

Abstract
A steady accumulation of experimental data argues that protein synthesis in neurons is not merely restricted to the somatic compartment, but also occurs in several discrete cellular micro-domains. Local protein synthesis is critical for the establishment of synaptic plasticity in mature dendrites and in directing the growth cones of immature axons, and has been associated with cognitive impairment in mice and humans. Although in recent years a number of important mechanisms governing this process have been described, it remains technically challenging to precisely monitor local protein synthesis in individual neuronal cell parts independent from the soma. This report presents the utility of employing microfluidic chambers for the isolation and treatment of single neuronal cellular compartments. Furthermore, it is demonstrated that a protein synthesis assay, based on fluorescent non-canonical amino acid tagging (FUNCAT), can be combined with this cell culture system to label nascent proteins within a discrete structural and functional domain of the neuron. Together, these techniques could be employed for the detection of protein synthesis within developing and mature neurites, offering an effective approach to elucidate novel mechanisms controlling synaptic maintenance and plasticity.
Keywords: neurons, axon, synapse, local protein synthesis, click assay, microfluidic chambers, FUNCAT
Introduction
Translation of synaptically localized mRNAs (local translation) plays an essential role in a number of important neuronal processes, including the consolidation of memory and activity-dependent forms of synaptic plasticity (Vanderklish and Edelman 2005). Moreover, inadequate or dysregulated protein synthesis in the brain can result in a number of neurodevelopmental disorders by altering synapse composition and plasticity, which are suspected to be causally related to some of the most devastating symptoms in schizophrenia, intellectual disability and autism spectrum disorders (ASD) (Bassell and Warren 2008; Liu-Yesucevitz et al. 2011).
Various mechanisms ensure the rapid transport of mature mRNAs, ribosomes and translational factors (e.g., Ef1α or eif2α) that co-localize at distant sites of translation (Akins et al. 2009). Moreover, protein synthesis is a multi-step process controlled by a subset of distinct processes, including non-coding microRNAs (miRNAs) (Schratt et al. 2006; Aschrafi et al. 2008), many different translation factors, and multiple mRNA-binding proteins (Abaza and Gebauer 2008; Jackson et al. 2010). Accumulating evidence also suggests that translation of proteins in distant synapses is regulated at several levels varying from transportation of essential factors to local regulation of their activity (Wang et al. 2009; Jung et al. 2014). Thus, local translation is a highly flexible process that facilitates fast and specific changes in individual compartments, ensuring a rapid response to local environmental changes. For example, the stabilization of several distinct forms of activity-dependent synaptic plasticity requires a wave of post-synaptic protein synthesis in the dendrites of neurons. Compelling data demonstrating the mechanisms connecting the processes of synaptic plasticity and memory consolidation comes from recent advances in our understanding of how neuronal protein synthesis is regulated by synaptic activity (Holt and Schuman 2013). Moreover, recent findings suggest that growing axons depend on the local synthesis of structural and cytoskeletal proteins. Extracellular cues direct growth cones by inducing rapid changes in local protein expression, and developing axons contain the necessary translational machinery and specific mRNAs to support local protein synthesis. Indeed, the proteins coordinating or comprising cytoskeletal elements are locally synthesized and contribute to the growth cone path finding and turning apparatus (Campbell and Holt 2001; Yoon et al. 2009). For example, some of the first identified functions of axonal protein synthesis were intricately linked to its ability to mediate the local translation of mRNAs that encode the cytoskeletal proteins in growing axons (Capano et al. 1987; Kaplan et al. 1992; Gioio et al. 1994).
Although important advances have been made in examining the intricate subprocesses occurring during translation at pre- and post-synaptic sites, direct detection of translation inside axons or dendrites of living neurons, using traditional methods, is challenging. Accordingly, there is a need to develop novel approaches to gain quantitative insight into local translational processes contributing to synapse formation and function. Previous research has suggested that the overall amount of proteins synthesized in distal segments of neurons is relatively low, and often neglected by overwhelming somal translation. Therefore, visualization of protein synthesis in small areas containing low numbers of active ribosomes requires a highly sensitive detection method. Such an experimental approach would facilitate the direct labeling of newly synthesized proteins, enabling the examination of functionally distinct neuronal compartments with low rates of translation. To achieve this, fluorescent non-canonical amino acid tagging (FUNCAT) was developed (Dieterich et al. 2010; Hinz et al. 2012), a click chemistry reaction that provides a powerful labeling approach to detect nascent proteins intracellularly (Tornoe et al. 2002; Hou et al. 2012). The concept behind this assay is the substitution of methionine with its homolog homopropargylglycine (HPG) in living cells, allowing the introduction of alkyne groups into nascent protein. These alkyne groups can be azide linked using the catalytic click reaction. The azide may be further “flavored” by additional functional groups, such as biotins for protein affinity purification experiments, or fluorophores for the direct visualization of nascent protein synthesis using confocal or fluorescence microscopy (Beatty et al. 2006; Dieterich et al. 2010; Tom Dieck et al. 2012). The distribution of the observed signal inside the cell may be used as a marker for active translation. Moreover, the intensity of the signal can be quantified, providing an estimate of the level of translation at a particular location (Tom Dieck et al. 2012).
The use of the click reaction to visualize local translation is largely hampered by the high level of proteins synthesized in the somata, many of which are transported to distal sites of neurons. Thus, the utility of microfluidic chambers to physically separate distal neuronal segments, such as axons from the somata and other proximal processes, is ideally suited to locally apply and fluidically isolate click reaction reagent to selectively examine local translation (Taylor et al. 2005). The combinatorial use of mircrofluidic chambers with fluorescent labeling of nascent proteins has previously been shown to allow for visualization of protein synthesis events within distinct neuronal sub-compartments, such as in growing axons (Ji and Jaffrey 2012; Tom Dieck et al. 2012). Here, we combined the utility of the microfluidic devices, to either establish neuronal connectivity between dissociated cortical neurons or to selectively grow axons and dendrites in parallel rows distal from their cell bodies, with FUNCAT to establish a qualitative nascent protein labeling approach in cortical neurons in vitro. The experimental paradigm employed here provides a versatile and highly sensitive assay for the detection of local translation in growing and mature neurons.
Materials & Methods
Animals
Wistar rat (Harlan laboratories B.V.; Boxmeer, The Netherlands) embryos were used as a resource for the isolation of primary neurons. Animals were housed 2 to 3 per cage using a 12-hr light cycle at controlled ambient temperature (21°C ± 1°C). All animal use, care and experiments were performed according to protocols approved by the Committee for Animal Experiments of the Radboud University Medical Centre, Nijmegen, The Netherlands.
Culture of Primary Neurons
All media and reagents used for cell culture were purchased from Life Technologies (Carlsbad, CA) unless stated otherwise. Primary cortical neurons were isolated from embryonic day 18 (E18) rat embryonic brains. The cortical region was isolated and placed in ice-cold HBSS washing buffer (Hanks’ Balanced Salt solution (HBSS) containing 2 mmol/L GlutaMAX and Pen/Strep antibiotics). The tissue was washed twice with HBSS buffer before the addition of 0.025% trypsin in HBSS solution, incubated for 15 min at 37°C. After incubation, the tissue was washed three times with HBSS before the addition of Neurobasal (NB) medium supplemented with 10% fetal bovine serum (FBS) and 2 mmol/L GlutaMAX. The tissue was titrated several times with a glass Pasteur pipette. This treatment was repeated with a flame-thinned tip glass Pasteur pipette to obtain fully dissociated cells. Separated cells were seeded in cell culture plates, which were coated overnight with 0.1 g/L, Poly-D-Lysine (PDL, molecular weight 70,000-150,000 Da) (Sigma-Aldrich; St. Louis, MO). For the first 5 hr, the cells were cultured in NB medium with 10% FBS and 2 mmol/L glutaMAX. Afterwards, the medium was replaced with culturing NB medium with serum-free neural supplement B27 and 2 mmol/L glutaMAX.
Microfluidic Chamber Assembly
Microfluidic axon and perfusion chambers were purchased from Xona microfluidics (Temecula, CA) and assembled according to the protocol provided by the manufacturer. In-depth descriptions for assembly and maintenance of microfluidic axon and perfusion chambers are also available (Park et al. 2006; Taylor et al. 2010). Briefly, microfluidic chambers were prepared by first cleaning with 70% ethanol. After the chambers were completely dry, they were placed bottom side down on PDL-coated coverglasses (Hecht Assitent, Sondheim von der Rhön, Germany). The chambers were subsequently filled with 100 µl seeding medium (NB medium containing 10% FBS and 2 mmol/L GlutaMAX) at least one day before the cells were seeded so that any formed bubbles would dissipate. Just before seeding the cells, all medium was removed from the wells to which cells were to be added without removing the medium from the channels. Primary neurons were diluted to 100,000 neurons (for the axon chambers, Fig. 3A) or 50,000 neurons (for the perfusion chambers, Fig. 4A) in 20 µl seeding medium, and the cells were added into one well for one channel. For the perfusion chamber, cells were added into both opposing channels. This would lead to axons growing from one channel while dendrites grow from the other resulting in synapse formation within the perfusion channel. After the cells had settled down (approximately 30 min after seeding), 100 µl seeding medium was added to all wells. After 24 hr, the seeding medium was removed and replaced with 150 µl culturing medium containing NB with B27 serum-free neural supplement and 2 mmol/L GlutaMAX. Medium was replaced every 3-4 days depending on the rate of evaporation.
Figure 3.

Nascent protein tagging in axons of cortical neurons growing in microfluidic chambers. (A) Schematic representation of a compartmentalized microfluidic chamber with the grey channel holding the cells from which the axons will originate. These will grow through the middle microgrooves towards the axon chamber (green). Right: A representative micrograph of growing axons from DIV 7 cortical neurons stained with phalloidin (green) and DAPI (blue). The white insert denotes the typical area in which the axonal HPG signal is measured. (B) Representative images of axons treated with HPG alone or combined with the translation inhibitor CHX; these compounds were added to the axon compartment and fluidically isolated. Neurons were stained for Tau (green) and HPG incorporation (inverted greyscale and red) and the overlay of the two fluorophores. (C) Quantification of the HPG signal in immature axons relative to axons treated with CHX. (D) Agarose gel electropherogram of the soma-enriched genes Scn3b and Glrb after RT-PCR amplification of RNA samples isolated from the soma or axon compartment including a H2O-negative RT-PCR control. (E) Representative images of cells within the soma compartment of the axon chamber in which only the axon compartment was treated with HPG. Neurons were stained for Tau (green) and HPG incorporation (inverted greyscale and red). (F) Quantification of the HPG signal in the soma compartment compared with cells treated with CHX. Data represent the mean ± SEM and single values shown for n=6 to 9 chambers collected from three independent experiments. CHX, cyclohexamide; DIV, days in vitro; HPG, homopropargylglycine. p-values are determined by two-tailed unpaired Students t-test. **p<0.01. Scale (A, B, E) 50 µm.
Figure 4.

Imaging of protein synthesis in neurons grown in microfluidic perfusion chambers. (A) Schematic overview of a microfluidic perfusion chamber with one region (green square) enlarged (inset of the perfusion channel on the right). On the left side of the chamber, there are three reservoirs. The two outer green wells contain a buffer to hold the perfusate inside the perfusion chamber preventing it from entering the microgrooves. The middle red well contains the desired perfusate. In the enlargement image, the red cells (lower) generate the axons growing towards the perfusion channel (presynaptic compartment), while the blue cells (upper) generate dendrites (postsynaptic compartment). (B) Representative confocal images of the HPG signal (red and greyscale) and Tau (blue) within the perfusion channel of chambers in which mature cortical neurons (DIV 14–16) were cultured. The neurons were treated locally within the perfusion channel with HPG alone or in combination with CHX. (C) Magnifications of the white square inserts depicted in (B) with the HPG shown in red and inverted greyscale. (D) Quantification of the relative intensity of the HPG signal inside the perfusion channel compared with neurons treated with CHX. (E) Representative confocal images of cells on the dendrite side (postsynaptic compartment) of the perfusion chamber with Tau (blue) and the HPG signal (greyscale). (F) Quantification of the relative intensity of the HPG signal of cells cultured on the dendrite side of the perfusion chamber. Data represent the mean ± SEM and single values shown for n=4 independent experiments. p-values are determined by two-tailed unpaired Students t-test. p<0.01. CHX, cyclohexamide; DIV, days in vitro; HPG, homopropargylglycine. **Scale (B and E) 50 µm; (C) 5 µm.
Click Reaction and Immunostaining
Labelling of proteins was performed using the Click-iT HPG Alexa Fluor 594 Protein Synthesis Assay Kit (Life Technologies), following the protocol provided by the manufacturer with some minor adjustments (Fig. 1). In brief, HPG was diluted in HBSS (Life Technologies) to a concentration of 2 mM. Before HPG application, the culture medium was replaced with HBSS to deplete methionine from the cells for 30 min. During this pre-incubation, the appropriate samples were also treated with 100 µM cycloheximide (Sigma-Aldrich) to inhibit protein synthesis. After methionine depletion, the cells were incubated with HPG in the following manner: For whole cell cultures, 100 µl HPG in HBSS solution was applied for 90 min under normal culture conditions. Some samples were treated with 50 mM KCl for the first 10 min of HPG incubation to enhance translation. Then, KCl was removed to minimize cytotoxicity. For microfluidic axon chambers, 100 µl HPG in HBSS solution was added to the “axon” side of the chamber with 200 µl HBSS on the opposite “soma” side of the chamber to achieve fluidic isolation of the HPG, and incubated for 120 min.
Figure 1.
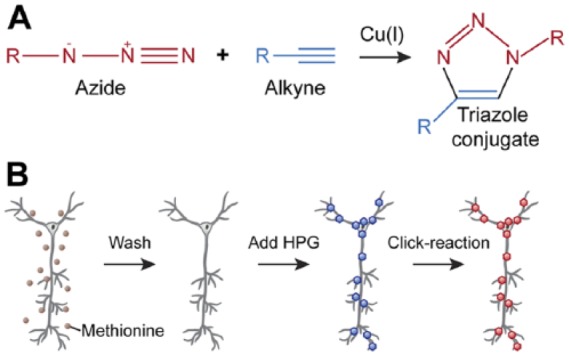
Protein labeling using the click reaction. (A) A scheme showing the copper-catalyzed click reaction in which two molecules containing either an alkyne (blue) or azide (red) group are covalently bound to form a stable triazole conjugate. (B) Click reaction workflow for labeling nascent proteins in neuronal cultures. First, cells are depleted of methionine (brown dots) and the medium replaced with HBSS buffer. Homopropargylglycine (HPG) is added to the cells, where it will be incorporated into newly synthesized proteins (blue hexagons). The incorporated HPG is then labeled with Alexa 594 containing an azide moiety (red hexagons). Thereafter, cells are subjected to immunostaining to label specific neuronal components.
For microfluidic perfusion chambers, HPG in HBSS was added to the perfusion well. In all other compartments, including the two wells next to the perfusion well, HBSS was added. HPG was perfused at an initial flow rate of 0.010 ml/hr and was slowed to 0.005 ml/hr after 10 min to facilitate HPG uptake into the cell. HPG was perfused for a total of 120 min. All HPG incubations were conducted in a cell culture incubator at 37°C and with 5% CO2. For microfluidic chambers, after HPG treatment, each of the following reagents were applied to the entire chamber. After HPG incubation, the cells were washed three times with pre-warmed PBS. Cells were then fixed by incubation with 4% paraformaldehyde (PFA; Sigma-Aldrich) and 16% sucrose in PBS for 15 min at room temperature. After fixation, cells were washed with PBS and incubated with 0.5% Triton-X 100 (Sigma-Aldrich) in PBS for 10 min at room temperature. Permeabilized cells were washed. The Alexa 594-azide containing click-reaction-cocktail was prepared according to the instructions of the assay kit provider. The reaction cocktail was added to the cells for 30 min at room temperature. The remaining click-reaction-cocktail was removed by washing the cells with PBS followed by removal of the silicone chambers. Samples were treated with 4% bovine serum albumin (BSA; ICN Biomedicals; Santa Ana, CA) in PBS blocking solution for 30 min at room temperature. Then, primary antibodies were diluted in blocking solution and added to the samples for an overnight incubation at 4°C. The next day, samples were washed with blocking solution and incubated with the Alexa-conjugated secondary antibodies diluted in 4% BSA-PBS for 30 min at room temperature. The samples were washed with PBS and then with distilled water to reduce the amount of potentially fluorescent salts in the samples. Finally, the samples were mounted in ProLong Gold anti-fade reagent (Life Technologies) and dried for at least 24 hr before image acquisition. The following primary antibodies were employed in the study: mouse anti-tau1 (MAB3420, Millipore, Billerica, MA; 1:1000); rabbit anti-MAP2 (ab32454, Abcam, Cambridge, UK; 1:2000); mouse anti-PSD95 (MA1-045, Thermo Scientific, Waltham, MA; 1:2000). Goat anti-rabbit Alexa 405 (Molecular Probes, Life Technologies, Grand Island, NY; 1:250) and goat anti-mouse Alexa 488 (Molecular Probes; 1:500) were used as secondary antibodies.
RNA Isolation and Reverse Transcription Polymerase Chain Reaction (RT-PCR)
Total RNA from microfluidic axon chambers was isolated as previously described (Taylor et al. 2009). RNA was isolated with the RNAeasy extraction kit (Qiagen; Germantown, MD), according to the manufacturer’s manual. RNA purity was determined using the Nanodrop ND1000 (Thermo Scientific) UV-spectrophotometer. The 260/280 nm ratios were measured and samples with ratios of 2.0 ± 0.05 were considered pure. cDNA was synthesized using the RevertAid RT reverse transcription kit (Thermo Scientific) from isolated RNA according to the protocol provided by the supplier. RT-PCR detection of mRNA transcripts (using 50 PCR cycles) was performed using the SensiFAST SYBR No-ROX kit (Bioline; London, UK). The following gene-specific primers were used: rat Scn3b, forward 5’- TGTG-GTGTGACTTGAGGTGAT, rat Scn3b, reverse 5’- TGTTGGCT-CTTCGGTTCAGG, rat RragB forward 5’- AAAGAACAGCGAGATGCCCA, rat RragB reverse 5’- GCTGCAGA-AGGAATGGATGG.
Microscopy and Image Analysis
Images were analyzed using the Fiji image processing software (Schindelin et al. 2012). All images from a single experiment ware taken using the same microscope settings to allow subsequent quantitative analysis of the fluorescence intensities. In all experiments, any residual background signal measured after blocking translation was subtracted from the data. For experiments performed in whole neuronal cultures (24 well), MAP2 staining was used to either define the somata or single neurites before the intensity of the HPG signal was measured. For the quantification of the HPG signal in dendrites or axons, MAP2 and Tau antibodies were used, respectively. The same selection threshold was used between images from the same experiment. In whole cell preparations, samples incubated with no HPG treatment served as a background control. The specificity of the click reaction in the absence of specific ligation azide binding partners was used to determine the baseline signal. Protein labelling in axons was quantified by measuring the average intensity of the HPG signal inside regions of interest as selected based on Tau staining. For measuring the HPG signal in the axon, Tau staining was used as a mask to select the region of interest, with the same selection threshold applied between images from the same experiment. For the microfluidic perfusion chambers, regions of interest were defined within the perfusion channel to measure the intensity of the HPG signal. For quantifying protein labelling in synapses, the average intensity of HPG was measured determining the fraction of HPG signal overlapping with the PSD-95 signal. This value was subsequently normalized for the area that was threshold selected for analysis. The same threshold was used across the different conditions. The same selection threshold was used between images from the same experiment. Fluorescence images of the axons were obtained using a Leica DMRA fluorescence microscope fitted with a DFC340 FX CCD camera (Leica; Wetzlar, Germany). A Leica TCS SP2 AOBS Confocal Laser Scanning Microscope (CLSM) was used to capture fluorescence images of whole neuronal cultures and of the microfluidic perfusion chambers.
Statistical Analysis
Quantitative data are presented as the mean ± SEM. Non-paired two-tailed Student’s t-test was used to determine significant differences between two groups. One-way ANOVA with Bonferroni’s multiple comparison testing was used to analyze significant differences between multiple groups. p≤ 0.05 was considered significant.
Results
Labelling of Nascent Proteins in Primary Neuronal Cultures
To examine protein synthesis in distal cellular compartments, such as axons and dendrites, we first confirmed that the click reaction can be used for the detection of nascent proteins in cortical neurons. To this end, rat primary cortical neurons [14 days in vitro (DIV)] were methionine-depleted and treated with HPG (Fig. 1). After HPG labelling, neurons were subjected to Tau and MAP2 staining to visualize axons and dendrites, respectively (Fig. 2A). Neurons treated with HPG alone showed a fluorescent signal representative of the baseline rate of translation in cortical neurons. To examine whether HPG incorporation is dependent on translation, we co-incubated HPG-treated cells with the protein synthesis inhibitor cycloheximide (CHX), which selectively inhibits eukaryotic translation elongation (Schneider-Poetsch et al. 2010). Translation inhibition resulted in a significant reduction in HPG incorporation, suggesting that HPG incorporation is specifically dependent on active mRNA translation (mean 1.913 ± 0.2035 HPG vs 0.8657 ± 0.1142 CHX relative intensity; p=0.0013). Furthermore, translation was strongly enhanced after neurons were depolarized with the addition of 50 mM KCl during the initial 10 min of HPG incubation. This increase could be blocked in cells treated with CHX (4.421 ± 0.3565 KCl vs 0.8624 ± 0.0917 KCl + CHX relative intensity; p≤0.0001) (Fig. 2B). Using MAP2 and Tau immunocytochemistry, selective HPG incorporation in the dendrites and axons of cortical neurons was detected, respectively. The results of this investigation revealed that KCl depolarization strongly enhanced the intensity of the HPG signal both in dendrites (0.1401 ± 0.0486 HPG vs 0.8106 ± 0.1319 KCl relative intensity; p≤0.0001) and in axons (mean 0.2885 ± 0.1535 HPG vs 1.509 ± 0.2817 KCl relative intensity; p<0.0001). This increase in translation could be completely blocked by the inclusion of CHX in dendrites (0.8106 ± 0.1319 KCl vs 0.1486 ± 0.0385 KCl + CHX relative intensity; p<0.0001) and axons (1.509 ± 0.2817 KCl vs 0.4866 ± 0.1388 KCl + CHX relative intensity; p≤0.0041) (Fig. 2C, 2D). These findings show the specificity of HPG and azide binding, as demonstrated by the general low background signal in the control and inhibited groups. However, under these conditions, it is not clear whether this signal is related to local protein synthesis in the neurites or whether it represents proteins that were produced in the somata and subsequently transported throughout the neuron.
Figure 2.

HPG incorporation into neurons is dependent on translational activity. (A) Representative images of HPG signal (inverted greyscale and red) in DIV 14 cortical neurons. Translation was stimulated by KCl-mediated neuronal potentiation and reduced by pre-incubation with the translation inhibitor cycloheximide (CHX). Immunostaining with anti-Tau (green) and anti-MAP2 (blue) distinguishes between axons and dendrites, respectively. (B) Quantification of the relative changes in somatic HPG signal intensity after treatment with CHX, KCl or CHX and KCl. (C-D) Shows the relative intensity of HPG in the (C) MAP2 positive dendrites and (D) Tau positive axons after treatment with CHX, KCl or CHX and KCl. Data represent the mean ± SEM and single values shown for n=11 to 15 samples collected from three independent experiments. DIV, days in vitro; HPG, homopropargylglycine; MAP2, microtubule-associated protein 2. One-way ANOVA with Bonferroni multiple comparison test; **p<0.01; ***p<0.001. Scale, 50 µm.
Labelling of Newly Synthesized Proteins in Growing Axons
Local translation in growing axons is essential for rapidly supplying the demand for newly synthesized proteins in these distal neuronal compartments. To visualize the fraction of proteins locally synthesized within distal neuronal compartments, we made use of microfluidic chambers allowing for fluidic isolation of growing axons (Taylor et al. 2005). Aiming at labelling locally synthesized proteins in growing axons, compounds were applied and fluidically isolated to the axonal compartment. Cortical neurons were cultured in microfluidic chambers in the “soma” compartment until their corresponding axons reached the adjacent unoccupied “axon” compartment (Fig. 3A). An absence of MAP2 within the axon compartment, using immunocytochemistry, suggested that this area is devoid of dendrites (Fig. 3B). Detection and quantification of HPG incorporation revealed that newly synthesized proteins can be detected in growing axons and that application of the translation inhibitor CHX significantly reduced the signal (mean 0.5238 ± 0.1515 HPG vs 0.0 ± 0.07834 CHX relative intensity; p=0.0050) (Fig. 3B, 3C). To demonstrate the absence of somata within the axon compartment, pure RNA samples prepared from the distal axons were assessed by RT-PCR, using gene-specific primer sets for mRNAs present in neuronal cell somata only. As shown in Fig. 3D, amplicons for Sodium Channel, Voltage Gated, Type III Beta Subunit (SCN3B) and Glycine Receptor, Beta (GLRB), were readily detected in RNA prepared from the neurons present in compartment harboring parental cell somata and proximal axons, but were not observed in RNA obtained from distal axons. These genes were selected as markers because, in a recent investigation using microarray and qPCR analyses of soma and axon RNAs, we found that SCN3B and GLRB mRNAs were present in the somata but were not localized in the axons (unpublished microarray data).
To investigate how the local treatment of axon compartments with HPG selectively incorporates this compound into axonal proteins without being introduced into parental cell soma proteins, we examined HPG incorporation into the parental somata compartment. Little HPG incorporation was detected in the parental somata of axons treated with this reagent. Moreover, the weak HPG signal detected in the somata was insensitive to CHX treatment (Fig. 3E, 3F). Images from the somatic area were captured at the center of the compartment, approximately 200 µm away from the 450-µm long microgroove barrier. These observations further demonstrate the feasibility of in situ tagging and the detection of relatively low-abundance, locally synthesized proteins in growing axons using microfluidic chambers.
Detection of Nascent Proteins in Postsynaptic Densities
The ability to detect relatively small amounts of newly produced proteins in defined cellular compartments, such as in growing neurites, led us to evaluate whether nascent proteins within synapses of mature neurons could be examined using click chemistry. To this end, the microfluidic perfusion chamber was utilized enabling the local perfusion of compounds through a channel in which neuronal synapses are formed (Fig. 4A). Cortical neurons were cultured in perfusion chambers for 14 to 16 days to allow for the formation and maturation of synapses inside the perfusion chamber. HPG alone or in combination with CHX was applied locally by pumping the solutions for 120 min through the perfusion channels harboring mature synapses (Fig. 4B, 4C). Quantification of incorporated HPG was performed to evaluate whether newly synthesized proteins could be imaged within the perfusion channel. This resulted in the detection of labelled HPG in the perfused area of the chamber. Importantly, the HPG signal was significantly diminished when translation was repressed by CHX (mean 0.4293 ± 0.09 HPG vs 0.0 ± 0.045 CHX relative intensity; p=0.0052) (Fig. 4D). We also examined whether perfusion of HPG resulted in its incorporation within the parental soma proteins. We chose to examine HPG incorporation in cell somata in the lateral compartment, which is connected to the perfusion chamber via the shorter microgroove barrier, enabling dendrites to project toward the central perfusion chamber. Investigation of HPG incorporation into the cell somata located in this compartment suggested that only a very small amount of HPG is incorporated in the parental cell somata of the postsynaptic compartment (Fig. 4E and 4F). Images were taken central of the dendrite-generating somatic compartment, at an approximate 200 µm away from the 75-µm long microgrooves.
To demonstrate that the perfused area of the chamber can be employed to label nascent proteins within mature synapses, we performed PSD-95 immunolabelling to mark the post-synaptic compartment of neurons grown in microfluidic perfusion chambers (Fig. 5A). The quantification of newly synthesized, labelled proteins colocalizing with postsynaptic densities visualized local translation at this site, which exhibited a significant reduction in HPG signal once CHX was applied (mean 0.3325 ± 0.03586 HPG vs 0.0 ± 0.04146 CHX relative intensity; p≤0.0001; Fig. 5B). In addition, a significant amount of HPG signal was observed outside the post-synaptic regions, indicating that nascent proteins are synthesized along the dendrite lengths. Collectively, these results demonstrate that the microfluidic perfusion chamber can be utilized to label and visualize nascent proteins in defined synaptic entities. Furthermore, these finding suggest that HPG can label translation in discrete neuronal compartments, demonstrating its general usefulness for investigating local translation.
Figure 5.

Visualization of nascent protein synthesis in postsynaptic densities from neurons grown in microfluidic perfusion chambers. (A) Representative confocal images of the perfusion channel from microfluidic perfusion chambers in which DIV 14-16 primary cortical neurons were grown. Neurons were treated with HPG alone or in combination with CHX and stained for HPG incorporation (inverted greyscale and red) or PSD-95 (green), the overlay shows the overlap between these two signals. (B) Quantified relative HPG signals inside PSD-95 puncta of neurons treated HPG alone or in which translation was inhibited using CHX, normalized for the total selected PSD-95-positive-area. Data represent the mean ± SEM and single values shown for n=10 to 11 culture chambers collected from three independent experiments. CHX, cyclohexamide; DIV, days in vitro; HPG, homopropargylglycine; PSD-95, postsynaptic density protein 95. p-values are determined by two-tailed unpaired Students t-test. ***p<0.0001. Scale, 5 µm.
Discussion
Previously, the majority of research to visualize local translation had been performed using genetic fluorophore labeling techniques, allowing for the visualization of specific individual proteins. This approach, however, is limited to monitoring only a few proteins at the same time. Here, it is demonstrated that the combination of amino acid tagging with microfluidic chambers can be harnessed for the labelling of the proteome within specific neuronal sub-compartments. This technique allowed for the detection of relatively low basal levels of nascent proteins synthesized within axons and synapses of neurons grown in microfluidic devices using the click amino acid labeling assay.
To achieve metabolic labeling of nascent proteins, HPG or azidohomoalanine (AHA) have been successfully employed as methionine analogs. Both molecules efficiently and abundantly label nascent proteins within the soma and neurites of neurons (Dieterich et al. 2010). It is important to note that both methionine surrogates have slightly different charging rates by methionyl-tRNA synthetase, with AHA incorporating faster into nascent polypeptides than HPG. This difference in incorporation rates can be harnessed for sequential pulse labeling of different protein populations within a single experiment. As demonstrated previously, during protein tagging, an initial pulse with HPG, followed by incubation with AHA offers the most optimal incorporation rates (Dieterich et al. 2010).
Studying the localized production of proteins within neuronal compartments is important for understanding processes such as axon development or synaptic plasticity. The combinatorial use of amino acid labeling and microfluidic cell culture chambers has been reported previously and facilitated the study of the local regulation of gene expression within neuronal processes (Ji and Jaffrey 2012; Tom Dieck et al. 2012). For example, combining this technique with the induction of long-term potentiation (LTP) or depression (LTD) enabled the investigation of the role of local translation in these physiological processes within synapses in vitro (Taylor et al. 2010). These forms of synaptic plasticity could either be achieved using specific pharmacological compounds such as DHPG, an agonist for the glutamate receptors Glu1 and Glu5 that induces LTD (Ito et al. 1992), or by the application of an alternative system, like the genetic introduction of opsins for the optogenetic control of depolarization (Kos et al. 2013). The advantage of this system is that the design of the perfusion chamber would permit the investigator to express the opsins exclusively in one cell population. In this manner, individual (sub-) populations could be stimulated in order to concomitantly quantify translation activity in neurons (Nagel et al. 2003; Zhang et al. 2010). Furthermore, this technique could also be applied to study the effect of the non-coding RNAs, such as miRNA, on global protein output within restricted neuronal compartments. For example, miR-16 was shown to locally control axonal eIF2B2 and eIF4G2 mRNA expression impacting axonal translation machinery (Kar et al. 2013).
In our experiments, we could detect nascent proteins under basal translation activity in cortical neurons. Our experiments furthermore showed that the click chemistry is sufficiently sensitive to detect relatively low amounts of locally produced proteins in growing axons and within synaptic densities. Previous reports showed similar findings in which neuronal stimulation resulted in enhanced mRNA translation. For example, neuronal stimulation using brain-derived neurotrophic factor (BDNF) resulted in a substantial enhancement of translation activity in the dendrites of hippocampal neurons (Dieterich et al. 2010). Furthermore, it was shown that LTD induction in microfluidic perfusion chambers by locally applying DHPG increased Arc levels within the stimulated synapse (Taylor et al. 2010). The detection of significant amounts of labeled proteins during basal translation suggests that, in growing axons, substantial local translation takes place during the experimental procedure without depolarization of the cells or the addition of growth factors. The benefit of using microfluidic chambers is that it allows for the isolated labeling and detection of proteins in axons and synapses, whereas, in traditional neuronal culture, the subtle translation activity within these neuronal components is possibly masked by vast amount of protein synthesis in the soma. Furthermore, our experiments showed that local application of the click chemistry did not lead to substantial labeling of nascent proteins within the somatic compartment. This suggests that the relatively short incubation period used during our experiments does not lead to intracellular diffusion of the HPG. However, it bears mentioning that we cannot exclude the possibility of diffusion occurring when longer incubation periods or higher concentrations of HPG are applied. Although CHX is a selective protein synthesis inhibitor, previous reports have shown that it can induce apoptosis when exposed to cells over extended periods of time (Borner et al. 1995). In our experiments, we exposed cells to CHX only for short periods of time and did not observe any apoptosis or necrosis in the neurons exposed to CHX.
In conclusion, here we report on the use of fluorescent amino acid labeling in microfluidic chambers to detect nascent, locally translated proteins in neuronal cultures. The premise of the presented technology is that it might help to identify novel regulatory mechanisms of local translation at nerve endings and will facilitate the illumination of novel pathways involved in neuronal development and synaptic plasticity.
Acknowledgments
The authors gratefully thank Ms. Jenna Gale (National Institute of Mental Health), Dr. Liesbeth Pierson and Dr. Stef Olsthoorn (General Instruments Department of the Radboud University) for their technical support and assistance.
Footnotes
Author Contributions: AK, KW, AEG, and AA conducted the studies; AK, KW, BBK, GJM and AA analyzed the data; AK, BBK and AA wrote the manuscript.
Competing Interests: The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The research of the authors is supported by grants from the Donders Center for Neuroscience fellowship award of the Radboudumc (to AA); the FP7-Marie Curie International Reintegration Grant (Grant 276868 to AA). This work was supported by the Division of Intramural Research Programs of the National Institute of Mental Health (MH002768).
References
- Abaza I, Gebauer F. (2008). Trading translation with RNA-binding proteins. RNA 14:404-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akins MR, Berk-Rauch HE, Fallon JR. (2009). Presynaptic translation: stepping out of the postsynaptic shadow. Front Neural Circuits 3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschrafi A, Schwechter AD, Mameza MG, Natera-Naranjo O, Gioio AE, Kaplan BB. (2008). MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci 28:12581-12590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassell GJ, Warren ST. (2008). Fragile X syndrome: loss of local mRNA regulation alters synaptic development and function. Neuron 60:201-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beatty KE, Liu JC, Xie F, Dieterich DC, Schuman EM, Wang Q, Tirrell DA. (2006). Fluorescence visualization of newly synthesized proteins in mammalian cells. Angew Chem Int Ed Engl 45:7364-7367. [DOI] [PubMed] [Google Scholar]
- Borner MM, Myers CE, Sartor O, Sei Y, Toko T, Trepel JB, Schneider E. (1995). Drug-induced apoptosis is not necessarily dependent on macromolecular synthesis or proliferation in the p53-negative human prostate cancer cell line PC-3. Cancer Res 55:2122-2128. [PubMed] [Google Scholar]
- Campbell DS, Holt CE. (2001). Chemotropic responses of retinal growth cones mediated by rapid local protein synthesis and degradation. Neuron 32:1013-1026. [DOI] [PubMed] [Google Scholar]
- Capano CP, Giuditta A, Castigli E, Kaplan BB. (1987). Occurrence and sequence complexity of polyadenylated RNA in squid axoplasm. J Neurochem 49:698-704. [DOI] [PubMed] [Google Scholar]
- Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. (2010). In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat Neurosci 13:897-905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gioio AE, Chun JT, Crispino M, Capano CP, Giuditta A, Kaplan BB. (1994). Kinesin mRNA is present in the squid giant axon. J Neurochem 63:13-18. [DOI] [PubMed] [Google Scholar]
- Hinz FI, Dieterich DC, Tirrell DA, Schuman EM. (2012). Non-canonical amino acid labeling in vivo to visualize and affinity purify newly synthesized proteins in larval zebrafish. ACS Chem Neurosci 3:40-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt CE, Schuman EM. (2013). The central dogma decentralized: new perspectives on RNA function and local translation in neurons. Neuron 80:648-657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J, Liu X, Shen J, Zhao G, Wang PG. (2012). The impact of click chemistry in medicinal chemistry. Expert Opin Drug Discov 7:489-501. [DOI] [PubMed] [Google Scholar]
- Ito I, Kohda A, Tanabe S, Hirose E, Hayashi M, Mitsunaga S, Sugiyama H. (1992). 3,5-Dihydroxyphenyl-glycine: a potent agonist of metabotropic glutamate receptors. Neuroreport 3:1013-1016. [PubMed] [Google Scholar]
- Jackson RJ, Hellen CU, Pestova TV. (2010). The mechanism of eukaryotic translation initiation and principles of its regulation. Nat Rev Mol Cell Biol 11:113-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji SJ, Jaffrey SR. (2012). Intra-axonal translation of SMAD1/5/8 mediates retrograde regulation of trigeminal ganglia subtype specification. Neuron 74:95-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Gkogkas CG, Sonenberg N, Holt CE. (2014). Remote control of gene function by local translation. Cell 157:26-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan BB, Gioio AE, Capano CP, Crispino M, Giuditta A. (1992). beta-Actin and beta-Tubulin are components of a heterogeneous mRNA population present in the squid giant axon. Mol Cell Neurosci 3:133-144. [DOI] [PubMed] [Google Scholar]
- Kar AN, MacGibeny MA, Gervasi NM, Gioio AE, Kaplan BB. (2013). Intra-axonal synthesis of eukaryotic translation initiation factors regulates local protein synthesis and axon growth in rat sympathetic neurons. J Neurosci 33:7165-7174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kos A, Loohuis NF, Glennon JC, Celikel T, Martens GJ, Tiesinga PH, Aschrafi A. (2013). Recent developments in optical neuromodulation technologies. Mol Neurobiol 47:172-185. [DOI] [PubMed] [Google Scholar]
- Liu-Yesucevitz L, Bassell GJ, Gitler AD, Hart AC, Klann E, Richter JD, Warren ST, Wolozin B. (2011). Local RNA translation at the synapse and in disease. J Neurosci (45):16086-16093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G, Szellas T, Huhn W, Kateriya S, Adeishvili N, Berthold P, Ollig D, Hegemann P, Bamberg E. (2003). Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc Natl Acad Sci U S A 100:13940-13945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JW, Vahidi B, Taylor AM, Rhee SW, Jeon NL. (2006). Microfluidic culture platform for neuroscience research. Nat Protoc 1:2128-2136. [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. (2012). Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676-682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. (2010). Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol 6:189-198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, Greenberg ME. (2006). A brain-specific microRNA regulates dendritic spine development. Nature 439:283-289. [DOI] [PubMed] [Google Scholar]
- Taylor AM, Blurton-Jones M, Rhee SW, Cribbs DH, Cotman CW, Jeon NL. (2005). A microfluidic culture platform for CNS axonal injury, regeneration and transport. Nat Methods 2:599-605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AM, Dieterich DC, Ito HT, Kim SA, Schuman EM. (2010). Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 66:57-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tom Dieck S, Muller A, Nehring A, Hinz FI, Bartnik I, Schuman EM, Dieterich DC. (2012). Metabolic labeling with noncanonical amino acids and visualization by chemoselective fluorescent tagging. Curr Protoc Cell Biol Chapter 7:Unit7 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tornoe CW, Christensen C, Meldal M. (2002). Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J Org Chem 67:3057-3064. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Edelman GM. (2005). Differential translation and fragile X syndrome. Genes Brain Behav 4:360-384. [DOI] [PubMed] [Google Scholar]
- Wang DO, Kim SM, Zhao Y, Hwang H, Miura SK, Sossin WS, Martin KC. (2009). Synapse- and stimulus-specific local translation during long-term neuronal plasticity. Science 324:1536-1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon BC, Zivraj KH, Holt CE. (2009). Local translation and mRNA trafficking in axon pathfinding. Results Probl Cell Differ 48:269-288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Gradinaru V, Adamantidis AR, Durand R, Airan RD, de Lecea L, Deisseroth K. (2010). Optogenetic interrogation of neural circuits: technology for probing mammalian brain structures. Nat Protoc 5:439-456. [DOI] [PMC free article] [PubMed] [Google Scholar]