

Abstract
Significant barriers, such as lack of professional guidelines, specialized training for interpretation of pharmacogenomics (PGx) data, and insufficient evidence to support clinical utility, prevent preemptive PGx testing from being widely clinically implemented. The current study, as a pilot project for the Right Drug, Right Dose, Right Time–Using Genomic Data to Individualize Treatment Protocol, was designed to evaluate the impact of preemptive PGx and to optimize the workflow in the clinic setting. We used an 84-gene next-generation sequencing panel that included SLCO1B1, CYP2C19, CYP2C9, and VKORC1 together with a custom-designed CYP2D6 testing cascade to genotype the 1013 subjects in laboratories approved by the Clinical Laboratory Improvement Act. Actionable PGx variants were placed in patient's electronic medical records where integrated clinical decision support rules alert providers when a relevant medication is ordered. The fraction of this cohort carrying actionable PGx variant(s) in individual genes ranged from 30% (SLCO1B1) to 79% (CYP2D6). When considering all five genes together, 99% of the subjects carried an actionable PGx variant(s) in at least one gene. Our study provides evidence in favor of preemptive PGx testing by identifying the risk of a variant being present in the population we studied.
The dosing recommendation for a medication is typically designed and optimized from an average drug response for the general population during clinical trials. In reality, there is a large individual variation in response to medications that varies from lack of efficacy to undesirable or sometimes life-threatening adverse drug response events (ADEs). Nearly 7 million emergency department visits are related to ADEs per year nationwide, which the CDC has estimated cost the United States a total of approximately $3.5 billion annually (Adults and Older Adult Adverse Drug Events. CDC, http://www.cdc.gov/MedicationSafety/Adult_AdverseDrugEvents.html, last accessed December 24, 2015). Therefore, ADEs represent a heavy burden for the US health care system with a greatly increased incidence of ADEs in the aging population. Genetics is among the factors that contribute to the large interindividual variation in response to drugs and pharmacogenomics (PGx)—the study to identify and define the role of inheritance in such variable drug response phenotypes.1 The ultimate clinical goals of PGx are to use genomics to guide therapy, that is, to avoid ADEs, maximize drug efficacy, and select responsive patients, all of which, if achieved, will reduce the burden for both the patients and the health care system.
Evidence is increasing to support the need to implement clinical PGx testing. Currently, >100 US Food and Drug Administration-approved drugs contain PGx biomarker information in their drug labels, which includes US Food and Drug Administration-recommended actions related to the detection of these biomarkers.2 Despite these recommendations, the pace of clinical PGx implementation has been slow. Barriers to preemptive PGx genotyping adoption include the lack of professional clinical guidelines for PGx phenotyping and reporting, the ability to return results to electronic medical records (EMRs), and linking clinical decision support (CDS) tools to assist clinicians in understanding, interpreting, and using PGx information.3, 4 The slow adoption of PGx testing also arises from limited expertise among health care providers because of a lack of training in clinical pharmacology and clinical molecular genetics. Therefore, accurate interpretation and reporting of PGx variants represent challenges for clinical diagnostics laboratories implementing PGx testing. Finally, preemptive PGx testing is still subject to ongoing debate on its clinical validity and clinical utility.
The Right Drug, Right Dose, Right Time–Using Genomic Data to Individualize Treatment (RIGHT) Protocol was designed as a proof-of-concept study for clinical implementation of preemptive PGx testing, including the use of CDS tools to assist clinicians by providing information related to actionable PGx variants at the point of care.5 Subjects were selected for RIGHT Protocol according to their likelihood of needing PGx information within a 3-year window, and 1013 patients were preemptively genotyped for CYP2C19, CYP2C9, VKORC1, and SLCO1B1 with the use of an 84-gene targeted next-generation sequencing (NGS) panel. The NGS panel makes it possible to identify novel sequence alterations and to perform future studies on the clinical utility of the other 80 candidate PGx gene targets not reported to the EMR during this study. In addition, because of difficulties in accurately interrogating the highly homologous CYP2D6 locus on chromosome 22 with the use of NGS technology, CYP2D6 was genotyped with a custom genotyping method. These PGx genes have well-established drug–gene interaction information such as clopidogrel (CYP2C19); warfarin (CYP2C9 and VKORC1); simvastatin (SLCO1B1); and codeine, tramadol, and tamoxifen (CYP2D6). Clinical NGS and genotyping pipelines have been built and validated for testing, phenotyping, and reporting for these five genes, followed by placing actionable PGx variants into the EMR which trigger CDS rules at the time when a prescription is entered. In addition to optimizing the clinical PGx pipeline for practice, the current study may provide evidence to support the value of preemptive PGx genotyping.
Materials and Methods
Study Subjects
All subjects enrolled in the Mayo Clinic Biobank who were at increased risk of initiating a statin drug within the next 3 years as predicted by a Cox proportional hazards model were eligible for the RIGHT Protocol.5 The multivariate predictive model included patient demographic characteristics (age, sex, and race/ethnicity) and six Clinical Classification Software codes, including lipid concentrations, diabetes, peripheral atherosclerosis, disease of blood-forming organs, coronary atherosclerosis, hypertension, and other coronary heart disease, as risk factors. Two thousand Mayo Biobank participants were identified and invited to participate; 1013 consented and provided a blood sample (Figure 1). The design and development of the RIGHT Protocol has been described in detail previously.5 The study was reviewed and approved by the Mayo Clinic Institutional Review Board.
Figure 1.
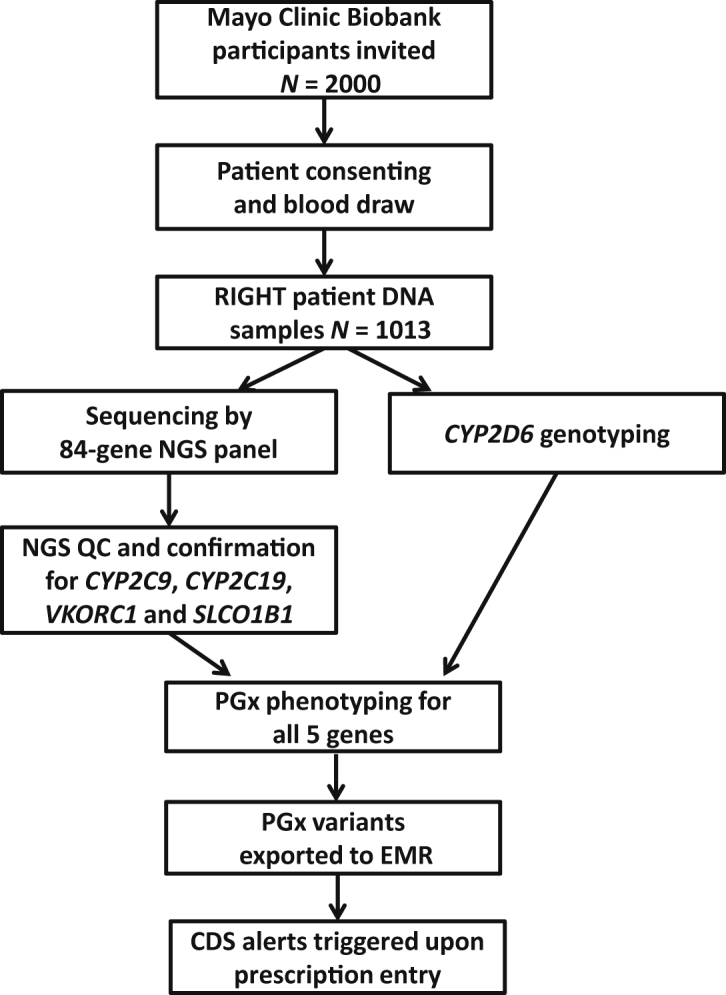
The RIGHT Protocol study design with a focus on five PGx genes. CDS, clinical decision support; EMR, electronic medical record; NGS, next-generation sequencing; PGx, pharmacogenomics; QC, quality control; RIGHT, Right Drug, Right Dose, Right Time–Using Genomic Data to Individualize Treatment.
PGx NGS and Genotyping
Preemptive sequencing and genotyping was conducted within two Clinical Laboratory Improvement Act-approved and College of American Pathologists-accredited clinical laboratories (Personalized Genomics Laboratory and Clinical Genome Sequencing Laboratory) with the use of the Mayo Clinic Department of Laboratory Medicine and Pathology's clinical validation standard operating procedures. The NGS method used the PGRN-Seq capture reagent5 designed and developed from a joint effort of the three Deep Sequencing Resources groups (University of Washington, the Genome Institute at Washington University, and the Human Genome Sequencing at Baylor College of Medicine) which are part of the nationwide Pharmacogenomics Research Network (PGRN; http://www.pgrn.org, last accessed December 24, 2015). The reagent captured 84 genes associated with PGx phenotypes, covering 968 kb of sequence that include 2 kb upstream and downstream of the coding region of these genes. KAPA HTP Library Preparation Kit (Kapa Biosystems, Inc., Wilmington, MA) and Bioo Scientific NEXTflex barcode adapters (Bioo Scientific Corporation, Austin, TX) were used for library preparation and precapture pooling, respectively. Genomic DNA samples of the 1013 RIGHT participants were randomly assigned to 14 96-well plates. One Coriell DNA sample (sample ID M04) was included in each pool of 48 samples as a positive control for comparison across the runs. The expected genotypes for this control DNA sample were as followed: CYP2C19 *2/*4, CYP2C9 *1/*2, heterozygous for VKORC1 C.1639G>A, and homozygous wild-type for SLCO1B1 c.521T>C (*5). One negative control sample with no DNA content is set up with each batch of 96 as a negative control. Two pools of 48 samples from each batch of 96 samples were loaded into two flow cells and were sequenced on the Illumina HiSeq2500 Sequencing System in the rapid run mode by using the TruSeq Rapid SBS Kit (Illumina, San Diego, CA) with the 200-cycle and 2 × 101 pair-end reads capability. A complete list of the 84 genes included on the PGRN-Seq capture panel was also reported previously.5
CYP2D6 was genotyped separately with the use of a custom-designed testing cascade, beginning with the Luminex Tag-It Mutation Detection Kit for Cytochrome P450 2D6 (CYP2D6 ASPE Kit v3) (Luminex Corporation, Austin, TX) and when needed, followed by a laboratory-developed copy number variation assay and/or sequencing assays.6 In addition, specific regions around the known CYP2C9 and CYP2C19 alleles (Table 1) were examined in the current study. Sanger sequencing was used to confirm variants in CYP2C19, CYP2C9, and VKORC1. A TaqMan Qualitative Assay (Life Technologies, Carlsbad, CA) was used to confirm SLCO1B1 variants. Variants were each confirmed up to a maximum of 10 times.
Table 1.
Predicted Enzyme Activity for CYP2C9, CYP2C19, and CYP2D6
| Predicted enzyme activity | Alleles |
||
|---|---|---|---|
| CYP2C9 | CYP2C19 | CYP2D6 | |
| Increased activity | NA | *17 | *2A |
| Normal activity | *1 | *1 | *1, *35 |
| Decreased activity | *2, *4, *5, *8, *11, *44 | *9 | *2, *9, *10, *14B, *17, *29, *41 |
| No activity or null alleles | *3 (minimum activity),*6 | *2, *3, *4, *5, *6, *7, *8, *10, *11 | *3, *4, *4N, *5, *6, *7, *8, *11, *12, *13, *14A, *15, *36, *68 |
Phenotyping was derived from the Human Cytochrome P450 (CYP) Allele Nomenclature Committee website (http://www.cypalleles.ki.se, last accessed December 24, 2015) and the PharmGKB website for the related Clinical Pharmacogenetics Implementation Consortium guidelines (https://www.pharmgkb.org/view/dosing-guidelines.do?source=CPIC, last accessed December 24, 2015). CYP2D6 *2A and *2 are as described in Black et al.6CYP2C19*11 is found in cis with the *2 variants; therefore, we have classified it as no activity or null allele.7
NA, not applicable.
The NGS bioinformatics methods enabled high-throughput variant interpretation, including an algorithm designed for PGx star (*) allele calling and delivery of clinical actionable variants to the EMR. Briefly, raw FASTQ sequence reads were generated by CASAVA (Illumina), aligned to the hg19 human reference sequence with the use of CLC Bio's Server software, version 4.1 (CLC Bio, Boston, MA), with single-nucleotide variants, small insertions and deletions called by CLC's Neighborhood Quality calling method. FASTQC was used to assess raw read quality. All results were loaded into the Mayo Clinic NGS interpretative environment, the NGS Workbench, for review of quality metrics and manual annotation of novel or ambiguous sequence alterations by laboratory technical staff and directors. The minimum targeted per-base read coverage for each region was 100 with regions containing <80 reads subject to confirmatory testing. The target variant frequency was 35% to 65% for heterozygous variants and >85% for homozygous variants. Variants with frequencies outside these ranges were subject to separate confirmatory testing. Consistency of variant calls between positive control samples that were included in each batch across all of the 14 sequencing runs was also calculated. Only variants meeting the NGS bioinformatics pipeline quality score cutoff were exported to the Oracle's Translational Research Platform database (Oracle, Redwood City, CA) and linked to the EMR for clinical use.
PGx Phenotype Derivation and Categorization
CYP2C9, CYP2C19, SLCO1B1, VKORC1, and CYP2D6 were approved for this study, for deposition into the EMR. The genotyping data were used to assign PGx phenotypes based on current understanding of the roles of these gene products in drug metabolizing and/or pharmacologic action and knowledge of the functional consequences of the identified sequence alterations. Specifically, star allele assignments and genotype-to-phenotype correlation (Table 1) for variants identified in CYP2C19, CYP2C9, and CYP2D6 were adapted from information available at the Human Cytochrome P450 (CYP) Allele Nomenclature Committee website (http://www.cypalleles.ki.se, last accessed December 24, 2015). The phenotyping algorithms were derived from the related Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines8, 9 at the Pharmacogenomics Knowledgebase (PharmGKB) website (https://www.pharmgkb.org, last accessed December 24, 2015) and are summarized in Table 2.
Table 2.
Method Used to Predict Drug Metabolizer Phenotype for CYP2C9, CYP2C19, and CYP2D6
| Predicted drug metabolizer phenotype | Actional PGx variant(s) | CYP2C9 | CYP2C19 |
CYP2D6 |
|
|---|---|---|---|---|---|
| Without gene duplication | With gene duplication | ||||
| UM | Yes | NA | Two increased activity alleles | Two increased activity alleles | Three normal and/or increased activity alleles |
| EM to UM | Yes | NA | Combination of 1 normal activity allele with 1 increased activity allele | Combination of 1 normal activity allele with 1 increased activity allele | Combination of 2 normal alleles with 1 decreased activity allele |
| EM | No | Two normal activity alleles | Two normal activity alleles | Two normal activity alleles; a combination of 1 increased activity allele with 1 decreased allele | Combination of 2 normal alleles with 1 null allele; a combination of 1 normal allele with 2 decreased activity alleles |
| IM to EM | Yes | Combination of 1 normal activity allele with 1 decreased activity allele | Combination of 1 increased activity allele with 1 null allele; a combination of 1 normal activity allele and a decreased activity allele | Combination of 1 normal activity allele with 1 decreased activity allele; a combination of 1 increased activity with 1 null allele | One increased activity allele with 2 null alleles, 3 decreased activity alleles |
| IM | Yes | Two decreased activity alleles; a combination of 1 normal activity allele with 1 null activity allele | Two decreased activity alleles; a combination of 1 normal activity allele with a null allele | One normal activity allele with 1 null activity allele; 2 decreased activity alleles | One normal allele with 2 or more null alleles, 2 decreased activity alleles with 1 null allele. |
| PM to IM | Yes | Combination of 1 decreased activity allele with 1 null allele | Combination of 1 decreased activity allele with 1 null allele | Combination of 1 decreased activity allele with 1 null allele | One decreased activity allele with 2 null alleles |
| PM | Yes | Two null alleles | Two null alleles | Two null alleles | Only null alleles detected |
See Table 1 for individual allele functions for these genes.
EM, extensive metabolizer; IM, intermediated metabolizer; NA, not applicable; PM, poor metabolizer; UM, ultrarapid metabolizer.
To calculate the total proportion of RIGHT participants carrying actionable PGx variants, we considered as normal (and consequently not actionable) only individuals with the extensive metabolizer (EM) status, based on our CYP phenotyping algorithms (Table 2). Individuals assigned as ultrarapid metabolizer (UM), intermediate metabolizer (IM), poor metabolizer (PM), or any other drug metabolizer status in between (ie, other than EM), were classified as carrying an actionable PGx variant(s). Table 2 has also included intermediate phenotype categories, for example, EM to UM, IM to EM, and PM to IM, with slight variation in definition among the three CYP enzymes.
For SLCO1B1, only the well-studied *5 allele (c.521T>C, p.Val174Ala or rs4149056) was investigated and reported. Individuals carrying both heterozygous (TC) and homozygous (CC) genotypes are considered to have an actionable PGx variant in this gene.10 For VKORC1, although the end point phenotype (ie, warfarin sensitivity) is determined by combined genotypes with the CYP2C9 *allele, the PGx actionable variants are determined according to the CPIC guideline for warfarin dosing.8 Specifically, individuals with either heterozygous GA or homozygous AA genotypes for VKORC1 rs9923231 (c.-1639G>A) are categorized as having an actionable PGx variant.
Delivery of PGx Information with EMR and CDS Rules
All sequence-verified and reportable variants for the five PGx genes interpreted in this study were transferred into Soft Biochemistry (SCC Soft Computer, Clearwater, FL) which links laboratory results to the EMR. CDS rules have been built for the five PGx genes and alerts are triggered when a clinician enters prescription(s) that encounters any of the actionable PGx variants for the individual patient. The alert contains recommendations and information for the ordering clinician to consider possible dose adjustment or to switch to alternative medication(s). The results can also be accessed directly by the patients through their Mayo Clinic online patient services accounts.
Functional and technical specifications of the PGx CDS rules were developed by a multidisciplinary team that consisted of clinical experts, clinical pharmacists and pharmacologists, informaticians, and information technology analysts. The specifications comprise all of the necessary information to develop, test, implement, and maintain the CDS rules, including knowledge translation, workflow analysis, data mapping, and log specifications. PGx alerts are integrated in the computerized physician order entry applications for inpatient and outpatient settings. The alerts are designed to deliver the necessary information and to facilitate further actions, including a link to additional related educational resources. In the design, alert fatigue is considered, and exclusion criteria are included in the rules to avoid unnecessary repetitive alerts. Transactional data associated with the alerts are collected to assess performance and clinical impact of the rules.
Results
Study Subjects
The baseline characteristics of the 1013 RIGHT patients were described in detail previously.5 Briefly, the majority of the 1013 subjects were non-Hispanic whites (86%) with slightly higher proportion of women (53%). The prescription history for these 1013 subjects showed many subjects had already been prescribed at least one medication referenced by the US Food and Drug Administration's list of PGx Biomarkers in Drug Labeling2 within the past 10 years. For example, approximately 75% of the RIGHT patients have had a prescription for tramadol and/or codeine.5
PGx NGS and Genotyping
Preemptive genotyping was completed for all 1013 participants in Clinical Laboratory Improvement Act-approved and College of American Pathologists-accredited clinical laboratories at the Mayo Clinic. For the NGS, expected genotype calls were 100% consistent between the duplicated positive control samples within runs and across the 14 sequencing batches. More than 96.3% of the samples met the minimum coverage cutoff quality control criteria. NGS on 37 samples were repeated from library preparation because of low sequencing quality (for all genes). One hundred twenty-four confirmation tests were performed on 81 samples with 54% of the confirmations because of either poor coverage or variant frequencies out of range in the regions of interest. The remaining samples (46% of the 81 samples) were subject to confirmation testing to verify variants observed by NGS.
Phenotyping and Proportion of Patients Carrying PGx Variant(s)
The percentage of patients in each metabolizer category among the total 1013 RIGHT subjects for each of the three CYP genes, CYP2D6, CYP2C9, and CYP2C19, is summarized in Figure 2, A–C. Note that 21%, 64%, and 40% of subjects are EM or normal metabolizer status for CYP2D6, CYP2C9, and CYP2C19, respectively, leaving the remainder of the population predicted to carry altered drug metabolizer status. The distribution of patients carrying different genotypes for SLCO1B1*5 and VKORC1 rs9923231 among the RIGHT participants is described in Figure 2D. When CYP2C9 and VKORC1 genotypes are considered together for warfarin sensitivity, 37% of the RIGHT participants were predicted to have altered warfarin sensitivity, which might require the adjustment of the initial dose of warfarin.9
Figure 2.

Distributions of RIGHT subjects in CYP2D6, CYP2C9, CYP2C19, SLCO1B1*5, and VKORC1 categories. Percentage of subjects among the total RIGHT cohort for each of the predicted CYP2D6 (A), CYP2C9 (B), and CYP2C19 (C) metabolizer groups; percentage of subjects among the total RIGHT cohort carrying various genotypes for SLCO1B1*5 (TT, TC, or TT) and VKORC1 SNP rs9923231 (GG, GA, and AA) (D). EM, extensive metabolizer; IM, intermediate metabolizer; PM, poor metabolizer; RIGHT, Right Drug, Right Dose, Right Time–Using Genomic Data to Individualize Treatment; UM, ultrarapid metabolizer.
The proportion of the RIGHT patients who carry actionable PGx variant(s) in the five genes studied is summarized in Figure 3. Of the 1013 RIGHT patients, 99% carry at least one actionable variant. Furthermore, 3% of participants carry actionable PGx variants in all of the five genes.
Figure 3.

Percentage of RIGHT subjects among groups carrying actionable PGx variants in zero to five of the PGx genes. PGx, pharmacogenomics; RIGHT, Right Drug, Right Dose, Right Time–Using Genomic Data to Individualize Treatment.
In addition to genomic variants linked to PGx phenotypes, multiple novel sequence alterations were also detected. There were a total of seven novel variants identified in CYP2C9, including six missense variants of uncertain significance (VUSs) and one frame shift mutation. The frame shift resulted from one base pair deletion that may cause a truncated CYP2C9 protein due to a subsequent early stop codon. There were nine VUSs detected in CYP2C19 of which seven were missense alterations and two were intronic alterations adjacent to the canonical splicing sites. VUS information was not available for SLCO1B1 and VKORC1 because sequence analysis for these two genes was targeted to a single variant position.
Six specific PGx CDS rules (drug–gene interaction) were developed and implemented in clinical practice: codeine–CYP2D6, tramadol–CYP2D6, tamoxifen–CYP2D6, simvastatin–SLCO1B1, clopidogrel–CYP2C19, and warfarin–CYP2C9/VKORC1. The alerts were designed to display specific recommendations for each drug–gene interaction. The alerts can display multiple interactions, including one gene with several drugs or several genes with several drugs. Hypothetical examples are shown for one drug-one gene (Figure 4A), two drugs-one gene (Figure 4B), and two genes-three drugs (Figure 4C). Specific PGx-CDS recommendations for each of the PGx variants tested in this study are summarized in Table 3.
Figure 4.

Examples of CYP2C19 CDS alerts triggered for the RIGHT subjects at the point of care. A: Patient predicted to carry one single actionable PGx variant-triggered alert for one medication, clopidogrel. B: Patient predicted to carry one single actionable PGx variant-triggered alerts for two medications, codeine and tramadol. C: Patient predicted to carry actionable PGx variants in two genes that triggered alerts for several relevant drugs. CDS, clinical decision support; MICS, Mayo Integrated Clinical Systems; PGx, pharmacogenomics; RIGHT, Right Drug, Right Dose, Right Time–Using Genomic Data to Individualize Treatment.
Table 3.
Specific PGx CDS Recommendations for Each of the PGx Variants Tested
| Drug–gene interaction | Phenotype | Recommendation |
|---|---|---|
| Simvastatin–SLCO1B1 | TT (normal activity) | Standard dosing |
| TC (intermediate) | Intermediate myopathy risk; reduce simvastatin dose or consider alternative therapy | |
| CC (poor) | High myopathy risk; avoid simvastatin and use alternative therapy | |
| Clopidogrel–CYP2C19 | Ultrarapid metabolizer | Standard dosing |
| Extensive to ultrarapid metabolizer | Standard dosing | |
| Extensive (normal) metabolizer | Standard dosing | |
| Intermediate to extensive metabolizer | Standard dosing | |
| Intermediate | Clopidogrel efficacy reduced; consider alternative therapy | |
| Poor to intermediate metabolizer | Clopidogrel efficacy reduced; consider alternative therapy | |
| Poor metabolizer | Clopidogrel efficacy significantly reduced; consider alternative therapy | |
| Warfarin–CYP2C9/VKORC1 | Initial warfarin dose is calculated and displayed | |
| Codeine–CYP2D6 Tramadol–CYP2D6 |
Ultrarapid metabolizer | Avoid codeine/tramadol because of potential for toxicity |
| Extensive to ultrarapid metabolizer | Increased toxicity such as respiratory depression may occur; caution is advised with codeine/tramadol | |
| Extensive metabolizer (normal) | Standard dosing | |
| Intermediate to ultrarapid metabolizer | Reduced efficacy may occur if an intermediate metabolizer or risk of toxicity may occur if an ultrarapid metabolizer; caution is advised, and alternative therapy other than codeine/tramadol might be considered | |
| Intermediate to extensive metabolizer | Standard dosing | |
| Intermediate metabolizer | Standard dosing | |
| Poor to intermediate metabolizer | Efficacy may be reduced for codeine/tramadol; other treatments should be considered | |
| Poor metabolizer | Avoid codeine/tramadol because of lack of efficacy |
CDS, clinical decision support; PGx, pharmacogenomics.
Discussion
The field of PGx has been established for more than half a century and has described several variations in individual genetic makeup that cause a change in drug response phenotypes.11 The ultimate clinical goal of PGx testing is to optimize treatment and to eliminate ADEs by treating the patient with the right drug, the right dose, at the right time. Considering the potential medical and financial benefits, preemptive PGx genotyping represents a promising component of precision medicine in the postgenomic era.
Despite barriers that affect the pace of PGx clinical implementation, significant progress has been made. Research consortia, such as PGRN, are addressing the challenges of clinical implementation through the PGRN-Translational Pharmacogenomics Program4 and developing drug–gene specific guidelines for PGx clinical implementation through the PGRN CPIC working groups and PharmGKB.8, 9 Currently, most PGx tests are ordered when a prescription is placed by a clinician (or even after the patient has been on the drug for several days) and are rarely performed preemptively. In some cases, the clinician may need to delay initiation of therapy until PGx results are returned or may go ahead and place the patient on the therapy in the absence of PGx results, which may lead to ADEs. The lack of data from large prospective clinical trials is partially responsible for the hesitance of clinical laboratories to implement preemptive PGx testing. The complexity of PGx interpretation and reporting, the clinician's engagement, and reimbursement policies from payers are also concerns.
At Mayo Clinic, an integrated local network, the PGx Task Force, consisting of leaders of the Mayo Clinic PGRN, the Mayo Clinic Center for Individualized Medicine, the Mayo Clinic Biobank, and the Personalized Genomics Laboratory was formed to initiate a preemptive PGx sequencing/genotyping project. We observed that among the 1013 RIGHT subjects tested for five well-characterized PGx genes (CYP2D6, CYP2C19, SLCO1B1, CYP2C9, and VKORC1), only 1% of subjects (10/1013) did not carry an actionable PGx variant. In other words, these 10 subjects were phenotyped as normal or EM for the three CYP genes and wild-type for the SLCO1B1*5 and VKORC1 rs9923231 variants (Figure 2). However, approximately 99% of RIGHT participants carry an actionable PGx variant(s) in at least one of the five studied genes. Although not all of the variants detected in these five genes would result in an ADE-related emergency department visit and there might be other PGx variants that exist but are outside the scope of the current study, our study provided additional evidence to support the need of preemptive PGx genotyping. Another preemptive PGx genotyping study performed by the Vanderbilt University also examined five currently implemented drug–gene interactions (clopidogrel–CYP2C19, simvastatin–SLCO1B1, warfarin–CYP2C9/VKORC1, thiopurines–TMPT, and tacrolimus–CYP3A5) and similarly showed the percentage of the study population carrying one or more actionable variants was 91%.12 The slight discrepancy between the numbers from the two preemptive PGx studies might have been due to the difference in the prevalence of actionable variants in CYP2D6 for our study, that is, approximately 79% of subjects carrying CYP2D6 variants, and less variable PGx genes (7% actionable variant carriers in TMPT and 24% for CYP3A5*1) included in the Vanderbilt study.
Although the PGRN-seq NGS panel was designed to capture the coding regions and a 2-kb sequence upstream and downstream of 84 PGx genes, subject phenotyping was only based on the genotypes of five genes with well-established PGx associations. As a result, one limitation of the current study is the possible overestimated prevalence of EM individuals predicted to have normal enzyme activity due to the lack of sequence information in other gene regions not captured and genes not analyzed on the NGS panel. Nevertheless, the use of NGS deep sequencing has made it possible for us to expand the mutation detection spectrum and to discover novel and rare sequence alterations.
Functional genomics studies are currently in progress to characterize the effect of VUSs identified in this study. The discovery-enabled capability of NGS-based PGx testing highlights one advantage over traditional single nucleotide polymorphism-based genotyping methods. As whole exome and whole genome sequencing, both of which are offered clinically, become more routine, further discovery of PGx actionable variants is likely. However, this increased discovery will come with new challenges associated with the interpretations of these novel variants identified in PGx genes.
In summary, we have demonstrated that 99% of this study population carries at least one actionable PGx variant, suggesting a high prevalence of actionable variants in the general population. Given this, preemptive PGx genotyping may benefit most individuals, with particular value in individuals taking multiple medications. As these study subjects are followed prospectively, additional data to define the utility of preemptive PGx testing will be gathered, as we track the number of CDS alerts triggered.
Acknowledgments
We thank the staff in the Personalized Genomics Laboratory and the Clinical Genome Sequencing Laboratory, Department of Laboratory Medicine and Pathology at the Mayo Clinic for their effort and contribution.
Footnotes
Supported in collaboration of the Mayo Clinic Center for Individualized Medicine, National Institutes of Health grants U19 GM61388 (The Pharmacogenomics Research Network), R01 GM28157, U01 HG005137, R01 CA138461, R01 AG034676 (The Rochester Epidemiology Project), and U01 HG06379 and U01 HG06379 Supplement (The Electronic Medical Record and Genomics or eMERGE Network).
Disclosures: J.L.B., III, holds stock and receives royalties from Oneome LLC and AssureX Health.
References
- 1.Wang L., McLeod H.L., Weinshilboum R.M. Genomics and drug response. N Engl J Med. 2011;364:1144–1153. doi: 10.1056/NEJMra1010600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.U.S. Food and Drug Administration. Table of pharmacogenomic biomarkers in drug labeling. US Department of Health and Human Services, 2015. Available at http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm (accessed December 24, 2015)
- 3.Farrugia G., Weinshilboum R.M. Challenges in implementing genomic medicine: the Mayo Clinic Center for Individualized Medicine. Clin Pharmacol Ther. 2013;94:204–206. doi: 10.1038/clpt.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shuldiner A.R., Relling M.V., Peterson J.F., Hicks J.K., Freimuth R.R., Sadee W. The Pharmacogenomics Research Network Translational Pharmacogenetics Program: overcoming challenges of real-world implementation. Clin Pharmacol Ther. 2013;94:207–210. doi: 10.1038/clpt.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bielinski S.J., Olson J.E., Pathak J., Weinshilboum R.M., Wang L., Lyke K.J., Ryu E., Targonski P.V., Van Norstrand M.D., Hathcock M.A., Takahashi P.Y., McCormick J.B., Johnson K.J., Maschke K.J., Rohrer Vitek C.R., Ellingson M.S., Wieben E.D., Farrugia G., Morrisette J.A., Kruckeberg K.J., Bruflat J.K., Peterson L.M., Blommel J.H., Skierka J.M., Ferber M.J., Black J.L., Baudhuin L.M., Klee E.W., Ross J.L., Veldhuizen T.L., Schultz C.G., Caraballo P.J., Freimuth R.R., Chute C.G., Kullo I.J. Preemptive genotyping for personalized medicine: design of the right drug, right dose, right time-using genomic data to individualize treatment protocol. Mayo Clin Proc. 2014;89:25–33. doi: 10.1016/j.mayocp.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black J.L., III, Walker D.L., O'Kane D.J., Harmandayan M. Frequency of undetected CYP2D6 hybrid genes in clinical samples: impact on phenotype prediction. Drug Metab Dispos. 2012;40:111–119. doi: 10.1124/dmd.111.040832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Skierka J.M., Black J.L., III Analysis of compound heterozygous CYP2C19 genotypes to determine cis and trans configurations. Pharmacogenomics. 2014;15:1197–1205. doi: 10.2217/pgs.14.72. [DOI] [PubMed] [Google Scholar]
- 8.Hicks J.K., Swen J.J., Thorn C.F., Sangkuhl K., Kharasch E.D., Ellingrod V.L., Skaar T.C., Muller D.J., Gaedigk A., Stingl J.C., Clinical Pharmacogenetics Implementation Consortium Clinical Pharmacogenetics Implementation Consortium guideline for CYP2D6 and CYP2C19 genotypes and dosing of tricyclic antidepressants. Clin Pharmacol Ther. 2013;93:402–408. doi: 10.1038/clpt.2013.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson J.A., Gong L., Whirl-Carrillo M., Gage B.F., Scott S.A., Stein C.M., Anderson J.L., Kimmel S.E., Lee M.T., Pirmohamed M., Wadelius M., Klein T.E., Altman R.B., Clinical Pharmacogenetics Implementation Consortium Clinical Pharmacogenetics Implementation Consortium Guidelines for CYP2C9 and VKORC1 genotypes and warfarin dosing. Clin Pharmacol Ther. 2011;90:625–629. doi: 10.1038/clpt.2011.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilke R.A., Ramsey L.B., Johnson S.G., Maxwell W.D., McLeod H.L., Voora D., Krauss R.M., Roden D.M., Feng Q., Cooper-Dehoff R.M., Gong L., Klein T.E., Wadelius M., Niemi M., Clinical Pharmacogenetics Implementation Consortium The clinical pharmacogenomics implementation consortium: CPIC guideline for SLCO1B1 and simvastatin-induced myopathy. Clin Pharmacol Ther. 2012;92:112–117. doi: 10.1038/clpt.2012.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vesell E.S., Page J.G. Genetic control of drug levels in man: antipyrine. Science. 1968;161:72–73. doi: 10.1126/science.161.3836.72. [DOI] [PubMed] [Google Scholar]
- 12.Van Driest S.L., Shi Y., Bowton E.A., Schildcrout J.S., Peterson J.F., Pulley J., Denny J.C., Roden D.M. Clinically actionable genotypes among 10,000 patients with preemptive pharmacogenomic testing. Clin Pharmacol Ther. 2014;95:423–431. doi: 10.1038/clpt.2013.229. [DOI] [PMC free article] [PubMed] [Google Scholar]