

Abstract
Choroidal neovascularization (CNV) occurs in a variety of chorioretinal diseases including age-related macular degeneration (AMD), and is the major cause of severe visual loss in patients with AMD. Oxidative stress has been thought to play an important role in the development of CNV. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase is one of the major intracellular sources of reactive oxygen species (ROS) in the vascular system. In this study, we examined the expression of p22phox, an integral subunit in the NADPH oxidase complex, in the mouse eye. We determined that p22phox is expressed in the retinal pigment epithelial (RPE) cells and inner retinal neurons. A small-interfering RNA (siRNA) designed against p22phox efficiently reduced the expression of the protein in the eye when delivered by means of recombinant adeno-associated virus (AAV) vector. Vector treatment inhibited CNV in the mouse when delivered into the subretinal space where RPE cells were transduced. These results suggest that NADPH oxidase–mediated ROS production in RPE cells may play an important role in the pathogenesis of neovascular AMD, and that this pathway may represent a new target for therapeutic intervention in AMD.
INTRODUCTION
Age-related macular degeneration (AMD) is the most common form of irreversible vision loss among the elderly in industrialized countries.1–3 The pathogenesis of AMD is complex, involving a variety of genetic and environmental factors. An increasing number of experimental and clinical findings indicate that pathogenic oxidative mechanisms contribute to the progression of AMD.4 The retina is particularly vulnerable to oxidative damage because it is associated with a greater oxygen consumption than any other tissue and is exposed to lifelong and accumulative exposure to light (thereby being prone to photo-oxidative damage); also, the retina is rich in polyunsaturated fatty acids and photosensitizers, and similar products are present in the adjacent retinal pigment epithelium (RPE).4 In addition, phagocytosis of photoreceptor outer segments by the RPE cells, a critical process in visual function, also results in the generation of reactive oxygen species (ROS).5–7 Despite the strong indirect evidence that oxidative damage contributes to AMD, direct evidence is lacking, and the molecular and cellular mechanisms for various functions of ROS in normal and pathological conditions leading to AMD are not well understood.
ROS, including free radicals such as superoxide anion (O2−), nitric oxide (NO−), hydroxyl (OH−), and oxidants such as H2O2,8 can be generated physiologically as byproducts of other biological reactions such as those occurring in mitochondria, peroxisomes, and the endoplasmic reticulum.9–11 The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex, however, is the only one that results in ROS generation not merely as a byproduct, but rather as the primary function of the enzyme system,12,13 and has been shown to be one of the main intracellular ROS sources in the vascular system.14,15 The active NADPH oxidase complex consists of two membrane-bound catalytic subunits, p22phox and gp91phox, and cytoplasmic proteins p40phox, p47phox, and p67phox that produce superoxide upon assembly of the active complex.16,17 NADPH oxidase was originally identified as a key component of human innate host defense. It is now well established, however, that NADPH oxidase and related enzymes are also present in many nonphagocytotic cells and tissues. Six additional homologs of the prototype gp91phox (also known as NOX2), have been identified in various tissues and cell types.18 The p22phox subunit forms heterodimers with various NOX enzymes and with the cytoplasmic components to form stable complexes that participate in many other important cellular processes including signal transduction and cell proliferation and apoptosis, and contribute to a multitude of physiological events.18
Understanding the role of oxidative stress in AMD pathogenesis is critically important for developing effective early therapeutic interventions. Although NADPH oxidase activity has been detected in human RPE cells in culture,5,6 the expression of its components and its potential role in AMD have not been investigated. In this study, we examined the expression of the p22phox subunit, an integral component of the NADPH oxidase multicomponent enzyme complex, in mouse eye. We show that the p22phox is normally expressed in RPE cells as well as in retinal neurons. A sequence-specific small-interfering RNA (siRNA) against p22phox efficiently reduced its expression in the mouse eye when delivered as a small hairpin RNA under the control of a H1 promoter by means of a recombinant adeno-associated virus (AAV). This AAV-siRNAp22phox inhibited choroidal neovascularization (CNV) in the rodent model. These results suggest that NADPH oxidase–mediated ROS production by RPE cells may play an important role in promoting the pathogenesis of AMD, and that this pathway may therefore represent a new target for therapeutic intervention in AMD.
RESULTS
Expression of p22phox in the rodent eye
NADPH oxidase is produced in phagocytic cells including those of the RPE, a monolayer of cells whose functions include turnover of rod photoreceptor outer segments. However, this enzyme has not been precisely localized in the neural retina. In order to detect the expression of p22phox protein in the mouse retina, we dissected eyes from C57BL/6J mice and separated the neural retina from the RPE choroid complex. Western blotting using a polyclonal antibody against p22phox detected a protein of relative molecular weight 22 kd in both retinal and RPE cell extracts (Figure 1). In neutrophils active p22phox is phosphorylated,19 and the more slowly migrating protein bands evident in the blot may represent phosphoproteins or/and dimers. These results indicate that NADPH is expressed both in the RPE and in the neural cells of the retina.
Figure 1. Western blotting analysis of p22phox expression in mouse eye.
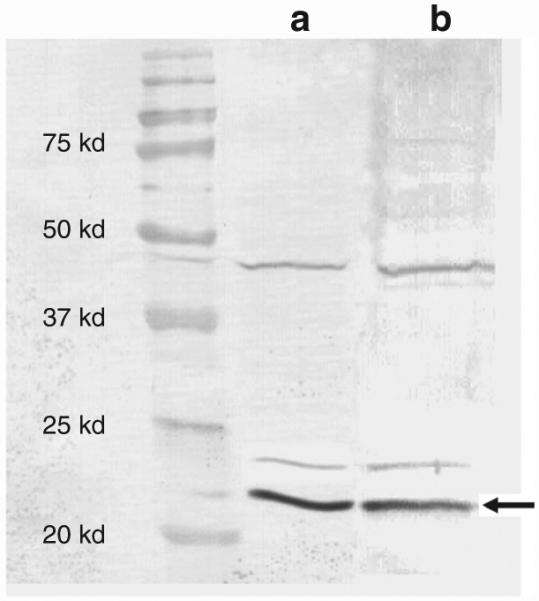
p22phox expression was detected using western blotting in (a) retinal as well as (b) retinal pigment epithelial (RPE) extracts. Protein extracts from retina and RPE/choroid were isolated by sonication, separated on 12% polyacrylamide gels, and transferred to a nitrocellulose membrane, which was then treated with a polyclonal antibody to p22phox. Antibody antigen complexes were detected using alkaline phosphatase-conjugated anti-rabbit immunoglobulin G antibody, and the filter was developed using NBT/BCIP.
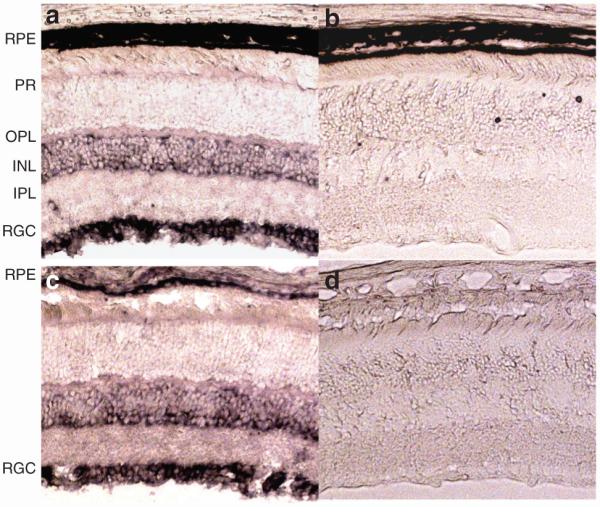
In order to make a more detailed examination of the cell types that express p22phox, we carried out indirect immunofluorescence staining of retinal tissue sections using the same antibody. Positive staining was detected in the retinal ganglion cell (RGC) and outer plexiform layer, a dense network of synapses between photoreceptor cells and the dendrites of secondary neurons (Figure 2a). The major part of the signal of p22phox staining co-localizes with the staining by an antibody against the NF-H, a neuronal marker (Figure 2b and c), suggesting that RGCs are the major site of p22phox expression in the retina. Fluorescent staining was also observed in the RPE of pigmented (C57 Bl/6) mice, but may have been quenched somewhat by the presence of pigment granules (Figure 2a). Staining was clearly evident in albino mouse eye (Figure 2d).
Figure 2. p22phox expression in the retina detected by immunofluorescence.

Cross-sections of pigmented C57BL/6J mouse eye double-labeled with antibody against p22phox (red) and HF-H (green). (a) p22phox expression in retina of the pigmented C57BL/6J mouse (red). (b) The same field stained with NF-H antibody (green). (c) merged image of a and b. (d) Cross-sections of an albino mouse eye. p22phox expression in the albino mouse eye showing clear expression in RPE cells in addition to the RGC and the IPL. RPE, retinal pigment epithelium; PR, photoreceptors; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; RGC, retinal ganglion cells.
In order to establish that p22phox is synthesized in the retinal cells in which we detected its accumulation, we employed in situ hybridization using an antisense probe to the mRNA. Hybridization confirmed p22phox expression in RGC and other neurons (Figure 3), as well as in RPE cells. Again, because of the background from melanin, hybridization was easier to detect in the eyes of albino mouse (Figure 3c).
Figure 3. p22phox mRNA expression in the retina detected by in situ hybridization.
(a) and (b) Cross-sections of the pigmented C57BL/6J mouse eye. (c) and (d) cross-sections of the albino mouse eye. (a,c) Antisense probe against p22phox complementary DNA. (b,d) Sense probe to p22phox (negative controls). In situ signal from p22phox mRNA was intense in RGCs, but seen in outer nuclear layer. Signal in the RPE is most evident in the albino mouse eye (c). RPE, retinal pigment epithelium; PR, photoreceptors; OPL, outer plexiform layer; INL, inner nuclear layer; IPL, inner plexiform layer; RGC, retinal ganglion cells.
Ocular injection of siRNA-p22phox-AAV leads to reduced p22phox protein levels
In order to investigate the role of p22phox in the eye, we designed a synthetic complementary DNA encoding siRNA against the p22phox gene and cloned it into an AAV2 vector under the control of the H1 promoter (Figure 4a). A GFP reporter gene under the control of the Chicken β-actin promoter was cloned upstream of the siRNA expression cassette in order to directly visualize expression from the vector after delivery. Vector was injected into the subretinal space of mouse eyes, and p22phox protein levels were analyzed on western blots 4–6 weeks after the injection. Protein extracts from this analysis were isolated from whole eyecups with the lens and cornea removed. As shown in Figure 4, the level of p22phox protein was reduced by >80% in eyes that received injections of AAV2-p22phox-siRNA.
Figure 4. Effects of subretinal AAV2-CBA-p22phox-siRNA vector.

(a) Map of the adeno-associated virus (AAV) vectors used. Green florescent protein (GFP) reporter gene is under the control of a chicken-β-actin promoter with a CMV enhancer (CBA), and the siRNA (either p22phox-specific or scrambled sequence) is under the control of the H1 promoter. (b) Western blots of retinal protein using antibody against p22phox. Protein was analyzed from three pairs of eyes, in which right eyes received AAV2-CBA-p22phox-siRNA while left eyes were untreated to show that p22phox protein is reduced in eyes receiving the vector. (c) The same blot as in a probed with an antibody against α-tubulin to normalize for sample loading in each lane. (d) Quantitative analysis of western blot data from extracts treated with siRNA-p22phox (n = 7). The protein levels were normalized to those of α-tubulin and the densitometric value from each of the seven siRNA-treated eyes was expressed as a percentage relative to the corresponding untreated contralateral controls (set at 100%). The P value was calculated based on Wilcoxon signed-rank test. siRNA, small-interfering RNA; SV40 pA, simian virus 40 poly A; TR, terminal repeat.
Subretinal injection of AAV2-p22phox-siRNA leads to reduced CNV in the mouse model
In order to investigate the role of p22phox in CNV (the major late complication of AMD) the AAV2-p22phox-siRNA and a control vector containing a scrambled siRNA under the control of the same promoter were tested in a murine model of CNV. In this model, the Bruch’s membrane, separating the RPE layer from the choroid layer, is disrupted at discrete sites using a laser. The proliferation of blood vessels from the wound, detected by fluorescein angiography, is taken as a measure of CNV. In our experiment, the right eyes of the animals were injected with AAV expressing the active or the scrambled siRNA, while the left eyes were uninjected. Both eyes were treated with the laser 4–6 weeks later, and CNV was evaluated 2 weeks thereafter. Representative CNV images from treated and untreated eyes are shown in Figure 6a–g, in which the extent of CNV is estimated by measuring the diameter of the vasculature emanating from the laser lesions (Figure 5).
Figure 6. Choroidal neovascularization (CNV) area measurements in treated and untreated eyes expressed as μm2.

SD is shown on each bar graph. IV, intravitreous injection; SR, subretinal injection. Ctrl, control (scrambled siRNA). The development of CNV in retinal whole mounts was quantitatively evaluated by measuring the vascular area at each laser lesion using the Zeiss AxioVision Software measurement tool. siRNA, small-interfering RNA.
Figure 5. siRNA-p22phox adeno-associated virus (AAV) treatment reduced choroidal neovascularization (CNV) lesions.

(a–f) Representative choroidal flatmounts and (g–l) cross-sections of (a–c, and g–i) untreated eyes and (d–f and j–l) eyes that received subretinal injections of siRNA-p22phox-AAV2. Retinal and choroidal vessels are labeled by CD31 antibody, an endothelial cell-specific marker (red). siRNA, small-interfering RNA.
CNV was inhibited in the eyes that had received subretinal injection of the siRNA-p22phox vector, but not in the eyes that had received intravitreal injection of the same vector, nor in eyes injected with vector expressing the scrambled siRNA, through either subretinal or intravitreal injection (Figure 6). Intravitreal injection with AAV2 leads to robust transduction of RGCs but does not lead to gene transfer to the outer layers of the retina or to the RPE.20 The development of CNV in animals that had received subretinal injections of AAV2-p22phox-siRNA was also evaluated by immunofluorescence staining of retinal sections through areas of CNV, using an antibody specific to CD31, an endothelial cell-specific marker. As shown in Figure 5, treated eyes showed much reduced vasculature density (reduced positive staining for CD31) at the site of CNV as compared to untreated eyes (Figure 5).
In a separate experiment, we also tested the same vector packaged into serotype 5 AAV capsids (AAV5-p22phox-siRNA). Serotype 5 vectors are associated with a faster onset of transgene expression and better tropism for RPE cells than serotype 2 vectors are (data not shown). Unlike in the original experiment, both eyes of test animals were laser-treated first, and the vector was subretinally injected into one of the eyes of each animal 24 hours thereafter. The development of CNV was evaluated again 2 weeks after laser treatment (13 days after subretinal injection). Again, the treated eyes showed significant reduction in CNV as compared to untreated eyes (Figure 6).
Subretinal injection of siRNA-p22phox leads to a decrease in the VEGF level
Since NADPH oxidase activation is known to stimulate angiogenesis by triggering vascular endothelial growth factor (VEGF), we measured VEGF levels in eyes that received either intravitreal or subretinal injections of siRNA-p22phox-AAV. Eyes in which p22phox was suppressed had significantly reduced levels of VEGF than the control eyes, whereas intravitreal injections (which did not suppress p22phox in the RPE) had no effect on the level of VEGF (Figure 7).
Figure 7. Enzyme-linked immunosorbent assay of vascular endothelial growth factor (VEGF) levels in eyes that received ocular administration of siRNA-p22phox-AAV2 through different routes.
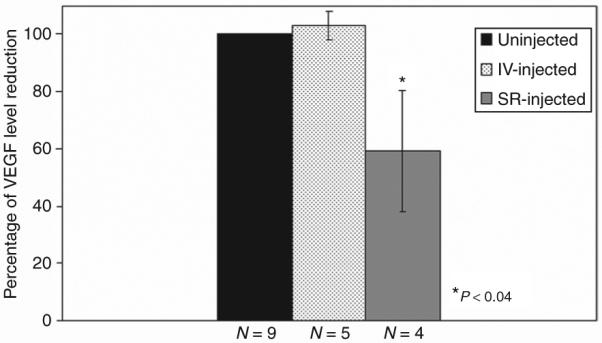
The average levels in uninjected eyes are normalized to a percentage. IV, intravitreal injection; SR, subretinal injection.
In order to further evaluate the oxidative status after downregulation of p22phox, we also carried out western blot analysis to determine the levels of 4-Hydroxy-2-nonenal and nitrotyrosine-modified proteins (markers of oxidative stress)21,22 in eyes receiving either intravitreal or subretinal injections of siRNA-p22phox-AAV. These indicators of long-term oxidative stress were similar after ocular injection of siRNA-p22phox through either of the routes (data not shown). This result suggests that the reduction in CNV in response to suppression of NADPH oxidase is not a consequence of changes in long-term oxidative burden, but may reflect short-term oxidative signaling from the RPE to the vasculature.
DISCUSSION
Accumulating evidence suggests that NAPDH oxidase plays an important role in redox-signaling pathways and contributes to age-related increases in oxidative stress in many vascular and neural degenerative diseases.18 In this study, we sought to characterize the expression of the small, membrane-bound catalytic subunit of NADPH oxidase, p22phox, in the retina, and to test what effect its downregulation by AAV-mediated delivery of siRNA against p22phox has on the development of laser-induced CNV in the mouse. We demonstrate that p22phox is expressed in the RPE cells and in inner retinal neurons, and that its downregulation in RPE cells in vivo efficiently inhibits the development of CNV, thereby suggesting that NADPH oxidase–derived ROS contributes to CNV development.
The RPE is a monolayer of pigmented cells that separates the neural retina from blood vessels of the choriocapillaris. This cell layer forms a part of the blood–retina barrier23,24 and provides multiple functions that are essential for normal visual functions, including nourishing the adjacent photoreceptor cell layer and removing their outer segments that are being continuously shed.23,24 Dysfunction of the RPE, mediated by ROS, has been suggested as a contributing factor in age-related macular degeneration.4 Although there are many other intracellular sources of ROS, NADPH oxidase is the only system that is “dedicated” to generating superoxide in a regulated manner, and participates in a variety of redox-sensing cellular functions ranging from cell growth/survival and death, to immune cell signaling. The importance of these functions of NADPH oxidase is also supported by the evolutionary conservation of members of NOX family in all multicellular organisms.25 The constitutive expression of the enzymatic p22phox subunit in RPE cells, as reported in this study, suggests that this NADPH oxidase complex also plays an important role in many cellular events that are dependent on redox-sensing in the RPE.
It has been well documented that, although low-level ROS production by NADPH oxidase is essential for cellular functions, overproduction of ROS through NADPH oxidase under pathophysiologic conditions contributes to many pathological vascular complications and neural degenerative diseases.18 NADPH oxidase activity in nonphagocytic cells is acutely increased by diverse pathophysiological stimuli including: (i) growth factors such as VEGF, thrombin, and insulin; (ii) G-protein-coupled receptor agonists such as angiotensin II and endothelin-1; (iii) cytokines such as tumor necrosis factor-α and transforming growth factor β; (iv) “metabolic” factors such as oxidized low-density lipoprotein, nonesterified (“free”) fatty acids, and glycated proteins; and (v) hypoxia–reoxygenation or ischemia–reperfusion.26,27 These conditions may also stimulate NADPH oxidase–derived ROS production in RPE cells and contribute to the pathogenesis of ocular diseases involving RPE.
Having shown here that downregulation of p22phox expression by siRNA in RPE cells reduces the development of CNV, it is worth considering possible mechanisms. It is well established that VEGF stimulates ROS production through NADPH oxidase, and the resultant ROS stimulates diverse redox signaling pathways leading to angiogenesis-related gene induction as well as to endothelial cell migration and proliferation, both being proangiogenic stimuli in vivo.28,29 However, it has also been established that an increase in ROS, particularly H2O2, stimulates production of VEGF through hypoxia-inducible factor-1-mediated transcriptional control.29 We have shown here that downregulation of p22phox significantly reduces the level of VEGF. It is known that VEGF is constitutively secreted from the basal side of RPE cells to maintain the normal choriocapillaris. Increased VEGF levels have been observed in the RPE overlying the macula, both in AMD patients30 and in animal models of CNV.31,32 It is also possible that alternative, nonmutually exclusive mechanisms are involved in the inhibition of CNV reported here. NADPH oxidase is also upregulated by several other growth factors and cytokines, many of which are secreted by RPE cells to maintain normalcy in retina and choriocapillaris, and by the immune-suppressive environment that protects the retina from acute inflammatory responses (reviewed in ref. 33). Although they are not yet well understood, several other mechanisms have been suggested in CNV development, including local hypoxia, wound healing processes, and inflammatory processes,34–36 and NADPH oxidase–derived ROS production has been implicated in these mechanisms in other tissues (see recent review ref. 18, and references therein). It appears, therefore, that NADPH oxidase–derived ROS is at the focus of multiple signaling pathways and cellular events that are redox dependent, and mechanisms to inhibit its activity provide promising targets for AMD therapy.
We also report p22phox expression in the RGCs and in a subset of other inner retinal neurons. This may have important implications for the treatment of retinal degenerative disorders involving ganglion cell loss, such as glaucoma. It has been shown that ischemia/reperfusion induces elevated production of ROS and results in neurotoxicity and neurodegeneration both in animal models and in humans. In the central nervous system, components of NADPH oxidase are present in many cell types including microglia, astrocytes, oligodendrocytes, and neurons (reviewed in refs. 18,37). Although NADPH oxidase–derived ROS do function as signaling molecules in normal tissue,38,39 ROS overproduction by NOX enzymes has been implicated in a variety of diseases of the central nervous system, including ischemic stroke, Alzheimer’s disease and Parkinson’s disease (reviewed in refs. 18,37). It is therefore conceivable that NADPH oxidase–derived ROS may play an equivalent role in retinal neuron pathology; e.g., under physiological stress such as increased intraocular pressure or ischemia/reperfusion, retinal neurons may experience oxidative damage–induced cell death mediated by NADPH oxidase. Further information on the specific mechanisms of the activation and regulation of NADPH should provide insights into designing effective general therapies to combat a range of vision-threatening diseases including glaucoma, optic neuritis, and retinitis pigmentosa.
Finally, endothelial NADPH oxidase is a major source of superoxide in blood vessels and has been implicated in the oxidative stress that accompanies vascular diseases, including atherosclerosis, hypertension, diabetic retinopathy, and heart failure.40,41 All components of phagocytic NADPH oxidase, including p22phox, gp91phox, p47phox, p67phox, and the small G-protein Rac1 are expressed in the cellular components of vascular systems (endothelial cells, vascular smooth muscle cells, adventitial and cardiac fibroblasts, and cardiomyocytes).18 Interestingly, we did not detect p22phox expression in retinal vessels either by immunolabeling or in situ hybridization. This discrepancy in the well documented expression of p22phox in the vascular system may be attributable to limitations in detection capacity inherent in the methods used in this study. It is likely that p22phox is expressed at low levels in the retinal vessels but is elevated in response to pathological conditions such as hyperglycemia. In fact, an NADPH oxidase–like enzyme was found to be expressed in retinal pericytes42,43 and was shown to be upregulated in response to angiotensin II, high glucose,43 or the saturated free-fatty acid palmitate.42 NADPH oxidase activity also shows an increase in the retinas of diabetic rats44 and in ischemic retinopathy.45,46 That is, low vascular expression levels coupled with the paucity of vascular cells available for antibody reaction in retinal sections may probably explain our inability to detect p22phox in normal retinal tissue.
In summary, our findings that the p22phox, an indispensable component of NADPH oxidase complex, is expressed abundantly in the inner retinal neurons and RPE suggest that NADPH oxidase–mediated ROS generation may play an important role in normal retinal function. Given that NADPH oxidase–derived ROS participate in diverse signaling pathways that are critical to many important biological processes, and the likely involvement of ROS in aging, vascular pathophysiology, and neurode-generative diseases, it is likely that this ROS-production pathway contributes to many degenerative ocular diseases such as glaucoma, diabetic retinopathy, and AMD. As shown in this study, downregulation of p22phox by AAV-mediated delivery of siRNA dramatically inhibits the development of CNV, suggesting a direct role for NADPH oxidase in the progression to pathogenic CNV. Therefore, targeting this pathway presents a new opportunity for therapeutic intervention against the CNV in patients with the exudative form of AMD. Since oxidative damage may also contribute to the pathogenesis of the atrophic form of AMD,47 the targeting of this pathway may also provide a novel strategy for treating nonexudative AMD as well. It should be pointed out that deficiency in phagocytotic NADPH oxidase complex results in chronic granulomatous disease, a rare genetic disorder of immunodeficiency disease characterized by the inability of phagocytes to kill micro-organisms by means of the production of ROS.48 Chorioretinal lesions have also been reported in patients with CDG.49,50 It is important, therefore, to investigate the long-term effects of suppressing p22phox (or other subunits of the NADPH oxidase complex) in the retina and RPE in order to design therapy targeting this pathway.
MATERIALS AND METHODS
Detection of p22phox protein by western blotting
Protein extracts from retina and RPE/choroid were isolated by sonication in the lysis buffer [Tris-buffered saline (0.05 mol/l Tris, 0.15 mol/l NaCl), 0.1 mmol/l EGTA, 1 mmol/l dithiothreitol, 1% Triton X-100, 0.3 mol/l sucrose, protease inhibitor cocktail], followed by centrifugation. Proteins were then separated by electrophoresis on a 12% sodium sodium dodecyl sulfate gel and transferred onto a nitrocellulose membrane. Nonspecific binding to the primary antibody was blocked with 5% dry milk in Tris-buffered saline for 1 hour at room temperature. The membrane was then incubated in the blocking buffer on a shaker at 4 °C overnight with the polyclonal anti-p22phox antibody (Santa Cruz Biotechnology, Santa Cruz, CA) diluted to 1:500. The membrane was washed in Tris-buffered saline and incubated with the alkaline phosphatase–conjugated anti-rabbit immunoglobulin G antibody, diluted at 1:2,000, for 2 hours at room temperature. The signals were detected using nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate, toluidine salt. Prestained protein standards were used (Precision Plus Protein Kaleidoscope standards, BioRad, Hercules, CA). For quantitative western analysis, infrared dye–conjugated secondary antibodies (IRDye 800, IRDye 700; Rockland Immunochemicals, Gilbertsville, PA) were used, in accordance with the manufacturer’s instructions. Visualization and quantification of the specific bands was performed using the Odyssey Infrared Fluorescence Imaging System (Li-Cor Biosciences, Lincoln, NE) in accordance with the manufacturer’s instructions. All chemicals and reagents used in this study were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise stated.
Immunofluorescent localization of p22phox
The mouse eyes were fixed in 4% paraformaldehyde/0.1 mol/l phosphate buffer (pH7.2) for 2 hours. After removal of the cornea and lens, the eyecups were cryoprotected in 30% sucrose in phosphate-buffered saline (PBS) for several hours or overnight prior to quick freezing in optical cutting temperature (OCT) compound. Then 12 μm thick sections were cut at −20 to −22 °C. A polyclonal antibody against p22phox (1:200 dilution; Santa Cruz Biotechnology, Santa Cruz, CA) was used. After removal of the primary antiserum and washing with PBS, the sections were incubated with Alexa594-conjugated secondary antibody (Molecular Probes/Invitrogen, Carlsbad, CA). Slides were coverslipped using VECTASHIELD (Vector Laboratories, Burlingame, CA), and antibody labeling was examined with a Zeiss (AxioVision Carl Zeiss MicroImaging, Thornwood, NY) microscope equipped with epifluorescence illumination and a high resolution digital camera.
In situ hybridization
p22phox mRNA was detected in the eyes of C57BL/6J and CD1 (albino) mice by in situ hybridization using a digoxigenin-labeled probe kit (Roche, Indianapolis, IN), in accordance with the manufacturer’s protocol. Briefly, the probe was produced by an in vitro transcription of a 256 base pair p22phox complementary DNA (position 182–437) that had been cloned in a TOPO II vector (Invitrogen). The eyes were fixed in 4% paraformaldehyde, cryoprotected, and embedded in OCT. Fourteen-micron sections were hybridized with the probe at 55°C overnight and detected with NBT/BCIP after incubation with anti-digoxigenin antibody. Levamisole was added to the sections to remove the endogenous phosphatase activity.
Animals and ocular injections
All the animals were treated in accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Adult C57Bl/6J and CD1 mice were purchased from Jackson Laboratory, and maintained in the University of Florida Health Science Center Animal Care Services Facilities at room temperature (20 °C) with illumination provided by standard fluorescent lighting on a 12 h/12 h light–dark cycle.
For ocular injections, the eyes of the mice were dilated by topical administration of 1% atrophine sulfate ophthalmic solution (Bausch & Lomb, Rochester, NY) and 2.5% phenylephrine hydrochloride ophthalmic solution (Akorn, Buffalo Grove, IL). The animals were then anesthetized with ketamine (72 mg/kg)/xylazine (4 mg/kg) intraperitoneal injection. For subretinal injection, a 33-gauge blunt needle equipped with a 10 μl Hamilton syringe (Hamilton Company, Reno, NV) was introduced through the corneal opening made with a disposable 30G1/2-gauge sharp needle, and 1 μl of vector suspension was slowly injected into the subretinal space under a dissecting microscope. For intravitreal injections, a 33-gauge beveled sharp needle was used, and the injections were made through sclera/choroids and retina into the vitreous cavity.
Laser-induced CNV
Adult mice were anesthetized, and the eyes were dilated as described earlier. A 532-nm-wavelength diode laser (Oculight SLx; Iridex, Mountain View, CA) was used. Laser parameters were 100 μm spot size, 0.1 second exposure, and 300-mW power. Laser photocoagulation was delivered through a slit lamp with a cover slide as a contact lens. A pattern of 4–6 lesions was placed concentrically around the optic nerve and focused on Bruch’s membrane. The formation of a bubble indicated rupture of Bruch’s membrane. The fundus was visualized using a KOWA Genesis fundus camera (Kowa Optimed, Torrance, CA).
Flatmount preparation quantitative evaluation of laser-induced CNV
Sclera/choroid/RPE flatmounts were prepared at 2 weeks after the laser treatment. The mice were deeply anesthetized and perfused with 1 ml of a PBS solution containing Rhodamine-labeled Dextran (50 mg/ml, Sigma, St. Louis, MO) and 4% paraformaldehyde. The eyes were then enucleated and fixed in 4% paraformaldehyde in PBS for 1–2 hours at room temperature and washed three times with PBS. Choroid and retinal flatmounts were prepared by carefully removing the cornea and lens, and four to five radial cuts were made to flatten the choroid and retina onto glass slides, with the sclera facing down. The retina was then carefully peeled off. An antifade mounting medium (VECTASHIELD; Vector Laboratories, CA) was used to preserve fluorescence. Choroidal and retinal flatmounts were imaged using a Zeiss fluorescence microscope (AxioVision; Carl Zeiss MicroImaging, NY). The development of CNV was quantitatively evaluated by measuring the vascular area at each laser lesion using a Zeiss AxioVision Software measurement tool. The data collected using this method were expressed as μm2.
ELISA for VEGF
Protein extracts from the retina and RPE/choroid were isolated as described earlier. The VEGF level in the eye was determined using a commercial kit (Quantikine Mouse VEGF Immunoassay; R & D System, Minneapolis, MN) in accordance with the manufacturer’s instructions.
ACKNOWLEDGMENTS
We acknowledge the National Institutes of Health grants EY13729, EY11123, NS36302, EY08571, EY016073, HL56921 and grants from the Macular Vision Research Foundation, Foundation Fighting Blindness, Juvenile Diabetes Research Foundation, and Research to Prevent Blindness, Inc. for partial support of this work. W.W.H. and the University of Florida have a financial interest in the use of adeno-associated virus (AAV) therapies, and own equity in a company (AGTC Inc.) that might, in the future, commercialize some aspects of this work. We thank Mike Daniel for help with statistical analysis, and Clay Smith for help with western blot analysis.
REFERENCES
- 1.Klein R. Overview of progress in the epidemiology of age-related macular degeneration. Ophthalmic Epidemiol. 2007;14:184–187. doi: 10.1080/09286580701344381. [DOI] [PubMed] [Google Scholar]
- 2.Smith W, Assink J, Klein R, Mitchell P, Klaver CC, Klein BE, et al. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology. 2001;108:697–704. doi: 10.1016/s0161-6420(00)00580-7. [DOI] [PubMed] [Google Scholar]
- 3.Seddon JM, Chen CA. The epidemiology of age-related macular degeneration. Int Ophthalmol Clin. 2004;44:17–39. doi: 10.1097/00004397-200404440-00004. [DOI] [PubMed] [Google Scholar]
- 4.Beatty S, Koh H, Phil M, Henson D, Boulton M. The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol. 2000;45:115–134. doi: 10.1016/s0039-6257(00)00140-5. [DOI] [PubMed] [Google Scholar]
- 5.Miceli MV, Liles MR, Newsome DA. Evaluation of oxidative processes in human pigment epithelial cells associated with retinal outer segment phagocytosis. Exp Cell Res. 1994;214:242–249. doi: 10.1006/excr.1994.1254. [DOI] [PubMed] [Google Scholar]
- 6.Yang D, Elner SG, Bian ZM, Till GO, Petty HR, Elner VM. Proinflammatory cytokines increase reactive oxygen species through mitochondria and NADPH oxidase in cultured RPE cells. Exp Eye Res. 2007;85:462–472. doi: 10.1016/j.exer.2007.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tate DJ, Jr., Miceli MV, Newsome DA. Phagocytosis and H2O2 induce catalase and metallothionein gene expression in human retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 1995;36:1271–1279. [PubMed] [Google Scholar]
- 8.Droge W. Free radicals in the physiological control of cell function. Physiol Rev. 2002;82:47–95. doi: 10.1152/physrev.00018.2001. [DOI] [PubMed] [Google Scholar]
- 9.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 2005;120:483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Schrader M, Fahimi HD. Mammalian peroxisomes and reactive oxygen species. Histochem Cell Biol. 2004;122:383–393. doi: 10.1007/s00418-004-0673-1. [DOI] [PubMed] [Google Scholar]
- 11.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–L1028. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 12.Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Cross AR, Segal AW. The NADPH oxidase of professional phagocytes— prototype of the NOX electron transport chain systems. Biochim Biophys Acta. 2004;1657:1–22. doi: 10.1016/j.bbabio.2004.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Babior BM. The NADPH oxidase of endothelial cells. IUBMB Life. 2000;50:267–269. doi: 10.1080/713803730. [DOI] [PubMed] [Google Scholar]
- 15.Gorlach A, Brandes RP, Nguyen K, Amidi M, Dehghani F, Busse R. A gp91phox containing NADPH oxidase selectively expressed in endothelial cells is a major source of oxygen radical generation in the arterial wall. Circ Res. 2000;87:26–32. doi: 10.1161/01.res.87.1.26. [DOI] [PubMed] [Google Scholar]
- 16.De Leo FR, Ulman KV, Davis AR, Jutila KL, Quinn MT. Assembly of the human neutrophil NADPH oxidase involves binding of p67phox and flavocytochrome b to a common functional domain in p47phox. J Biol Chem. 1996;271:17013–17020. doi: 10.1074/jbc.271.29.17013. [DOI] [PubMed] [Google Scholar]
- 17.Segal AW, Abo A. The biochemical basis of the NADPH oxidase of phagocytes. Trends Biochem Sci. 1993;18:43–47. doi: 10.1016/0968-0004(93)90051-n. [DOI] [PubMed] [Google Scholar]
- 18.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 19.Regier DS, Greene DG, Sergeant S, Jesaitis AJ, McPhail LC. Phosphorylation of p22phox is mediated by phospholipase D-dependent and -independent mechanisms. Correlation of NADPH oxidase activity and p22phox phosphorylation. J Biol Chem. 2000;275:28406–28412. doi: 10.1074/jbc.M004703200. [DOI] [PubMed] [Google Scholar]
- 20.Mori K, Gehlbach P, Yamamoto S, Duh E, Zack DJ, Li Q, et al. AAV-mediated gene transfer of pigment epithelium-derived factor inhibits choroidal neovascularization. Invest Ophthalmol Vis Sci. 2002;43:1994–2000. [PubMed] [Google Scholar]
- 21.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 22.Ischiropoulos H, Zhu L, Chen J, Tsai M, Martin JC, Smith CD, et al. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch Biochem Biophys. 1992;298:431–437. doi: 10.1016/0003-9861(92)90431-u. [DOI] [PubMed] [Google Scholar]
- 23.Bok D. The retinal pigment epithelium: a versatile partner in vision. J Cell Sci Suppl. 1993;17:189–195. doi: 10.1242/jcs.1993.supplement_17.27. [DOI] [PubMed] [Google Scholar]
- 24.Rizzolo LJ. Polarity and the development of the outer blood-retinal barrier. Histol Histopathol. 1997;12:1057–1067. [PubMed] [Google Scholar]
- 25.Kawahara BT, Quinn MT, Lambeth JD. Molecular evolution of the reactive oxygen-generating NADPH oxidase (Nox/Duox) family of enzymes. BMC Evol Biol. 2007;7:109. doi: 10.1186/1471-2148-7-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Griendling KK, Sorescu D, Ushio-Fukai M. NAD(P)H oxidase: role in cardiovascular biology and disease. Circ Res. 2000;86:494–501. doi: 10.1161/01.res.86.5.494. [DOI] [PubMed] [Google Scholar]
- 27.Ushio-Fukai M, Alexander RW. Reactive oxygen species as mediators of angiogenesis signaling: role of NAD(P)H oxidase. Mol Cell Biochem. 2004;264:85–97. doi: 10.1023/b:mcbi.0000044378.09409.b5. [DOI] [PubMed] [Google Scholar]
- 28.Ushio-Fukai M. Redox signaling in angiogenesis: role of NADPH oxidase. Cardiovasc Res. 2006;71:226–235. doi: 10.1016/j.cardiores.2006.04.015. [DOI] [PubMed] [Google Scholar]
- 29.Ushio-Fukai M. VEGF signaling through NADPH oxidase-derived ROS. Antioxid Redox Signal. 2007;9:731–739. doi: 10.1089/ars.2007.1556. [DOI] [PubMed] [Google Scholar]
- 30.Kliffen M, Sharma HS, Mooy CM, Kerkvliet S, de Jong PT. Increased expression of angiogenic growth factors in age-related maculopathy. Br J Ophthalmol. 1997;81:154–162. doi: 10.1136/bjo.81.2.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryan SJ. Subretinal neovascularization. Natural history of an experimental model. Arch Ophthalmol. 1982;100:1804–1809. doi: 10.1001/archopht.1982.01030040784015. [DOI] [PubMed] [Google Scholar]
- 32.Yi X, Ogata N, Komada M, Yamamoto C, Takahashi K, Omori K, et al. Vascular endothelial growth factor expression in choroidal neovascularization in rats. Graefes Arch Clin Exp Ophthalmol. 1997;235:313–319. doi: 10.1007/BF01739641. [DOI] [PubMed] [Google Scholar]
- 33.Strauss O. The retinal pigment epithelium in visual function. Physiol Rev. 2005;85:845–881. doi: 10.1152/physrev.00021.2004. [DOI] [PubMed] [Google Scholar]
- 34.Kent D, Sheridan C. Choroidal neovascularization: a wound healing perspective. Mol Vis. 2003;9:747–755. [PubMed] [Google Scholar]
- 35.Das A, McGuire PG. Retinal and choroidal angiogenesis: pathophysiology and strategies for inhibition. Prog Retin Eye Res. 2003;22:721–748. doi: 10.1016/j.preteyeres.2003.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Mousa SA, Lorelli W, Campochiaro PA. Role of hypoxia and extracellular matrix-integrin binding in the modulation of angiogenic growth factors secretion by retinal pigmented epithelial cells. J Cell Biochem. 1999;74:135–143. [PubMed] [Google Scholar]
- 37.Infanger DW, Sharma RV, Davisson RL. NADPH oxidases of the brain: distribution, regulation, and function. Antioxid Redox Signal. 2006;8:1583–1596. doi: 10.1089/ars.2006.8.1583. [DOI] [PubMed] [Google Scholar]
- 38.Kamsler A, Segal M. Hydrogen peroxide as a diffusible signal molecule in synaptic plasticity. Mol Neurobiol. 2004;29:167–178. doi: 10.1385/MN:29:2:167. [DOI] [PubMed] [Google Scholar]
- 39.Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, et al. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 40.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 41.Li JM, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 42.Cacicedo JM, Benjachareowong S, Chou E, Ruderman NB, Ido Y. Palmitate-induced apoptosis in cultured bovine retinal pericytes: roles of NAD(P)H oxidase, oxidant stress, and ceramide. Diabetes. 2005;54:1838–1845. doi: 10.2337/diabetes.54.6.1838. [DOI] [PubMed] [Google Scholar]
- 43.Manea A, Raicu M, Simionescu M. Expression of functionally phagocytetype NAD(P)H oxidase in pericytes: effect of angiotensin II and high glucose. Biol Cell. 2005;97:723–734. doi: 10.1042/BC20040107. [DOI] [PubMed] [Google Scholar]
- 44.Ellis EA, Grant MB, Murray FT, Wachowski MB, Guberski DL, Kubilis PS, et al. Increased NADH oxidase activity in the retina of the BBZ/Wor diabetic rat. Free Radic Biol Med. 1998;24:111–120. doi: 10.1016/s0891-5849(97)00202-5. [DOI] [PubMed] [Google Scholar]
- 45.Al-Shabrawey M, Bartoli M, El-Remessy AB, Platt DH, Matragoon S, Behzadian MA, et al. Inhibition of NAD(P)H oxidase activity blocks vascular endothelial growth factor overexpression and neovascularization during ischemic retinopathy. Am J Pathol. 2005;167:599–607. doi: 10.1016/S0002-9440(10)63001-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saito Y, Geisen P, Uppal A, Hartnett ME. Inhibition of NAD(P)H oxidase reduces apoptosis and avascular retina in an animal model of retinopathy of prematurity. Mol Vis. 2007;13:840–853. [PMC free article] [PubMed] [Google Scholar]
- 47.Rodrigues EB. Inflammation in dry age-related macular degeneration. Ophthalmologica. 2007;221:143–152. doi: 10.1159/000099293. [DOI] [PubMed] [Google Scholar]
- 48.Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. 2003;15:578–584. doi: 10.1016/s0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 49.Goldblatt D, Butcher J, Thrasher AJ, Russell-Eggitt I. Chorioretinal lesions in patients and carriers of chronic granulomatous disease. J Pediatr. 1999;134:780–783. doi: 10.1016/s0022-3476(99)70299-4. [DOI] [PubMed] [Google Scholar]
- 50.Kim SJ, Kim JG, Yu YS. Chorioretinal lesions in patients with chronic granulomatous disease. Retina. 2003;23:360–365. doi: 10.1097/00006982-200306000-00012. [DOI] [PubMed] [Google Scholar]