

Abstract
CYP2W1 is a recently discovered human cytochrome P450 enzyme with a distinctive tumor-specific expression pattern. We show here that CYP2W1 exhibits tight binding affinities for retinoids, which have low nanomolar binding constants, and much poorer binding constants in the micromolar range for four other ligands. CYP2W1 converts all-trans retinoic acid (atRA) to 4-hydroxy atRA and all-trans retinol to 4-OH all-trans retinol, and it also oxidizes retinal. The enzyme much less efficiently oxidizes 17β-estradiol to 2-hydroxy-(17β)-estradiol and farnesol to a monohydroxylated product; arachidonic acid is, at best, a negligible substrate. These findings indicate that CYP2W1 probably plays an important role in localized retinoid metabolism that may be intimately linked to its involvement in tumor development.
Introduction
Human cytochrome P450 (CYP or P450) enzymes play an important role in the metabolism of steroids, vitamins, eicosanoids, fatty acids, and xenobiotics. The human genome encodes 57 full-length cytochrome P450 enzymes, several of which are denoted as orphan enzymes because their properties, substrate specificities, and physiologic functions are obscure (Guengerich, 2015). The partial cDNA sequence of CYP2W1, one of these orphan P450 enzymes (GenBank Accession No. AK000366), was first found in a cDNA library from the human hepatoma cell line HepG2, and the full gene sequence was later obtained from the Celera sequence database (Karlgren et al., 2005, 2006).
Unlike most cytochrome 2 family members, CYP2W1 is not expressed in adult human liver or human primary hepatocytes (Choudhary et al., 2005; Girault et al., 2005; Karlgren et al., 2006; Karlgren and Ingelman-Sundberg, 2007). The expression level is also low or undetectable in adult extrahepatic tissues (Choudhary et al., 2005; Karlgren et al., 2006; Karlgren and Ingelman-Sundberg, 2007). Higher levels of CYP2W1 mRNA were observed in some human fetal tissues, such as kidney, liver, and lung, although the levels were still very low compared with CYP2W1 expression in human hepatoma cell line HepG2 cells (Choudhary et al., 2005; Karlgren et al., 2006). Additionally, CYP2W1 expression is strongly upregulated in differentiating human keratinocytes, and this differentiation-induced upregulation of CYP2W1 is inhibited by retinoic acid (Du et al., 2006a,b). CYP2W1 is also significantly expressed in late stage mouse fetal tissues (Choudhary et al., 2005), and high expression of CYP2W1 is observed in rat fetal tissues, with the highest expression in the colon at day 21 just before delivery. Decreasing amounts of CYP2W1 mRNA have been seen in the colon of 8- to 14- week old rats, indicating that the CYP2W1 gene is gradually “silenced” after birth (Karlgren et al., 2006; Karlgren and Ingelman-Sundberg, 2007).
In contrast to its absence or low expression in normal tissues, CYP2W1 shows a distinctive tumor-related expression pattern. CYP2W1 mRNA has been detected in many tumor samples, especially colon and adrenal tumors. High expression of CYP2W1 in some human colon tumors was revealed by Western blots (Karlgren et al., 2006). Subsequent studies showed that CYP2W1 expression is a prognostic marker for colon cancer (Edler et al., 2009; Stenstedt et al., 2012). CYP2W1 was also expressed at much higher levels in gastric cancer samples than in normal tissues as shown by serial analysis of gene expression (Karlgren and Ingelman-Sundberg, 2007). Furthermore, CYP2W1 gene expression has been detected in both subconfluent and confluent MCF10A human breast epithelial cells (Thomas et al., 2006). More recently, CYP2W1 was shown to be highly expressed in both normal and neoplastic adrenal glands, but it was absent or low in normal nonadrenal tissues (Ronchi et al., 2014).
The distinctive expression pattern of CYP2W1, with high expression in fetal and tumor tissues, but no expression in normal adult tissues, suggests that cancer-specific expression of P450s might be related to reversion to more primitive, embryonic cell types in which it had specific physiologic roles. Exploration of the endogenous substrates for CYP2W1 will help to clarify the physiologic roles that CYP2W1 may play in fetal development and cancer progression.
CYP2W1 has been expressed in human embryonic kidney cells (Karlgren et al., 2006), and modified CYP2W1 enzymes with an N-terminal truncation have been heterologously expressed in Escherichia coli (Wu et al., 2006; Yoshioka et al., 2006). Limited studies have been published regarding the substrate selectivities of CYP2W1, some of which are contradictory. CYP2W1 has been reported to oxidize arachidonic acid at a slow rate (Karlgren et al., 2006; Wu et al., 2006) and the metabolites were identified as 14,15-DHET, 11,12-DHET, and 8,9-DHET by tandem mass spectrometry (MS/MS) analysis (Karlgren et al., 2006); however, Yoshioka et al. (2006) reported that CYP2W1 showed no activity toward fatty acids, including arachidonic acid. Metabolomics analysis reported that CYP2W1 oxidizes lysophospholipids (Xiao and Guengerich, 2012). CYP2W1 has also been reported to turn over a variety of exogenous chemicals, including benzphetamine, several polycyclic aromatic hydrocarbon dihydrodiols (Wu et al., 2006), the prodrug AQ4N [1,4-bis{[2-(dimethylamino-N-oxide)ethyl]amino}-5,8-dihydroxyanthracene-9,10-dione] (Nishida et al., 2010), indoles (Yoshioka et al., 2006), indolines (2-methyl-5-nitroindoline or 5-bromoindoline) (Gomez et al., 2010), and fluorinated 2-aryl-benzothiazole antitumor agents (Wang and Guengerich, 2012).
Here we evaluated the catalytic activities of CYP2W1 toward physiologically relevant substrates, including the retinoids, steroids, and fatty acids. Our results indicate that CYP2W1 oxidizes retinoic acid to 4-hydroxy retinoic acid and retinol to 4-hydroxy retinol, and it also oxidizes retinal. Furthermore, CYP2W1 oxidizes 17β-estradiol and farnesol, but the binding affinities for these substrates are much lower than those of the retinoids and their oxidation by CYP2W1 is unlikely to be physiologically relevant.
Materials and Methods
Materials.
All-trans retinoic acid (atRA), retinal and retinol, lanosterol, cholesterol, progesterone, cortisone, hydrocortisone, 17β-estradiol, testosterone, ergocalciferol, cholecalciferol, farnesol, geraniol, geranylgeraniol, arachidonic acid, and lauric acid were purchased from Sigma-Aldrich (St. Louis, MO). The authentic standard 4-hydroxy atRA was purchased from Santa Cruz Biotechnologies (Santa Cruz, CA). The CYP2W1 pCWori plasmid and pGro12 expression plasmid encoding the chaperone proteins GroEL/ES were generous gifts from Dr. F. P. Guengerich (Vanderbilt University, Nashville, TN). Recombinant human nicotinamide adenine dinucleotide phosphate (NADPH)-cytochrome P450 reductase (CPR) was expressed in E. coli and purified as previously described (Dierks et al., 1998). Baculovirus-insect cell microsomes expressing CYP3A7, human reductase, and cytochrome b5, and baculovirus-insect cell microsomes expressing CYP2C8, human reductase and cytochrome b5 were purchased from Corning Life Sciences (Tewksbury, MA).
Bacterial Expression and Purification of CYP2W1.
The N-terminal hydrophobic sequence of CYP2W1 before the well-conserved proline-rich region was truncated and modified, as reported previously (Wu et al., 2006). The N-terminal amino acid sequence was MAKKTSSKGKL PPGPRPLP. The experimental procedures were adapted from previous reports (Wu et al., 2006; Nishida et al., 2010). E. coli DH5α competent cells were transformed with a pCwori plasmid encoding CYP2W1 and a pGro12 plasmid encoding the chaperone proteins GroES/L. Ampicillin and kanamycin-resistant colonies were picked and used to inoculate 100 ml overnight small-scale LB cultures. A 15-ml aliquot of the overnight LB culture was transferred to 1 liter of TB medium containing 100 μg/ml of ampicillin, 50 μg/ml of kanamycin, and 0.1% L-arabinose for the induction of GroES/L expression. The cultures were grown at 37°C and 220 rpm. After approximately 4 hours, when the optical density at 600 nm reached 0.8, 0.5 mM 5-aminolevulinic acid and 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) were added to induce P450 expression. The cultures were continued at 29°C at 190 rpm for 40 hours before harvesting. The cell cultures were harvested by centrifugation at 6000g for 15 minutes, and the cell pellets were frozen and stored at −80°C. Subsequent steps were performed at 4°C. The frozen pellet was broken into smaller pieces and mixed with 5 ml of lysis buffer [100 mM potassium phosphate buffer, pH 7.4, with 20% (v/v) glycerol, 0.1 mM dithiothreitol (DTT), 1 mM phenylmethylsulfonyl fluoride, and 50 μg/ml lysozyme] per 1 mg of pellet dry weight. The cells were lysed by sonication, and the cell lysate was centrifuged at 10,000g for 20 minutes. The supernatant was transferred to a chilled tube and centrifuged at 100,000g for 1 hour. The pellet containing the P450 membrane fractions was resuspended in 15 ml/1 g of wet-weight membrane fractions of 50 mM Tris-acetate buffer, pH 7.4, containing 250 mM sucrose and 0.25 mM EDTA. The mixture was then diluted with four parts of 300 mM potassium phosphate buffer, pH 7.6, containing 20% (v/v) glycerol, 1.25% (w/v) CHAPS, 0.1 mM DTT, and 0.1 mM EDTA. This mixture was stirred for 2 hours and then centrifuged at 100,000g for 1 hour. The supernatant generated above was collected, and 20 mM imidazole was added before loading onto a HisTrap HP column (GE Healthcare Life Sciences, Pittsburgh, PA) that had been equilibrated with eight column volumes of buffer A [300 mM potassium phosphate buffer, pH 7.6 with 20% (v/v) glycerol, 1% (w/v) CHAPS, 0.1 mM DTT, and 0.1 mM EDTA] followed by one column volume of buffer A with 20 mM imidazole. After washing with eight column volumes of buffer A with 20 mM imidazole, two column volumes of 300 mM potassium phosphate buffer, pH 7.6, containing 20% (v/v) glycerol, 0.1 mM DTT, and 0.1 mM EDTA were added to remove CHAPS. The P450 protein was eluted at a flow rate of 0.5 ml/min on an eight-column volume gradient from 10% to 100% buffer B [300 mM potassium phosphate buffer, pH 7.6, containing 20% (v/v) glycerol, 0.1 mM DTT, 0.1 mM EDTA, and 200 mM imidazole] followed by 100% buffer B for two column volumes. The eluted P450 fractions were pooled and loaded onto an SP-Sepharose column that had been equilibrated with column volumes of buffer C (100 mM potassium phosphate, pH 7.4 containing 20% glycerol) and 50 mM NaCl. After washing with three column volumes of buffer C with 50 mM NaCl then three column volumes of buffer C with 150 mM NaCl, the P450 protein was eluted at a flow rate of 0.5 ml/min on a four-column volume gradient from 20% to 100% buffer D (buffer C with 500 mM NaCl) followed by 100% buffer D for two additional column volumes. The typical yield for CYP2W1 expression using this method was ∼25 nmol/liter. More than one purified enzyme preparation was used in this study. The concentration of CYP2W1 in each purified preparation was determined from the absorbance at 450 nm in the ferrous-CO difference spectrum using an extinction coefficient of 91,000 M−1 cm−1 (Guengerich et al., 2009).
Spectroscopic Analysis of Ligand Binding to CYP2W1.
To measure retinoid binding to CYP2W1, purified CYP2W1 (2 μM or 200 nM) in 100 mM potassium phosphate buffer was added to the sample cuvette, and the same buffer without the protein was added to the reference cuvette. The retinoid compound (10 μM) was added in 0.4-μl increments to both cuvettes to normalize the absorption of the retinoid, and the absolute spectrum was recorded by scanning from 500 nm to 350 nm. The difference spectrum was obtained by subtracting the ligand-free baseline spectrum from the ligand-added spectra. The difference in absorbance between 390 nm and 420 nm was used for data analysis. Data were fitted to the Morrison eq. 1 using OriginLab, where Aobs is the observed absorption shift, Amax is the maximal shift, KS is the apparent dissociation constant, [Et] is the enzyme concentration, [S] is the ligand concentration, and n is the Hill coefficient. To measure the binding of all the other ligands, purified CYP2W1 (2 μM) in 100 mM potassium phosphate buffer was added to both the sample and reference cuvettes. The test compound (10 mM) was added in 0.4-μl increments to the sample cuvette, and the same amount of ethanol was added to the reference cuvette. The resulting difference spectrum was recorded by scanning from 500 nm to 350 nm by UV-vis spectrometry. The difference in absorbance between 390 nm and 420 nm was used for data analysis. Data were fitted to the Hill eq. 2 using OriginLab.
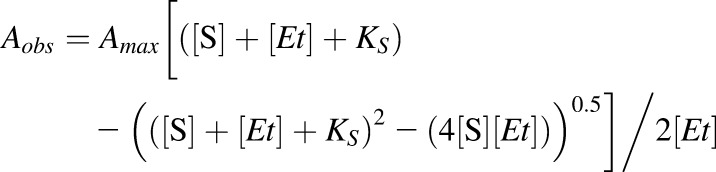 |
(1) |
| (2) |
The binding experiments for all test compounds were repeated at least twice. The binding constants were calculated as mean ± S.D. of values obtained from data fitting of individual experiments.
HPLC and LC-MS Analysis of Retinoid Metabolites.
CYP2W1 and CYP3A7 catalyzed reactions were carried out in 1.5-ml amber-colored Eppendorf tubes. The retinoid was added at a final concentration of 10 μM to 500 μl of reaction mixture. CYP2W1 reactions contained 100 nM purified CYP2W1, 200 nM purified human CPR in 100 mM potassium phosphate buffer (pH 7.4) with 1 mM deferoxamine and an NADPH regenerating system (5 mM glucose 6-phosphate, 4 U/ml glucose-6-phosphate dehydrogenase and 5 mM MgCl2). CYP3A7 and CYP2C8 reactions were carried out in 500 μl of reaction mixture containing 100 nM baculovirus-insect cell microsomes expressing human CYP3A7/CYP2C8 and cytochrome b5, supplemented with recombinant human CPR, 1 mM deferoxamine, and an NADPH regenerating system (5 mM glucose 6-phosphate, 4 U/ml glucose-6-phosphate dehydrogenase and 5 mM MgCl2). The mixtures were preincubated at 37°C for 5 minutes before the reaction was initiated by addition of 1 mM NADPH and further incubated at 37°C for 30 minutes.
The reaction (500 μl) was terminated by adding 1 ml of ethyl acetate. After vortexing, the mixture was centrifuged at 1,000g for 5 minutes, and the organic layer was separated and evaporated under nitrogen. The reaction was extracted twice by repeating this step. The dried sample residue was reconstituted with 100 μl of a solution consisting of 50% 50 mM aqueous ammonium acetate and 50% methanol. The retinoid metabolites were separated on an Agilent 1200 series HPLC system with an Agilent reverse phase Eclipse XDB-C18 column (5 µm, 4.6 ×150 mm; Agilent Technologies, Santa Clara, CA). The retinoids and their metabolites were detected using their absorbance at 340 nm. The mobile phase at a flow rate of 1 ml/min consisted of a linear gradient from an initial 35:65 20 mM ammonium acetate/methanol mixture to 0:100 20 mM ammonium acetate/methanol over 30 minutes. It was then held isocratic for 5 minutes before returning to the initial condition over 0.5 minute. The system was then re-equilibrated for another 10 minutes before the next sample injection.
Identification of the retinoid metabolites was accomplished by liquid chromatography-mass spectrometry (LC-MS) on a Waters Micromass ZQ (Waters, Milford, MA) coupled to a Waters Alliance HPLC equipped with a 2695 separation module, a Waters 2487 dual λ absorbance detector, and an Agilent reverse-phase Eclipse XDB-C18 column (5 µm, 4.6×150 mm). The MS was operated in the positive-ion mode for retinol metabolite detection or in the negative-ion mode for retinoic acid metabolite detection using electrospray (ES) to monitor the precursor-to-product ion transition of m/z 269 → 285 for retinol oxidation and 299 → 315 for retinoic acid oxidation. The retinoid metabolites were separated with an Agilent reverse phase Eclipse XDB-C18 column (5 µm, 4.6 × 150 mm). The mobile phase at a flow rate of 0.3 ml/min consisted of a linear gradient from an initial 35:65 20 mM ammonium acetate/methanol mixture to 0:100 20 mM ammonium acetate/methanol over 40 minutes. It was then held isocratic for 5 minutes before returning to the initial condition over 0.5 minute. The system was then re-equilibrated for another 10 minutes before the next sample injection. Retinoids and their metabolites were analyzed using the total ion current signal and the extracted ion for the specific mass. The MS settings for retinol metabolite detection were as follows: mode, ES+; capillary voltage, 3.5 kV; cone voltage, 30 V; extractor voltage, 3 V; radiofrequency voltage, 0.3 V; desolvation temperature, 250°C; source temperature, 120°C; desolvation flow, 350 liter/h; cone flow, 65 liter/hour. The MS settings for retinoic acid metabolite detection were as follows: mode, ES−; capillary voltage, 2.8 kV; cone voltage, 16 V; extractor voltage, 3 V; radiofrequency voltage, 0 V; desolvation temperature, 200°C; source temperature, 149°C; desolvation flow, 778 liter/h; cone flow, 112 liter/h. The UV absorbance at 340 nm and MS ES+/ES− spectra were recorded simultaneously.
Determination of Kinetic Parameters for CYP2W1-Catalyzed atRA Metabolism.
The reactions (1 ml) were carried out for 30 minutes at 37°C at atRA concentrations of 1, 2, 3, 4, 6, 8, 10, and 15 μM, and the products were analyzed by HPLC as described already herein. The amount of the 4-hydroxy atRA product was determined by peak area using the standard curve of the authentic 4-OH atRA standard. To determine the KM and Vmax values, the data points were fitted to the Hill eq. 3 using OriginLab, where, V is the product forming rate determined at any ligand concentration, Vmax is the maximal rate, Km is the substrate concentration at which the half maximal rate is achieved, [Et] is the total enzyme concentration used, [S] is the ligand concentration, and n is the Hill coefficient:
| (3) |
The experiments were repeated twice and the Km and Vmax were calculated as mean ± S.D. of values obtained from data fitting of individual experiments.
GC-MS Analysis of CYP2W1 17β-Estradiol/Farnesol Metabolism.
Reactions were carried out in 1.5-ml amber-colored Eppendorf tubes. The substrate was added at a final concentration of 100 μM to 500 μl of reaction mixture containing 1 μM purified CYP2W1, 2 μM purified human CPR in 100 mM potassium phosphate buffer (pH 7.4) with 1 mM deferoxamine, and an NADPH regenerating system (5 mM glucose 6-phosphate, 4 U/ml glucose-6-phosphate dehydrogenase and 5 mM MgCl2). The mixtures were preincubated at 37°C for 5 minutes before the reaction was initiated by addition of 1 mM NADPH and further incubated at 37°C. The reaction (0.5 ml) was terminated with 5 ml of methyl tert-butyl ether (MTBE). After vortexing, the mixture was centrifuged at 1000g for 5 minutes, and the organic layer was separated and evaporated under nitrogen. The dried sample residue was then trimethylsilylated to give the trimethylsilyl derivatives by resuspension in 100 μl of N,O-Bis(trimethylsilyl)trifluoroacetamide (Pierce Biotechnology, Rockford, IL) for 1 hour at 37°C. The reaction mixtures were analyzed using an HP5790 gas chromatography system fitted with a DB5-MS column (30 m × 0.25 mm × 0.25 μm). Separation was achieved by using a column temperature of 70°C for 2 minutes, then increasing by 10°C/min to 300°C, and finally held at 300°C for 5 minutes.
LC-MS/MS Analysis of CYP2W1 Arachidonic Acid Metabolism.
Arachidonic acid metabolites were analyzed with a quantitative profiling method using liquid chromatography and ES ionization tandem MS (MS/MS) (Yang et al., 2009). The reaction was terminated with an equal volume of CUDA solution in methanol. The LC system used for analysis was an Agilent 1200 SL (Agilent Corporation, Palo Alto, CA) equipped with a 2.1 × 50 mm Kinetex C18 column with a 3.0 µm particle size (Agilent Corporation). Mobile phase A consisted of water and 0.1% glacial acetic acid and mobile phase B of acetonitrile/methanol (84:16) with 0.1% glacial acetic acid. Chromatography was optimized to separate all analytes in 3 minutes. Gradient elution was performed at a flow rate of 1000 µl/min. The gradient program was started with 60% B. After 0.65 minute, B was increased linearly to 90% in 2 minutes. Finally, the eluent was shifted back to the starting condition with 0.1 minute and kept for 0.9 minute. The column was connected to a 4000 QTrap tandem MS (Applied Biosystems Instrument Corporation, Foster City, CA) equipped with an ES source (Turbo V). The instrument was operated in negative multiple reaction monitor (MRM) mode. The optimized conditions and the MRM transition were reported previously (Yang et al., 2009). Analyst software 1.5 was used to quantify the peaks according to the standard curves.
Results
Ligand-Induced Binding Spectra and Spectral Binding Constants.
The constants for the binding to CYP2W1 of a series of endogenous hormone molecules, including retinoids, steroids, vitamin Ds, fatty acids, and isoprenoids, were assessed by difference UV-vis spectroscopy. The test compounds were added to 2 µM CYP2W1 in the sample cuvette, and an equal amount of ethanol, which was used to dissolve the test compounds, was added to the same enzyme solution in the reference cuvette. Typically, a type I difference spectrum with a peak at about 390 nm and a trough at about 420 nm is observed if the ligand displaces the water ligand coordinated to the ferric ion, which shifts the ferric ion from a 6-fold to a 5-fold coordination state. Some ligands may also induce a reverse type I difference spectrum upon binding to P450, which features a trough at about 390 nm and a peak at about 420 nm. The absence of a detectable spectroscopic change indicates that the ligand does not bind or, if bound, fails to alter the binding of the water ligand. The binding constants and the types of binding spectrum induced by ligand binding to CYP2W1 are summarized in Table 1.
TABLE 1.
Spectroscopic binding constants and nature of difference binding spectrum induced by ligand binding to CYP2W1
| Compounds | Binding Constant (Ks) | Binding Mode |
|---|---|---|
| All-trans retinol | 40.3 ± 7.0 nM | Type 1 |
| All-trans retinal | 55.2 ± 14.0 nM | Type 1 |
| All-trans retinoic acid (atRA) | 63.7 ± 5.9 nM | Type 1 |
| Lanosterol | nd | nd |
| Cholesterol | nd | nd |
| Progesterone | nd | nd |
| Cortisone | nd | nd |
| Hydrocortisone (cortisol) | nd | nd |
| 17β-Estradiol | 30.1 ± 11.2 μM | Reverse type 1 |
| Testosterone | nd | nd |
| Ergocalciferol (vitamin D2) | nd | nd |
| Cholecalciferol (vitamin D3) | nd | nd |
| Farnesol | 23.8 ± 9.0 μM | Reverse type 1 |
| Geraniol | nd | nd |
| Geranylgeraniol | 25.0 ± 0.3 μM | Type 1 |
| Arachidonic acid | 64.5 ± 39.1 μM | Type 1 |
| Lauric acid | nd | nd |
nd, not detectable.
The assessment of the binding of retinoids to CYP2W1 was slightly modified compared with other substrates. As retinoid has an absorbance maximum at 360 nm, to normalize the retinoid absorbance, retinoid was added to both the enzyme solution in the sample cuvette and the buffer solution in the reference cuvette, and the P450 difference spectrum was obtained by manually subtracting the ligand-free spectrum from the ligand-added spectrum. AtRA binding to CYP2W1 induced a type 1 difference spectrum, with a characteristic spectral minimum at 420 nm (Fig. 1A). The difference spectrum induced by atRA binding showed an isosbestic point at ∼403 nm. The absorbance difference between 390 nm and 420 nm depended on the retinoic acid concentration, and the data were fitted with the tight-binding Morrison equation since the plot showed atRA had a high binding affinity for CYP2W1. The spectral binding constant (Ks) for atRA binding to CYP2W1 was determined to be 63.7 ± 5.9 nM (Fig. 1B). The absorbance difference saturated at ∼500 nM of atRA concentration, despite the fact that 2 µM CYP2W1 was present in the titration. As the critical micelle concentration of atRA is estimated to be lower than 1 µM, atRA possibly formed micelles beyond the critical micelle concentration and was no longer available for CYP2W1 binding. The binding of atRA was also assessed by titrating atRA to 200 nM CYP2W1. Although the absolute absorbance of 200 nM CYP2W1 was lower than 0.1, which was out of the linear absorbance range, spectral changes were still significant enough to be fitted with the Morrison equation, yielding a Ks of 115.9 ± 33.4 nM (Supplemental Fig. 1, K and L). All-trans retinol and all-trans retinal also induced type 1 difference spectra (Supplemental Fig. 1, A and C). The Ks of all-trans retinol was 40.3 ± 7.0 nM (Supplemental Fig. 1B), and the Ks of all-trans retinal was 55.2 ± 14.0 nM (Supplemental Fig. 1D). All three retinoids exhibited tight binding affinities with low nanomolar Ks values.
Fig. 1.

Representative difference in binding spectra induced by ligand binding to CYP2W1. (A) Type 1 difference in binding spectrum induced by titration of 2 µM CYP2W1 with atRA. The atRA absorbance was normalized by adding atRA to both the sample and reference cuvettes. The difference spectra were obtained by subtracting the ligand-free absolute spectrum from the ligand-bound spectra. (B) The difference between absorbance at 390 nm and 420 nm was plotted against the atRA concentration, and data were fitted to the Morrison equation to obtain a binding constant. (C) Reverse type 1 difference in binding spectrum induced by titration of 2 µM CYP2W1 with 17β-estradiol. (D) The difference between the absorbance at 390 nm and 420 nm was plotted against the 17β-estradiol concentration. The data were fitted to the Hill equation to obtain a binding constant.
The binding of arachidonic acid also induced a type 1 difference spectrum (Supplemental Fig. 1E). The spectral binding constant was determined by plotting the absorbance difference between peak and trough versus substrate concentration, and the data were fitted with the Hill equation. Ks of arachidonic acid was determined to be 64.5 ± 39.1 µM (Supplemental Fig. 1F). Arachidonic acid is a polyunsaturated fatty acid with four cis-double bonds, which have structural similarity to the isoprenoid chain of retinoids (Fig. 2). The saturated lauric acid did not induce any spectral change (Supplemental Fig. 2J), indicating the unsaturated hydrocarbon chain was crucial for binding to CYP2W1.
Fig. 2.

Structures of endogenous ligands that induced spectral changes upon binding to CYP2W1.
The binding of 17β-estradiol induces a reverse type I difference spectrum, which is characterized by a peak with an absorbance maximum at 420 nm and a trough with an absorbance minimum at 390 nm (Fig. 1C). The difference spectrum induced by 17β-estradiol showed an isosbestic point at ∼402 nm, except at the three highest concentrations (40, 45, and 50 µM), which might be due to the increased scattering observed at these three concentrations. Among all the steroids screened in this study, 17β-estradiol was the only one that induced spectral changes with CYP2W1. The differences between the absorbance at 390 nm and the absorbance at 420 nm were plotted against the 17β-estradiol concentration, and the data were fitted to the Hill equation. The Ks of 17β-estradiol was determined to be 30.1 ± 11.2 µM (Fig. 1D). None of the other steroids tested in this study, including lanosterol, cholesterol, progesterone, testosterone, cortisone, and hydrocortisone, induced any spectral changes (Supplemental Fig. 2, A–F). The binding of lanosterol and cholesterol was assessed in the presence of 0.05% methyl-β-cyclodextrin to enhance the solubility of these two ligands. Neither lanosterol nor cholesterol induced any spectrum change when added to CYP2W1 (Supplemental Fig. 2, A and B). The addition of vitamin D2 and vitamin D3 also did not induce any detectable difference spectrum (Supplemental Fig. 2, G and H). Farnesol and geranylgeraniol have structural similarity with the retinoid chain, as they both consist of isoprene units (Fig. 2). Farnesol is made of three isoprene units and geranylgeraniol of four such units. Both farnesol and geranylgeraniol induced CYP2W1 spectral changes. Farnesol induced a reverse type 1 spectrum (Supplemental Fig. 1G), whereas geranylgeraniol induced a type 1 binding spectrum (Supplemental Fig. 1I). The binding constant for farnesol was 23.8 ± 9.0 μM (Supplemental Fig. 1H) and that for geranylgeraniol was 25.0 ± 0.3 μM (Supplemental Fig. 1J). Geraniol, which is made of only two isoprenoid units, failed to induce any spectral changes (Supplemental Fig. 2I). The difference spectra induced by at-retinol, at-retinal, arachidonic acid, farnesol, and geranylgeraniol did not show clear isosbestic points, presumably due to the scattering observed at higher concentrations (Supplemental Fig. 1, A, C, E, G, and I).
AtRA Metabolism by CYP2W1.
The catalytic activity of CYP2W1 was tested with retinoids as substrates, using purified, reconstituted human CYP2W1 and human CPR. The retinoids examined were atRA, all-trans retinol, and all-trans retinal. The retinoids were incubated with CYP2W1, and the products of the reactions were analyzed by HPLC and LC-MS. A metabolite peak eluted at 22.0 minutes was detected after incubating atRA with CYP2W1, and this peak was not present in the absence of CYP2W1 (Fig. 3A). This metabolite yielded a deprotonated molecular ion ([M-H]-) of m/z 315, which corresponds to the addition of 16 Da to the deprotonated molecular ion (m/z 299) of atRA, indicating formation of a singly oxygenated product (Fig. 3, C and E). The metabolite had the same UV-vis spectrum with a maximum at 335 nm as the substrate atRA (Fig. 4), indicating that oxidation had not altered the conjugated polyene chromophore. Comparison of the chromatographic retention time and MS of the metabolite with those of an authentic sample established that the metabolite was 4-hydroxy atRA (Fig. 3, B, D, and F). The identity of the metabolite was further confirmed by comparison with the known atRA metabolites generated by CYP3A7, which was previously shown to be the most active human P450 form in generating 4-hydroxy atRA, 4-oxo-atRA, and 18-hydroxy atRA (Marill et al., 2000). Incubation of atRA with microsomes containing CYP3A7, cytochrome b5, and CPR generated three metabolites that eluted at 19.9, 22.5, and 25.7 minutes, respectively (Fig. 3G). The metabolite eluting at 19.9 minutes was characterized mass spectrometrically by a deprotonated molecular ion at m/z 313, indicating it was 4-oxo atRA. The metabolites eluting at 22.5 and 25.7 minutes were both characterized by MS-deprotonated molecular ions at m/z 315 (Fig. 3H). As previously reported, the three metabolites were eluted in the order of 4-oxo, 4-hydroxy, and 18-hydroxy atRA under the same HPLC conditions used in this study (Marill et al., 2000). The 22.5-minute metabolite is therefore 4-hydroxy atRA, and that at 25.7 minute is 18-hydroxy atRA. The 4-hydroxy atRA metabolite generated by CYP3A7 eluted with the same retention time as the atRA metabolite generated by CYP2W1 (Fig. 3, A and G), confirming that CYP2W1 oxidizes atRA to 4-hydroxy atRA.
Fig. 3.

LC-MS analysis of CYP2W1 oxidation of atRA. (A) LC chromatogram with the detector set at 340 nm of the metabolites of atRA generated by incubation with CYP2W1. The red trace is incubation without CYP2W1, and the black trace is complete incubation with the enzyme present; (B) LC chromatogram with the detector set at 340 nm of authentic 4-hydroxy atRA; (C) Extracted ion (m/z = 315) chromatogram in negative ES mode of atRA metabolites generated by incubation with CYP2W1; (D) extracted ion (m/z = 315) chromatogram of authentic 4-hydroxy atRA; (E) EI-mass spectrum of the peak eluted at 22.0 minutes in Fig. 3C; and (F) EI-mass spectrum of the peak eluted at 21.7 minutes in Fig. 3D; LC-MS analysis of the CYP3A7-catalyzed oxidation of atRA. (G) LC chromatogram with detector at 340 nm. The red trace is incubation without enzyme, and the black trace is complete incubation with CYP3A7 present; (H) extracted ion (m/z = 315) chromatogram in negative ES mode of atRA metabolites generated by incubation with baculovirus-insect cell microsomes expressing CYP3A7 and cytochrome b5 supplemented with recombinant CPR.
Fig. 4.

UV-vis spectrum of (A) atRA, (B) atRA metabolite by CYP2W1, and (C) 4-hydroxy atRA standard.
The second peak eluting at 29.0 minutes also exhibited a deprotonated molecular ion of m/z 315, but it was present both in the presence and in the absence of CYP2W1 (Fig. 3, A and C). This peak is presumably an air oxidation product of atRA. It has been specifically shown not to be 4-hydroxy atRA, 18-hydroxy atRA, or 5,6-epoxy atRA by comparing it with authentic standards and/or with the previously identified metabolites of CYP3A7. The two peaks that eluted slightly earlier than atRA were the 13-cis and 9-cis isomers of retinoic acid, respectively, as confirmed by coelution experiments with authentic standards. Isomerization of the substrate occurred during incubation and was independent of CYP2W1, as these two peaks also appeared in the absence of CYP2W1.
The formation of 4-hydroxy atRA was dependent on the concentration of atRA (Fig. 5). Kinetic studies indicate that CYP2W1 oxidizes atRA with Km = 5.6E-6 ± 6.0E-7 M and Vmax = 0.000085 ± 0.00004 second−1. The Vmax/Km for atRA oxidation by CYP2W1 was determined to be 15.2 M−1 s−1.
Fig. 5.

Kinetics of formation of 4-hydroxy atRA. Initial atRA concentrations ranged from 1 to 15 µM, and the data were fitted to the Hill equation to obtain the kinetic parameters.
Oxidation of All-Trans Retinol and Retinal by CYP2W1.
CYP2W1 also oxidizes all-trans retinol and all-trans retinal. Incubating all-trans retinol with CYP2W1 generated a metabolite that eluted at 34.7 minutes (Fig. 6A). In the MS, this metabolite exhibited a protonated molecular ion after water loss ([MH − H2O]+) at m/z 285, which is 16 Da higher than that of the corresponding ([MH − H2O]+) of retinol itself at m/z 269 (Fig. 6B). CYP2W1 thus oxidizes retinol into a product with one additional oxygen atom. Incubating all-trans retinol with CYP3A7 and CYP2C8 produced the same metabolites eluting at 34.7 minutes (Fig. 6, C and E), and these metabolites also gave the same molecular ion of m/z 285 (Fig. 6, D and F). CYP2C8 has been reported to metabolize all-trans retinol to 4-OH all-trans retinol (Leo et al., 1989). Therefore, the metabolite generated by CYP2W1 is also likely to be 4-OH all-trans retinol, as it eluted at the same retention time as the retinol metabolite generated by CYP2C8.
Fig. 6.

LC-MS analysis of CYP2W1, CYP3A7, and CYP2C8, all-trans retinol metabolites. In the plots, the red trace is from a control incubation without a P450 enzyme, and the black trace is from the complete incubation with the indicated P450 enzyme. (A) LC chromatogram with the detector at 340 nm and (B) extracted ion (m/z = 285) chromatogram in positive ES mode of all-trans retinol metabolites generated by incubation with and without CYP2W1 enzyme; (C) LC chromatogram with the detector at 340 nm and (D) extracted ion (m/z = 285) chromatogram of all-trans retinol metabolites generated by incubation with and without baculovirus-insect cell microsomes expressing CYP3A7 and cytochrome b5 supplemented with recombinant CPR; (E) LC chromatogram with the detector at 340 nm; and (F) extracted ion (m/z = 285) chromatogram of all-trans retinol metabolites generated by incubation with and without baculovirus-insect cell microsomes expressing CYP2C8 and cytochrome b5 supplemented with recombinant CPR.
The metabolism of all-trans retinal by CYP2W1 generates multiple products. Similar to the incubation with all-trans retinol and atRA, incubation of all-trans retinal in the absence of CYP2W1 also generated new peaks that are probably substrate isomers. Four new product peaks were observed in the presence of CYP2W1 that did not appear in the absence of CYP2W1 (Supplemental Fig. 3); however, we were not able to determine the precise identities of these peaks.
Arachidonic Acid Metabolism by CYP2W1.
The metabolism of arachidonic acid catalyzed by CYP2W1 was studied by LC-ESI MS/MS in negative MRM mode (Yang et al., 2009). The hydroxyl (hydroxyeicosatetraenoic acid, HETE) and epoxy (epoxyeicosatetraenoic acid) metabolites of arachidonic acid were quantified along with the corresponding internal standards. The hydroxyl and epoxy metabolites were also present in the control sample without CYP2W1, presumably due to air oxidation of arachidonic acid. The products were identified by comparing the concentration of each metabolite in the full reaction sample versus the control sample lacking CYP2W1 (Fig. 7, A and B). The concentration of 8,9-diHETrE was approximately 2.5-fold higher in the full reaction sample than that in the control sample, indicating that 8,9-diHETrE was the major product in the CYP2W1-catalyzed oxidation of arachidonic acid. The second most abundant metabolites were 14,15-diHETrE and 11,12-diHETrE, both of which showed a 2-fold increase in the full reaction compared with the control incubation. The concentrations of 15-HETE, 11-HETE, and 8-HETE were also slightly higher in the complete reaction in comparison with the control reaction. The concentrations of all the other metabolites were not significantly different between the reaction and control incubations. The concentrations of 5,6-diHETrE and 8,9-EpETrE metabolites were approximately 1.5-fold higher in the reaction sample than the control sample (Fig. 7B), but the differences were not significant when the reactions were compared directly (Fig. 7A). The most abundant three metabolites, 8,9-diHETrE, 11,12-diHETrE, and 14,15-diHETrE, were previously reported as the products of CYP2W1-catalyzed arachidonic acid metabolism (Karlgren et al., 2006), but these earlier results were contradicted in a second article (Yoshioka et al., 2006). Our findings indicate that arachidonic acid is, at best, a very poor substrate for CYP2W1 and should not be a physiologically relevant substrate.
Fig. 7.

Quantification of the arachidonic acid metabolites (A) in the presence of CYP2W1 (red) and in the absence of CYP2W1 (blue); (B) the ratio of the concentration of the indicated metabolite in the full incubation with CYP2W1 over the concentration of the metabolite in the absence of the enzyme.
Oxidation of 17β-Estradiol and Farnesol by CYP2W1.
The catalytic activity of CYP2W1 on the metabolism of 17β-estradiol was tested in reconstituted reactions using purified CYP2W1 and human CPR. The reaction products were converted to trimethylsilyl derivatives and analyzed by GC-MS. A trace of a 17β-estradiol oxidation product was detected by extracted ion monitoring at m/z 504, the value expected for a monohydroxylated product. The m/z 504 extracted ion peak was present only in the full reaction sample (Supplemental Fig. 4A) and was absent in the control sample without CYP2W1 (Supplemental Fig. 4C). The metabolite was identified as 2-OH 17β-estradiol by the identity of its retention time (25.1 minutes) with that of an authentic 2-hydroxy 17β-estradiol standard (Supplemental Fig. 4D).
The metabolism of farnesol catalyzed by CYP2W1 was also examined by GC-MS. A extracted ion (m/z 382) peak for a monohydroxylated product was detected at 32.2 minutes only in the full reaction sample, but not in the control sample without CYP2W1 (Supplemental Fig. 5A). No metabolites were found in incubations with geranylgeraniol, the homolog of farnesol with one additional isoprene unit.
Discussion
To identify the endogenous substrates for CYP2W1, the binding affinities of CYP2W1 for retinoids, steroids, vitamin Ds, fatty acids, and isoprenoids were assessed spectroscopically by measuring ligand-induced binding spectrum changes. CYP2W1 exhibited tight binding toward retinoids with nanomolar binding constants. Among all the steroids tested in this study, 17β-estradiol was the only sterol that detectably bound to CYP2W1, but its binding constant was 30.1 ± 11.2 µM, nearly 1000-fold weaker than for the retinoids. CYP2W1 also showed a weak binding affinity for arachidonic acid, which had a binding constant of 64.5 ± 39.1 µM. Furthermore, CYP2W1 also weakly bound farnesol and geranylgeraniol (Table 1). The CYP2W1 ligands identified here, except 17β-estradiol, contained polyunsaturated hydrocarbon chains (Fig. 2). The unsaturated polyene structure appears to be important for binding to CYP2W1, as CYP2W1 bound to arachidonic acid, but not the saturated lauric acid. The length of the isoprene chain was also crucial for CYP2W1 binding, as the enzyme bound to farnesol and geranylgeraniol, but it did not detectably bind to geraniol; however, the binding constants (Table 1) clearly establish that the retinoids are by far the best ligands for CYP2W1.
CYP2W1 catalyzed the oxidation of atRA to 4-hydroxy atRA. The identity of the 4-hydroxy metabolite was confirmed by comparing with an authentic 4-hydroxy atRA standard and with the known metabolites of atRA formed by CYP3A7. The kinetic studies indicate that CYP2W1 oxidized atRA with Km = 5.6 ± 0.6 µM and Vmax = 0.000085 ± 0.00004 second−1. CYP2W1 also oxidized retinol to 4-hydroxy-retinol, and retinal to a mixture of products whose identities were not established.
Trace amounts of the 2-hydroxy metabolite were observed in the oxidation of 17β-estradiol by CYP2W1. CYP2W1 also slowly oxidized farnesol to an unidentified singly-hydroxylated product.
The physiologic concentration of retinoic acid in human plasma was reported to be 1–3 ng/ml (approximately 3 to 10 nM) (De Ruyter et al., 1979). The CYP2W1 Ks for retinoic acid was 63.7 ± 5.9 nM, and CYP2W1 oxidized retinoic acid with Km = 5.6 ± 0.6 µM. The Ks value was approximately 6- to 20-fold higher than the physiologic concentration, whereas the Km value was more than 600-fold higher than the physiologic concentration; however, the unbound tumor retinoic acid concentration may be higher than the unbound plasma concentration. Given that CYP2W1 is highly expressed in tumors, the metabolism of retinoic acid by CYP2W1 may still be physiologically important. Alternatively, considering the relative lower catalytic efficiency (Vmax/Km = 15.2 M−1 S−1), retinoic acid might participate in a tumor regulation pathway by acting as potent inhibitor for CYP2W1.
The concentration of plasma 17β-estradiol has more variability, depending on the age and time in the menstrual cycle in women. Younger women tend to have higher 17β-estradiol levels, ranging from 50–500 pg/ml (approximately 0.1 to 1 nM), and postmenopausal women have 17β-estradiol values of 10–20 pg/ml (0.02–0.04 nM) (Santanam et al., 1998). Under physiologic conditions, arachidonic acid is mostly present in an albumin-bound form. In the presence of albumin, the concentration of free arachidonic acid (unbound) is typically below 0.1 μM (Brash, 2001). The metabolism of retinoic acid, 17β-estradiol, and arachidonic acid have all been found to be related to tumor development (Schneider et al., 1982; Marks et al., 2000; Tang and Gudas, 2011); however, the physiologic concentrations of 17β-estradiol and arachidonic acid are much lower than their dissociation constants for binding to CYP2W1, which precludes a role for them as important physiologic substrates for this enzyme. Retinoic acid thus appears to be the only relevant physiologic substrate for CYP2W1 among a broad range of potential endogenous substrates.
The role of CYP2W1 in retinoid metabolism implied by our results may well be related to the high expression of this enzyme in tumor tissues. Retinoids exert many important functions in cells, most notably in vision, cell proliferation, cell differentiation, immune function, neural function, and the establishment of the body plan during early development. Previous studies have shown that retinoid signaling is often compromised early in carcinogenesis, indicating that impaired retinoid signaling might be required for tumor development. Additionally, retinoids are used in cancer treatment, as they can induce cell differentiation and arrest proliferation (Tang and Gudas, 2011). Retinoid metabolism is usually altered in tumor cells as a result of: 1) a decreased uptake of retinol; 2) impairment of the metabolism of retinol to retinoic acid; and 3) enhancement of the conversion of retinoic acid to metabolites with lower bioactivity owing to increased expression of retinoic acid catabolic enzymes such as CYP26A1 (Mongan and Gudas, 2007). These features of retinoid biology indicate that P450 enzymes with catalytic properties toward retinoids are likely players in tumor development.
The CYP26 family, which consists of the three highly conserved enzymes—CYP26A1, CYP26B1, and CYP26C1—are major enzymes in the metabolism of retinoic acid; CYP26A1 is the most important among them. CYP26A1 has a nanomolar spectroscopic binding affinity (Ks = 529 nM) for retinoic acid and efficiently oxidizes retinoic acid (depletion Km = 9.4 ± 3.3 nM and Vmax = 11.3 ± 4.3 pmoles min−1 pmole P450−1) (Lutz et al., 2009). Like CYP26A1, retinoic acid is a tight-binding ligand for CYP2W1 (Ks = 58.5 nM). Retinoic acid is oxidized by CYP2W1 to 4-hydroxy atRA but less efficiently than by CYP26. The Vmax/Km for atRA oxidation by CYP26A1 is 2.0E7 M−1 s−1, whereas the Vmax/Km for atRA oxidation by CYP2W1 was 15.2 M−1 s−1. Considering the binding affinity and relatively low catalytic efficiency to retinoic acid, CYP2W1 is more likely associated with regulation of retinoic acid localization than depletion of this substrate.
In conclusion, in this study, we have identified five substrates of CYP2W1, including retinoic acid, retinol, retinal, 17β-estradiol, and farnesol. The retinoids are tight-binding ligands for CYP2W1 with nanomolar binding affinity, whereas the other three substrates only have micromolar binding affinities. CYP2W1 was shown to catalyze the metabolism of all five compounds, but the only physiologically relevant substrates are the retinoids, with retinoic acid being oxidized to the 4-hydroxy derivative. The results imply that CYP2W1 is likely to play a role in localized retinoid metabolism, and this function is related to its distinct tumor-specific expression pattern.
Supplementary Material
Abbreviations
- atRA
all-trans-retinoic acid
- CPR
cytochrome P450 reductase
- CUDA
1-cyclohexylureido, 3-dodecanoic acid
- CYP or P450
cytochrome P450
- DTT
dithiothreitol
- 8,9-diHETrE
(5Z,11Z, 14Z)-8,9-dihydroxyeicosatrienoic acid
- 11,12-diHETrE
(5Z,8Z,14Z)-11,12-dihydroxyeicosatrienoic acid
- ES
electrospray
- 14,15-diHETrE
(5Z,8Z,11Z)-14,15-dihydroxyeicosatrienoic acid
- HETE
hydroxyeicosatetraenoic acid
- Ks
spectral binding constant
- LC-MS
liquid chromatography-mass spectrometry
- MBTE
methyl tert-butyl ether
- MRM
multiple reaction monitor
- MS/MS
mass spectrometry
- NADPH
nicotinamide adenine dinucleotide phosphate
Authorship Contributions
Participated in research design: Zhao, Wan, Yang, Hammock, Ortiz de Montellano.
Conducted experiments: Zhao, Wan, Yang.
Performed data analysis: Zhao, Wan, Yang, Hammock, Ortiz de Montellano.
Wrote or contributed to the writing of the manuscript: Zhao, Yang, Hammock, Ortiz de Montellano.
Footnotes
This research was supported by National Institutes of Health National Institute of General Medical Sciences [Grant GM25515], the National Institute of Environmental Health Sciences [Grant R01 EX002710 and P42 Superfund Program], and the West Coast Central Comprehensive Metabolomic Research Core [U24 DK097154].
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Brash AR. (2001) Arachidonic acid as a bioactive molecule. J Clin Invest 107:1339–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary D, Jansson I, Stoilov I, Sarfarazi M, Schenkman JB. (2005) Expression patterns of mouse and human CYP orthologs (families 1-4) during development and in different adult tissues. Arch Biochem Biophys 436:50–61. [DOI] [PubMed] [Google Scholar]
- De Ruyter MG, Lambert WE, De Leenheer AP. (1979) Retinoic acid: an endogenous compound of human blood. Unequivocal demonstration of endogenous retinoic acid in normal physiological conditions. Anal Biochem 98:402–409. [DOI] [PubMed] [Google Scholar]
- Dierks EA, Davis SC, Ortiz de Montellano PR. (1998) Glu-320 and Asp-323 are determinants of the CYP4A1 hydroxylation regiospecificity and resistance to inactivation by 1-aminobenzotriazole. Biochemistry 37:1839–1847. [DOI] [PubMed] [Google Scholar]
- Du L, Neis MM, Ladd PA, Keeney DS. (2006a) Differentiation-specific factors modulate epidermal CYP1-4 gene expression in human skin in response to retinoic acid and classic aryl hydrocarbon receptor ligands. J Pharmacol Exp Ther 319:1162–1171. [DOI] [PubMed] [Google Scholar]
- Du L, Neis MM, Ladd PA, Lanza DL, Yost GS, Keeney DS. (2006b) Effects of the differentiated keratinocyte phenotype on expression levels of CYP1-4 family genes in human skin cells. Toxicol Appl Pharmacol 213:135–144. [DOI] [PubMed] [Google Scholar]
- Edler D, Stenstedt K, Öhrling K, Hallström M, Karlgren M, Ingelman-Sundberg M, Ragnhammar P. (2009) The expression of the novel CYP2W1 enzyme is an independent prognostic factor in colorectal cancer: a pilot study. Eur J Cancer 45:705–712. [DOI] [PubMed] [Google Scholar]
- Girault I, Rougier N, Chesné C, Lidereau R, Beaune P, Bieche I, de Waziers I. (2005) Simultaneous measurement of 23 isoforms from the human cytochrome P450 families 1 to 3 by quantitative reverse transcriptase-polymerase chain reaction. Drug Metab Dispos 33:1803–1810. [DOI] [PubMed] [Google Scholar]
- Gomez A, Nekvindova J, Travica S, Lee MY, Johansson I, Edler D, Mkrtchian S, Ingelman-Sundberg M. (2010) Colorectal cancer-specific cytochrome P450 2W1: intracellular localization, glycosylation, and catalytic activity. Mol Pharmacol 78:1004–1011. [DOI] [PubMed] [Google Scholar]
- Guengerich FP (2015) Human cytochrome P450 enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry, 4th ed. (Ortiz de Montellano PR, ed) pp 523–785, Springer, New York. [Google Scholar]
- Guengerich FP, Martin MV, Sohl CD, Cheng Q. (2009) Measurement of cytochrome P450 and NADPH-cytochrome P450 reductase. Nat Protoc 4:1245–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlgren M, Gomez A, Stark K, Svärd J, Rodriguez-Antona C, Oliw E, Bernal ML, Ramón y Cajal S, Johansson I, Ingelman-Sundberg M. (2006) Tumor-specific expression of the novel cytochrome P450 enzyme, CYP2W1. Biochem Biophys Res Commun 341:451–458. [DOI] [PubMed] [Google Scholar]
- Karlgren M, Ingelman-Sundberg M. (2007) Tumour-specific expression of CYP2W1: its potential as a drug target in cancer therapy. Expert Opin Ther Targets 11:61–67. [DOI] [PubMed] [Google Scholar]
- Karlgren M, Miura S, Ingelman-Sundberg M. (2005) Novel extrahepatic cytochrome P450s. Toxicol Appl Pharmacol 207(2, Suppl):57–61. [DOI] [PubMed] [Google Scholar]
- Leo MA, Lasker JM, Raucy JL, Kim CI, Black M, Lieber CS. (1989) Metabolism of retinol and retinoic acid by human liver cytochrome P450IIC8. Arch Biochem Biophys 269:305–312. [DOI] [PubMed] [Google Scholar]
- Lutz JD, Dixit V, Yeung CK, Dickmann LJ, Zelter A, Thatcher JE, Nelson WL, Isoherranen N. (2009) Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem Pharmacol 77:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marill J, Cresteil T, Lanotte M, Chabot GG. (2000) Identification of human cytochrome P450s involved in the formation of all-trans-retinoic acid principal metabolites. Mol Pharmacol 58:1341–1348. [DOI] [PubMed] [Google Scholar]
- Marks F, Müller-Decker K, Fürstenberger G. (2000) A causal relationship between unscheduled eicosanoid signaling and tumor development: cancer chemoprevention by inhibitors of arachidonic acid metabolism. Toxicology 153:11–26. [DOI] [PubMed] [Google Scholar]
- Mongan NP, Gudas LJ. (2007) Diverse actions of retinoid receptors in cancer prevention and treatment. Differentiation 75:853–870. [DOI] [PubMed] [Google Scholar]
- Nishida CR, Lee M, de Montellano PR. (2010) Efficient hypoxic activation of the anticancer agent AQ4N by CYP2S1 and CYP2W1. Mol Pharmacol 78:497–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronchi CL, Sbiera S, Volante M, Steinhauer S, Scott-Wild V, Altieri B, Kroiss M, Bala M, Papotti M, Deutschbein T, et al. (2014) CYP2W1 is highly expressed in adrenal glands and is positively associated with the response to mitotane in adrenocortical carcinoma. PLoS One 9:e105855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santanam N, Shern-Brewer R, McClatchey R, Castellano PZ, Murphy AA, Voelkel S, Parthasarathy S. (1998) Estradiol as an antioxidant: incompatible with its physiological concentrations and function. J Lipid Res 39:2111–2118. [PubMed] [Google Scholar]
- Schneider J, Kinne D, Fracchia A, Pierce V, Anderson KE, Bradlow HL, Fishman J. (1982) Abnormal oxidative metabolism of estradiol in women with breast cancer. Proc Natl Acad Sci USA 79:3047–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenstedt K, Hallstrom M, Johansson I, Ingelman-Sundberg M, Ragnhammar P, Edler D. (2012) The expression of CYP2W1: a prognostic marker in colon cancer. Anticancer Res 32:3869–3874. [PubMed] [Google Scholar]
- Tang X-H, Gudas LJ. (2011) Retinoids, retinoic acid receptors, and cancer. Annu Rev Pathol 6:345–364. [DOI] [PubMed] [Google Scholar]
- Thomas RD, Green MR, Wilson C, Weckle AL, Duanmu Z, Kocarek TA, Runge-Morris M. (2006) Cytochrome P450 expression and metabolic activation of cooked food mutagen 2-amino-1-methyl-6-phenylimidazo[4,5-b]pyridine (PhIP) in MCF10A breast epithelial cells. Chem Biol Interact 160:204–216. [DOI] [PubMed] [Google Scholar]
- Wang K, Guengerich FP. (2012) Bioactivation of fluorinated 2-aryl-benzothiazole antitumor molecules by human cytochrome P450s 1A1 and 2W1 and deactivation by cytochrome P450 2S1. Chem Res Toxicol 25:1740–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu ZL, Sohl CD, Shimada T, Guengerich FP. (2006) Recombinant enzymes overexpressed in bacteria show broad catalytic specificity of human cytochrome P450 2W1 and limited activity of human cytochrome P450 2S1. Mol Pharmacol 69:2007–2014. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Guengerich FP. (2012) Metabolomic analysis and identification of a role for the orphan human cytochrome P450 2W1 in selective oxidation of lysophospholipids. J Lipid Res 53:1610–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Schmelzer K, Georgi K, Hammock BD. (2009) Quantitative profiling method for oxylipin metabolome by liquid chromatography electrospray ionization tandem mass spectrometry. Anal Chem 81:8085–8093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshioka H, Kasai N, Ikushiro S, Shinkyo R, Kamakura M, Ohta M, Inouye K, Sakaki T. (2006) Enzymatic properties of human CYP2W1 expressed in Escherichia coli. Biochem Biophys Res Commun 345:169–174. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.