

Abstract
Background
Studies have detected mis-assemblies in genomes of the species Corynebacterium pseudotuberculosis. These new discover have been possible due to the evolution of the Next-Generation Sequencing platforms, which have provided sequencing with accuracy and reduced costs. In addition, the improving of techniques for construction of high accuracy genomic maps, for example, Whole-genome mapping (WGM) (OpGen Inc), have allow high-resolution assembly that can detect large rearrangements.
Results
In this work, we present the resequencing of Corynebacterium pseudotuberculosis strain 1002 (Cp1002). Cp1002 was the first strain of this species sequenced in Brazil, and its genome has been used as model for several studies in silico of caseous lymphadenitis disease. The sequencing was performed using the platform Ion PGM and fragment library (200 bp kit). A restriction map was constructed, using the technique of WGM with the enzyme KpnI. After the new assembly process, using WGM as scaffolder, we detected a large inversion with size bigger than one-half of genome. A specific analysis using BLAST and NR database shows that the inversion occurs between two homology RNA ribosomal regions.
Conclusion
In conclusion, the results showed by WGM could be used to detect mismatches in assemblies, providing genomic maps with high resolution and allow assemblies with more accuracy and completeness. The new assembly of C. pseudotuberculosis was deposited in GenBank under the accession no. CP012837.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-2673-7) contains supplementary material, which is available to authorized users.
Keywords: Genomics, Sequencing, Optical mapping, Mis-assembly
Background
Corynebacterium pseudotuberculosis (Cp) is a Gram-positive, pleomorphic, facultative intracellular pathogenic bacteria that belongs to the group Corynebacterium, Mycobacterium, Nocardia and Rhodococcus (CMNR) [1]. Cp can be classified into two biovars: equi and ovis. Biovar equi is characterized by its capacity to nitrate-reductase production, while the biovar ovis, cannot [2]. Genomic plasticity analysis using 15 Cp strains demonstrates that the group of strains belonging to the ovis biovar are highly similar [3]. Cp is the etiological agent of the caseous lymphadenitis (CLA) disease, that affects mainly sheep and goat causing huge economic losses by affecting meet and wool production [4, 5]. It is also capable to cause diseases in cattle and humans. However, so far there is no proper diagnosis method or effective treatment available for Cp infection.
With the advent of next-generation sequencing (NGS) platforms [6–8], so far 37 Cp genomes have been completely sequenced of which Cp1002 is the first sequenced genome [3, 9–14]. Sequencing of several new strains are ongoing in our laboratory.
Recently the Cp31 strain that was originally sequenced using the SOLiD v3 platform and mate-pair library [9], was re-sequenced using Ion PGM platform [15]. This new sequencing discovered a new ~91 Kbp fragment in the Cp31 genome that is not present in NCBI. Therefore, there are possibilities that some of the available Cp genomes in NCBI may be incomplete and warns resequencing, reassembly, and minimization or closing gaps.
Due to the presence of highly repetitive regions that code for phage sequences, transposons, plasmid, and ribosomal RNA (rRNA) [16] in genomes and lack of good assemble software, finishing of assemblies is most critical step in genome assembly process [17]. Several strategies have been used to perform the scaffold based assemble process, for example: (i) scaffolding by reference, (ii) scaffolding by mate-pair libraries, or (iii) scaffolding by optical maps.
In the reference strategy, the contigs are oriented and positioned based on similar regions in a reference genome. This is a cost effective and a totally in silico method that can be executed through scaffolding software such as CONTIGuator [18] or Mauve [19], in addition to closing gaps software, like MapRepeat [20]. However, this strategy is not able to detect large sequence modifications, e.g., large inversions detected between operons rRNA [21] or large chromosomal rearrangement [22] among others. The scaffolding by mate-pair libraries uses the distance of paired reads present in the contigs extremities to detect their orders. SSPACE [23] and GapFiller [24] like software can perform scaffolding and gap closing using paired data. The typical values for paired distances are 3 Kbp, 6 Kbp, 8 Kbp or 20 Kbp. However, if the length of the repetitive regions is bigger than the paired reads distance, the software cannot perform the scaffolding process [25].
On the other hand, whole-genome mapping (WGM), also known as optical mapping, uses images of unique DNA molecules immobilized in a polarized glass surface. The molecules are digested in situ by restriction enzymes, fragments sizes are calculated, and the high-resolution physical restriction map are used to determine the fragments order [26, 27]. Thus, optical mapping is considered one of the most accurate techniques to perform contigs scaffolding and it has been used to finishing several bacterial genomes [28]. The WGM technique uses Argus system (OpGen Inc, Gaithersburg, MD) that can be divided into four steps: (i) Extraction of chromosomal DNA, (ii) immobilization and in situ restriction digestion, (iii) image capture and measurement, and (iv) map assembly and analysis [26].
Recently, optical mapping has been largely used with success to detect genetic inversions in bacterial genomes. For example, WGM was used to detect a large genetic inversion between two Methicillin-resistant Staphylococcus aureus strains [29]. In a long-term evolution experiment, WGM was combined with genome sequencing (WGS) and PCR to analyze rearrangements in twelve Escherichia coli populations propagated in a glucose-limited environment for over 25 years [22]. In this experiment, they detected 19 inversions where three inversions found to have sizes larger than one-half of the chromosome. Thus, WGM can be considered to detect large rearrangements and mismatches in assemblies.
Corynebacterium pseudotuberculosis strain 1002
Corynebacterium pseudotuberculosis strain 1002 (Cp1002) was isolated from a Caprine caseosus in Curaça county, state of Bahia (Brazil) in 1971 [30]. Cp1002 was the first strain of this species sequenced in Brazil and its genome is used as a model for several studies of caseous lymphadenitis. Thus, this strain is considered to be representative for the ovis biovar and important for caseous lymphadenitis researches in Brazil.
The first sequencing of Cp1002 was performed using 454 Roche and Sanger that showed a circular genome with ~2.35 Mbp, G + C content of 52.2 %, 12 rRNA, 48 tRNA, 2,095 CDS, and 47 pseudogenes [13]. To finish the Cp1002 assembly, it was used the genetic order of Corynebacterium species with high similarity [13]. None experimental strategy was used to contigs scaffolding. Therefore, it is possible that mis-assemblies remained in the submitted genome of Cp1002 available in NCBI. Because of its importance in studies of caseous lymphadenitis, and after the results obtained previous studies [15], we consider Cp1002 as the candidate for a new sequencing in order to detect possible mis-assemblies.
In this work, we perform a resequencing of Cp1002 using the platform Ion PGM. We also construct a restriction map using the WGM technique (OpGen Inc, Gaithersburg, MD), and new assembly and annotation are performed. We also compared the newly obtained genome sequence with the first genome available at NCBI.
Methods
Strain and DNA isolation
Cp1002 was grown in brain-heart-infusion (BHI-HiMedia Laboratories Pvt. Ltd., India) at 37 °C under rotation. Extraction of chromosomal DNA was performed using 30 mL of 48–72 h culture of C. pseudotuberculosis, centrifuged at 4 °C and 4000 rpm for 15 minutes. Re-suspension of cell pellets was done in 600 μL Tris/EDTA/NaCl [10 mM Tris/HCl (pH 7.0), 10 mM EDTA (pH 8.0), and 300 mM NaCl], and transferred to tubes with beads for cell lysis using Precellys (2 cycles of 15 seconds at 6500 rpm with 30 seconds between them). Purification of DNA with phenol/chloroform/isoamyl alcohol (25:24:1) was followed by precipitation with ethanol/NaCl/glycogen (2.5 v, 10 % NaCl and 1 % glycogen). The DNA was re-suspended in 30 μL MilliQ water, the concentration was determined by spectrophotometer, and the DNA was visualized using 1 % agarose gel electrophoresis.
Optical mapping
First, the DNA was extracted and isolated using Argus Sample Preparation Kit and Agencourt Genfind v2 DNA Isolation Kit. The DNA was immobilized and digested in situ in a MapCard Processor using the restriction enzyme (KpnI). Thereafter, the molecules were imaged by fluorescence microscopy, and processed to detect restriction sites using the image acquisition software of Argus WGM system (OpGen Inc). Lastly, the Argus assembly software (OpGen Inc) was used to calculate a consensus of a restriction map and Argus MapSolver™ software (OpGen Inc, Gaithersburg, MD) was employed to import the DNA sequence and converted to in silico data.
Sequencing, assembly and annotation
The genome of Cp1002 was sequenced using Ion Torrent PGM System with 200 bp sequencing kit. The analysis of quality of the reads was performed using the FastQC software (http://www.bioinformatics.babraham.ac.uk/projects/fastqc) and showed a Phred value, in most cases, greater than 20. Hence, it was not applied trimming or quality steps to raw reads before assembly. The de novo assembly was performed using Mira 3.9.18 [31] applying the parameters “-GE:not = 16 IONTOR_SETTINGS -AS:mrpc = 100”. The scaffolding and gap closing were performed with SIMBA software (http://ufmg-simba.sourceforge.net) using the report generated by the software MapSolver™ (http://opgen.com/genomic-services/softwares/mapsolver) as reference to the scaffolder. The finishing of the genome was done using CLC Genomics Workbench 7.0 (Qiagen, USA) and the Website BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi). The annotation was performed using in-house scripts to fetch the annotations of a manually curated C. pseudotuberculosis genome annotation database obtained in the UniProt database (http://uniprot.org). Finally, the pseudogenes were curated manually using the Artemis software [32] and the UniProt database.
Comparing assemblies
To validate and to compare the new assembly (we called as Cp1002B) with the old genome of C. pseudotuberculosis 1002 available at NCBI (NC_017300) (we termed as Cp1002A), we performed the alignment between the experimental restriction map (obtained by WGM) of C. pseudotuberculosis 1002 with Cp1002B and with Cp1002A using MapSolver™ software (default parameters were used).
Thereafter, we used a modified version of the software CONTIGuator [18] to generate a syntenic comparison between Cp1002A and Cp1002B. For this comparison, we used the complete genome in a FASTA format for both the assemblies. Additionally, the annotation file (GenBank file) of Cp1002, the Website BLAST and NR database were used to detect repetitive regions that could be involved in possible genomic rearrangements.
Results
De novo assembly and annotation
The new assembly Cp1002B on Mira showed 9 contigs through 731,481 reads, with a N50 value of 402,955 bp and a deep coverage of ~58-fold (Table 1). The genome represents a circular chromosome of 2,335,107 bp, 52.2 % of G + C content, 12 rRNA, 48 tRNA, 2,071 CDS, and 43 pseudogenes.
Table 1.
Statistics of the C. pseudotuberculosis 1002 new assembly
| Assembler | Mira 3.9.18 |
|---|---|
| Reads assembled | 731,481 |
| Contigs | 9 |
| Shortest contig | 4,133 |
| Largest contig | 542,891 |
| N50 | 402,955 |
| N90 | 218,254 |
| N95 | 147,989 |
| Total coverage | 58.63 |
Comparison between assemblies of Cp1002
The alignment between the experimental restriction map of Cp1002 (obtained by WGM) and the in silico restriction map of Cp1002B (obtained by MapSolver™) shows that the new assembly presents a high accuracy (Fig. 1). On the other side, the alignment between the experimental restriction map of Cp1002 and the in silico restriction map of Cp1002A shows a large inversion with a size larger than one-half of the genome (Fig. 2).
Fig. 1.
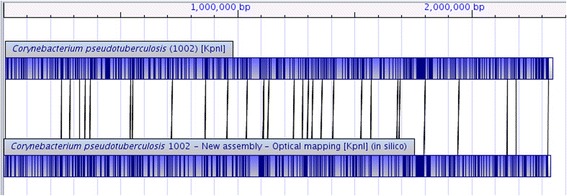
Alignment between the restriction map of C. pseudotuberculosis 1002 (above) and the in silico map of the new assembly of C. pseudotuberculosis 1002 (below). Both restriction maps were generated using the restriction enzyme KpnI. The alignment shows a high similarity between the two restriction maps, indicating a high probability of a correct assembly
Fig. 2.
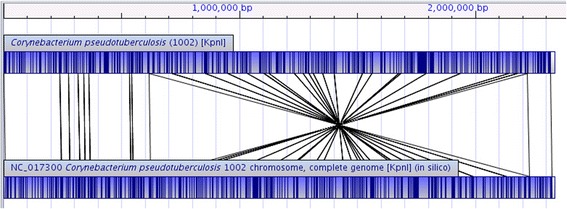
Alignment between the restriction map of C. pseudotuberculosis 1002 (above) and the in silico map of the complete genome of C. pseudotuberculosis 1002 (NC_017300) obtained from NCBI database (below). Both the restriction maps were generated using the restriction enzyme KpnI. The alignment shows a large inversion between the two restriction maps. A detailed analysis using CLC Genomics Workbench 7, BLAST and NR database shows that the inversion occurs between two rRNA regions
The syntenic comparison between Cp1002A and Cp1002B (Fig. 3) shows a genetic inversion that occurs between two regions encoding ribosomal RNA. The inversion occurs between the first rRNA operon (Fig. 3c) and the last rRNA operon (Fig. 3d), both highlighted in blue color in the figures.
Fig. 3.
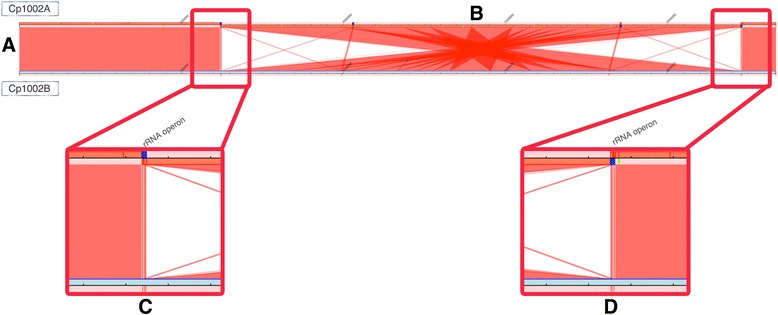
Syntenic comparison between the first assembly (Cp1002A) and the new assembly (Cp1002B). a The genome of Cp1002A is showed above, while the genome of Cp1002B in shown below. Red lines linking the line above and the line below indicate syntenic regions. The annotation of Cp1002A was used to insert color targets in the graph that detect repetitive regions: blue for rRNA operons, light blue for transposons, yellow for plasmids and green for phages. b The genomes are highly similar, except by a genetic inversion larger than 1 Mbp between two rRNA operons. c rRNA operon in the left side of the genetic inversion. It is possible to detect a change in the sense strand after the rRNA operon that indicates an inversion. d rRNA operon in the right side of the inversion sequence
Discussion
Our results showed that, in the new assembly, the number of CDS and pseudogenes are less in number as compared to the first assembly (Table 2). However, we believe that the new annotations are more accurate since bigger and improved databases are used. For instance, in Cp1002A we detected 592 CDS as hypothetical proteins, with an average length of 617 bp. However, in Cp1002B we detected 551 hypothetical proteins, with an average length of 632 bp; thus improving the annotation. In some cases, we observed that two small hypothetical proteins join to form one large hypothetical protein. The results also showed that there is only 6 bp difference between these two assembled genomes Cp1002A and Cp1002B. Although, this value can be considered insignificant, this difference can be due to the homopolymer errors undetected in the manual frameshift curation.
Table 2.
Comparison between the assemblies of C. pseudotuberculosis 1002: Cp1002A (first assembly) and Cp1002B (new assembly)
| Cp1002A | Cp1002B | |
|---|---|---|
| Genome length | 2,335,113 bp | 2,335,107 bp |
| CDS | 2,095 | 2,071 |
| Hypothetical proteins | 592 | 551 |
| Pseudogenes | 47 | 43 |
| Depth coverage | 31x | 58x |
| GC % | 52.2 % | 52.2 % |
| rRNAs | 12 | 12 |
| tRNAs | 48 | 48 |
Previously, it was predicted that the Cp1002 genome presented high similarity in genomic architecture, gene content and genetic order when compared to other Corynebacterium species [13]. Indeed, the assembly of Cp1002A was performed using reference-based assemblies techniques with short reads as well as other Cp strains [14]. The large inversion detected here is a mis-assembly caused by the limitations of the reference-based assembly strategies. Although genomes of the same specie tend to show high synteny, reference-based strategies cannot detect large inversions, as the mis-assembly detected in this work. Mis-assemblies in Cp genomes have been detected previously using mate-pair libraries [15], however it is the first time that WGM was used to correct Cp genome assemblies. The WGM technique is efficient to provide high accurate assemblies [22, 28, 29], and in this work, it was important to correct the assembly of Cp1002.
Furthermore, we detected a large inversion between two operons that encodes rRNA. The genome of Cp1002A presents a high synteny with other Cp strains [13]. However, Cp1002B shows a large inversion. Occurrences of large inversions are reported in several bacterial species [21, 22, 29]. Before the age of modern techniques for constructions of optical mapping, it was established the genome map of Salmonella paratyphi A using four endonucleases, XbaI, I-CeuI, AvrII (BlnI), and SpeI to generate fragments that could be compared [21]. They also compare the results with maps of other Salmonella species, and detect an inversion of half the genome between rRNA operons rrnH and rrnG. They postulated that the presence of this inversion is due to homologous recombination between the ribosomal genes. Another work proposed that the mechanism of producing chromosomal rearrangements is recombinational exchanges between homologous sequences, as found in ribosomal operon, similar to our observation here [33]. The large inversion detected between two rRNA operons in Cp1002 is not reported in Cp genome strains belong to ovis biovar.
Conclusions
Our new assembly (GenBank accession no. CP012837) was performed through a de novo strategy validated by experimental evidence (WGM), while the older assembly was performed by reference strategy. Thus, the new assembly corrected a large mis-assemble in Cp1002 genome that was not detected in the previous sequencing and assembly projects. Our optical mapping detected a large inversion between two rRNA operons in Corynebacterium pseudotuberculosis strain 1002. Inversion in Cp genome strains belong to ovis biovar are not reported so far but may be detected if we use WGM technique. However, the real effects of such major changes in the bacterial DNA need further evaluation.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
The genome sequence for C. pseudotuberculosis 1002 (Cp1002B) has been deposited in the GenBank database (accession no. CP012837).
The WGM dataset used to the Cp1002B sequence placements by MapSolver™ is included within the article (Additional file 1).
Acknowledgements
The authors thank the Ministério da Pesca e Aquicultura da Republica Federativa do Brasil and the funding agencies: Coordenação de Aperfeiçoamento de Pessoal de Pessoal de Nível Superior (CAPES), Fundação Amazônia de Amparo a Estudos e Pesquisas do Pará (FAPESPA), Fundação de Amparo a Pesquisa do Estado de Minas Gerais (FAPEMIG) and Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq).
Abbreviations
- CLA
caseous lymphadenitis
- CDS
coding sequence
- Cp
Corynebacterium pseudotuberculosis
- Cp1002
Corynebacterium pseudotuberculosis strain 1002
- Cp1002A
Corynebacterium pseudotuberculosis strain 1002 (first assembly)
- Cp1002B
Corynebacterium pseudotuberculosis strain 1002 (new assembly)
- Cp31
Corynebacterium pseudotuberculosis strain 31
- NCBI
National Center for Biotechnology Information
- PCR
polymerase chain reaction
- WGM
whole-genome mapping
- WGS
whole-genome sequencing
Additional file
C. pseudotuberculosis 1002 sequence placement. This XML file contains the restriction site positions used to the sequence placements by MapSolver™. (XML 88 kb)
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DCBM: wrote the manuscript; TJS, FLP, FA, DB, ACP, SSH, TDLS, AFC, CAGL, HCPF, AS, RTJR and VACA gave insights about the manuscript; FR and FAD performed the experiments; DCBM, TJS and FLP: performed bioinformatics analysis; VACA, RTJR, AS and HCPF: designed and coordinate of experiments; all authors read and approved the final manuscript.
References
- 1.Dorella FA, Carvalho Pacheco L, Oliveira SC, Miyoshi A, Azevedo V. Corynebacterium pseudotuberculosis: microbiology, biochemical properties, pathogenesis and molecular studies of virulence. Vet Res. 2006;37:201–218. doi: 10.1051/vetres:2005056. [DOI] [PubMed] [Google Scholar]
- 2.Aleman M, Spier SJ, Wilson WD, Doherr M. Corynebacterium pseudotuberculosis infection in horses: 538 cases (1982–1993) J Am Vet Med Assoc. 1996;209:804–809. [PubMed] [Google Scholar]
- 3.Soares SC, Silva A, Trost E, Blom J, Ramos R, Carneiro A, Ali A, Santos AR, Pinto AC, Diniz C, Barbosa EGV, Dorella FA, Aburjaile F, Rocha FS, Nascimento KKF, Guimarães LC, Almeida S, Hassan SS, Bakhtiar SM, Pereira UP, Abreu VAC, Schneider MPC, Miyoshi A, Tauch A, Azevedo V. The Pan-Genome of the Animal Pathogen Corynebacterium pseudotuberculosis Reveals Differences in Genome Plasticity between the Biovar ovis and equi Strains. PLoS One. 2013;8:e53818. doi: 10.1371/journal.pone.0053818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paton M, Walker S, Rose I, Watt G. Prevalence of caseous lymphadenitis and usage of caseous lymphadenitis vaccines in sheep flocks. Aust Vet J. 2003;81:91–95. doi: 10.1111/j.1751-0813.2003.tb11443.x. [DOI] [PubMed] [Google Scholar]
- 5.Williamson L. Caseous lymphadenitis in small ruminants. Vet Clin North Am Food Anim Pract. 2001;17:359–71. doi: 10.1016/S0749-0720(15)30033-5. [DOI] [PubMed] [Google Scholar]
- 6.El-Metwally S, Hamza T, Zakaria M, Helmy M. Next-Generation Sequence Assembly: Four Stages of Data Processing and Computational Challenges. PLoS Comput Biol. 2013;9:e1003345. doi: 10.1371/journal.pcbi.1003345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Harismendy O, Ng PC, Strausberg RL, Wang X, Stockwell TB, Beeson KY, Schork NJ, Murray SS, Topol EJ, Levy S, et al. Evaluation of next generation sequencing platforms for population targeted sequencing studies. Genome Biol. 2009;10:R32. doi: 10.1186/gb-2009-10-3-r32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Metzker ML. Emerging technologies in DNA sequencing. Genome Res. 2005;15:1767–1776. doi: 10.1101/gr.3770505. [DOI] [PubMed] [Google Scholar]
- 9.Silva A, Ramos RTJ, Ribeiro Carneiro A, Cybelle Pinto A, de Castro Soares S, Rodrigues Santos A, Silva Almeida S, Guimaraes LC, Figueira Aburjaile F, Vieira Barbosa EG, Alves Dorella F, Souza Rocha F, Souza Lopes T, Kawasaki R, Gomes Sa P, da Rocha Coimbra NA, Teixeira Cerdeira L, Silvanira Barbosa M, Cruz Schneider MP, Miyoshi A, Selim SAK, Moawad MS, Azevedo V. Complete Genome Sequence of Corynebacterium pseudotuberculosis Cp31, Isolated from an Egyptian Buffalo. J Bacteriol. 2012;194:6663–6664. doi: 10.1128/JB.01782-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sousa TJ, Mariano D, Parise D, Parise M, Viana MVC, Guimarães LC, Benevides LJ, Rocha F, Bagano P, Ramos R, Silva A, Figueiredo H, Almeida S, Azevedo V. Complete Genome Sequence of Corynebacterium pseudotuberculosis Strain 12C. Genome Announc. 2015;3:e00759–15. doi: 10.1128/genomeA.00759-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baraúna RA, Guimarães LC, Veras AAO, de Sá PHCG, Graças DA, Pinheiro KC, Silva ASS, Folador EL, Benevides LJ, Viana MVC, Carneiro AR, Schneider MPC, Spier SJ, Edman JM, Ramos RTJ, Azevedo V, Silva A. Genome Sequence of Corynebacterium pseudotuberculosis MB20 bv. equi Isolated from a Pectoral Abscess of an Oldenburg Horse in California. Genome Announc. 2014;2:e00977–14. doi: 10.1128/genomeA.00977-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Håvelsrud OE, Sørum H, Gaustad P. Genome Sequences of Corynebacterium pseudotuberculosis Strains 48252 (Human, Pneumonia), CS_10 (Lab Strain), Ft_2193/67 (Goat, Pus), and CCUG 27541. Genome Announc. 2014;2:e00869–14. doi: 10.1128/genomeA.00869-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz JC, D’Afonseca V, Silva A, Ali A, Pinto AC, Santos AR, Rocha AAMC, Lopes DO, Dorella FA, Pacheco LGC, Costa MP, Turk MZ, Seyffert N, Moraes PMRO, Soares SC, Almeida SS, Castro TLP, Abreu VAC, Trost E, Baumbach J, Tauch A, Schneider MPC, McCulloch J, Cerdeira LT, Ramos RTJ, Zerlotini A, Dominitini A, Resende DM, Coser EM, Oliveira LM, et al. Evidence for Reductive Genome Evolution and Lateral Acquisition of Virulence Functions in Two Corynebacterium pseudotuberculosis Strains. PLoS One. 2011;6:e18551. doi: 10.1371/journal.pone.0018551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cerdeira LT, Carneiro AR, Ramos RTJ, de Almeida SS, D’Afonseca V, Schneider MPC, Baumbach J, Tauch A, McCulloch JA, Azevedo VAC, Silva A. Rapid hybrid de novo assembly of a microbial genome using only short reads: Corynebacterium pseudotuberculosis I19 as a case study. J Microbiol Methods. 2011;86:218–223. doi: 10.1016/j.mimet.2011.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Ramos RTJ, Carneiro AR, de Castro SS, Barbosa S, Varuzza L, Orabona G, Tauch A, Azevedo V, Schneider MP, Silva A. High efficiency application of a mate-paired library from next-generation sequencing to postlight sequencing: Corynebacterium pseudotuberculosis as a case study for microbial de novo genome assembly. J Microbiol Methods. 2013;95:441–447. doi: 10.1016/j.mimet.2013.06.006. [DOI] [PubMed] [Google Scholar]
- 16.Bashir A, Klammer AA, Robins WP, Chin C-S, Webster D, Paxinos E, Hsu D, Ashby M, Wang S, Peluso P, Sebra R, Sorenson J, Bullard J, Yen J, Valdovino M, Mollova E, Luong K, Lin S, LaMay B, Joshi A, Rowe L, Frace M, Tarr CL, Turnsek M, Davis BM, Kasarskis A, Mekalanos JJ, Waldor MK, Schadt EE. A hybrid approach for the automated finishing of bacterial genomes. Nat Biotechnol. 2012;30:701–707. doi: 10.1038/nbt.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ribeiro FJ, Przybylski D, Yin S, Sharpe T, Gnerre S, Abouelleil A, Berlin AM, Montmayeur A, Shea TP, Walker BJ, Young SK, Russ C, Nusbaum C, MacCallum I, Jaffe DB. Finished bacterial genomes from shotgun sequence data. Genome Res. 2012;22:2270–2277. doi: 10.1101/gr.141515.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galardini M, Biondi EG, Bazzicalupo M, Mengoni A. CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomes. Source Code Biol Med. 2011;6. [DOI] [PMC free article] [PubMed]
- 19.Darling AC, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mariano DC, Pereira FL, Ghosh P, Barh D, Figueiredo HC, Silva A, Ramos RT, Azevedo VA. MapRepeat: an approach for effective assembly of repetitive regions in prokaryotic genomes. Bioinformation. 2015;11:276. doi: 10.6026/97320630011276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu S-L, Sanderson KE. The chromosome of Salmonella paratyphi A is inverted by recombination between rrnH and rrnG. J Bacteriol. 1995;177:6585–6592. doi: 10.1128/jb.177.22.6585-6592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Raeside C, Gaffe J, Deatherage DE, Tenaillon O, Briska AM, Ptashkin RN, Cruveiller S, Medigue C, Lenski RE, Barrick JE, Schneider D. Large Chromosomal Rearrangements during a Long-Term Evolution Experiment with Escherichia coli. mBio. 2014;5:e01377–14–e01377–14. doi: 10.1128/mBio.01377-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27:578–579. doi: 10.1093/bioinformatics/btq683. [DOI] [PubMed] [Google Scholar]
- 24.Boetzer M, Pirovano W. Toward almost closed genomes with GapFiller. Genome Biol. 2012;13:R56. doi: 10.1186/gb-2012-13-6-r56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loman NJ, Constantinidou C, Chan JZ, Halachev M, Sergeant M, Penn CW, Robinson ER, Pallen MJ. High-throughput bacterial genome sequencing: an embarrassment of choice, a world of opportunity. Nat Rev Microbiol. 2012;10:599–606. doi: 10.1038/nrmicro2850. [DOI] [PubMed] [Google Scholar]
- 26.Onmus-Leone F, Hang J, Clifford RJ, Yang Y, Riley MC, Kuschner RA, Waterman PE, Lesho EP. Enhanced De Novo assembly of high throughput pyrosequencing data using whole genome mapping. PLoS One. 2013;8:e61762. doi: 10.1371/journal.pone.0061762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neely RK, Deen J, Hofkens J. Optical mapping of DNA: single-molecule-based methods for optical mapping of D. Wiley Online Libr. 2011;95:298–311. doi: 10.1002/bip.21579. [DOI] [PubMed] [Google Scholar]
- 28.Latreille P, Norton S, Goldman BS, Henkhaus J, Miller N, Barbazuk B, Bode HB, Darby C, Du Z, Forst S, Gaudriault S, Goodner B, Goodrich-Blair H, Slater S. Optical mapping as a routine tool for bacterial genome sequence finishing. BMC Genomics. 2007;8:321. doi: 10.1186/1471-2164-8-321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shukla SK, Kislow J, Briska A, Henkhaus J, Dykes C. Optical Mapping Reveals a Large Genetic Inversion between Two Methicillin-Resistant Staphylococcus aureus Strains. J Bacteriol. 2009;191:5717–5723. doi: 10.1128/JB.00325-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Meyer R, Carminati R, Cerqueira RB, Vale V, Viegas S, Martinez T, Nascimento I, Schaer R, Silva JA, Ribeiro M, et al. Evaluation of the goats humoral immune response induced by the Corynebacterium pseudotuberculosis lyophilized live vaccine. Rev Ciênc Médicas E Biológicas. 2002;1:42–48. [Google Scholar]
- 31.Chevreux B, Wetter T, Suhai S. German conference on bioinformatics. 1999. Genome sequence assembly using trace signals and additional sequence information; pp. 45–56. [Google Scholar]
- 32.Carver T, Harris SR, Berriman M, Parkhill J, McQuillan JA. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics. 2012;28:464–469. doi: 10.1093/bioinformatics/btr703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson P, Roth J. Spontaneous tandem genetic duplications in Salmonella typhimurium arise by unequal recombination between rRNA (rrn) cistrons. Proc Natl Acad Sci. 1981;78:3113–3117. doi: 10.1073/pnas.78.5.3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The genome sequence for C. pseudotuberculosis 1002 (Cp1002B) has been deposited in the GenBank database (accession no. CP012837).
The WGM dataset used to the Cp1002B sequence placements by MapSolver™ is included within the article (Additional file 1).