

Abstract
Cytochrome P450 (CYP) 26A1 and 26B1 are heme-containing enzymes responsible for metabolizing all-trans retinoic acid (at-RA). No crystal structures have been solved, and therefore homology models that provide structural information are extremely valuable for the development of inhibitors of cytochrome P450 family 26 (CYP26). The objectives of this study were to use homology models of CYP26A1 and CYP26B1 to characterize substrate binding characteristics, to compare structural aspects of their active sites, and to support the role of CYP26 in the metabolism of xenobiotics. Each model was verified by docking at-RA in the active site and comparing the results to known metabolic profiles of at-RA. The models were then used to predict the metabolic sites of tazarotenic acid with results verified by in vitro metabolite identification experiments. The CYP26A1 and CYP26B1 homology models predicted that the benzothiopyranyl moiety of tazarotenic acid would be oriented toward the heme of each enzyme and suggested that tazarotenic acid would be a substrate of CYP26A1 and CYP26B1. Metabolite identification experiments indicated that CYP26A1 and CYP26B1 oxidatively metabolized tazarotenic acid on the predicted moiety, with in vitro rates of metabolite formation by CYP26A1 and CYP26B1 being the highest across a panel of enzymes. Molecular analysis of the active sites estimated the active-site volumes of CYP26A1 and CYP26B1 to be 918 Å3 and 977 Å3, respectively. Overall, the homology models presented herein describe the enzyme characteristics leading to the metabolism of tazarotenic acid by CYP26A1 and CYP26B1 and support a potential role for the CYP26 enzymes in the metabolism of xenobiotics.
Introduction
Cytochrome P450 26A1 and 26B1 (CYP26A1 and CYP26B1, respectively) are members of the cytochrome P450 superfamily that are responsible for the metabolism of retinoic acid (Ray et al., 1997; Guengerich, 2006; Lutz et al., 2009; Thatcher and Isoherranen, 2009; Ross and Zolfaghari, 2011). They are widely expressed throughout the adult human body, though CYP26A1 is the primary isoform expressed in the adult liver, with little to no hepatic expression of CYP26B1 observed (Xi and Yang, 2008; Thatcher and Isoherranen, 2009; Thatcher et al., 2010; Topletz et al., 2012). CYP26B1 mRNA expression has been reported to be highest in the adult brain (White et al., 2000; Thatcher and Isoherranen, 2009; Topletz et al., 2012). Additional sites of expression for CYP26A1 mRNA include the skin, testes, kidney, and lung, whereas CYP26B1 mRNA has been identified in skin, lung, testes, placenta, ovaries, and intestine (Xi and Yang, 2008; Osanai and Lee, 2011; Topletz et al., 2012). Although the enzymes generally exhibit the highest degree of activity toward all trans-retinoic acid (at-RA), they also metabolize other retinoic acid isomers, albeit with lower activity levels (White et al., 1996, 2000; Taimi et al., 2004; Ross and Zolfaghari, 2011). CYP26A1 and CYP26B1 also metabolize retinoic acid metabolites 4-hydroxy-at-RA, 4-oxo-at-RA, and 18-hydroxy-at-RA (Lutz et al., 2009; Shimshoni et al., 2012; Topletz et al., 2012, 2014).
Retinoic acid signaling and altered retinoic acid concentrations play a role in various disease states (Miller, 1998; Kuenzli and Saurat, 2001; Njar, 2002; Ahmad and Mukhtar, 2004; Njar et al., 2006; Verfaille et al., 2008). As a result, the design and synthesis of inhibitors of CYP26 activity has received great interest in recent years. While inhibitors of CYP26 have been identified, the role of these enzymes in the metabolism of xenobiotics remains unclear. Of special interest are the synthetic retinoic acid receptor agonists that are structurally similar to retinoic acid and have moieties amenable to oxidative metabolism. One such retinoid is tazarotene, an acetylene-containing compound that is administered topically for stable plaque psoriasis or mild acne (Tang-Liu et al., 1999). Tazarotene is a prodrug whose activity is attributed to an active metabolite, tazarotenic acid, which binds with high affinity to retinoic acid receptors (Chandraratna, 1996). The active metabolite shares key structural features with at-RA and has been reported to be metabolized by a number of drug-metabolizing enzymes, including CYP2C8, CYP3A4, FMO1, and FMO3 (Madhu et al., 1997; Tang-Liu et al., 1999; Attar et al., 2003, 2005). Whether tazarotenic acid is a substrate of CYP26A1 and CYP26B1 is currently unknown.
In the absence of crystal structures, homology modeling is a commonly applied computational technique used to predict protein structure and function (Hillisch et al., 2004; Cavasotto and Phatak, 2009). The hypotheses generated by a homology model can be used to assess target druggability, to aid in the rational design of ligands and to predict drug metabolism and toxicity, all of which can then be used to iteratively refine the model (Hillisch et al., 2004). The flexible and hydrophobic nature of cytochrome P450 active sites often presents a challenge when attempting to use a homology model to accurately predict the site of metabolism for a given substrate (Williams et al., 2000; Lewis, 2002; Eksterowicz et al., 2014). Several models have been published for CYP26A1 and CYP26B1 and have been successfully used to rationalize the stereoselective product formation of 4-OH-at-RA by CYP26A1 or the binding of triazole- or imidazole-containing inhibitors within the active site of each enzyme (Gomaa et al., 2006, 2008, 2011a, b; Karlsson et al., 2008; Shimshoni et al., 2012; Sun et al., 2015). There are currently no homology models that compare the structure and function of CYP26A1 and CYP26B1 on the basis of the metabolism of a xenobiotic compound, and attempts to crystallize either isoform have been largely unsuccessful.
The aim of this work was to evaluate and characterize the active sites of CYP26A1 and CYP26B1 using homology modeling supported by xenobiotic metabolism data. Models were constructed for each enzyme and compared for structural similarities and differences. at-RA and tazarotenic acid were docked into the active sites and the predicted sites of metabolism evaluated. Metabolite identification experiments were used to confirm the hypotheses generated by the homology models. Finally, in vitro experiments were carried out to compare the metabolism of tazarotenic acid across a panel of drug-metabolizing enzymes and to determine the kinetic parameters for the formation of metabolites from tazarotenic acid by CYP26A1 and CYP26B1.
Materials and Methods
Materials.
Tazarotenic acid and all metabolite standards were obtained from Tocris Chemicals (Bristol, United Kingdom). CYP26A1 was expressed and characterized as previously described (Lutz et al., 2009). All other reagents were obtained as noted below. Solvents were from Sigma-Aldrich (St. Louis, MO) and were of the highest grade available.
Sequence Verification and Expression of CYP26B1.
To express recombinant CYP26B1, the human CYP26B1 cDNA was obtained from OriGene Technologies (Rockville, MD). Upon sequencing of the obtained clone, two single-nucleotide polymorphisms (SNPs) were discovered that differed from the sequence reported in NCBI (Q9NR63). The two SNPs were an A191G conversion resulting in an H>R amino acid change and a G788A conversion resulting in a G>S amino acid change (CYP26B1*1, Fig. 1). To determine which of the possible SNPs would be reflective of the CYP26B1 sequence in the human population, genomic DNA was extracted from 12 human livers from the University of Washington human liver bank and the two sections of the CYP26B1 gene were sequenced in all 12 donors. In brief, genomic DNA (50 ng) was amplified by polymerase chain reaction (PCR) using either forward (5′-TCTTTGAGGGCTTGGATCTG-3′) and reverse (5′-GGCAGAGAGGGAAGG-3′) primers for the A191G SNP or forward (5′-GACAAAGGGGAGAGGTGTCA-3′) and reverse (5′-GTAGAAATGGCTGGGCACAT-3′) primers for the G778A SNP at concentrations of 400 nM. The primers and template DNA together with a puReTaq Ready-to-Go PCR bead (Amersham Biosciences, Piscataway, NJ) were mixed in a final volume of 25 μl, and PCR amplification was done as follows: After an initial denaturing step at 94°C for 4 minutes, amplification was performed for 32 cycles of denaturation (94°C for 30 seconds), annealing (55°C for 20 seconds), and extension (72°C for 30 seconds), followed by a final extension at 72°C for 30 seconds. PCR products were analyzed by gel electrophoresis, spin column–purified to remove unincorporated nucleotides and primers, using the QIAquick PCR Purification Kit (Qiagen Inc., Hilden, Germany), and sequenced for the forward and reverse direction on an ABI Prism 377Xl DNA Sequencer (Applied Biosystems, Foster City, CA) with the ABI Prism BigDye Terminator Cycle Sequencing Ready Reaction Kit (PerkinElmer, Waltham, MA). After the wild-type sequence was confirmed, the CYP26B1 coding sequence from the original clone was amplified as 6xHis tag with a tobacco etch virus (TEV) cleavage site added to maintain similarity with the commercially available clone, as previously described (Topletz et al., 2012). CYP26B1 protein was expressed using the Bac-to-Bac baculovirus expression system (Life Technologies/ThermoFisher Scientific, Grand Island, NY) in Sf9 cells according to the manufacturer’s instructions, as described previously (Topletz et al., 2012). Sf-900 II SFM liquid media (Life Technologies) supplemented with 2.5% fetal bovine serum was used, and during protein expression ferric citrate (0.2 mM) and δ-aminolevulinic acid (0.3 mM) were added to the media 24 hours postinfection to facilitate heme synthesis. The cells were harvested 48 hours postinfection, washed once in phosphate buffered saline with 1 mM phenylmethylsulfonyl fluoride, pelleted, and stored at –80°C. Membrane fractions containing CYP26B1 were prepared by centrifugation as described previously (Topletz et al., 2012) and P450 content determined via CO-difference spectra.
Fig. 1.

Multiple sequence alignment of CYP120, CYP26A1, CYP26B1*1, and CYP26B1 amino acid sequences. The sequence identity between CYP26A1 and CYP26B1, CYP26B1*1, and CYP120 was 44.47%, 44.26%, and 33.26%, respectively. For CYP26B1*1, a 99.61% and 34.69% sequence identity was observed with CYP26B1 and CYP120, respectively. Amino acids are represented in red (small/hydrophobic), blue (acidic), purple (basic), and green (hydroxyl/sulfhydryl/amine). Sequence consensus is indicated by an asterisk (residue is fully conserved across all sequences), a colon (consensus group contains very similar properties), or a period (consensus group contains weakly similar properties).
IC50 Determination for Retinoic Acid Receptor Agonists.
Six retinoic acid receptor agonists were assessed for in vitro inhibition of CYP26A1- and CYP26B1-catalyzed 9-cis-4-hydroxyretinoic acid formation. Various concentrations of each inhibitor (0–100 μM) were incubated with 5 pmol of CYP26A1 or CYP26B1, 10 pmol of cytochrome P450 reductase, and 100 nM 9-cis-retinoic acid in 100 mM potassium phosphate buffer (pH 7.4). Incubations were initiated by the addition of 1 mM NADPH (final concentration) and quenched after 2 minutes (CYP26A1) or 5 minutes (CYP26B1) with 5 volumes of ethyl acetate containing acitretin as an internal standard. All samples were evaporated to dryness under a gentle stream of N2, reconstituted in methanol, and assayed for 9-cis-4-hydroxyretinoic acid concentrations by high-performance liquid chromatography with UV detection, as previously described (Thatcher et al., 2011). All IC50 determinations were conducted in triplicate.
Homology Modeling.
Homology models of CYP26A1 and CYP26B1 were constructed using Prime (Schrödinger LLC, New York, NY). The amino acid sequence of human CYP26A1 was obtained from the NCBI protein server (GenBank ID: 2688846), and the CYP26B1 amino acid sequence was obtained as described above. CYP120 (crystal structure, pdb 2VE3) was used as the template for both homology models. Compared with CYP120, CYP26A1 had 33% sequence identity and 53% positive sequence coverage, and CYP26B1 had 34% sequence identity and 54% positive sequence coverage. The heme prosthetic group was added to each homology model and ligated to Cys442 (CYP26A1) or Cys441 (CYP26B1), followed by energy minimization prior to ligand docking using OPLS_2005 force-field constraints as defined within the MacroModel algorithm (Schrödinger). To flexibly dock at-RA, tazarotenic acid, and tazarotenic acid sulfoxide, a ligand grid (12 × 12 × 12 Å) to which the center of mass of each ligand would be constrained was defined and centered approximately 2–3 Å above the heme iron using Glide (Schrödinger). Structural rationalization of each homology model was performed through the evaluation of Ramachandran plots and model assessment of odd bond lengths and angles. Determination of model flexibility was assessed by comparison of helical versus loop motifs and by prediction of two-degrees-of-freedom structure characteristics using PSIPRED (University College London, UK) and SSPro (Schrödinger). Ligand structures were also minimized using the OPLS_2005 force-field constraints within LigPrep (Schrödinger). GlideScore and eModel scoring algorithms were used to assess the docking poses of the ligands within the active site of each enzyme (Friesner et al., 2004, 2006). The use of the eModel scoring algorithm allowed for selection of the best docking pose on the basis of GlideScore, grid score, and ligand score of each docked ligand (Perola et al., 2004). The hydrogen atoms nearest the heme iron for the at-RA and tazarotenic acid docking poses with the highest GlideScores and eModel scores were used as the predicted sites of oxidative metabolism for each compound. The volume of the active site within each homology model was estimated using DoGSiteScorer (Universität Hamburg, Hamburg, Germany; Volkamer et al., 2012).
Metabolic Profiling.
Owing to its relative potency in the CYP26A1 and CYP26B1 inhibition assays, tazarotenic acid was selected for further evaluation. In vitro experiments to elucidate the metabolic pathways of tazarotenic acid in CYP26A1 and CYP26B1 recombinant preparations were carried out using previously optimized conditions of 20 nM recombinant enzyme, 200 nM purified human reductase, and tazarotenic acid (10 μM) in 100 mM potassium phosphate buffer (pH 7.4; final volume 100 μl). Incubations were initiated with the addition of 1 mM NADPH (final concentration) and incubated at 37°C for 30 minutes. Control incubations were performed in the absence of NADPH. Subsequent incubations to confirm the sequential metabolism of tazarotenic acid by CYP26A1 and CYP26B1 used 10 μM tazarotenic acid sulfoxide (final concentration) as the substrate in the incubation. Upon completion, in vitro incubations were extracted into 4 volumes of ethyl acetate, dried under a gentle stream of N2 at 40°C, reconstituted in 100 μl methanol prior to liquid chromatography–tandem mass spectrometry (LC-MS/MS) analysis, and compared with synthetic standards of each metabolite, if available. LC-MS/MS analysis was performed as described below.
Enzyme Kinetics.
In vitro enzyme kinetic parameters were determined for tazarotenic acid metabolite formation using 5 nM CYP26A1 or CYP26B1, 25 nM purified human reductase, and 0–10 μM tazarotenic acid. Incubations were carried out for 10 minutes at 37°C to ensure product linearity with regard to time and protein concentration. Additional experiments to determine the kinetic parameters for the sequential metabolism of tazarotenic acid metabolite sulfoxide used substrate concentrations ranging from 0 to 50 μM. Samples were prepared as described for in vitro metabolic profiling experiments.
Identification of the Enzymes Responsible for the Oxidative Metabolism of Tazarotenic Acid.
To assess the relative contribution of CYP26A1 and CYP26B1 to the in vitro oxidative metabolism of tazarotenic acid, metabolite formation was monitored across a panel of drug-metabolizing enzymes. Previously reported studies that characterized the enzymes responsible for the metabolism of tazarotenic acid were conducted at substrate concentrations of 1–200 μM (Attar et al., 2003). As total circulating plasma concentrations of tazarotenic acid are approximately 1–280 nM following typical doses of tazarotene, current studies were conducted using clinically relevant substrate concentrations. In vitro incubations consisted of 5 nM recombinant enzyme, 50 nM purified human reductase, and 100 nM tazarotenic acid (final concentrations) in 100 mM potassium phosphate buffer (pH 7.4). Following a 3-minute preincubation at 37°C, reactions were initiated with the addition of 1 mM NADPH (final concentration). For incubations utilizing flavin-containing mono-oxygenase enzymes (FMOs), the preincubation step consisted of enzyme and NADPH followed by initiation with substrate because of the known instability of FMOs at 37°C in the absence of cofactor (Foti and Fisher, 2004). Incubations (50 μl, final volume) were carried out for 30 minutes at 37°C before being quenched with 3 volumes (v/v) of ice-cold acetonitrile containing tolbutamide as an internal standard. Samples were mixed and sedimented at 1240g for 10 minutes before being transferred for LC-MS/MS analysis. Data were expressed as the percentage of total metabolite formed across the panel of enzymes for each individual metabolite.
LC-MS/MS Analysis.
Analysis of tazarotenic acid and its metabolites was conducted using LC-MS/MS. The analytical platform was composed of an Applied Biosystems API4000 fitted with an electrospray ionization source (Applied Biosystems, Foster City, CA). Liquid chromatography and sample introduction was achieved using two LC-20AD binary pumps with an in-line DGU-20A5 solvent degasser (Shimadzu, Columbia, MD) and a LEAP CTC HTS PAL autosampler (CTC Analytics, Carrboro, NC). An injection volume of 10 μl was used for all analyses. For enzyme kinetic experiments, chromatographic separation was achieved using 0.1% formic acid (v/v) in water (mobile phase A) and 0.1% formic acid in methanol/acetonitrile (1:1; mobile phase B) on a Synergi 2.5-μm Hydro RP 100 Å (50 × 2.0 mm) column (Phenomenex, Torrance, CA). Gradient conditions consisted of 2.5% B (0–0.4 minutes), 2.5% B–95% B (from 0.4–1.4 minutes), 95% B (from 1.4 to 2.5 minutes), and re-equilibration at 2.5% B for 0.5 minutes. For metabolite identification experiments, the same mobile phase system was used with a Kinetex 2.6-μm C18 100-Å (100 × 2.1 mm) column (Phenomenex). A gradient of 2.5% B (0–3 minutes), 2.5% B–95% B (from 3 to 14 minutes), 95% B (from 14 to17 minutes) followed by re-equilibration at 2.5% B for 3 minutes was used to achieve chromatographic separation of all analytes. Initial metabolite identification experiments used full-scan analysis from 100 to 800 amu followed by analysis of the corresponding product ion spectra for each observed analyte. Subsequent LC-MS/MS analyses used multiple-reaction monitoring (MRM) for each analyte. MRM transitions (positive ionization mode) were as follows: tazarotenic acid (m/z 324.2 / 294.3), tazarotenic acid sulfoxide and hydroxytazarotenic acid (m/z 340.3 / 280.3), tazarotenic acid sulfone (m/z 356.3 / 276.3) and the internal standard tolbutamide (m/z 271.2 / 91.1). Generic parameters applied to all MS analyses included the curtain gas (12 arbitrary units), collisionally activated dissociation (CAD) gas (medium), ion spray voltage (5000 V), source temperature (500°C), and ion source gas 1 and gas 2 (30 arbitrary units, each).
Data Analysis.
Mass spectrometry data were evaluated using Analyst (version 1.5; Applied Biosystems). Analyte concentrations were determined by comparing peak areas in unknown samples to those obtained from standard curves with analytical standards (dynamic range: 1–2000 nM; weighting: 1/x). Parameter fitting for IC50 and enzyme kinetic data were performed using GraphPad Prism as described below (version 6.03; GraphPad Software Inc., San Diego, CA).
IC50 values for retinoic acid receptor agonists in the 9-cis-4-hydroxyretinoic acid assay were determined by nonlinear regression using eq. 1. In the following equation, 100%*(vi/v) represents the percentage activity remaining for a given inhibitor concentration, [I], (vi/v)max*100% is the maximum observed activity with no inhibitor present, and (vi/v)min is the remaining enzyme activity at infinitely high concentrations of inhibitor.
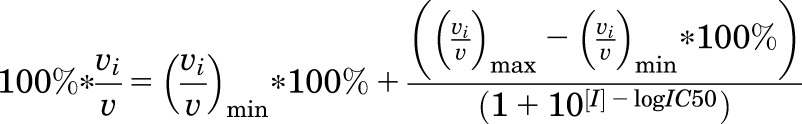 |
(1) |
Enzyme kinetic parameters (Km and Vmax) were estimated through nonlinear regression analysis using the Michaelis-Menten model as shown in eq. 2. In the equation below, Km denotes half the substrate concentration ([S]) at maximal reaction velocity (Vmax).
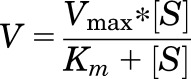 |
(2) |
Results
Homology Modeling.
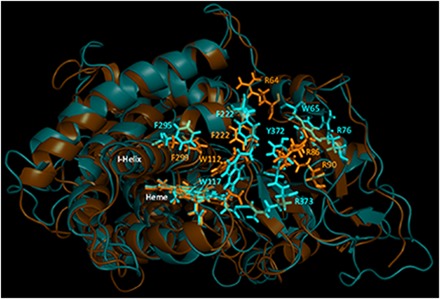
Characterization of CYP26B1 from a commercially available clone (OriGene) and a set of 12 human livers identified two amino acids that differed from the currently accepted amino acid sequence of the enzyme (Fig. 1). The two sequencing differences were an A191G substitution resulting in an H64R amino acid change and a G778A substitution resulting in a G260S amino acid change. All 12 human donors sequenced had a CYP26B1 sequence identical to the clone obtained from OriGene and did not have the two sequencing differences observed in the previously reported clone of CYP26B1 (UniProtKB Q9NR63). Therefore, the sequence with arginine at position 64 and serine in residue 260 was accepted as the wild-type sequence of CYP26B1 (CYP26B1*1). The sequence of CYP26B1*1 is shown in Fig. 1 and was used to build the subsequent CYP26B1 homology model. To assess the active sites of CYP26A1 and CYP26B1, homology models of each enzyme were constructed using the crystal structure of CYP120 (pdb 2VE3), which showed the highest degree of sequence similarity with CYP26A1 or CYP26B1 in a BLAST search. Sequence analysis indicated CYP26A1 had a sequence identity of 44.47% with CYP26B1 (Q9NR63), 44.26% with CYP26B1*1, and 33.26% with CYP120 (2VE3). CYP26B1*1 had a 99.61% sequence identity with CYP26B1 (Q9NR63) and a 34.69% sequence identity with CYP120. Superimposition of the CYP26A1 or CYP26B1 homology models with the template structure (CYP120) resulted in root-mean-square deviation (RMSD) values of 1.038 and 1.168, respectively. Superimposition of the CYP26A1 and CYP26B1 homology models with each other resulted in a RMSD value of 1.651. Similarities in hydrophobic binding residues were observed for CYP26A1 and CYP26B1, with W112, Phe222, and Phe299 occupying analogous positions in the CYP26A1 active site as W117, Phe222, and Phe295 did in the CYP26B1 active site (Fig. 2). A greater divergence was observed for those amino acid residues potentially capable of stabilizing the carboxylate moiety of at-RA in the active site of CYP26A1 (Arg64, Arg86, and Arg90) and CYP26B1 (W65, Arg76, Tyr372, and Arg373). Parameters detailing the structural evaluation of the template and the model are shown in Table 1 with the corresponding Ramachandran plots shown in Supplemental Fig. 1. The active-site volumes of CYP26A1 and CYP26B1 were estimated to be 918 Å3 and 977 Å3, respectively.
Fig. 2.

Structural alignment of CYP26A1 (orange) and CYP26B1 (cyan) homology models. Sequence alignment of the two homology models indicated a structural identity of 44.26% and an root-mean-square deviation value of 1.651. Structural similarity was observed for the portion of the active site of each enzyme that may contribute to hydrophobic binding interactions with a given ligand (Trp112, Phe222, and Phe299 for CYP26A1; Trp117, Phe222, and Phe295 for CYP26B1). The active-site volumes of CYP26A1 and CYP26B1 were estimated to be 918 Å3 and 977 Å3, respectively.
TABLE 1.
Estimated parameters for CYP26A1 and CYP26B1 homology models
| Parameter | CYP26A1 Model | CYP26B1 Model |
|---|---|---|
| Template | CYP120 (pdb 2VE3) | CYP120 (pdb 2VE3) |
| Template sequence identity | 33% | 34% |
| Template positive sequence coverage | 53% | 54% |
| BLAST query coverage | 89% | 89% |
| BLAST E-Value | 2e-82 | 6e-91 |
| RMSD versus template | 1.038 | 1.168 |
| Favorable bond angles | 86.2% | 95.0% |
| Allowable and favorable bond angles | 98.9% | 98.3% |
| I-Helix residues | M287–L318 | M283–A304 |
| Active-site volume | 918.01 Å3 | 976.86 Å3 |
| at-RA docking score (4-(S)-OH) | −9.552 | −4.999 |
| at-RA distance from heme (4-(S)-OH) | 3.85 Å | 2.99 Å |
| at-RA docking score (4-(R)-OH) | N/A | −4.128 |
| at-RA distance from heme (4-(R)-OH) | N/A | 4.06 Å |
| at-RA docking score (16-OH) | −9.552 | −4.488 |
| at-RA distance from heme (16-OH) | 3.49 Å | 2.77 Å |
| at-RA docking score (18-OH) | −9.552 | −4.435 |
| at-RA distance from heme (18-OH) | 5.10 Å | 3.10 Å |
| Tazarotenic acid docking score | −11.016 | −9.172 |
| Tazarotenic acid distance from heme | 4.21 Å (to sulfur) | 4.11 Å (to sulfur) |
| Tazarotenic acid sulfoxide docking score | −11.912 | −9.843 |
| Tazarotenic acid sulfoxide distance from heme | 4.38 (to sulfur) | 3.58 Å (to sulfur) |
To further assess the validity of the homology models, at-RA was docked into the active site of the CYP26A1 and CYP26B1 homology models. In both models, the β-ionone ring of at-RA was oriented toward the heme iron, with the models confirming that the hydrogen atoms at the 4-, 16-, or 18-position of the β-ionone ring of at-RA were positioned toward the heme iron. As with previously published homology model results, a single docking orientation of at-RA in the active site of CYP26A1 was able to account for oxidation at the 4-, 16-, and 18- positions (Shimshoni et al., 2012). Further, only the hydrogen atom, which when abstracted would lead to formation of the 4-(S)-hydroxyretinoic acid metabolite, was directed toward the heme iron at a distance of 3.85 Å, with the hydrogen atoms at the 16- and 18- positions approximately 3.49 Å and 5.10 Å away from the heme iron, respectively (Fig. 3A and Supplemental Fig. 2). In this orientation, the corresponding hydrogen to form 4-(R)-hydroxyretinoic acid was directed away from the heme iron in a metabolically unfavorable position at a distance of 5.59 Å. The CYP26A1 homology model was unable to resolve an orientation of at-RA in the active site conducive with abstraction of the hydrogen in the 4-(R)-position. Conversely, docking orientations where either hydrogen atom on the carbon atom at the 4-position of the β-ionone ring was oriented toward the heme iron were identified using the CYP26B1 homology model (Fig. 3B), suggesting that formation of 4-hydroxyretinoic acid by CYP26B1 would not be stereoselective. For formation of 4-(R)-hydroxyretinoic acid (Fig. 3B orange structure and Supplemental Fig. 2), the hydrogen atom was located approximately 4.06 Å from the heme iron. When docked in such a way that the resulting product would be 4-(S)-hydroxyretinoic acid (Fig. 3B cyan structure and Supplemental Fig. 2), the hydrogen atom to be abstracted was positioned approximately 2.99 Å from the heme iron. Docking of at-RA in the CYP26B1 homology model such that 16-hydroxyretinoic acid (Fig. 3C and Supplemental Fig. 2) or 18-hydroxyretinoic acid (Fig. 3D and Supplemental Fig. 2) would be the expected products positioned the sites of metabolism approximately 2.77 Å and 3.10 Å from the heme iron, respectively. The CYP26B1 docking score for the at-RA orientation leading to formation of 4-(S)-hydroxyretinoic acid was slightly more favorable than that leading to formation of 4-(R)-hydroxyretinoic acid and was similar to the docking scores observed when the 16- or 18-position was oriented toward the heme iron (Table 1). Arg90 (CYP26A1) and Ser369 or Arg373 (CYP26B1) were located within 3 Å of the carboxylic acid moiety of at-RA. Amino acid residues depicted in Fig. 3 and Supplemental Fig. 2 are located within 3 Å of the docked at-RA ligand.
Fig. 3.

CYP26A1 (A) and CYP26B1 (B–D) homology models with at-RA docked in the active site. A single docking orientation of at-RA in the CYP26A1 homology model accounted for 4-, 16-, and 18-hydroxylation of at-RA and suggested that abstraction of the hydrogen atom leading to formation of 4-(S)-hydroxy-at-RA was the preferred binding orientation for CYP26A1, supporting the reported stereoselective metabolism of at-RA by CYP26A1 (A). Alternatively, docking of at-RA in the active site of CYP26B1 suggested that orientation of either hydrogen atom at the prochiral 4-position of the beta-ionone ring toward the heme iron was equally favorable, in agreement with the observed formation of both 4-(R)-hydroxy- (orange structure) and 4-(S)-hydroxy-at-RA (cyan structure) by CYP26B1 (B). The CYP26B1 model was also capable of docking at-RA such that the 16-hydroxy- or 18-hydroxyretinoic acid metabolites would be the predicted metabolite products (C and D).
To select a nonendogenous retinoid-like molecule to include in the homology model analysis, a panel of retinoic acid receptor agonists was screened for inhibitory potency against CYP26A1 and CYP26B1. IC50 values ranged from 3.7 to 18 μM for CYP26A1 and from 0.13 to 31 μM for CYP26B1 (Table 2). Tazarotenic acid was the second most potent inhibitor from the panel against CYP26A1, the most potent inhibitor against CYP26B1, and the only inhibitor to exhibit low- to submicromolar inhibition potency in both assays. No evidence of time-dependent inhibition was observed with tazarotenic acid. As such, it was selected for further evaluation in the CYP26A1 and CYP26B1 homology models.
TABLE 2.
CYP26A1 and CYP26B1 IC50 values for retinoic acid receptor agonists and their derivatives (95% confidence interval shown in parentheses)
| Retenoic Acid Receptor Agonist | CYP26A1 IC50 | CYP26B1 IC50 |
|---|---|---|
| μM | μM | |
| AM80 | 12 (8.1–18) | 6.6 (2.3–19) |
| AM580 | 5.6 (3.0–10) | 2.2 (1.4–3.2) |
| BMS753 | 18 (4.4–76) | 28 (21–37) |
| BMS961 | 14 (9.6–20) | 31 (15–63) |
| Tazarotenic Acid | 6.1 (3.2–12) | 0.13 (0.09–0.19) |
| TTNPB | 3.7 (1.4–9.8) | 3.4 (2.2–5.2) |
A single binding orientation was observed for tazarotenic acid in the CYP26A1 homology model with the heme iron located approximately 4.21 Å from the sulfur atom of the benzothiopyranyl ring and 5.15 Å from the adjacent aromatic hydrogen (Fig. 4A). Key amino acids involved in ligand binding with CYP26A1 included Met60, Arg90, Trp112, Leu120, Phe222, Phe299, Thr304, Val370, Pro371, Gly372, Thr476, Pro478, and Thr479 (Supplemental Fig. 3). A hydrogen bonding interaction was predicted between the carboxylic acid moiety of tazarotenic acid and Arg90. The observed docking pose would predict either sulfoxidation or hydroxylation to occur on the benzothiopyranyl ring of tazarotenic acid. Similar active-site interactions were predicted when tazarotenic acid sulfoxide was docked in the active site of CYP26A1, with the sulfur atom and adjacent aromatic hydrogen atom 4.38 Å and 3.16 Å away from the heme iron, respectively (Fig. 4B). Hydrogen bonding was predicted to occur between the carboxylate group of tazarotenic acid sulfoxide and Arg90 and between the pyridinyl nitrogen and Gly372 (Supplemental Fig. 3). Docking of tazarotenic acid in the active site of CYP26B1 resulted in a pose similar to that identified for 16-hydroxylation of at-RA, with hydrogen bonding interactions between the substrate and Arg373 and Ile396 stabilizing the carboxylic acid moiety of tazarotenic acid. In this orientation, the sulfur and adjacent aromatic hydrogen atom were located approximately 4.11 Å and 4.07 Å from the heme iron, respectively (Fig. 4C and Supplemental Fig. 3). Tazarotenic acid sulfoxide bound in a similar manner in the active site of CYP6B1, with the sulfur and adjacent aromatic hydrogen atom located approximately 3.58 Å and 3.53 Å from the heme iron (Fig. 4D). Hydrogen bonds were predicted between the carboxylate of tazarotenic acid sulfoxide and Arg373 and Ile396, as well as between the pyridinyl nitrogen of tazarotenic acid sulfoxide and Asp398 (Fig. Supplemental Fig. 3). As with the docking poses obtained with CYP26A1, results from the CYP26B1 homology model would predict metabolism to occur on the benzothiopyranyl moiety of tazarotenic acid or tazarotenic acid sulfoxide, with the sulfur atom generally in closest proximity to the heme iron.
Fig. 4.

CYP26A1 and CYP26B1 homology models with tazarotenic acid docked in the active site. Docking of tazarotenic acid in the active sites of CYP26A1 (A) and CYP26B1 (C) suggested metabolism at or near the sulfur atom of the benzothiopyranyl ring system was the preferred site of metabolism. Similar binding orientations were observed for tazarotenic acid sulfoxide in the active sites of CYP26A1 (B) or CYP26B1 (D).
Metabolic Profile.
The oxidative metabolites of tazarotenic acid, whose formation are catalyzed by CYP26A1 and CYP26B1, were characterized using recombinant enzymes. Metabolism of tazarotenic acid by CYP26A1 and CYP26B1 resulted in the formation of tazarotenic acid sulfoxide, tazarotenic acid sulfone, and a hydroxylated metabolite of tazarotenic acid (Fig. 5A). Comparison of the electrospray ionization fragmentation patterns of the tazarotenic acid and tazarotenic acid sulfoxide synthetic standards with the hydroxylated metabolite of tazarotenic acid suggested that the location of the hydroxyl moiety was an aromatic hydroxylation on the benzothiopyranyl ring system (Supplemental Figs. 4 and 5). Formation of all tazarotenic acid metabolites was NADPH-dependent. To assess the sequential metabolism of tazarotenic acid by CYP26A1 and CYP26B1, incubations were conducted using tazarotenic acid sulfoxide as the starting material. Both CYP26A1 and CYP26B1 catalyzed the metabolism of tazarotenic acid sulfoxide to tazarotenic acid sulfone (Fig. 5B). No activation of the acetylene moiety was observed in incubations with either enzyme. A proposed metabolic scheme is shown in Fig. 6.
Fig. 5.

Extracted ion chromatograms for the metabolic profile of tazarotenic acid (A) and tazarotenic acid sulfoxide (B) by CYP26A1 and CYP26B1. Tazarotenic acid sulfoxide (TA-sulfoxide) and an additional hydroxylated metabolite of tazarotenic acid (hydroxy-TA) were identified as the primary metabolites of tazarotenic acid by CYP26A1 and CYP26B1, with subsequent conversion of tazarotenic acid sulfoxide to tazarotenic acid sulfone also observed. Data sets are offset for clarity and control traces represent incubations conducted in the absence of NADPH.
Fig. 6.

Proposed metabolic scheme of tazarotenic acid by CYP26A1 and CYP26B1. CYP26A1 or CYP26B1 contributes to each of the metabolic steps identified in the metabolism of tazarotenic acid.
In Vitro Enzyme Kinetics.
The enzyme kinetic parameters describing the formation of tazarotenic acid sulfoxide and hydroxytazarotenic acid were determined for CYP26A1 and CYP26B1. Enzyme kinetic parameters were determined using Michaelis-Menten kinetics and are reported in Table 3 and shown in Fig. 7. In general, incubations with CYP26B1 resulted in slightly higher Km and kcat values compared with incubations conducted with CYP26A1. Intrinsic clearance values (calculated as Vmax / Km) were slightly higher for CYP26A1 compared with CYP26B1 owing primarily to the difference in Km values between the two enzymes. Both enzymes appeared to favor formation of the hydroxylated metabolite of tazarotenic acid. Formation of tazarotenic acid sulfone from tazarotenic acid sulfoxide was linear through a substrate concentration of 50 μM, and as such no kinetic parameters were determined for this metabolic pathway.
TABLE 3.
Enzyme kinetic parameters for NADPH-dependent metabolism of tazarotenic acid to tazarotenic acid sulfoxide and hydroxytazarotenic acid (95% confidence interval shown in parentheses)
| CYP26A1 |
CYP26B1 |
||||
|---|---|---|---|---|---|
| Metabolite | Km | Clint | Km | Clint | kcat,B1 kcat,A1 |
| μM | ml/min per nanomole | μM | ml/min per nanomole | ||
| TA-Sulfoxide | 0.24 (0.18–0.27) | 1.03 (0.87–1.18) | 1.01 (0.85–1.18) | 0.38 (0.34–0.44) | 1.60 (1.52–1.68) |
| Hydroxy-TAa | 0.39 (0.31–0.42) | 4.01 (3.52–4.48) | 0.56 (0.42–0.63) | 3.67 (3.22–4.14) | 1.32 (1.27–1.37) |
Metabolite standard not available; concentrations derived from tazarotenic acid sulfoxide standard curve.
Fig. 7.

Enzyme kinetic plots for the formation of tazarotenic acid sulfoxide and hydroxytazarotenic acid by CYP26A1 (circles) and CYP26B1 (squares). Formation was characterized by Michaelis-Menten kinetics with CYP26B1 having kcat values that were approximately 1.3- to 1.6-fold higher than those observed for CYP26A1.
Identification of the Enzymes Responsible for the Oxidative Metabolism of Tazarotenic Acid.
Previous reports evaluating the enzymes responsible for tazarotenic acid metabolism in vitro have implicated CYP2C8, FMO1, and FMO3 in the formation of tazarotenic acid sulfoxide (Attar et al., 2003). Using an expanded drug-metabolizing enzyme panel and clinically relevant concentrations of tazarotenic acid, additional enzymes were identified that may contribute to the metabolism of tazarotenic acid. The highest rates of tazarotenic acid sulfoxide formation were observed for CYP26A1 and CYP26B1, followed by CYP2C8 and CYP3A7 (Fig. 8A). Formation of the sulfoxide metabolite was also observed in incubations with CYP2C9, CYP2J2, CYP3A4, CYP3A5, and aldehyde oxidase. Minor contributions were noted for CYP1A2 and CYP2B6. No metabolite formation was observed in incubations with FMO1, FMO3, or FMO5. The hydroxylated metabolite of tazarotenic acid was formed primarily by CYP26A1 and CYP26B1, with additional contributions from CYP2C8, CYP3A5, and CYP3A7 (Fig. 8B). As with the formation of tazarotenic acid sulfoxide, trace amounts of the hydroxylated metabolite were also observed in incubations with the majority of the enzymes evaluated in the panel.
Fig. 8.

Formation of tazarotenic acid sulfoxide and hydroxytazarotenic acid in a panel of recombinant enzymes. Formation of both enzymes was predominantly catalyzed by CYP26A1 and CYP26B1 with additional contributions from CYP3A and CYP2C isozymes.
Discussion
The CYP26 family of cytochrome P450 (CYP26A1, CYP26B1, and CYP26C1) has been identified as being responsible for the metabolism of at-RA and its metabolites (Ray et al., 1997; Taimi et al., 2004; Guengerich, 2006; Lee et al., 2007; Lutz et al., 2009; Thatcher and Isoherranen, 2009; Helvig et al., 2011; Ross and Zolfaghari, 2011; Nelson et al., 2013). To date, however, no known xenobiotic compounds have been identified as substrates of CYP26A1 or CYP26B1, the two most characterized CYP26 isoforms. As both enzymes are expressed in human skin and many retinoid-based treatments are administered topically, the potential exists for these CYP26s to contribute to the metabolism and elimination of these compounds (Heise et al., 2006; Osanai and Lee, 2011; Topletz et al., 2012). Furthermore, a major focal point in the development of synthetic retinoids is to overcome the pharmacokinetic shortcomings of at-RA, such as the observed autoinduction of its clearance pathways. Tazarotene is an acetylenic retinoid that is readily converted via hydrolysis to tazarotenic acid upon topical administration (Duvic, 1997; Madhu et al., 1997; Tang-Liu et al., 1999; Menter, 2000; Yu et al., 2003; Attar et al., 2005; Talpur et al., 2009). It is prescribed for the treatment of abnormal keratinocyte proliferation, as is observed in patients with stable plaque psoriasis, mild-to-moderate acne and basal cell carcinoma (Tang-Liu et al., 1999; Talpur et al., 2009). The pharmacological mechanism of action described for tazarotene involves metabolism to tazarotenic acid, which subsequently binds to retinoic acid receptors, primarily retinoic acid receptor (RAR)-β and RAR-γ (Chandraratna, 1996). As an inhibitor of CYP26A1 and CYP26B1, tazarotenic acid may also serve to locally increase concentrations of retinoic acid in the skin, a mechanism that may hold potential in treating dermatological disorders. As such, a greater understanding of the structural characteristics of CYP26A1 and CYP26B1 that mediate interactions with tazarotenic acid may have significant clinical relevance in terms of developing the next generation of topical pharmaceutics.
Homology models were designed to characterize the active site and substrate binding characteristics of CYP26A1 and CYP26B1. Prior to designing the homology models, the wild-type gene sequence of CYP26B1 was verified from a panel of human livers, as the initial clone differed in two amino acid residues from the currently available sequence of the enzyme (NCBI Reference Sequence: NP_063938.1; UniProtKB Q9NR63), which had been isolated from human retinal cDNA (White et al., 2000). Consistent with the commercially available clone, the amino acid sequence analysis showed that the amino acid residues at positions 64 and 260 in the CYP26B1 sequence are an arginine and a serine residue, respectively, as opposed to the histidine and glycine originally reported from the retinal cDNA of a single human donor (Fig. 1). However, as neither of these residues appears to be involved in substrate binding, it is unlikely that they influenced the results of previously published CYP26B1 homology models (Karlsson et al., 2008; Saenz-Mendez et al., 2012). Estimates of the active-site volume for CYP26A1 (918 Å3) and CYP26B1 (977 Å3) suggest that each enzyme can bind ligands such as at-RA and tazarotenic acid with molecular volumes of approximately 300 Å3 as well as much larger ligands, likewise to the ligand binding profiles of other cytochrome P450 enzymes. Structural comparison of the two homology models suggests that selectivity between CYP26A1 and CYP26B1 may be dependent upon interactions with acidic binding residues, as the active-site amino acids involved in enzyme-ligand hydrophobic interactions appear to be fairly well conserved between the two enzymes (Fig. 2).
The importance of being able to corroborate homology model data with supporting “site-of-metabolism” data cannot be understated. While successfully predicting the ability of an enzyme to bind an inhibitor suggests that the general active-site characteristics of the homology model are representative of the actual enzyme, being able to predict the correct orientation of the ligand within the active site of the enzyme imparts an additional level of rigor to the homology model. As an initial attempt to validate the homology models, at-RA was docked into the active sites of CYP26A1 and CYP26B1. The mechanism for the formation of 4-hydroxyretinoic acid by CYP26A1 and CYP26B1 involves hydrogen atom abstraction at a prochiral center, resulting in formation of either 4-(S)- or 4-(R)-at-RA. Previous in vitro data suggests that CYP26A1 preferentially catalyzes the stereoselective metabolism of at-RA to 4-(S)-hydroxy-at-RA, whereas CYP26B1 catalyzes the formation of both 4-hydroxy-at-RA enantiomers, results which have also been rationalized through homology modeling of CYP26A1(Shimshoni et al., 2012; Topletz, 2013). In the case of the CYP26A1 homology model presented here, all docking attempts resulted in the hydrogen atom that leads to formation of 4-(S)-hydroxy-at-RA being oriented toward the heme, with the model unable to orient at-RA in the active site of CYP26A1 in a manner conducive with formation of 4-(R)-hydroxy-at-RA. Conversely, when at-RA was docked into the active site of CYP26B1 docking poses, with either hydrogen atom at the 4-position toward the heme iron were observed. Taken together, this validation of the stereoselective metabolism of at-RA by the homology models suggests that the critical structural differences between the two enzymes which impart the stereoselective properties of at-RA metabolism by CYP26A1 and CYP26B1 are sufficiently captured by the models.
To apply the model to a xenobiotic ligand, the sites of metabolism of tazarotenic acid, an inhibitor of CYP26, were predicted. When tazarotenic acid was docked in the active sites of CYP26A1 or CYP26B1, a single orientation was observed with the benzothiopyranyl moiety directed toward the heme (Fig. 4, A and C). A number of the residues that appear to be important in orienting tazarotenic acid in the active sites of CYP26A1 and CYP26B1 have also been reported to be involved in the binding of at-RA in the active sites of these enzymes. For example, Trp112, Phe222, Phe299, Thr304, Pro369, and Val370 have been proposed to be involved in CYP26A1 binding of both at-RA as well as retinoic acid–metabolism blocking agents (RAMBAs) that are able to inhibit the activity of CYP26A1 (Gomaa et al., 2008, 2011a, b; Karlsson et al., 2008). Similarly, Trp65, Trp117, Thr121, Phe222, Phe295, Ser369, Val370, and Pro371 have been suggested to be key residues in binding at-RA and other ligands of CYP26B1 (Karlsson et al., 2008; Saenz-Mendez et al., 2012). Results from metabolite identification studies confirmed the ability of CYP26A1 and CYP26B1 to contribute to the metabolism of tazarotenic acid and tazarotenic acid sulfoxide only at the benzothiopyranyl end of the molecule, and a proposed metabolic scheme is shown in Fig. 6. Enzyme kinetic experiments suggest that the metabolism of tazarotenic acid by CYP26A1 and CYP26B1 represents a rare example of a substrate having higher kcat values for CYP26B1 compared with CYP26A1.
Finally, previously reported efforts to identify the enzymes responsible for the metabolism of tazarotenic acid have implicated CYP2C8, FMO1, and FMO3 in the metabolism of tazarotenic acid to a sulfoxide metabolite (Attar et al., 2003). As recent literature reports have highlighted the importance of characterizing contributions of drug-metabolizing enzymes at clinically relevant concentrations (Filppula et al., 2011; VandenBrink et al., 2011; Karonen et al., 2012), we have re-evaluated the enzymatic pathways responsible for the metabolism of tazarotenic acid. When in vitro experiments were conducted to identify the drug-metabolizing enzymes responsible for the oxidative metabolism of tazarotenic acid at the clinically relevant concentration of 100 nM, CYP26A1 and CYP26B1 were identified as the major cytochrome P450 isoforms involved in the formation of tazarotenic acid sulfoxide and hydroxytazarotenic acid in vitro (Fig. 8). Other enzymes that contributed to the formation of the sulfoxide metabolite included CYP2C8, CYP2C9, CYP2J2, CYP3A4, CYP3A5, and CYP3A7 and aldehyde oxidase, though metabolite formation rates by the CYP26 isoforms were at least 2.5-fold higher than those observed for any other cytochrome P450 enzymes. No formation of the sulfoxide by FMO1, FMO3, or FMO5 was observed, suggesting that these enzymes do not play a role in the formation of the metabolite at submicromolar concentrations of tazarotenic acid.
To our knowledge, our report describes the first known homology models of CYP26A1 and CYP26B1 that incorporate metabolic data from both endogenous and xenobiotic substrates. It also details the first known contributions of the CYP26 family of cytochrome P450 enzymes to the metabolism of a xenobiotic compound and provides additional computational analyses of the characteristics of each enzyme at its active site. Analysis of the active-site features of the CYP26A1 and CYP26B1 homology models suggests that the greatest site of structural divergence is in the carboxylate-binding region of the active site and that the enzymes may be capable of binding much larger ligands as well, a focus of ongoing research in our laboratory. Further understanding of the active-site characteristics of CYP26A1 and CYP26B1 that play a role in their substrate binding properties should serve to increase the probability of identifying CYP26-selective inhibitors that may ultimately prove useful in the treatment of various disease states.
Supplementary Material
Abbreviations
- at-RA
all trans–retinoic acid
- CYP
cytochrome p450
- FMO
flavin-containing mono-oxygenase enzyme
- LC-MS/MS
liquid chromatography–tandem mass spectrometry
- PCR
polymerase chain reaction
- RMSD
root-mean-square deviation
- SNP
single-nucleotide polymorphism
Authorship Contributions
Participated in research design: Foti, Isoherranen, Diaz, Douguet.
Conducted experiments: Foti, Dickmann, Buttrick.
Contributed new reagents or analytic tools: Foti, Zelter, Dickmann.
Performed data analysis: Foti, Dickmann, Buttrick.
Wrote or contributed to the writing of the manuscript: Foti, Isoherranen, Diaz, Douguet.
Footnotes
This research was supported in part by Amgen Inc. (Thousand Oaks, CA), by l’Institut National de la Santé et de la Recherche Médicale (INSERM), by the National Institutes of Health National Institute of General Medical Sciences [Grants R01-GM081569, R01-GM111772], by the National Institutes of Health National Institute of Aging [Grant R41AG046987], and by a RRIA award from the Michael J. Fox Foundation for Parkinson’s Research. Philippe Diaz is a cofounder and chief scientific officer of DermaXon. The authors declare no additional competing financial interests.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Ahmad N, Mukhtar H. (2004) Cytochrome p450: a target for drug development for skin diseases. J Invest Dermatol 123:417–425. [DOI] [PubMed] [Google Scholar]
- Attar M, Dong D, Ling KH, Tang-Liu DD. (2003) Cytochrome P450 2C8 and flavin-containing monooxygenases are involved in the metabolism of tazarotenic acid in humans. Drug Metab Dispos 31:476–481. [DOI] [PubMed] [Google Scholar]
- Attar M, Yu D, Ni J, Yu Z, Ling KH, Tang-Liu DD. (2005) Disposition and biotransformation of the acetylenic retinoid tazarotene in humans. J Pharm Sci 94:2246–2255. [DOI] [PubMed] [Google Scholar]
- Cavasotto CN, Phatak SS. (2009) Homology modeling in drug discovery: current trends and applications. Drug Discov Today 14:676–683. [DOI] [PubMed] [Google Scholar]
- Chandraratna RA. (1996) Tazarotene--first of a new generation of receptor-selective retinoids. Br J Dermatol 135 (Suppl 49):18–25. [DOI] [PubMed] [Google Scholar]
- Duvic M. (1997) Tazarotene: a review of its pharmacological profile and potential for clinical use in psoriasis. Expert Opin Investig Drugs 6:1537–1551. [DOI] [PubMed] [Google Scholar]
- Eksterowicz J, Rock DA, Rock BM, Wienkers LC, Foti RS. (2014) Characterization of the active site properties of CYP4F12. Drug Metab Dispos 42:1698–1707. [DOI] [PubMed] [Google Scholar]
- Filppula AM, Laitila J, Neuvonen PJ, Backman JT. (2011) Reevaluation of the microsomal metabolism of montelukast: major contribution by CYP2C8 at clinically relevant concentrations. Drug Metab Dispos 39:904–911. [DOI] [PubMed] [Google Scholar]
- Foti RS, Fisher MB. (2004) Impact of incubation conditions on bufuralol human clearance predictions: enzyme lability and nonspecific binding. Drug Metab Dispos 32:295–304. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 47:1739–1749. [DOI] [PubMed] [Google Scholar]
- Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. (2006) Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem 49:6177–6196. [DOI] [PubMed] [Google Scholar]
- Gomaa MS, Yee SW, Milbourne CE, Barbera MC, Simons C, Brancale A. (2006) Homology model of human retinoic acid metabolising enzyme cytochrome P450 26A1 (CYP26A1): active site architecture and ligand binding. J Enzyme Inhib Med Chem 21:361–369. [DOI] [PubMed] [Google Scholar]
- Gomaa MS, Armstrong JL, Bobillon B, Veal GJ, Brancale A, Redfern CP, Simons C. (2008) Novel azolyl-(phenylmethyl)]aryl/heteroarylamines: potent CYP26 inhibitors and enhancers of all-trans retinoic acid activity in neuroblastoma cells. Bioorg Med Chem 16:8301–8313. [DOI] [PubMed] [Google Scholar]
- Gomaa MS, Bridgens CE, Aboraia AS, Veal GJ, Redfern CP, Brancale A, Armstrong JL, Simons C. (2011a) Small molecule inhibitors of retinoic acid 4-hydroxylase (CYP26): synthesis and biological evaluation of imidazole methyl 3-(4-(aryl-2-ylamino)phenyl)propanoates. J Med Chem 54:2778–2791. [DOI] [PubMed] [Google Scholar]
- Gomaa MS, Bridgens CE, Veal GJ, Redfern CP, Brancale A, Armstrong JL, Simons C. (2011b) Synthesis and biological evaluation of 3-(1H-imidazol- and triazol-1-yl)-2,2-dimethyl-3-[4-(naphthalen-2-ylamino)phenyl]propyl derivatives as small molecule inhibitors of retinoic acid 4-hydroxylase (CYP26). J Med Chem 54:6803–6811. [DOI] [PubMed] [Google Scholar]
- Guengerich FP (2006) Human cytochrome P450 enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano PR ed) pp 377–574, Kluwer Academic/Plenum Publishers, New York. [Google Scholar]
- Heise R, Mey J, Neis MM, Marquardt Y, Joussen S, Ott H, Wiederholt T, Kurschat P, Megahed M, Bickers DR, et al. (2006) Skin retinoid concentrations are modulated by CYP26AI expression restricted to basal keratinocytes in normal human skin and differentiated 3D skin models. J Invest Dermatol 126:2473–2480. [DOI] [PubMed] [Google Scholar]
- Helvig C, Taimi M, Cameron D, Jones G, Petkovich M. (2011) Functional properties and substrate characterization of human CYP26A1, CYP26B1, and CYP26C1 expressed by recombinant baculovirus in insect cells. J Pharmacol Toxicol Methods 64:258–263. [DOI] [PubMed] [Google Scholar]
- Hillisch A, Pineda LF, Hilgenfeld R. (2004) Utility of homology models in the drug discovery process. Drug Discov Today 9:659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson M, Strid Å, Sirsjö A, Eriksson LA. (2008) Homology models and molecular modeling of human retinoic acid metabolizing enzymes cytochrome P450 26A1 (CYP26A1) and P450 26B1 (CYP26B1). J Chem Theory Comput 4:1021–1027. [DOI] [PubMed] [Google Scholar]
- Karonen T, Neuvonen PJ, Backman JT. (2012) CYP2C8 but not CYP3A4 is important in the pharmacokinetics of montelukast. Br J Clin Pharmacol 73:257–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuenzli S, Saurat JH. (2001) Retinoids for the treatment of psoriasis: outlook for the future. Curr Opin Investig Drugs 2:625–630. [PubMed] [Google Scholar]
- Lee SJ, Perera L, Coulter SJ, Mohrenweiser HW, Jetten A, Goldstein JA. (2007) The discovery of new coding alleles of human CYP26A1 that are potentially defective in the metabolism of all-trans retinoic acid and their assessment in a recombinant cDNA expression system. Pharmacogenet Genomics 17:169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DF. (2002) Molecular modeling of human cytochrome P450-substrate interactions. Drug Metab Rev 34:55–67. [DOI] [PubMed] [Google Scholar]
- Lutz JD, Dixit V, Yeung CK, Dickmann LJ, Zelter A, Thatcher JE, Nelson WL, Isoherranen N. (2009) Expression and functional characterization of cytochrome P450 26A1, a retinoic acid hydroxylase. Biochem Pharmacol 77:258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhu C, Duff S, Baumgarten V, Rix P, Small D, Tang-Liu D. (1997) Metabolic deesterification of tazarotene in human blood and rat and human liver microsomes. J Pharm Sci 86:972–974. [DOI] [PubMed] [Google Scholar]
- Menter A. (2000) Pharmacokinetics and safety of tazarotene. J Am Acad Dermatol 43:S31–S35. [DOI] [PubMed] [Google Scholar]
- Miller WH., Jr (1998) The emerging role of retinoids and retinoic acid metabolism blocking agents in the treatment of cancer. Cancer 83:1471–1482. [DOI] [PubMed] [Google Scholar]
- Nelson CH, Buttrick BR, Isoherranen N. (2013) Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr Top Med Chem 13:1402–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Njar VC. (2002) Cytochrome p450 retinoic acid 4-hydroxylase inhibitors: potential agents for cancer therapy. Mini Rev Med Chem 2:261–269. [DOI] [PubMed] [Google Scholar]
- Njar VC, Gediya L, Purushottamachar P, Chopra P, Vasaitis TS, Khandelwal A, Mehta J, Huynh C, Belosay A, Patel J. (2006) Retinoic acid metabolism blocking agents (RAMBAs) for treatment of cancer and dermatological diseases. Bioorg Med Chem 14:4323–4340. [DOI] [PubMed] [Google Scholar]
- Osanai M, Lee GH. (2011) Enhanced expression of retinoic acid-metabolizing enzyme CYP26A1 in sunlight-damaged human skin. Med Mol Morphol 44:200–206. [DOI] [PubMed] [Google Scholar]
- Perola E, Walters WP, Charifson PS. (2004) A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 56:235–249. [DOI] [PubMed] [Google Scholar]
- Ray WJ, Bain G, Yao M, Gottlieb DI. (1997) CYP26, a novel mammalian cytochrome P450, is induced by retinoic acid and defines a new family. J Biol Chem 272:18702–18708. [DOI] [PubMed] [Google Scholar]
- Ross AC, Zolfaghari R. (2011) Cytochrome P450s in the regulation of cellular retinoic acid metabolism. Annu Rev Nutr 31:65–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saenz-Méndez P, Elmabsout AA, Sävenstrand H, Awadalla MK, Strid Å, Sirsjö A, Eriksson LA. (2012) Homology models of human all-trans retinoic acid metabolizing enzymes CYP26B1 and CYP26B1 spliced variant. J Chem Inf Model 52:2631–2637. [DOI] [PubMed] [Google Scholar]
- Shimshoni JA, Roberts AG, Scian M, Topletz AR, Blankert SA, Halpert JR, Nelson WL, Isoherranen N. (2012) Stereoselective formation and metabolism of 4-hydroxy-retinoic Acid enantiomers by cytochrome p450 enzymes. J Biol Chem 287:42223–42232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Song S, Hao CZ, Huang WX, Liu CC, Xie HL, Lin B, Cheng MS, Zhao DM. (2015) Molecular recognition of CYP26A1 binding pockets and structure-activity relationship studies for design of potent and selective retinoic acid metabolism blocking agents. J Mol Graph Model 56:10–19. [DOI] [PubMed] [Google Scholar]
- Taimi M, Helvig C, Wisniewski J, Ramshaw H, White J, Amad M, Korczak B, Petkovich M. (2004) A novel human cytochrome P450, CYP26C1, involved in metabolism of 9-cis and all-trans isomers of retinoic acid. J Biol Chem 279:77–85. [DOI] [PubMed] [Google Scholar]
- Talpur R, Cox K, Duvic M. (2009) Efficacy and safety of topical tazarotene: a review. Expert Opin Drug Metab Toxicol 5:195–210. [DOI] [PubMed] [Google Scholar]
- Tang-Liu DD, Matsumoto RM, Usansky JI. (1999) Clinical pharmacokinetics and drug metabolism of tazarotene: a novel topical treatment for acne and psoriasis. Clin Pharmacokinet 37:273–287. [DOI] [PubMed] [Google Scholar]
- Thatcher JE, Isoherranen N. (2009) The role of CYP26 enzymes in retinoic acid clearance. Expert Opin Drug Metab Toxicol 5:875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher JE, Zelter A, Isoherranen N. (2010) The relative importance of CYP26A1 in hepatic clearance of all-trans retinoic acid. Biochem Pharmacol 80:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thatcher JE, Buttrick B, Shaffer SA, Shimshoni JA, Goodlett DR, Nelson WL, Isoherranen N. (2011) Substrate specificity and ligand interactions of CYP26A1, the human liver retinoic acid hydroxylase. Mol Pharmacol 80:228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topletz AR (2013) The Relative Importance of CYP26A1 and CYP26B1 in Mediating Retinoid Homeostasis: Studies on the Formation, Elimination and Biological Activity of All-Trans-Retinoic Acid Metabolites. Doctoral dissertation, University of Washington, Seattle, WA. [Google Scholar]
- Topletz AR, Thatcher JE, Zelter A, Lutz JD, Tay S, Nelson WL, Isoherranen N. (2012) Comparison of the function and expression of CYP26A1 and CYP26B1, the two retinoic acid hydroxylases. Biochem Pharmacol 83:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topletz AR, Tripathy S, Foti RS, Shimshoni JA, Nelson WL, Isoherranen N. (2015) Induction of CYP26A1 by metabolites of retinoic acid: evidence that CYP26A1 is an important enzyme in the elimination of active retinoids. Mol Pharmacol 87:430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VandenBrink BM, Foti RS, Rock DA, Wienkers LC, Wahlstrom JL. (2011) Evaluation of CYP2C8 inhibition in vitro: utility of montelukast as a selective CYP2C8 probe substrate. Drug Metab Dispos 39:1546–1554. [DOI] [PubMed] [Google Scholar]
- Verfaille CJ, Borgers M, van Steensel MA. (2008) Retinoic acid metabolism blocking agents (RAMBAs): a new paradigm in the treatment of hyperkeratotic disorders. J Dtsch Dermatol Ges 6:355–364. [DOI] [PubMed] [Google Scholar]
- Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. (2012) Combining global and local measures for structure-based druggability predictions. J Chem Inf Model 52:360–372. [DOI] [PubMed] [Google Scholar]
- White JA, Guo YD, Baetz K, Beckett-Jones B, Bonasoro J, Hsu KE, Dilworth FJ, Jones G, Petkovich M. (1996) Identification of the retinoic acid-inducible all-trans-retinoic acid 4-hydroxylase. J Biol Chem 271:29922–29927. [DOI] [PubMed] [Google Scholar]
- White JA, Ramshaw H, Taimi M, Stangle W, Zhang A, Everingham S, Creighton S, Tam SP, Jones G, Petkovich M. (2000) Identification of the human cytochrome P450, P450RAI-2, which is predominantly expressed in the adult cerebellum and is responsible for all-trans-retinoic acid metabolism. Proc Natl Acad Sci USA 97:6403–6408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. (2000) Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell 5:121–131. [DOI] [PubMed] [Google Scholar]
- Xi J, Yang Z. (2008) Expression of RALDHs (ALDH1As) and CYP26s in human tissues and during the neural differentiation of P19 embryonal carcinoma stem cell. Gene Expr Patterns 8:438–442. [DOI] [PubMed] [Google Scholar]
- Yu Z, Sefton J, Lew-Kaya D, Walker P, Yu D, Tang-Liu DD. (2003) Pharmacokinetics of tazarotene cream 0.1% after a single dose and after repeat topical applications at clinical or exaggerated application rates in patients with acne vulgaris or photodamaged skin. Clin Pharmacokinet 42:921–929. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.