

Abstract
Whether dramatic or modest, recovery of neurological function after spinal cord injury (SCI) is greatly due to neuroplasticity — the process by which the nervous system responds to injury by establishing new synaptic connections or by altering the strength of existing synapses. However, the same neuroplasticity that allows locomotor function to recover also produces negative consequences such as pain and dysfunction of organs controlled by the autonomic nervous system. In this review we focus specifically on structural neuroplasticity (the growth of new synaptic connections) after SCI and on the consequent development of pain and autonomic dysreflexia, a condition of episodic hypertension. Neuroplasticity after SCI is stimulated by the deafferentation of spinal neurons below the lesion and by the expression of growth-promoting neurotrophins such as nerve growth factor (NGF). A broad range of therapeutic strategies that affect neuroplasticity is being developed for the treatment of SCI. At one end of the spectrum are therapeutic strategies that directly or indirectly increase NGF in the injured spinal cord, and have the most robust effects on neuroplasticity. At the other end of the spectrum are neuroprotective strategies focused on supporting and rescuing uninjured, or partially injured, axons; these might limit the deafferentation stimulus for neuroplasticity. In the middle of this spectrum are strategies that block axon growth inhibitors without necessarily providing a growth stimulus. The literature supports the view that the negative consequences of neuroplasticity develop more commonly with therapies that directly stimulate nerve growth than they develop in the untreated injured cord. Compared to these conditions, neuroplasticity with negative outcomes is less prevalent after treatments that that neutralize axon growth inhibitors, and least apparent after strategies that promote neuroprotection.
Keywords: Neuroplasticity, Spinal cord injury, Review, Autonomic dysreflexia, Pain
Introduction
Neuroplasticity after spinal cord injury (SCI) can encompass a variety of responses within the injured cord, the brain and the ganglia outside the central nervous system. They range from structural neuroplasticity (regenerative growth responses of injured motor and sensory projections, and collateral sprouting of nearby intact fibers), to changes in gene expression (altered expression profiles of neurotransmitters, neuropeptides, growth factors and their associated receptors) (Gris et al., 2009), to changes in membrane ion channels (Bareyre et al., 2004; Deumens et al., 2008; Frigon and Rossignol, 2006; Ghosh et al., 2010; Hains et al., 2003; Steward et al., 2003). The primary injury leads to secondary sequellae such as excitotoxicity, tissue ischemia and inflammation (Tator, 1995; Young, 1993) that further exacerbate the injury, a process termed secondary injury. Both the primary and secondary aspects of the SCI can trigger neuroplasticity that potentially promotes recovery from the injury but it also may have deleterious consequences. Furthermore, as presented elsewhere in this volume, even post-injury training and rehabilitation can induce plastic changes in the nervous system; these too may have yet unknown negative consequences. This chapter will focus on negative aspects of neuroplasticity as it concerns growth of axons and primary afferent fibers that can be induced by primary and secondary SCI and by therapies designed to improve neurological recovery by increasing nerve growth.
SCI partially denervates spinal neurons by disrupting ascending and descending pathways to and from the brain as well as inputs from the dorsal root ganglia. This denervation provides a stimulus for intact axons of interneurons, white matter tracts and sensory neurons to replace lost inputs to the spinal neurons that have empty synapses. Moreover, the loss of connections of intact ascending sensory systems may prompt collateral sprouting in search of new targets caudal to the injury. Collateral or regenerative sprouting of intraspinal axons after SCI may allow them to make new connections or to strengthen existing synapses to partially denervated or other spinal neurons. Indeed, some recovery of motor control after cord hemisection in cats has been attributed to collateral sprouting of primary afferent axons (Goldberger et al., 1993; Helgren and Goldberger, 1993) and early studies demonstrating enlarged excitatory postsynaptic potentials in chronic spinal cats attributed part of the increase to sprouting of primary afferent fibers (Nelson and Mendell, 1979). Therefore SCI itself sets the stage for a growth response within the injured cord, and neuroplasticity is a key feature in spontaneous recovery from SCI. However, maladaptive neuroplasticity is a feature of many of the negative outcomes of SCI. Examples of this are muscle spasticity, neuropathic pain, autonomic dysreflexia, urinary bladder dyssynergia, bowel dysfunction, cardiac arrhythmias and sexual dysfunction (Christensen and Hulsebosch, 1997a; Collins et al., 2006; de Groat and Yoshimura, 2006; Johnson, 2006; Mathias, 2006; Nout et al., 2006). These dysfunctions are caused, at least in part, by an imbalance of inhibitory and excitatory synaptic inputs to spinal neurons and several relate to a loss of coordination of autonomic and somatic control.
Two conditions, neuropathic pain and autonomic dysreflexia, are common adverse outcomes of SCI that appear to involve neuroplasticity as part of their etiology. Neuropathic pain after SCI has many underlying factors such as increases in neuronal excitability due to products of microglial activation, changes in sodium channel expression, and changes in glutamate receptor expression (Deumens et al., 2008) but this discourse will address primarily two factors that involve neural growth, namely collateral sprouting of calcitonin gene-related peptide (CGRP)-containing primary afferent fibers in the spinal cord dorsal horn (Christensen and Hulsebosch, 1997b) and aberrant growth of descending serotonergic axons in the dorsal horn rostral to the cord lesion site (Oatway et al., 2005). This growth can occur both rostral and caudal to the injury, in parallel with the neuropathic pain that can be evoked above, at and below the injury site. Autonomic dysreflexia is an abnormality of blood pressure control characterized by extreme hypertension accompanied by a pounding headache and slow heart rate [for review see (Weaver et al., 2006), (Mathias, 2006)]. This hypertension occurs in response to sensory input entering the spinal cord below the level of the lesion. This input leads to exaggerated spinal reflex sympathetic (autonomic) responses that can be associated with an increased CGRP-containing primary afferent arbor in the dorsal horn (Krenz and Weaver, 1998; Krenz et al., 1999) or, in the presence of a normal arbor, with loss of descending inhibitory influences on spinal sympathetic reflexes (Gris et al., 2005). In this condition, the key changes occur below the site of injury.
Structural neuroplasticity in pain and autonomic dysreflexia after SCI
Collateral sprouting of CGRP-immunoreactive small diameter primary afferent fibers into the laminae III–V of the dorsal horn after SCI has been linked with the development of chronic neuropathic pain and autonomic dysreflexia (Christensen and Hulsebosch, 1997a, 1997b; Jacob et al., 2001; Krenz and Weaver, 1998; Krenz et al., 1999; Weaver et al., in press; Wong et al., 2000) (Fig. 1A). Sprouting of larger diameter fibers has also been noted after SCI (Krenz and Weaver, 1998) and may contribute to dysreflexia as this condition can also be induced by non-noxious stimuli such as light touch (Marsh and Weaver, 2004). Primary afferent sprouting may be important near the site of injury, for example in the case of at-level neuropathic pain, or elsewhere in the cord, for example in the region of input from pelvic afferent projections that initiate autonomic dysreflexia after a high thoracic lesion. A caveat that must be introduced is that neuropathic pain and autonomic dysreflexia are not totally dependent upon this type of primary afferent sprouting. Mechanical allodynia and hyperalgesia after low thoracic clip compression SCI are not always accompanied by increased distribution or density of CGRP-immunoreactive afferent arbors (Bruce et al., 2002) and a study of autonomic dysreflexia in different strains of mutant mice showed robust dysreflexia in some strains that had no changes in their CGRP-immunoreactive arbor (Jacob et al., 2003). Because of its robust expression in the dorsal horn of the spinal cord, CGRP is a ready target for assessment of changes in the primary afferent arbor. However, CGRP is expressed in less than half of nociceptive dorsal root sensory neurons and this peptide is also expressed in small myelinated and unmyelinated primary afferent neurons that are not associated with nociception, demonstrating that changes in CGRP-immunoreactive populations cannot be associated solely with sensations of pain (Lawson et al., 2002, 1996). After SCI or dorsal rhizotomy, enlargements can be detected within the arbors of Aδ, Aβ and C primary afferent fibers and CGRP is expressed by many but not all of the neurons in these populations (Krenz and Weaver, 1998; McNeill and Hulsebosch, 1987; Wong et al., 2000). A subset of CGRP-immunoreactive C-fiber primary afferent neurons expresses substance P and is closely associated with nociception (Ju et al., 1987; Price, 1985). Changes in this afferent population were not detected after a high thoracic compression SCI (Marsh and Weaver, 2004), but injury of another type or at another spinal cord location may trigger involvement of this phenotype of neuron. Moreover, loss of small diameter myelinated primary afferent neurons and changes in the functions of C-fiber afferent neurons in bladder pathways occur after SCI (de Groat and Yoshimura, 2006). Similar changes in sensory pathways affecting other targets could contribute to maladaptive plasticity after SCI but have not been examined systematically. Much remains unknown about plasticity within primary afferent populations after SCI. Although sprouting of CGRP-immunoreactive dorsal horn sensory fibers clearly is not entire story about plasticity in sensory systems after SCI, this phenomenon is well documented, can contribute to pathological conditions and is a valid therapeutic target (see below).
Fig. 1.

Photomicrographs of CGRP-immunoreactive fibers in the dorsal horn of a control intact rat (A1) and of rats 2 weeks after cord transection at the 4th thoracic (T4) segment (A2-6 and B1-3). A) Cord-injured rats received an intrathecal infusion of nonimmune rabbit IgG or an intrathecal infusion of anti-NGF Ab. Transverse tissue sections are from the T6 segment. Panels 4–6 are magnifications of laminae III–V from panels 1–3 (boxed area). Scale bars: 1–3, 100 μm; 3–6, 50 μm. The anti-NGF treatment markedly decreased the size of the CGRP+ arbor in the deeper laminae. [Adapted, with permission, from (Krenz et al., 1999)]. B) Immediately after cord injury adenoviral vectors encoding green fluorescent protein (GFP adts, B1), or NGF (NGF Adts, B2) or Semaphorin 3A (sema3A adts, B3) were injected into the lumbosacral spinal cord. These sections of the L6/S1 cord show that NGF overexpression induced CGRP+ fiber sprouting throughout the dorsal horns and even dorsal columns. Conversely, Sema3A reduced post-traumatic aberrant CGRP fiber sprouting into the dorsal horn. Scale bar: 200 μm. [Adapted, with permission, from (Cameron et al., 2006)].
For decades investigators have been designing therapeutic manipulations with a goal of fostering regenerative or collateral growth, pushing the innate recovery systems further. How does it sometimes go wrong? We will consider three broad categories of treatment and consider if some types of therapies are more likely than others to lead to the development of negative consequences associated with neuroplasticity. The first targets injured or spared axons and promotes regenerative sprouting in the hopes of improving neurological recovery. The second category of treatment targets the spinal lesion’s microenvironment that actively inhibits axon growth and includes treatments that remove or block inhibitors of axon growth. The third category of treatments is broadly termed neuroprotective because they focus on limiting secondary injury that can be a stimulus for maladaptive plasticity.
SCI treatment strategies that promote axon growth
Many therapeutic strategies for SCI focus on encouraging the growth and regenerative sprouting of spared and injured axons through the non-permissive environment of the injured CNS. These strategies include the direct administration of neurotrophins or transplantation of various cell types that by nature or experimental manipulation express neurotrophins. For example, transplanting fibroblasts engineered to express NGF into the acute or chronically injured rat spinal cord induces robust sprouting of primary sensory dorsolateral fasciculus axons as well as cerulospinal and putative ventral horn motor axons (Grill et al., 1997; Tuszynski et al., 1996). Whereas this group did not assess possible effects of the increased growth of CGRP-expressing fibers in the injured spinal cord on neurological outcomes, others have shown that virally-mediated NGF expression in the uninjured (Romero et al., 2000) and injured (Cameron et al., 2006) spinal cord correlates with increased CGRP fiber collateral sprouting and the development of hyperalgesia and increased autonomic dysreflexia.
NGF expression after SCI has been the target of strategies to reduce the development of autonomic dysreflexia and chronic pain syndromes in rat models of SCI. NGF expression does increase after SCI, particularly near the lesion site as shown by immunocytochemical detection of the growth factor protein and mRNA (Brown et al., 2004, 2007; Krenz and Weaver, 2000). This NGF is upregulated within ramified microglia, astrocytes, intermediate grey neurons, pial cells, and leptomeningeal and Schwann cells of the injured cord (Fig. 2). Furthermore intrathecal delivery of anti-NGF antibodies for weeks after SCI decreases the size of the CGRP-immunoreactive afferent arbor in models of neuropathic pain (Christensen and Hulsebosch, 1997a, 1997b) and of autonomic dysreflexia (Krenz et al., 1999) (Fig. 1A). An antibody to NGF and a trkA-IgG fusion protein both blocked the development of autonomic dysreflexia in rat models of thoracic SCI (Krenz et al., 1999; Marsh et al., 2002) (Fig. 3). Moreover, a treatment that inhibits growth of C-fibers, semaphorin 3A, when delivered into the injured cord also blocked the enlargement of the CGRP-immunoreactive arbor and reduced autonomic dysreflexia (Cameron et al., 2006) (Fig. 1B). Finally, in addition to primary afferent sprouting after SCI, a significant increase in the axon density of lumbosacral dorsal grey commissural propriospinal neurons has been reported, demonstrating plasticity of the propriospinal neuronal systems as well (Hou et al., 2008). This plasticity corroborates electrophysiological evidence for propriospinal plasticity in the etiology of autonomic dysreflexia (Krassioukov et al., 2002). The propriospinal plasticity occurs in concert with the responses of the primary afferent arbor but its relationship to the increased intraspinal NGF after SCI has not been established. All of the above studies suggest that treatments that might increase NGF within the injured spinal cord generate the risk of augmenting the development of neuropathic pain and autonomic dysfunction. Furthermore, increased endogenous NGF in the spinal cord and dorsal root ganglia are implicated in plasticity that contributes to lower urinary tract dysfunction after SCI (Seki et al., 2002, 2004).
Fig. 2.

Photomicrographs of NGF immunoreactive cells in longitudinal sections of the T5 spinal cord at 7 days after T4 spinal cord injury. A–C) NGF is expressed in cells at and below the pial surface of the cord (A) and is co-localized with the low affinity neurotrophin receptor p75NTR (B) in cells adjacent to the pia that may be leptomeningeal cells (C, arrow head). D–F) NGF is expressed in large stellate cells in the white matter (D) that are immunoreactive for glial acidic fibrillary protein (E, GFAP), identifying them as astrocytes (F, arrow head). Scale bar in F is 50 μm and refers to all panels.
Adapted, with permission, from Brown et al. (2004).
Fig. 3.

Mean arterial pressure (MAP), pulsatile arterial pressure (AP), and heart rate (HR) in two rats 2 weeks after T4 clip compression spinal cord injury: after intrathecal treatment with control IgG (A) and after intrathecal treatment with an anti-NGF treatment (trkA-IgG) (B). Colon distension for 60 s (onset and duration marked with a thick line) stimulated an increase in AP and MAP and a decrease in HR in the control treated rat. In contrast, colon distension caused only a modest increase in AP and MAP after anti-NGF treatment (B).
Adapted, with permission, from Marsh et al. (2002).
Strategies to promote neural plasticity also include various cell therapies including olfactory ensheathing cell and neural progenitor cell transplantation. Olfactory neurons are continuously renewed thoughout life and extend processes that grow long distances from their origins in the olfactory mucosa up to their targets in the olfactory bulb. The remarkable growth of these nerve fibers is attributed to specialized glia, the olfactory ensheathing cells that enwrap the growing fibers and extend processes that accompany the growing fibers to their targets (Raisman, 2001). The ability of olfactory ensheathing cells to promote nerve growth into the adult CNS suggested that these cells may be able to encourage axonal growth in the injured spinal cord. Many groups have shown that olfactory ensheathing cells do support axonal growth and recovery after transplantation (Keyvan-Fouladi et al., 2003; Lu et al., 2002; Ramon-Cueto et al., 2000). However, olfactory ensheathing cell transplantation has been associated with increased CGRP fiber sprouting and the development of autotomy in animals (Richter et al., 2005).
Neural precursor cell transplantation has been suggested to improve recovery in models of SCI by cell replacing cells lost to the injury (Cummings et al., 2005; Hofstetter et al., 2005; Karimi-Abdolrezaee et al., 2006) and/or by providing trophic support that promotes tissue sparing (Teng et al., 2002) and neuroplasticity (Lu et al., 2003). Interestingly, whether neural precursor cell transplantation leads to maladaptive neuroplasticity may depend on their differentiation in the injured spinal cord. Hofstetter et al. (Hofstetter et al., 2005) found that neural precursor cell transplantation into the injured rat spinal cord led to increased CGRP fiber sprouting that correlated tightly with the development of allodynia in these mice. However this negative effect of neural precursor cell transplantation was avoided when the neural precursor cells were transduced to express the neurogenin-2 transcription factor before transplantation. The transplantation of neurogenin-2-transduced neural precursor cells did not result in CGRP fiber sprouting or allodynia. The authors attributed the different behavior of transplanted naïve neural precursor cells and neurogenin-2-transduced neural precursor cells to the fact that the naïve cells largely differentiated into astrocytes while the neurogenin-2-expressing cells largely differentiated into neurons and oligodendrocytes but not into astrocytes. The authors further argued that, as astrocytes are known to secrete various growth factors including NGF, this trophic effect may account for the CGRP fiber sprouting and hence allodynia in transplanted animals. This explanation is corroborated by the demonstration that C17.2 neural stem cells produce NGF after transplantation into spinal cord injured rats that correlates with CGRP fiber sprouting (Lu et al., 2003). Furthermore, when neural precursor cell transplantation into the injured rat spinal cord was coupled with PDGF-AA administration to encourage oligodendrocyte differentiation, approximately 50% of the transplanted neural precursor cells expressed oligodendrocyte lineage markers and led to improved locomotor recovery without accompanying neuropathic pain (Karimi-Abdolrezaee et al., 2006, 2010).
SCI treatment strategies that block axon growth inhibitors
Two classes of molecules inhibit axon growth and regeneration after SCI. First, several components of central myelin inhibit axon growth including myelin-associated glycoprotein (MAG), NOGO, and oligodendrocyte myelin glycoprotein (Omgp). All three of these inhibitors bind to and act through the same receptor complex consisting of the Nogo receptor (NgR), p75 and lingo (Zhou and Li, 2007). Blocking Nogo action by the administration of a neutralizing anti-Nogo antibody increased neuroplasticity and improved locomotor recovery without the development of hypersensitivity or allodynia (Liebscher et al., 2005) or muscle spasms (Gonzenbach et al., 2010). A study comparing neurological recovery in spinal cord injured rats receiving an anti-Nogo antibody or treadmill training or both demonstrated that two therapies that improve locomotor recovery after SCI may do so through encouraging different forms of neuroplasticity. This study showed that different types of neuroplasticity may counteract each other when used in combination (Maier et al., 2009). The second class is made up of ECM (extracellular matrix) proteins in the scar, of which chondroitin sulfate proteoglycans are probably the most important (Asher et al., 2001; Fawcett and Asher, 1999; Galtrey and Fawcett, 2007). CSPGs are a class of ECM macromolecules that share a common structure comprising a central core protein with a number of chondroitin sulfate side chains (Morgenstern et al., 2002). CSPGs are present in the adult CNS (Bignami et al., 1992) and following injury their expression increases greatly (Lemons et al., 1999; McKeon et al., 1991). Enzymatic digestion of the chondroitin sulfate side chains found on all CSPGs using the enzyme chondroitinase ABC in spinal cord-injured rats increases collateral or regenerative sprouting of descending projections and improves locomotor recovery (Barritt et al., 2006). Whereas chondroitinase treatment in these rats also increased sprouting of CGRP fibers adjacent to the lesion, this sprouting was mostly found in the dorsal columns, as opposed to in the grey matter, and was not associated with an increase in mechanical allodynia or thermal hyperalgesia.
Of the other axon-repelling molecules in the scar Ephs and ephrins are probably amongst the best known. Eph Receptor tyrosine kinases form the largest family of receptor tyrosine kinases and are known to mediate repulsive interactions between cells and projecting axons when activated by their ephrin ligands. Several studies have shown an increased expression of Ephs and ephrins after SCI. To test the role of increased EphA4 expression in the injured spinal cord an anti-EphA4 oligonucleotide was administered to rats intrathecally after SCI. Behavioral testing in treated rats demonstrated that knocking down EphA4 expression in the injured spinal cord did not improve locomotor recovery but did lead to the development of mechanical allodynia (Cruz-Orengo et al., 2006). A second group that downregulated EphA4-ephrin signaling in the injured spinal cord using a peptide antagonist was able to demonstrate increased locomotor recovery and corticospinal tract sprouting (Fabes et al., 2007). The different finding of these two groups probably reflects differences in how the anti-EphA4 oligonucleotide and blocking peptide work. The intrathecal delivery of the anti-EphA4 oligonucleotide would be expected to reduce EphA4 expression in spinal neurons and hence affect only propriospinal axons. The EphA4 blocking peptide would be predicted to antagonize EphA4 signaling throughout the spinal cord affecting both propriospinal and descending corticospinal projections. These studies demonstrate that blocking inhibitors of axon growth has the potential to promote both regenerative and maladaptive sprouting and that the resolution to this problem may lie in the development of strategies that target specific neuronal populations or axon projections.
SCI treatment strategies that promote neuroprotection
Anti-inflammatory treatments dedicated to neuroprotection in the acute days after SCI have been investigated for decades. Indeed the intended action of the well-studied methylprednisolone treatment for SCI was reduction of inflammation and neuroprotection (Bracken et al., 1990). The outcomes after SCI are highly dependent upon a fine balance of growth and death within the injured cord as well as the equilibrium between excitatory and inhibitory control systems. Our laboratories have promoted an anti-inflammatory treatment that limits the influx of activated neutrophils and monocytes into the injured spinal cord with the use of intravenously delivered blocking antibodies that target a key leukocyte integrin, CD11d/CD18. This treatment limits intraspinal inflammation, reduces oxidative damage and lesion size and leads to significant improvement in motor function and reduction of autonomic dysreflexia and neuropathic pain (Bao et al., 2004a, 2004b; Gris et al., 2004; Oatway et al., 2005; Saville et al., 2004). We anticipated that this treatment would also avert primary afferent sprouting, as NGF, the likely trigger of this sprouting, is found in the injured cord in areas of high inflammation (Brown et al., 2004). Unexpectedly, the anti-CD11d monoclonal antibody treatment did not block the increased arbor of CGRP-immunoreactive fibers in laminae III–V of the lower thoracic and lumbar spinal cord after clip compression injury at the 4th thoracic segment (Gris et al., 2005). Despite this, the treatment markedly reduced lesion size and, in this study, significantly reduced the magnitude of autonomic dysreflexia. We concluded that the neuroprotection actually permitted the primary afferent sprouting response, but the reduction of secondary damage with sparing of descending pathways was of greater importance in the overall control of blood pressure. Therefore the neuroplasticity did not yield its possible negative outcome perhaps because a significant component of supraspinal control of the spinal autonomic neurons was maintained, sufficient to limit the development of autonomic dysreflexia. Either sufficient inhibitory influences on the spinal circuits were maintained, or the opportunity for synapse formation by this exaggerated CGRP-immunoreactive input may have been minimized. In this study methylprednisolone also permitted the increased arbor of CGRP-immunoreactive fibers in the dorsal horn, but did not lead to significant tissue sparing near the lesion and only transiently limited autonomic dysreflexia. These findings suggest that the possible negative aspects of neuroplasticity depend upon the milieu within the injured cord, with its specific balance of losses and excesses.
Although the anti-CD11d integrin treatment did not alter primary afferent sprouting in the dorsal horn, it led to increased projections of serotonergic axons into and through the spinal cord lesion and it abolished an accumulation of serotonergic [5-hydroxytryptamine (5-HT)] axons that develops in the dorsal horn and in deeper layers rostral to a SCI site (Oatway et al., 2005) (Figs. 4A–D). In an older study we noted clumps of disorganized axons rostral to a SCI that stained for growth-associated protein-43, suggesting an upregulated growth response in this region (Weaver et al., 1997). The serotonergic accumulations found more recently may be part of that growth pattern.
Fig. 4.

Serotonin (5-HT) immunoreactivity in laminae I–IV of the dorsal horn rostral to a T12 spinal cord injury 4 weeks after the injury. A–C) Photomicrographs of transverse sections of the dorsal horn in sham-injured, vehicle-treated, and anti-CD11d mAb-treated rats. After SCI, the distribution and density of 5-HTimmunoreactive fibers were increased significantly, primarily in the superficial laminas, with punctate fibers in laminas III and IV. Anti-CD11d mAb treatment normalized the distribution of 5-HT-immunoreactive fibers toward patterns observed in sham-injured animals. Scale bar, 100 μm. D) The area of 5-HT-immunoreactivity in the dorsal horn rostral to the injury at T12–13 presented as the mean area±SE of 5-HT-immunoreactivity in the sham injured (n=6), vehicle-treated (n=5), and anti-CD11d mAb-treated (n=6) groups. *, P<0.05 compared with vehicle-treated rats; +, P<0.05 compared with sham-injured rats. E) Photomicrograph of 5-HT3-R-immunoreactivity in a transverse section from the 9th thoracic segment of a rat injured at T12. Immunoreactivity is primarily localized within the superficial laminae I and II of the dorsal horn with sparse amounts in the deeper laminae. Scale bar=100 μm (A) refers to all sections.
Adapted, with permission, from Oatway et al. (2005).
SCI leads to loss of descending serotonergic control of several populations of spinal neurons. Serotonergic projections from the nucleus raphe magnus to the spinal cord synapse in the dorsal horn, causing inhibition or facilitation of pain signaling that depends on the receptor stimulated (Bardin et al., 2000; Calejesan et al., 1998). Thus, loss of descending serotonergic inputs caudal to the lesion site contributes to neuropathic pain (Hains et al., 2002). Serotonergic projections to spinal cord autonomic and motor nuclei are also key regulators of these functions (Chalmers et al., 1985; Saruhashi et al., 1996). The serotonergic projections are often monitored as a means of determining the efficacy of a treatment in promoting regeneration or sparing of important descending pathways. Indeed the anti-CD11d antibody treatment did promote some sparing and projection of serotonergic axons through the cord lesion site in our study (Oatway et al., 2005). But we speculated that the mechanism by which this treatment probably blocked neuropathic pain rostral to the injury site was by limiting the excessive growth of serotonergic axons rostral to the lesion. Similarly, Inman and Steward (Inman and Steward, 2003) reported a dense accumulation of 5-HT-immunoreactive fibers rostral to a spinal cord lesion site that were “disorganized and meandering” and unable to extend into the central lesion. They termed this response “aberrant regenerative sprouting,” defined as the axonal growth that does not culminate in reconnection of the injured axon with its normal target. The increased density of serotonergic axons rostral to the SCI in our study appeared to promote at-level neuropathic pain through actions of the serotonin on pro-nociceptive 5-HT3 receptors (Oatway et al., 2004) (Fig. 4E and Fig. 5). The anti-CD11d mAb treatment reduced the aberrant growth response of serotonergic axons rostral to the injury, perhaps because of white matter sparing at the lesion borders that suggests diminished axon injury, and gray matter sparing that would increase the availability of target neurons for connection (Gris et al., 2004). Therefore, whereas regeneration and sprouting of serotonergic pathways through the lesion into the more distal spinal cord is an important objective, the sprouting immediately rostral to the lesion is a form of neuroplasticity associated with maladaptive conditions. Perhaps by reducing the hostility of the lesion environment, and fostering projections of serotonergic axons through or next to the injury, the anti-CD11d treatment indirectly limited the excess growth rostral to the lesion. Alternatively, the reduction of inflammation rostral to the injury may have directly reduced stimuli for excess growth such as upregulated growth factors produced by inflammatory cells and glia. In the same vein, neurotrophin-induced sprouting of descending corticospinal tract projections has recently been shown to depend on immune cell activation (Chen et al., 2008). Thus reduced serotonergic fiber sprouting rostral to the spinal lesion in CD11d mAb-treated rats and reduction of their neuropathic pain (Fig. 5) may be partly due to the decreased immune cell activation that normally synergizes with neurotrophins to facilitate structural plasticity. These examples demonstrate that neuroprotective treatments may avert the negative consequences of neuroplasticity by a variety of mechanisms.
Fig. 5.
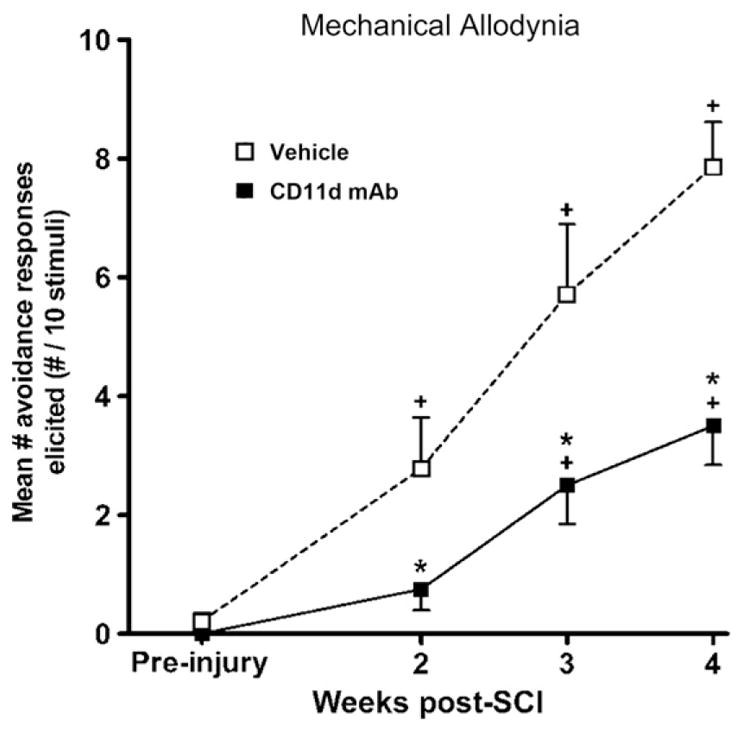
The development of mechanical allodynia at the level of a T12 spinal cord injury site. Testing sessions consisted of 10 stimulations with an innocuous, monofilament stimulus 1 week before injury and at 2, 3, and 4 weeks after injury. Each data point represents the mean±SEM number of avoidance responses made to 10 stimulations. The number of avoidances made in response to dorsal trunk stimulation was significantly reduced after anti-CD11d mAb treatment (filled squares) (n=7) when compared with vehicle-treated animals (open squares) (n=8), suggesting the reduction of at-level mechanical allodynia. *, P<0.05 compared with vehicle-treated rats; +, P<0.05 compared with mean responses before injury.
Adapted, with permission, from Oatway et al. (2005)
Summary and conclusions
In summary, neuroplasticity is at the root of both the neurological recovery and the neuropathology that develops after SCI. The number of instances where neuroplasticity leads to improved locomotor function after SCI is truly remarkable and suggests that axons stimulated to grow after injury do possess an intrinsic ability to rewire spinal circuits in a manner that produces functional recovery. The dark side of neuroplasticity is evidenced by the many cases in which the re-wiring also leads to pain, autonomic dysreflexia and other negative sequellae of SCI. The literature suggests that we might expect these negative effects of neuroplasticity under some conditions more than others. For example, treatments or conditions that directly or indirectly increase neurotrophins, such as NGF, in the spinal cord seem to be most highly correlated with the incidence of pain and autonomic dysreflexia probably because of the very strong growth-promoting effects of these molecules. On the other hand treatment strategies that focus on blocking growth inhibitors seem to be less associated with negative consequences of neuroplasticity perhaps because they have an indirect and more moderate effect on axon growth by removing impediments to neuroplasticity. Finally neuroprotective strategies may be the safest of the treatment strategies with respect to the development of negative consequences of neuroplasticity as they focus on sparing descending and ascending tracts and thus may limit the deafferentation that may act as a stimulus to promote neuroplasticity.
As the field of SCI research matures and therapeutic strategies progress from preclinical studies to clinical trials it is important to keep in mind that achieving neurological recovery by promoting neuroplasticity comes with a price. Increasing neuroplasticity to improve neurological outcomes after SCI should not be abandoned as a strategy, but we must be mindful that the stronger the drive for neuroplasticity, the greater the potential for negative consequences.
References
- Asher RA, Morgenstern DA, Moon LD, Fawcett JW. Chondroitin sulphate proteoglycans: inhibitory components of the glial scar. Prog Brain Res. 2001;132:611–619. doi: 10.1016/S0079-6123(01)32106-4. [DOI] [PubMed] [Google Scholar]
- Bao F, Chen Y, Dekaban GA, Weaver LC. An anti-CD11d integrin antibody reduces cyclooxygenase-2 expression and protein and DNA oxidation after spinal cord injury in rats. J Neurochem. 2004a;90:1194–1204. doi: 10.1111/j.1471-4159.2004.02580.x. [DOI] [PubMed] [Google Scholar]
- Bao F, Chen Y, Dekaban GA, Weaver LC. Early anti-inflammatory treatment reduces lipid peroxidation and protein nitration after spinal cord injury in rats. J Neurochem. 2004b;88:1335–1344. doi: 10.1046/j.1471-4159.2003.02240.x. [DOI] [PubMed] [Google Scholar]
- Bardin L, Lavarenne J, Eschalier A. Serotonin receptor subtypes involved in the spinal antinociceptive effect of 5-HT in rats. Pain. 2000;86:11–18. doi: 10.1016/s0304-3959(99)00307-3. [DOI] [PubMed] [Google Scholar]
- Bareyre FM, Kerschensteiner M, Raineteau O, Mettenleiter TC, Weinmann O, Schwab ME. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nat Neurosci. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- Barritt AW, Davies M, Marchand F, Hartley R, Grist J, Yip P, McMahon SB, Bradbury EJ. Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. J Neurosci. 2006;26:10856–10867. doi: 10.1523/JNEUROSCI.2980-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignami A, Asher R, Perides G. The extracellular matrix of rat spinal cord: a comparative study on the localization of hyaluronic acid, glial hyaluronate-binding protein, and chondroitin sulfate proteoglycan. Exp Neurol. 1992;117:90–93. doi: 10.1016/0014-4886(92)90115-7. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J, et al. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- Brown A, Ricci MJ, Weaver LC. NGF message and protein distribution in the injured rat spinal cord. Exp Neurol. 2004;188:115–127. doi: 10.1016/j.expneurol.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Brown A, Ricci MJ, Weaver LC. NGF mRNA is expressed in the dorsal root ganglia after spinal cord injury in the rat. Exp Neurol. 2007;205:283–286. doi: 10.1016/j.expneurol.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Bruce JC, Oatway MA, Weaver LC. Chronic pain after clip-compression injury of the rat spinal cord. Exp Neurol. 2002;178:33–48. doi: 10.1006/exnr.2002.8026. [DOI] [PubMed] [Google Scholar]
- Calejesan AA, Ch’ang MH, Zhuo M. Spinal serotonergic receptors mediate facilitation of a nociceptive reflex by subcutaneous formalin injection into the hind-paw in rats. Brain Res. 1998;798:46–54. doi: 10.1016/s0006-8993(98)00394-1. [DOI] [PubMed] [Google Scholar]
- Cameron AA, Smith GM, Randall DC, Brown DR, Rabchevsky AG. Genetic manipulation of intraspinal plasticity after spinal cord injury alters the severity of autonomic dysreflexia. J Neurosci. 2006;26:2923–2932. doi: 10.1523/JNEUROSCI.4390-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalmers JP, Kapoor V, Macrae IM, Minson JB, Pilowsky P, West MJ. New approaches to the study of bulbospinal (B3) serotonergic neurons in the control of blood pressure. J Hypertens Suppl. 1985;3:S5–S9. [PubMed] [Google Scholar]
- Chen Q, Smith GM, Shine HD. Immune activation is required for NT-3-induced axonal plasticity in chronic spinal cord injury. Exp Neurol. 2008;209:497–509. doi: 10.1016/j.expneurol.2007.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen M, Hulsebosch C. Chronic central pain after spinal cord injury. J Neurotrauma. 1997a;14:517–537. doi: 10.1089/neu.1997.14.517. [DOI] [PubMed] [Google Scholar]
- Christensen MD, Hulsebosch CE. Spinal cord injury and anti-NGF treatment results in changes in CGRP density and distribution in the dorsal horn in the rat. Exp Neurol. 1997b;147:463–475. doi: 10.1006/exnr.1997.6608. [DOI] [PubMed] [Google Scholar]
- Collins HL, Rodenbaugh DW, DiCarlo SE. Spinal cord injury alters cardiac electrophysiology and increases the susceptibility to ventricular arrhythmias. Prog Brain Res. 2006;152:275–288. doi: 10.1016/S0079-6123(05)52018-1. [DOI] [PubMed] [Google Scholar]
- Cruz-Orengo L, Figueroa JD, Velazquez I, Torrado A, Ortiz C, Hernandez C, Puig A, Segarra AC, Whittemore SR, Miranda JD. Blocking EphA4 upregulation after spinal cord injury results in enhanced chronic pain. Exp Neurol. 2006;202:421–433. doi: 10.1016/j.expneurol.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Cummings BJ, Uchida N, Tamaki SJ, Salazar DL, Hooshmand M, Summers R, Gage FH, Anderson AJ. Human neural stem cells differentiate and promote locomotor recovery in spinal cord-injured mice. Proc Natl Acad Sci U S A. 2005;102:14069–14074. doi: 10.1073/pnas.0507063102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Groat WC, Yoshimura N. Mechanisms underlying the recovery of lower urinary tract function following spinal cord injury. Prog Brain Res. 2006;152:59–84. doi: 10.1016/S0079-6123(05)52005-3. [DOI] [PubMed] [Google Scholar]
- Deumens R, Joosten EA, Waxman SG, Hains BC. Locomotor dysfunction and pain: the scylla and charybdis of fiber sprouting after spinal cord injury. Mol Neurobiol. 2008;37:52–63. doi: 10.1007/s12035-008-8016-1. [DOI] [PubMed] [Google Scholar]
- Fabes J, Anderson P, Brennan C, Bolsover S. Regeneration-enhancing effects of EphA4 blocking peptide following corticospinal tract injury in adult rat spinal cord. Eur J Neurosci. 2007;26:2496–2505. doi: 10.1111/j.1460-9568.2007.05859.x. [DOI] [PubMed] [Google Scholar]
- Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377–391. doi: 10.1016/s0361-9230(99)00072-6. [DOI] [PubMed] [Google Scholar]
- Frigon A, Rossignol S. Functional plasticity following spinal cord lesions. Prog Brain Res. 2006;157:231–260. doi: 10.1016/s0079-6123(06)57016-5. [DOI] [PubMed] [Google Scholar]
- Galtrey CM, Fawcett JW. The role of chondroitin sulfate proteoglycans in regeneration and plasticity in the central nervous system. Brain Res Rev. 2007;54:1–18. doi: 10.1016/j.brainresrev.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Haiss F, Sydekum E, Schneider R, Gullo M, Wyss MT, Mueggler T, Baltes C, Rudin M, Weber B, Schwab ME. Rewiring of hindlimb corticospinal neurons after spinal cord injury. Nat Neurosci. 2010;13:97–104. doi: 10.1038/nn.2448. [DOI] [PubMed] [Google Scholar]
- Goldberger ME, Murray M, Tessler A. Sprouting and regeneration in the spinal cord. Their roles in recovery of function after spinal cord injury. In: Gorio A, editor. Neuroregeneration. Raven Press; New York: 1993. pp. 241–264. [Google Scholar]
- Gonzenbach RR, Gasser P, Zorner B, Hochreutener E, Dietz V, Schwab ME. Nogo-A antibodies and training reduce muscle spasms in spinal cord-injured rats. Ann Neurol. 2010;68:48–57. doi: 10.1002/ana.22009. [DOI] [PubMed] [Google Scholar]
- Grill RJ, Blesch A, Tuszynski MH. Robust growth of chronically injured spinal cord axons induced by grafts of genetically modified NGF-secreting cells. Exp Neurol. 1997;148:444–452. doi: 10.1006/exnr.1997.6704. [DOI] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Oatway MA, Chen Y, Hamilton EF, Dekaban GA, Weaver LC. Transient blockade of the CD11d/CD18 integrin reduces secondary damage after spinal cord injury, improving sensory, autonomic, and motor function. J Neurosci. 2004;24:4043–4051. doi: 10.1523/JNEUROSCI.5343-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gris D, Marsh DR, Dekaban GA, Weaver LC. Comparison of effects of methylprednisolone and anti-CD11d antibody treatments on autonomic dysreflexia after spinal cord injury. Exp Neurol. 2005;194:541–549. doi: 10.1016/j.expneurol.2005.03.016. [DOI] [PubMed] [Google Scholar]
- Gris P, Tighe A, Thawer S, Hemphill A, Oatway M, Weaver L, Dekaban GA, Brown A. Gene expression profiling in anti-CD11d mAb-treated spinal cord-injured rats. J Neuroimmunol. 2009;209:104–113. doi: 10.1016/j.jneuroim.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Hains BC, Everhart AW, Fullwood SD, Hulsebosch CE. Changes in serotonin, serotonin transporter expression and serotonin denervation supersensitivity: involvement in chronic central pain after spinal hemisection in the rat. Exp Neurol. 2002;175:347–362. doi: 10.1006/exnr.2002.7892. [DOI] [PubMed] [Google Scholar]
- Hains BC, Klein JP, Saab CY, Craner MJ, Black JA, Waxman SG. Upregulation of sodium channel Nav1.3 and functional involvement in neuronal hyperexcitability associated with central neuropathic pain after spinal cord injury. J Neurosci. 2003;23:8881–8892. doi: 10.1523/JNEUROSCI.23-26-08881.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgren M, Goldberger M. The recovery of postural reflexes and locomotion following low thoracic hemisection in adult cats involves compensation by undamaged primary afferent pathways. Exp Neurol. 1993;123:17–34. doi: 10.1006/exnr.1993.1137. [DOI] [PubMed] [Google Scholar]
- Hofstetter CP, Holmstrom NA, Lilja JA, Schweinhardt P, Hao J, Spenger C, Wiesenfeld-Hallin Z, Kurpad SN, Frisen J, Olson L. Allodynia limits the usefulness of intraspinal neural stem cell grafts; directed differentiation improves outcome. Nat Neurosci. 2005;8:346–353. doi: 10.1038/nn1405. [DOI] [PubMed] [Google Scholar]
- Hou S, Duale H, Cameron AA, Abshire SM, Lyttle TS, Rabchevsky AG. Plasticity of lumbosacral propriospinal neurons is associated with the development of autonomic dysreflexia after thoracic spinal cord transection. J Comp Neurol. 2008;509:382–399. doi: 10.1002/cne.21771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Steward O. Ascending sensory, but not other long-tract axons, regenerate into the connective tissue matrix that forms at the site of a spinal cord injury in mice. J Comp Neurol. 2003;462:431–449. doi: 10.1002/cne.10768. [DOI] [PubMed] [Google Scholar]
- Jacob JE, Pniak A, Weaver LC, Brown A. Autonomic dysreflexia in a mouse model of spinal cord injury. Neuroscience. 2001;108:687–693. doi: 10.1016/s0306-4522(01)00436-5. [DOI] [PubMed] [Google Scholar]
- Jacob JE, Gris P, Fehlings MG, Weaver LC, Brown A. Autonomic dysreflexia after spinal cord transection or compression in 129Sv, C57BL, and Wallerian degeneration slow mutant mice. Exp Neurol. 2003;183:136–146. doi: 10.1016/s0014-4886(03)00161-4. [DOI] [PubMed] [Google Scholar]
- Johnson RD. Descending pathways modulating the spinal circuitry for ejaculation: effects of chronic spinal cord injury. Prog Brain Res. 2006;152:415–426. doi: 10.1016/S0079-6123(05)52028-4. [DOI] [PubMed] [Google Scholar]
- Ju G, Hokfelt T, Brodin E, Fahrenkrug J, Fischer JA, Frey P, Elde RP, Brown JC. Primary sensory neurons of the rat showing calcitonin gene-related peptide immunoreactivity and their relation to substance P-, somatostatin-, galanin-, vaso-active intestinal polypeptide- and cholecystokinin-immunoreactive ganglion cells. Cell Tissue Res. 1987;247:417–431. doi: 10.1007/BF00218323. [DOI] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Morshead CM, Fehlings MG. Delayed transplantation of adult neural precursor cells promotes remyelination and functional neurological recovery after spinal cord injury. J Neurosci. 2006;26:3377–3389. doi: 10.1523/JNEUROSCI.4184-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi-Abdolrezaee S, Eftekharpour E, Wang J, Schut D, Fehlings MG. Synergistic effects of transplanted adult neural stem/progenitor cells, chondroitinase, and growth factors promote functional repair and plasticity of the chronically injured spinal cord. J Neurosci. 2010;30:1657–1676. doi: 10.1523/JNEUROSCI.3111-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyvan-Fouladi N, Raisman G, Li Y. Functional repair of the corticospinal tract by delayed transplantation of olfactory ensheathing cells in adult rats. J Neurosci. 2003;23:9428–9434. doi: 10.1523/JNEUROSCI.23-28-09428.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krassioukov AV, Johns DG, Schramm LP. Sensitivity of sympathetically correlated spinal interneurons, renal sympathetic nerve activity, and arterial pressure to somatic and visceral stimuli after chronic spinal injury. J Neurotrauma. 2002;19:1521–1529. doi: 10.1089/089771502762300193. [DOI] [PubMed] [Google Scholar]
- Krenz N, Weaver L. Sprouting of primary afferent fibers after spinal cord transection in the rat. Neuroscience. 1998;85:443–458. doi: 10.1016/s0306-4522(97)00622-2. [DOI] [PubMed] [Google Scholar]
- Krenz NR, Weaver LC. Nerve growth factor in glia and inflammatory cells of the injured rat spinal cord. J Neurochem. 2000;74:730–739. doi: 10.1046/j.1471-4159.2000.740730.x. [DOI] [PubMed] [Google Scholar]
- Krenz NR, Meakin SO, Krassioukov AV, Weaver LC. Neutralizing intraspinal nerve growth factor blocks autonomic dysreflexia caused by spinal cord injury. J Neurosci. 1999;19:7405–7414. doi: 10.1523/JNEUROSCI.19-17-07405.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson SN, McCarthy PW, Prabhakar E. Electrophysiological properties of neurones with CGRP-like immunoreactivity in rat dorsal root ganglia. J Comp Neurol. 1996;365:355–366. doi: 10.1002/(SICI)1096-9861(19960212)365:3<355::AID-CNE2>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- Lawson SN, Crepps B, Perl ER. Calcitonin gene-related peptide immunoreactivity and afferent receptive properties of dorsal root ganglion neurones in guinea-pigs. J Physiol. 2002;540:989–1002. doi: 10.1113/jphysiol.2001.013086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemons ML, Howland DR, Anderson DK. Chondroitin sulfate proteoglycan immunoreactivity increases following spinal cord injury and transplantation. Exp Neurol. 1999;160:51–65. doi: 10.1006/exnr.1999.7184. [DOI] [PubMed] [Google Scholar]
- Liebscher T, Schnell L, Schnell D, Scholl J, Schneider R, Gullo M, Fouad K, Mir A, Rausch M, Kindler D, Hamers FP, Schwab ME. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Ann Neurol. 2005;58:706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- Lu J, Feron F, Mackay-Sim A, Waite PM. Olfactory ensheathing cells promote locomotor recovery after delayed transplantation into transected spinal cord. Brain. 2002;125:14–21. doi: 10.1093/brain/awf014. [DOI] [PubMed] [Google Scholar]
- Lu P, Jones LL, Snyder EY, Tuszynski MH. Neural stem cells constitutively secrete neurotrophic factors and promote extensive host axonal growth after spinal cord injury. Exp Neurol. 2003;181:115–129. doi: 10.1016/s0014-4886(03)00037-2. [DOI] [PubMed] [Google Scholar]
- Maier IC, Ichiyama RM, Courtine G, Schnell L, Lavrov I, Edgerton VR, Schwab ME. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain. 2009;132:1426–1440. doi: 10.1093/brain/awp085. [DOI] [PubMed] [Google Scholar]
- Marsh DR, Weaver LC. Autonomic dysreflexia, induced by noxious or innocuous stimulation, does not depend on changes in dorsal horn substance p. J Neurotrauma. 2004;21:817–828. doi: 10.1089/0897715041269605. [DOI] [PubMed] [Google Scholar]
- Marsh DR, Wong ST, Meakin SO, MacDonald JI, Hamilton EF, Weaver LC. Neutralizing intraspinal nerve growth factor with a trkA-IgG fusion protein blocks the development of autonomic dysreflexia in a clip-compression model of spinal cord injury. J Neurotrauma. 2002;19:1531–1541. doi: 10.1089/089771502762300201. [DOI] [PubMed] [Google Scholar]
- Mathias CJ. Orthostatic hypotension and paroxysmal hypertension in humans with high spinal cord injury. Prog Brain Res. 2006;152:231–243. doi: 10.1016/S0079-6123(05)52015-6. [DOI] [PubMed] [Google Scholar]
- McKeon RJ, Schreiber RC, Rudge JS, Silver J. Reduction of neurite outgrowth in a model of glial scarring following CNS injury is correlated with the expression of inhibitory molecules on reactive astrocytes. J Neurosci. 1991;11:3398–3411. doi: 10.1523/JNEUROSCI.11-11-03398.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNeill DL, Hulsebosch CE. Intraspinal sprouting of rat primary afferents after deafferentation. Neurosci Lett. 1987;81:57–62. doi: 10.1016/0304-3940(87)90340-5. [DOI] [PubMed] [Google Scholar]
- Morgenstern DA, Asher RA, Fawcett JW. Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res. 2002;137:313–332. doi: 10.1016/s0079-6123(02)37024-9. [DOI] [PubMed] [Google Scholar]
- Nelson SG, Mendell LM. Enhancement in Ia-motoneuron synaptic transmission caudal to chronic spinal cord transection. J Neurophysiol. 1979;42:642–654. doi: 10.1152/jn.1979.42.3.642. [DOI] [PubMed] [Google Scholar]
- Nout YS, Leedy GM, Beattie MS, Bresnahan JC. Alterations in eliminative and sexual reflexes after spinal cord injury: defecatory function and development of spasticity in pelvic floor musculature. Prog Brain Res. 2006;152:359–372. doi: 10.1016/S0079-6123(05)52024-7. [DOI] [PubMed] [Google Scholar]
- Oatway MA, Chen Y, Weaver LC. The 5-HT3 receptor facilitates at-level mechanical allodynia following spinal cord injury. Pain. 2004;110:259–268. doi: 10.1016/j.pain.2004.03.040. [DOI] [PubMed] [Google Scholar]
- Oatway MA, Chen Y, Bruce JC, Dekaban GA, Weaver LC. Anti-CD11d integrin antibody treatment restores normal serotonergic projections to the dorsal, intermediate, and ventral horns of the injured spinal cord. J Neurosci. 2005;25:637–647. doi: 10.1523/JNEUROSCI.3960-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J. An immunohistochemical and quantitative examination of dorsal root ganglion neuronal subpopulations. J Neurosci. 1985;5:2051–2059. doi: 10.1523/JNEUROSCI.05-08-02051.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raisman G. Olfactory ensheathing cells — another miracle cure for spinal cord injury? Nat Rev Neurosci. 2001;2:369–375. doi: 10.1038/35072576. [DOI] [PubMed] [Google Scholar]
- Ramon-Cueto A, Cordero MI, Santos-Benito FF, Avila J. Functional recovery of paraplegic rats and motor axon regeneration in their spinal cords by olfactory ensheathing glia. Neuron. 2000;25:425–435. doi: 10.1016/s0896-6273(00)80905-8. [DOI] [PubMed] [Google Scholar]
- Richter MW, Fletcher PA, Liu J, Tetzlaff W, Roskams AJ. Lamina propria and olfactory bulb ensheathing cells exhibit differential integration and migration and promote differential axon sprouting in the lesioned spinal cord. J Neurosci. 2005;25:10700–10711. doi: 10.1523/JNEUROSCI.3632-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero MI, Rangappa N, Li L, Lightfoot E, Garry MG, Smith GM. Extensive sprouting of sensory afferents and hyperalgesia induced by conditional expression of nerve growth factor in the adult spinal cord. J Neurosci. 2000;20:4435–4445. doi: 10.1523/JNEUROSCI.20-12-04435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saruhashi Y, Young W, Perkins R. The recovery of 5-HT immunoreactivity in lumbosacral spinal cord and locomotor function after thoracic hemisection. Exp Neurol. 1996;139:203–213. doi: 10.1006/exnr.1996.0094. [DOI] [PubMed] [Google Scholar]
- Saville LR, Pospisil CH, Mawhinney LA, Bao F, Simedrea FC, Peters AA, O’Connell PJ, Weaver LC, Dekaban GA. A monoclonal antibody to CD11d reduces the inflammatory infiltrate into the injured spinal cord: a potential neuroprotective treatment. J Neuroimmunol. 2004;156:42–57. doi: 10.1016/j.jneuroim.2004.07.002. [DOI] [PubMed] [Google Scholar]
- Seki S, Sasaki K, Fraser MO, Igawa Y, Nishizawa O, Chancellor MB, de Groat WC, Yoshimura N. Immunoneutralization of nerve growth factor in lumbosacral spinal cord reduces bladder hyperreflexia in spinal cord injured rats. J Urol. 2002;168:2269–2274. doi: 10.1016/S0022-5347(05)64369-8. [DOI] [PubMed] [Google Scholar]
- Seki S, Sasaki K, Igawa Y, Nishizawa O, Chancellor MB, De Groat WC, Yoshimura N. Suppression of detrusor-sphincter dyssynergia by immunoneutralization of nerve growth factor in lumbosacral spinal cord in spinal cord injured rats. J Urol. 2004;171:478–482. doi: 10.1097/01.ju.0000088340.26588.74. [DOI] [PubMed] [Google Scholar]
- Steward O, Zheng B, Tessier-Lavigne M. False resurrections: distinguishing regenerated from spared axons in the injured central nervous system. J Comp Neurol. 2003;459:1–8. doi: 10.1002/cne.10593. [DOI] [PubMed] [Google Scholar]
- Tator CH. Update on the pathophysiology and pathology of acute spinal cord injury. Brain Pathol. 1995;5:407–413. doi: 10.1111/j.1750-3639.1995.tb00619.x. [DOI] [PubMed] [Google Scholar]
- Teng YD, Lavik EB, Qu X, Park KI, Ourednik J, Zurakowski D, Langer R, Snyder EY. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc Natl Acad Sci U S A. 2002;99:3024–3029. doi: 10.1073/pnas.052678899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuszynski MH, Gabriel K, Gage FH, Suhr S, Meyer S, Rosetti A. Nerve growth factor delivery by gene transfer induces differential outgrowth of sensory, motor, and noradrenergic neurites after adult spinal cord injury. Exp Neurol. 1996;137:157–173. doi: 10.1006/exnr.1996.0016. [DOI] [PubMed] [Google Scholar]
- Weaver L, Cassam A, Krassioukov A, Llewellyn-Smith I. Changes in immunoreactivity for growth associated protein-43 suggest reorganization of synapses on spinal sympathetic neurons after cord transection. Neuroscience. 1997;81:535–551. doi: 10.1016/s0306-4522(97)00151-6. [DOI] [PubMed] [Google Scholar]
- Weaver LC, Marsh DR, Gris D, Brown A, Dekaban GA. Autonomic dysreflexia after spinal cord injury: central mechanisms and strategies for prevention. Prog Brain Res. 2006;152:245–263. doi: 10.1016/S0079-6123(05)52016-8. [DOI] [PubMed] [Google Scholar]
- Weaver L, Verghese P, Bruce J, Fehlings M, Krenz NR, Marsh D. Autonomic dysreflexia and primary afferent sprouting after clip compression injury of the rat spinal cord. J Neurotrauma. 2001;18:1107–1119. doi: 10.1089/08977150152693782. [DOI] [PubMed] [Google Scholar]
- Wong ST, Atkinson BA, Weaver LC. Confocal microscopic analysis reveals sprouting of primary afferent fibres in rat dorsal horn after spinal cord injury. Neurosci Lett. 2000;296:65–68. doi: 10.1016/s0304-3940(00)01601-3. [DOI] [PubMed] [Google Scholar]
- Young W. Secondary injury mechanisms in acute spinal cord injury. J Emerg Med. 1993;11 (Suppl 1):13–22. [PubMed] [Google Scholar]
- Zhou XF, Li HY. Roles of glial p75NTR in axonal regeneration. J Neurosci Res. 2007;85:1601–1605. doi: 10.1002/jnr.21220. [DOI] [PubMed] [Google Scholar]