

Abstract
Purpose
To report a case of Batten disease due to a previously unreported mutation in PPT1.
Methods
A nine-year old female presented with classic clinical findings of Batten Disease.
Results
Genetic testing for the mutations in the most common Batten disease gene, CLN3, was negative. Evaluation of a panel of genes known to be implicated in neuronal ceroid lipofuscinoses revealed disease causing mutations in PPT1, one of which was novel.
Conclusion
Mutations in PPT1 typically cause the infantile form of neuronal ceroid lipofuscinosis. Clinical diagnosis of the juvenile form of neuronal ceroid lipofuscinosis, Batten disease, should still be considered in cases with negative CLN3 genetic testing. Batten disease can occur due to genetic heterogeneity. Testing of other members of the neuronal ceroid lipofuscinosis gene family can lead to confirmation of the correct diagnosis.
Keywords: juvenile neuronal ceroid lipofuscinosis, Batten disease, PPT1, CLN3
CASE REPORT
A nine-year old female initially presenting in 2013 had experienced vision problems over the preceding twelve to eighteen months. She noted night-blindness. Her mother reported that she was having progressive difficulties in school. Review of systems was remarkable for recent poor balance. She had no relevant family history. Over the preceding six months her visual acuity deteriorated from 20/30 to hand motions in both eyes. Examination was notable for optic nerve pallor, a bull’s eye maculopathy, severe attenuation of the retinal arterioles and patchy atrophy of the retinal pigment epithelium throughout the mid periphery without bone spicule-like pigmentation.
Optical Coherence Tomography (OCT) revealed profound loss of the outer retinal laminar structures (Figure 1). Electroretinography demonstrated diffusely decreased sensitivities in both rod and cone responses. Electron microscopy of a skin biopsy revealed no fingerprint profiles. Genetic testing demonstrated no sequence alterations in the 15 exons and adjacent intronic regions of the CLN3 gene. Expanded testing that was obtained a month after initial presentation of genes implicated in the various forms of neuronal ceroid lipofuscinosis that included PPT1 (CLN1), TPP1 (CLN2), CLN3, CLN5, CLN6, MFSD8 (CLN7), CLN8, CTSD (CLN10), and KCTD7 (CLN14) revealed two mutations in the PPT1 gene: a missense mutation, Thr75Pro, and a novel splice site mutation, IVS2+1 G>A. The two mutations were on opposite alleles. Magnetic resonance imaging (MRI) of the brain revealed normal orbits but diffuse volume loss most prominent in the cerebellum.
Figure 1.
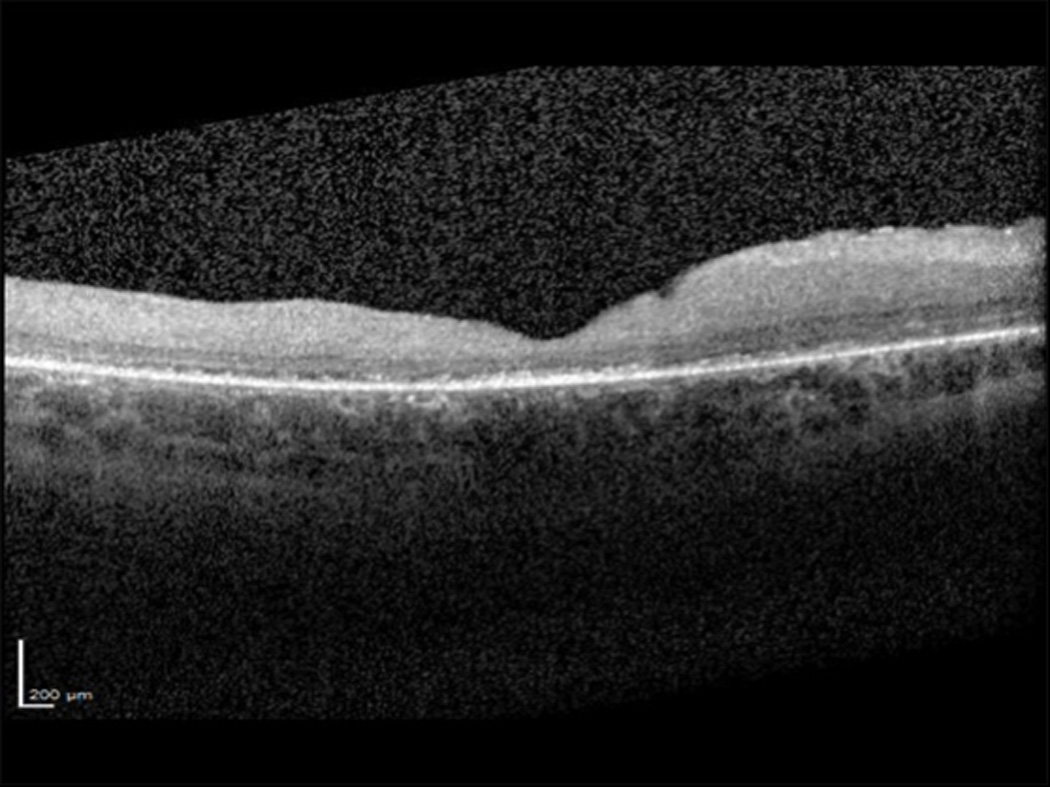
Spectral domain optical coherence tomograph (SD-OCT) of the macula of the right eye showing irregular foveal contour, epiretinal membrane, and retinal thinning with subfoveal attenuation and loss of outer retinal laminar structures that include the external limiting membrane, ellipsoid zone and interdigitation zone.
DISCUSSION
The neuronal ceroid lipofuscinoses are a large group of neurodegenerative syndromes characterized by the accumulation of autofluorescent storage material in neurons and other cell types by electron microscopy (EM) examination of tissue material [1]. Classic symptoms of neuronal ceroid lipofuscinosis include mental and motor deterioration, epilepsy, visual loss, ataxia and reduced life span [1]. Classification of NCL is based on age of onset (congenital, infantile, late infantile, juvenile and adult onset), the neurologic phenotype, ultrastructural morphology of the storage material, and, more recently, the underlying genetic defect [2]. Mutations in thirteen genes — PPT1, TPP1, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, DNAJC5, CTSF, ATP13A2, GRN, KCTD7, are known to be implicated in the pathogenesis of neuronal ceroid lipofuscinoses [3].
The juvenile form of neuronal ceroid lipofuscinosis, most commonly referred to as Batten disease, is a fatal, autosomal recessive lysosomal storage disorder of the nervous system that typically begins in childhood. Early symptoms usually appear between the ages of four and eight years, with the mean age of five. Progressive loss of vision over a period of months often is the first and only clinical sign of the disease for several years. Other early symptoms are subtle and oftentimes are overlooked or misdiagnosed. They include personality and behavioral changes, tantrums, speech disturbances, slow learning, loss of recent memory and clumsiness [2]. Severe visual impairment occurs two to four years after the onset of the initial visual problems. Over time, mental impairment becomes more profound with worsening seizures, progressive loss of motor skills, ataxia, dementia, and paralysis. Progressive neurologic decline ensues with death by the late teens or early twenties.
In the initial stages of the disease, when visual loss is the dominating symptom, these patients who usually present to eye care providers, can be misdiagnosed with Stargardt’s disease or cone-rod dystrophy. Ophthalmic examination early in the course of the disease may reveal macular changes only, such as mild retinal pigment epithelium disturbances. Subsequently a bull’s-eye type macular atrophy develops, followed by progressive alterations in the retinal pigment epithelium throughout the fundus with narrowing of retinal vessels and optic atrophy [4]. Vision loss is profound and progressive over months. Visual fields show steadily severe global constriction. Electroretinography reveals loss of photoreceptor function early [5] with a markedly reduced B:A ratio in the single flash photopic ERG with additional A-wave delay [6].
Batten’s disease is typically caused by mutations in CLN3 which includes a common 1 kb deletion but also a spectrum of point mutations. Genetic heterogeneity for Batten’s disease is known, however, with mutations in several genes described as a cause of juvenile neuronal ceroid lipofuscinosis. Our patient had two disease causing mutations in PPT1 demonstrating the genetic heterogeneity of this condition. The Thr75Pro missense substitution in the PPT1 gene has previously been reported in individuals with juvenile onset of the infantile form of neuronal ceroid lipofuscinosis [7–9]. To the best of our knowledge, the other mutation, a splice-site variant, has not been previously reported. It is predicted to cause abnormal splicing of the PPT1 gene by altering the canonical splice donor site of intron 2. This likely results in either nonsense mediated mRNA decay or an abnormal protein product if the aberrant message is translated. The Batten disease term was originally reserved only for the juvenile onset of neuronal ceroid lipofuscinosis. This case demonstrates the overlapping phenotypic spectrum of the genetic forms of neuronal ceroid lipofuscinosis. Although mutations in CLN3 may be the most common in juvenile neuronal ceroid lipofuscinosis, when this testing is negative assessment of other neuronal ceroid lipofuscinosis genes should be considered. The PPT1 gene is typically associated with infantile presentation of severe neurologic manifestations that initially include seizures, but phenotypic variability exists and some cases present later only with visual decline, more consistent with juvenile onset Batten’s disease.
It can be difficult to confirm a diagnosis of neuronal ceroid lipofuscinosis given the genetic and phenotypic heterogeneity. Tissue biopsy and electron microscopy can show reliable and characteristic findings but they may not be present in every case, so a negative biopsy cannot be used to exclude neuronal ceroid lipofuscinosis. For instance, in this patient electron microscopy of a skin biopsy was interpreted as having no ultrastructural evidence of Batten disease. Although no fingerprint profiles, which are classically seen in juvenile neuronal ceroid lipofuscinosis were evident, the biopsy did reveal “ill-defined granular material within the cytoplasm of fibroblasts and endothelial cells.” This finding is consistent with the infantile form of neuronal ceroid lipofuscinosis due to mutations in PPT1, where granular osmiophilic deposits (GROD) are a characteristic electron-microscopy pattern [10]. The frequent lack of definitive pathologic findings in these cases may add uncertainty as to the accuracy of the diagnosis. In these cases genetic testing is often a reliable way to confirm the diagnosis.
CONCLUSION
Patients with Batten disease often initially present to eye care providers with vision loss that precedes any other neurologic symptoms. Genetic testing is typically the most reliable modality for confirmation of the diagnosis. It is not unreasonable to target CLN3 as an initial step in the diagnostic algorithm. When no disease causing variations are identified in CLN3, then evaluation of other neuronal ceroid lipofuscinosis genes especially PPT1 should be considered due to the genetic heterogeneity of this condition. .
Summary Statement.
We report a case of Batten disease due to a previously unreported mutation in PPT1. Mutations in PPT1 typically cause the infantile form of neuronal ceroid lipofuscinosis. Clinical diagnosis of the juvenile form of neuronal ceroid lipofuscinosis, Batten disease, should still be considered in cases with negative CLN3 genetic testing. Batten disease can occur due to genetic heterogeneity.
Acknowledgments
The authors would like to thank Mohammed Zeina, the patient and her family. This study was supported by funding from Hope for Vision, Foundation Fighting Blindness, Search for Vision, Research to Prevent Blindness, and NEI core grant EY001792
Footnotes
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- 1.Haltia M. The neuronal ceroid-lipofuscinoses: from past to present. Biochim Biophys Acta. 2006;1762(10):850–856. doi: 10.1016/j.bbadis.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Mink JW, et al. Classification and natural history of the neuronal ceroid lipofuscinoses. J Child Neurol. 2013;28(9):1101–1105. doi: 10.1177/0883073813494268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mole SE, Williams RE, Goebel HH. Correlations between genotype, ultrastructural morphology and clinical phenotype in the neuronal ceroid lipofuscinoses. Neurogenetics. 2005;6(3):107–126. doi: 10.1007/s10048-005-0218-3. [DOI] [PubMed] [Google Scholar]
- 4.Hainsworth DP, et al. Funduscopic and angiographic appearance in the neuronal ceroid lipofuscinoses. Retina. 2009;29(5):657–668. doi: 10.1097/IAE.0b013e31819b0542. [DOI] [PubMed] [Google Scholar]
- 5.Weleber RG. The dystrophic retina in multisystem disorders: the electroretinogram in neuronal ceroid lipofuscinoses. Eye (Lond) 1998;12(Pt 3b):580–590. doi: 10.1038/eye.1998.148. [DOI] [PubMed] [Google Scholar]
- 6.Collins J, et al. Batten disease: features to facilitate early diagnosis. Br J Ophthalmol. 2006;90(9):1119–1124. doi: 10.1136/bjo.2006.091637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchison HM, et al. Mutations in the palmitoyl-protein thioesterase gene (PPT; CLN1) causing juvenile neuronal ceroid lipofuscinosis with granular osmiophilic deposits. Hum Mol Genet. 1998;7(2):291–297. doi: 10.1093/hmg/7.2.291. [DOI] [PubMed] [Google Scholar]
- 8.Kousi M, et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain. 2009;132(Pt 3):810–819. doi: 10.1093/brain/awn366. [DOI] [PubMed] [Google Scholar]
- 9.MRC Laboratory for Molecular Cell Biology, U.C.L. [cited 2015 03/16/2015];NCL Mutation and Patient Database. 2014 Available from: http://www.ucl.ac.uk/ncl/mutation.shtml.
- 10.Zeman W, Donahue S. Fine Structure of the Lipid Bodies in Juvenile Amaurotic Idiocy. Acta Neuropathol. 1963;3:144–149. doi: 10.1007/BF00687063. [DOI] [PubMed] [Google Scholar]