 5′ end) of the uniquely mapped read was defined as the ribosome “P-site” position. The RPF density was computed after mapping uniquely mapped reads to each individual mRNA transcript according to the NCBI Refseq gene annotation. Uniquely mapped reads of RNA-seq and Ribo-seq in the mRNA coding region were used to calculate the RPKM values for estimating mRNA expression and translation levels respectively. For m6A-seq, uniquely mapped reads in the 5′UTR were used to calculate the RPKM values for estimating the m6A levels.
Identification of m6A sites
We used a similar scanning strategy reported previously to identify m6A peaks in the immunoprecipitation sample as compared to the input sample7. In brief, for NCBI RefSeq genes whose maximal read coverage are greater than 15 in the input (RNA-seq), a sliding window of 80 nucleotides with step size of 40 nucleotides was employed to scanning the longest isoform (based on CDS length; in the case of equal CDS, the isoform with longer 5′ UTR was selected). For each window, a Peak-Over-Median score (POM) was derived by calculating the ratio of mean read coverage in the window to the median read coverage of the whole gene body. Windows scoring higher than 3 in the IP sample were obtained and all the resultant overlapping m6A peak windows in the IP sample were iteratively clustered to infer the boundary of m6A enriched region as well as peak position with maximal read coverage. Finally, a Peak-Over-Input (POI) score was assigned to each m6A enriched region by calculating the ratio of POM in the IP sample to that in the input sample. A putative m6A site was defined if the POI score was higher than 3. The peak position of each m6A site was classified into five mutually exclusive mRNA structural regions including TSS (the first 200 nucleotides of mRNA), 5′ UTR, CDS, stop codon (a 400 nt window flanking mRNA stop codon) and 3′ UTR.
m6A motif analysis
The m6A peaks with POI score higher than 10 were selected for consensus motif finding. We used MEME Suite for motif analysis33. In brief, the flanking sequences of m6A peaks (±40 nt) with POI scores were retrieved from mouse transcritpome and were used as MEME input.
Extended Data
Extended Data Figure 1. Subcellular localization of the m6A machinery in cells before and after heat shock stress.

a, MEF cells before or 2 h after heat shock (42°C, 1 h) were immunostanied with antibodies indicated. DAPI was used for nuclear staining. b, MEF (left panel) and HeLa cells (right panel) were subject to heat shock stress (42°C, 1 h) followed by recovery at 37°C for various times. Anti-YTHDF2 immunostaining was counter stained by DAPI. Representative of at least 50 cells.
Extended Data Figure 2. mRNA stability and induction in response to heat shock stress.

a, Effects of heat shock stress on mRNA stability. MEF cells without heat shock stress (No HS), immediately after heat shock stress (42°C, 1 h) (Post HS 0h), or 2 h recovery at 37°C (Post HS 2h) were subject to further incubation in the presence of 5 μg/ml ActD. At the indicated times, mRNA levels were determined by qPCR. Error bars, mean ± s.e.m. n=3. b, MEF cells were collected at indicated times after heat shock stress (42°C, 1 h) followed by RNA extraction and real-time PCR. Relative levels of indicated transcripts are normalized to β-actin. Error bars, mean ± s.e.m. n=3, biological replicates. c, HSF1 WT and KO cells were subject to heat shock stress (42°C, 1 h) followed by recovery at 37°C for various times. Real-time PCR was conducted to quantify transcripts encoding Hsp70 and YTHDF2. Relative levels of transcripts are normalized to β-actin. Error bars, mean ± s.e.m. *, p < 0.05, **, p < 0.01, unpaired two-tailed t-test; n=3, biological replicates.
Extended Data Figure 3. Characterization of m6A sites in MEF cells with or without heat shock stress.

m6A profiling was conducted on MEF cells before (a) or 2 h after heat shock (42°C, 1 h) (b). Left, pie chart presenting fractions of m6A peaks in different transcript segments. Right, sequence logo representing the consensus motif relative to m6A.
Extended Data Figure 4. m6A profiling of HSPA8 in MEF cells with or without heat shock stress.

An example of constitutively expressed transcript HSPA8 in MEF cells with or without heat shock stress. Coverage of m6A IP and control reads (input) are indicated in red and gray, respectively. The transcript architecture is shown below the x-axis.
Extended Data Figure 5. Dynamic m6A modification of HSPA1A by YTHDF2 and FTO.

An example of stress-induced transcript HSPA1A in post-stressed MEF cells with either YTHDF2 or FTO knockdown. Coverage of m6A IP and control reads (input) are indicated in red and blue, respectively. The transcript architecture is shown below the x-axis.
Extended Data Figure 6. Direct competition between YTHDF2 and FTO in m6A binding.

a, Synthesized mRNA with m6A was incubated with FTO (2 μg) in the presence of increasing amount of YTHDF2 (0, 0.5, 1, 2 μg), followed by RNA pulldown and immunoblotting. b, Synthesized mRNA with m6A was incubated with FTO (1 μg in top panel and 2 μg in bottom panel) in the absence of presence of YTHDF2 (4 μg), followed by m6A dot blotting.
Extended Data Figure 7. YTHDF2 knockdown does not affect Hsp70 transcription after stress.

MEF cells with or without YTHDF2 knockdown were subject to heat shock stress (42°C, 1 h) followed by recovery at 37°C for various times. Real-time PCR was conducted to quantify Hsp70 mRNA levels. Error bars, mean ± s.e.m.; n=3, biological replicates.
Extended Data Figure 8. FTO knockdown promotes Hsp70 synthesis.

a, m6A blotting of purified HSPA1A in MEF with or without FTO knockdown. mRNAs synthesized by in vitro transcription in the absence or presence of m6A were used as control. RNA staining is shown as loading control. Representative of two biological replicates. b, MEF cells with or without FTO knockdown were collected at indicated times after heat shock stress (42°C, 1 h) followed by immunoblotting using antibodies indicated. N: no heat shock. Representative of three biological replicates.
Extended Data Figure 9. m6A modification promotes cap-independent translation.

a, Fluc reporter mRNAs with or without 5′UTR was synthesized in the absence or presence of m6A. The transfected MEFs were incubation in the presence of 5 μg/ml ActD. At the indicated times, mRNA levels were determined by qPCR. Error bars, mean ± s.e.m.; n=3, biological replicates. b, Fluc reporter mRNAs with or without Hsp70 5′UTR was synthesized in the absence of presence of m6A, followed by addition of a non-functional cap analog ApppG. Fluc activity in transfected MEF cells was recorded using real-time luminometry. c, Constructs expressing Fluc reporter bearing 5′UTR from Hsc70 or Hsp105 are depicted on the top. Fluc activities in transfected MEF cells were quantified and normalized to the control containing normal A. Error bars, mean ± s.e.m.; *p < 0.05, unpaired two-tailed t-test; n=3, biological replicates.
Extended Data Figure 10. Site-specific detection of m6A modification on HSPA1A.

a, Sequences of HSPA1A template and the DNA primer used for site-specific detection. Synthesized mRNAs containing single site m6A (red) are used as positive control. Autoradiogram shows primer extension of controls (left panel) and endogenous HSPA1A (right panel). b, Fluc mRNAs with or without m6A incorporation were incubated in the RRL at 30°C for up to 60 min. mRNA levels were determined by qPCR. Error bars, mean ± s.e.m.; n=3, biological replicates.
Supplementary Material
Acknowledgments
We'd like to thank Qian lab members for helpful discussion and Dr. Samie Jaffrey for critical reading of the manuscript. We thank Cornell University Life Sciences Core Laboratory Center for performing deep sequencing. This work was supported by grants to to S.-B.Q. from US National Institutes of Health (DP2 OD006449, R01AG042400) and US Department of Defense (W81XWH-14-1-0068).
Footnotes
Online Content Methods, along with additional Extended Data display items and Source Data are available in the online version of the paper.
Author Information: Sequencing data have been deposited at NCBI Sequence Read Archive under accession number SRA061617.
The authors declare no competing financial interests.
Author Contributions: J.Z. and S.-B.Q. conceived the project. J.Z. performed most experiments. J.W. analyzed the data. X.G. performed Ribo-seq. X.Z. assisted heat shock assays. S.-B.Q. wrote the manuscript. All authors discussed results and edited the manuscript.
References
- 1.Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15:313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m(6)A RNA methylation. Nat Rev Genet. 2014;15:293–306. doi: 10.1038/nrg3724. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 2014;505:117–120. doi: 10.1038/nature12730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu N, et al. N(6)-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature. 2015;518:560–564. doi: 10.1038/nature14234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 2015;161:1388–1399. doi: 10.1016/j.cell.2015.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer KD, et al. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149:1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dominissini D, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 8.Jia G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7:885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zheng G, et al. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49:18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qian SB, McDonough H, Boellmann F, Cyr DM, Patterson C. CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature. 2006;440:551–555. doi: 10.1038/nature04600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz S, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8:284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schwartz S, et al. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155:1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anckar J, Sistonen L. Regulation of HSF1 function in the heat stress response: implications in aging and disease. Annu Rev Biochem. 2011;80:1089–1115. doi: 10.1146/annurev-biochem-060809-095203. [DOI] [PubMed] [Google Scholar]
- 14.Mendillo ML, et al. HSF1 drives a transcriptional program distinct from heat shock to support highly malignant human cancers. Cell. 2012;150:549–562. doi: 10.1016/j.cell.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol. 2010;11:113–127. doi: 10.1038/nrm2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hinnebusch AG. The scanning mechanism of eukaryotic translation initiation. Annu Rev Biochem. 2014;83:779–812. doi: 10.1146/annurev-biochem-060713-035802. [DOI] [PubMed] [Google Scholar]
- 17.Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–237. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 18.Panniers R. Translational control during heat shock. Biochimie. 1994;76:737–747. doi: 10.1016/0300-9084(94)90078-7. [DOI] [PubMed] [Google Scholar]
- 19.Richter K, Haslbeck M, Buchner J. The heat shock response: life on the verge of death. Mol Cell. 2010;40:253–266. doi: 10.1016/j.molcel.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 20.McGarry TJ, Lindquist S. The preferential translation of Drosophila hsp70 mRNA requires sequences in the untranslated leader. Cell. 1985;42:903–911. doi: 10.1016/0092-8674(85)90286-7. [DOI] [PubMed] [Google Scholar]
- 21.Klemenz R, Hultmark D, Gehring WJ. Selective translation of heat shock mRNA in Drosophila melanogaster depends on sequence information in the leader. EMBO J. 1985;4:2053–2060. doi: 10.1002/j.1460-2075.1985.tb03891.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rubtsova MP, et al. Distinctive properties of the 5′-untranslated region of human hsp70 mRNA. J Biol Chem. 2003;278:22350–22356. doi: 10.1074/jbc.M303213200. [DOI] [PubMed] [Google Scholar]
- 23.Sun J, Conn CS, Han Y, Yeung V, Qian SB. PI3K-mTORC1 attenuates stress response by inhibiting cap-independent Hsp70 translation. J Biol Chem. 2011;286:6791–6800. doi: 10.1074/jbc.M110.172882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang X, et al. Translational control of the cytosolic stress response by mitochondrial ribosomal protein L18. Nat Struct Mol Biol. 2015;22:404–410. doi: 10.1038/nsmb.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Harcourt EM, Ehrenschwender T, Batista PJ, Chang HY, Kool ET. Identification of a selective polymerase enables detection of N(6)-methyladenosine in RNA. J Am Chem Soc. 2013;135:19079–19082. doi: 10.1021/ja4105792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kershaw CJ, O'Keefe RT. Splint ligation of RNA with T4 DNA ligase. Methods Mol Biol. 2012;941:257–269. doi: 10.1007/978-1-62703-113-4_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stark MR, Pleiss JA, Deras M, Scaringe SA, Rader SD. An RNA ligase-mediated method for the efficient creation of large, synthetic RNAs. RNA. 2006;12:2014–2019. doi: 10.1261/rna.93506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelletier J, Sonenberg N. Internal initiation of translation of eukaryotic mRNA directed by a sequence derived from poliovirus RNA. Nature. 1988;334:320–325. doi: 10.1038/334320a0. [DOI] [PubMed] [Google Scholar]
- 29.Hellen CU, Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules. Genes Dev. 2001;15:1593–1612. doi: 10.1101/gad.891101. [DOI] [PubMed] [Google Scholar]
- 30.Kunkel TA, Erie DA. DNA mismatch repair. Annu Rev Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 31.Maroney PA, Chamnongpol S, Souret F, Nilsen TW. Direct detection of small RNAs using splinted ligation. Nat Protoc. 2008;3:279–287. doi: 10.1038/nprot.2007.530. [DOI] [PubMed] [Google Scholar]
- 32.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bailey TL, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009;37:W202–208. doi: 10.1093/nar/gkp335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.