

Abstract
Little is known about histone modifiers and histone marks in colorectal cancers (CRC). The present study therefore addressed the role of histone acetylation and histone deacetylases (HDAC) in CRCs in situ and in vitro. Immunohistochemistry of primary CRCs (n=47) revealed that selected histone marks were frequently present (H3K4me3: 100%; H3K9me3: 77%; H3K9ac: 75%), partially displayed intratumoral heterogeneity (H3K9me3; H3K9ac) and were significantly linked to higher pT category (H3K9me3: p=0.023; H3K9ac: p=0.028). Furthermore, also HDAC1 (62%), HDAC2 (100%) and HDAC3 (72%) expression was frequent, revealing four CRC types: cases expressing 1) HDAC1, HDAC2 and HDAC3 (49%), 2) HDAC2 and HDAC3 (30%), 3) HDAC1 and HDAC2 (10.5%) and 4) exclusively HDAC2 (10.5%). Correlation to clinico-pathological parameters (pT, pN, G, MSI status) revealed that heterogeneous HDAC1 expression correlated with lymph node status (p=0.012). HDAC expression in situ was partially reflected by six CRC cell lines, with similar expression of all three HDACs (DLD1, LS174T), preferential HDAC2 and HDAC3 expression (SW480, Caco2) or lower HDAC2 and HDAC3 expression (HCT116, HT29). HDAC activity was variably higher in HCT116, HT29, DLD1 and SW480 compared to LS174T and Caco2 cells. Treatment with broad (SAHA) and specific (MS-275; FK228) HDAC inhibitors (HDACi) caused loss of cell viability in predominantly MSIpositive CRC cells (HCT116, LS174T, DLD1; SAHA, MS-275 and in part FK228). In contrast, MSI-negative CRC cells (Caco2, HT29, SW480) were resistant, except for high doses of FK228 (Caco2, HT29). Cell viability patterns were not linked to different efficacies of HDACi on reduction of HDAC activity or histone acetylation, p21 expression and/or induction of DNA damage (γH2A-X levels). In summary, this study reveals inter- and intra-tumoral heterogeneity of histone marks and HDAC expression in CRCs. This is reflected by diverse HDACi responses in vitro, which do not follow known modes of action. Together, this implies further exploitation of histone alterations in CRC for molecular classification and/or novel treatment options.
Keywords: Epigenetic, colorectal carcinoma, class I HDACs, histone marks, heterogeneity, HDACi
Introduction
Colorectal carcinomas (CRCs) are a phenotypic and molecular heterogeneous group of lower gastrointestinal tract tumors [1]. Carcinogenesis, progression and treatment outcome of CRC has so far been mainly investigated from the genetic point of view, yielding valuable clinico-pathological routine diagnostic biomarkers for prognostic sub-classification and treatment prediction [2-4]. Recently, also epigenetic alterations were similarly addressed, with specific DNA methylation patterns linked to the carcinogenesis of a specific molecular and phenotypic group of CpG Island Methylator Phenotype (CIMP) CRC [5-8]. CIMP-type CRCs, which undergo the serrated pathway of carcinogenesis, are associated with acquired microsatellite instability (MSI) via MLH1 promoter hypermethylation and exhibit BRAF mutations in about 70%. These CRCs show a worse prognosis and appear to have limited responses to 5FU-based therapies [8,9].
Whether similar clinico-pathological relevant associations exist for epigenetic alteration of CRC at the level of histones is still an evolving field. Indeed, increased expression of histone deacetylases (HDACs) has been reported before for CRC [10-13]. One study comprehensively analyzed HDAC class I expression in tissue microarrays of CRCs, revealing frequent HDAC2 and HDAC3 protein expression (>50% of cases). In addition, HDAC patterns were associated with tumor cell proliferation, differentiation and worse prognosis [10]. HDAC1 [12] and HDAC3 [13] were also examined in smaller series of CRCs. In fact, only one study [14] clearly associated a specific HDAC, the class III HDAC SIRT1, with the phenotypic and molecular subclass of MSI and CIMP-type CRCs. Thus, previous studies point towards the involvement of histones and histone modifiers in the clinico-pathological behavior of CRC. That histone marks and modifiers may be supportive for cancer sub-classification, prognostication and/or even HDAC therapeutic targeting is suggested by studies of HDAC expression, histone acetylation and HDAC inhibitor application to other cancer entities [15-17]. However, a comprehensive examination of histone marks and histone modifiers as well as their potential as therapeutic targets in selective clinico-pathological groups of CRCs in situ and in vitro is still lacking.
Clearly, patterns of histone modifiers and histone marks induce active gene transcription (H3K4me3, histone acetylation) or gene silencing (H3K9me3, H3K27me3) [18] and hence influence distinct cellular processes [19]. By modulation of histone acetylation, HDAC inhibitors (HDACi) may influence gene expression, thereby e.g. 1) restoring expression of silenced tumor suppressor genes (such as CDKN1A/p21) leading to growth inhibition [16] or 2) causing suppression of RAD51 and MRE11 expression leading to increased DNA damage and γH2A-X marked DNA double strand breaks [17].
Indeed, treatment of CRC cell lines by HDACi induced cell death and further sensitized these cells to radiotherapy [20] or to 5FU-based chemotherapy [21,22]. Moreover, a recent study [23] revealed the inhibitory effect of a broad HDACi in combination with Selumetinib - a MEK inhibitor - in a KRAS mutant CRC cell line model. These studies hence underline the potential therapeutic benefit of histone modifier inhibitors in (RAS mutant) CRCs.
Nevertheless, despite these previous studies so far none of the in vitro studies evaluated the potential diverse effects of broad or specific HDACi in a spectrum of cell lines reflecting the distinct clinico-pathological and molecular subclasses of CRCs.
Therefore, the present study focused on examination of three class I HDACs as well as H3K9ac and H3K4me3 (active) and H3K9me3 (repressive) histone marks in entire serial sections of tissue specimens of primary CRCs. This was accompanied by in vitro analyses of six CRC cell lines mirroring at least in part the clinico-pathological and molecular spectrum CRC cases in situ [24]. Here, the cellular (cell viability) and molecular (HDAC activity, p21 induction, DNA damage) effects of three HDACi (broad: SAHA/Vorinostat; HDAC class I specific: Entinostat/MS-275; HDAC1, HDAC2 specific: Romidepsin/FK228) [17] were comprehensively evaluated.
Materials and methods
Patients and tissue specimens
The study included archival, Formalin-fixed and Paraffin-embedded (FFPE) tissue specimens of 47 cases with primary CRCs treated at the University Medical Center, Freiburg, Germany without neoadjuvant radio/chemotherapy. All tissue specimens were re-evaluated by a pathologist according to the current WHO [1] and TNM [25] classification. Clinico-pathological parameters (age, sex, location, tumor classification, tumor grading, histological subtype) are provided in Table 1. MSI status was determined by staining for MLH1 (ready to use antibody IR079, DakoCytomation, Hamburg, Germany), MSH2 (ready to use antibody IR085, DakoCytomation, Glostrup, Denmark), MSH6 (ready to use antibody IR086, DakoCytomation, Hamburg, Germany) and PMS2 (ready to use antibody IR087, DakoCytomation, Hamburg, Germany) according to standard procedures and expression loss of at least one marker was considered indicative of MSI [26,27]. In 13 cases, adjacent non-dysplastic mucosa was analyzed in addition to the tumors. The use of tissue specimens was approved by the local ethics institution (#251/04/07/09, Ethik-Kommission, Albert-Ludwigs-Universität, Freiburg, Germany).
Table 1.
Clinico-pathological parameters of colorectal cancer patients investigated
| Cases (n) | Percent (%, n/47) | |
|---|---|---|
| Age | ||
| <50 | 8 | 17.0 |
| ≥50 | 39 | 83.0 |
| Sex | ||
| Male | 29 | 61.7 |
| Female | 18 | 38.3 |
| Location | ||
| Colon | 44 | 93.6 |
| Rectum* | 3 | 6.4 |
| pT | ||
| pT1 | 1 | 2.1 |
| pT2 | 9 | 19.1 |
| pT3 | 31 | 66.0 |
| pT4 | 6 | 12.8 |
| pN | ||
| pN0 | 29 | 61.7 |
| pN1 | 10 | 21.3 |
| pN2 | 7 | 14.9 |
| pNx | 1 | 2.1 |
| Tumour grading | ||
| G1 | 1 | 2.1 |
| G2 | 40 | 85.1 |
| G3 | 6 | 12.8 |
| Histological subtype | ||
| NOS | 44 | 93.6 |
| Mucinous | 3 | 6.4 |
| Microsatellite status | ||
| MSS | 42 | 89.4 |
| MSI | 5 | 10.6 |
The table summarizes the major clinico-pathological parameters of the investigated 47 cases.
No neoadjuvant radio-/chemotherapy.
Immunohistochemistry
Immediate serial sections of FFPE tissue specimens (n=47) were subjected to deparaffinization, antigen retrieval (pH6.1) and subsequent staining with primary antibodies for H3K9ac (polyclonal rabbit, 1:200, #9671, Cell Signaling Technologies, Danvers, USA), H3K4me3 (polyclonal rabbit, 1:1000, #ab8580, abcam, Cambridge, UK), H3K9me3 (polyclonal rabbit, 1:1000, #ab8898, abcam, Cambridge, UK), HDAC1 (polyclonal rabbit, 1:500, #ab19845, abcam, Cambridge, UK), HDAC2 (mouse monoclonal, HDAC-62, 1:12500, #ab12169, abcam, Cambridge, UK) and HDAC3 (mouse monoclonal, 40/HDAC3, 1:500, #611125, BD Biosciences, San Jose, USA) was performed. After washing all reactions were continued using the Dako REAL Detection System (alkaline phosphatase/RED, rabbit/mouse) on the Autostainer System (all DakoCytomation now Agilent, Glostrup, Denmark).
The expected nuclear histone mark levels and HDAC expression were evaluated in invasive tumor cells by semi-quantitative scoring of intensity of staining (score 0-3) and the percentage of cells stained (score 0-4). Multiplication of the scores resulted in an immuno reactive score (IRS) scoring system ranging from 0 to 12. An IRS of 0-6 was defined as “negative” and an IRS of 7-12 was defined as “positive”, as previously described [10,28]. In addition, adjacent non-dysplastic colonic mucosa (n=13) were similarly evaluated for HDAC1, HDAC2 and HDAC3.
Cell culture and treatment protocols
Six previously described [24] colorectal cancer cell lines (HCT116, DLD1, LS174T, HT29, SW480, Caco2) were examined. HCT116 and HT29 were cultivated in McCoys´5A, DLD1 in RPMI 1640 medium and SW480, LS174T and Caco2 in DMEM all from GIBCO™ (Life Technologies, Carlsbad, USA), each supplemented with 10% GIBCO™ Fetal Bovine Serum and 1% GIBCO™ L-Glutamin at 37°C in a 5% CO2 atmosphere [29]. All cell lines were verified at the Leibniz-Institut DSMZ-Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH.
Three HDAC inhibitors (HDACi) with different chemical structure and different specificity [17] were used: suberoylanilide hydroxamic acid (SAHA or Vorinostat) a hydroxamat and broad HDACi; MS-275 (Entinostat) a benzamide directed against HDAC class I and FK228 (Romidepsin) a cyclic peptide directed against HDAC1 and HDAC2. HDACi were all purchased from Selleckchem (Munich, Germany) and diluted in DMSO. For in vitro experiments, dose response curves with 0.1 µM, 2.5 µM and 5 µM (SAHA, MS-275) and 0.1 nM, 2.5 nM and 5 nM (FK228) were initially performed. Thereafter, doses were set for all other experiments as specified in Table 2.
Table 2.
HDACi doses selected from cell viability analyses for further experiments
| SAHA | MS-275 | FK228 | ||||
|---|---|---|---|---|---|---|
|
|
||||||
| Dose (µM) | % cell viability | Dose (µM) | % cell viability | Dose (nM) | % cell viability | |
| HCT116 | 2.5 | 52 | 2.5 | 44 | 2.5 | 55 |
| HT29 | 5.0 | 92 | 5.0 | 96 | 2.5 | 58 |
| DLD1 | 2.5 | 54 | 5.0 | 55 | 5.0 | 101 |
| SW480 | 5.0 | 79 | 5.0 | 82 | 5.0 | 83 |
| LS174T | 5.0 | 70 | 5.0 | 69 | 2.5 | 54 |
| Caco2 | 5.0 | 80 | 5.0 | 92 | 5.0 | 65 |
The table presents the HDACi doses - selected from cell viability analysis - used for activity assay as well as western blots for p21, p.H2A.X and histone acetylation.
MTS assay
Cell viability was measured 72 h post inhibitor treatment with increasing doses (see cell culture and treatment protocols) by CellTiter 96 Aqueous One Solution Reagent (Promega, Madison, USA) according to the manufacturer’s protocol and as previously described [30].
Immunoblotting
Immunoblotting was performed according to established protocols [30] with primary antibodies against HDAC1 (polyclonal rabbit, 1:500 #ab19845, abcam, Cambridge, UK), HDAC2 (mouse monoclonal, HDAC2-62, 1:1000, #ab12169, abcam, Cambridge, UK), HDAC3 (mouse monoclonal, 40/HDAC3, 1:1000, #611125, BD Biosciences, San Jose, USA), β-Actin (mouse monoclonal, AC-15, 1:2000, #A5441, Sigma-Aldrich, St. Louis, USA), Anti-acetyl H3 (polyclonal rabbit, 1:2000, #06-599, Millipore, Hessen, Germany), p21 (polyclonal rabbit, 1:1000, #2947, CST, Denver, USA) and γH2A-X (polyclonal rabbit, 1:1000, #9718, CST Denver, USA). Bands were quantified by densitometry using Image J software.
HDAC activity assay
Measurement of HDAC activity was performed using the HDAC Cell-Based Activity Assay Kit (Cayman) according to the manufacturer’s protocol and as previously described [28]. Cultured cells were incubated for 24 h with the HDACi in a 96- Well Clear Bottom Black Culture Plate at 37°C in a 5% CO2 atmosphere. The fluorescent intensity of each well was read with a fluorometer (excitation 350 nm, emission 450 nm).
Statistical analyses
Statistical evaluation of immunohistochemistry was done for experimental data (histone marks, HDACs) and clinico-pathological parameters (pT, pN, grading, MSI status; see Table 1). Correlation between parameters was performed using the Fisher’s Exact Test for nominal scaled parameters and according to development of metric scaled parameters the Mann-Whitney Test or Kruskall Wallis Test. In case of a significant Kruskall Wallis Test, a pairwise comparison with the Mann-Whitney Test was subsequently performed. To compare HDAC expression of tumor tissue versus adjacent non-dysplastic tissue, the Wilcoxon Test was performed. A P-value of <0.05 (exact 2-sided) was considered to be statistically significant. Statistical analyses were done with SPSS 22.0.
For in vitro analyses, significance levels were calculated using the t-test at all times of three independent experiments. P-value of <0.05 was considered significant (*, ** and *** indicate significant levels in a range from 0.01-0.05, 0.01-0.001 and <0.001 respectively).
Results
Active and repressive histone marks are frequent in CRC
High levels of histone marks were frequently observed for H3K9ac (75%), H3K4me3 (100%) and H3K9me3 (77%) in CRCs.
The majority of cases (63.5%) exhibited all three histone marks (Figure 1A, sample case 1), whereas 13% of cases exhibited exclusively the active histone marks H3K9ac and H3K4me3, but not the repressive histone mark H3K9me3 (Figure 1A, sample case 3). In addition, 10.5% of cases showed a combination of active and repressive histone marks, being positive for H3K4me3, H3K9me3, but heterogeneous for H3K9ac (Figure 1A, sample case 4, arrow). Finally, 13% of cases were only positive for H3K4me3 (Figure 1A, sample case 2).
Figure 1.
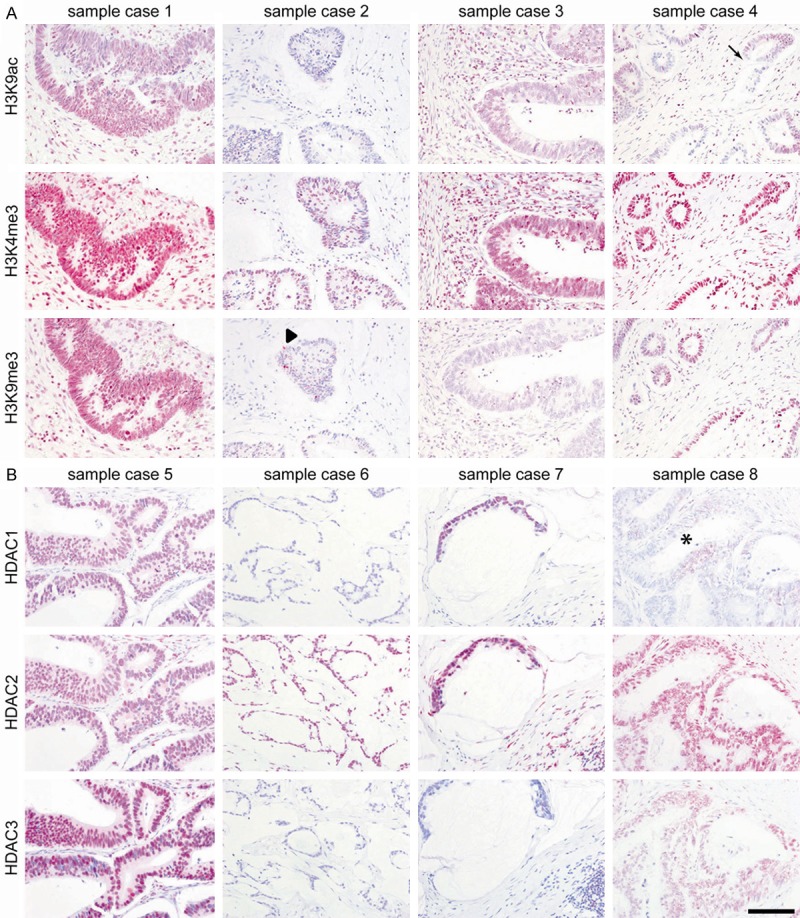
Colorectal carcinomas are characterized by four patterns of histone alterations. The figure shows expression patterns of H3K9ac, H3K4me3, H3K9me3 (A) and HDAC1, HDAC2, HDAC3 (B). The arrow in sample case 4 (A) indicates intraglandular heterogeneity for H3K9ac (see also Figure 2). The arrowhead in sample case 2 (A) indicates an intraepithelial lymphocyte with positive immunoreactivity for H3K9me3, whereas the glandular epithelium shows not any positive immunoreactivity. The * in sample case 4 (B) indicates intraglandular heterogeneity for HDCA1. Immunohistochemistry was quantified by IRS. Scale bar 100 µm.
Distinct patterns and intratumoral heterogeneity of HDAC1, HDAC2 and HDAC3 expression in CRCs
A positive HDAC expression (IRS >6) was found in 62% (HDAC1), 100% (HDAC2) and 72% (HDAC3) of all investigated CRCs.
HDAC2 expression was significant higher in tumor cells compared to adjacent non-dysplastic colon mucosa (p=0.02). In contrast, HDAC1 and HDAC3 expression in tumor cells compared to adjacent non-dysplastic colon mucosa were indiscriminative.
Four distinct groups of CRCs were defined according to HDAC expression patterns (Figure 1B): Group 1 (49% of cases) showed positive expression for all HDACs (e.g. case 5); group 2 (30% of cases) with positive HDAC2 and HDAC3, but negative HDAC1 expression (e.g. case 8); group 3 (10.5% of cases) with positive HDAC2, but negative HDAC1 and HDAC3 expression (e.g. case 6); and group 4 (10.5% of cases) with positive HDAC1 and HDAC2, but negative HDAC3 expression (e.g. case 7).
Importantly, whilst HDAC2 was distributed homogeneously within each expressing case, HDAC1 showed intratumoral heterogeneity, even within the same tumor gland (Figures 1B, 2A-C asterisk). This was also seen for H3K9ac (Figure 1A, sample case 4, arrow). Besides this localized intratumoral heterogeneity in more well differentiated CRCs, poorly differentiated CRCs showed general area-wide heterogeneous HDAC1 expression, which was in fact reflected by the percentage of positive tumor cells of the IRS score (Figure 2F-H).
Figure 2.
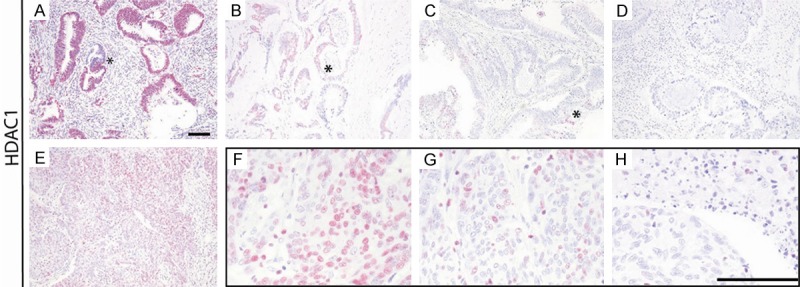
Intratumoral and intraglandular heterogeneity of HDAC1 expression. The figure shows heterogeneity of HDAC1 expression in different CRC cases with expression levels >80% (A), 10-50% (B), <10% (C) and 0% (D), even within the same tumor gland (A-C; asterisks), and within one case with expression levels >80% (E), >50-80% (F), 10-50% (G) and <10% (H). Scale bars 100 µm.
Correlation of histone marks and HDAC expression with clinico-pathological parameters
The distinct patterns of histone marks and HDACs were next correlated to clinico-pathological parameters (Table 1). This revealed significant lower levels of the repressive histone mark H3K9me3 in higher tumor stages (pT; p=0.023). No significant associations were found for H3K9ac, H3K4me3, HDAC1, HDAC2 and HDAC3 upon considering general positivity (IRS >6).
However, upon taking into account the observed intratumoral heterogeneity (percentage of positive tumor cells, with score 0=0%, 1<10%, 2=10-50%, 3 >50-80%, 4 >80%) additional significant associations were observed: The active histone mark H3K9ac was significantly more heterogeneous expressed (shown in Figure 1A, arrow) in higher tumor stages (pT; p=0.028) particular with more negative areas. Moreover, HDAC1 was significantly correlated with lymph node status (pN; p=0.012), whereby a more heterogeneous expression - as also reflected by the percent of positive tumor cells (Figure 2A-C) - was linked to lymph node metastasis.
HDAC expression and activity in CRC cell lines in vitro
To validate CRC cell lines for further investigation of HDAC inhibition, first six CRC cell lines (HCT116, DLD1, LS174T, HT29, SW480, and Caco2) were analyzed for HDAC expression and HDAC activity (Figure 3).
Figure 3.
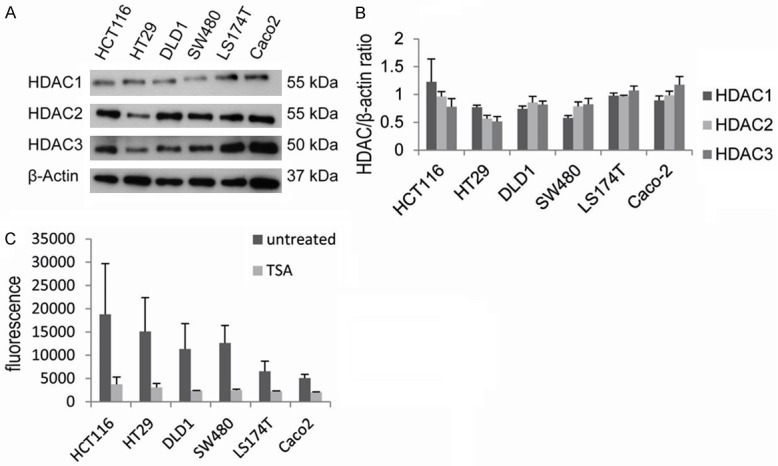
HDACs are frequently expressed in CRC cell lines. HDAC levels were analyzed by immunoblotting, shown is one representative blot (A) and the quantification by densitometry (B). HDAC activity was measured in all six CRC cell lines (C), with TSA as control (C). Diagrams show the mean ± SEM of three independent experiments.
As observed in situ, all cell lines showed frequent expression of HDAC1, HDAC2 and HDAC3 (Figure 3A, 3B). Thereby, HDAC1 (range HDAC/ß-actin ratio: 0.58-1.23) and HDAC3 (range HDAC/ß-actin ratio: 0.52-1.18) levels were more variable than HDAC2 expression levels (range HDAC/ß-actin ratio: 0.79-0.99), as measured upon quantification of immunoblots. Furthermore, as seen in situ, also in vitro specific groups of HDAC expressing cell lines were observed (Figure 3B): LS174T and DLD1 cells had similar HDAC1, HDAC2 and HDAC3 expression (“group 1” of CRCs). HCT116 and HT29 showed lower HDAC2 and HDAC3 expression as compared to HDAC1 (partial “group 4” CRCs), although HDAC1 expression was rather variable in HCT116 cells. SW480 and Caco2 cells had higher HDAC2 and HDAC3 than HDAC1 expression (“group 2” CRCs). Finally, upon comparison of HDAC expression across cell lines the lowest levels of HDAC1 were observed in SW480 cells, lowest HDAC2 and HDAC3 levels in HT29 cells.
Next, the general level of HDAC activity was measured in all cell lines under normal cell culture conditions (Figure 3C). This revealed that HDAC activity was lower in LS174T and Caco2 cells and appeared higher in HCT116, HT29, DLD1 and SW480 cells. However, the latter four cell lines showed quite variable, high HDAC activity levels.
The data hence show that HDAC expression in cell lines is quite heterogeneous within and between cell lines-mirroring the situation in human tissue specimens of CRCs - and that this is also reflected by the HDAC activity levels.
CRC cell lines exhibit distinct responses to broad and specific HDAC inhibitors
To investigate how the CRC cells with distinct patterns of HDAC expression and activity respond to HDACi, one broad (SAHA) and two more specific (MS-275: HDAC class I; FK228: HDAC1, HDAC2) HDACi were tested in dose response experiments (0.1 µM-5 µM (SAHA, MS-275), and 0.1 nM-5 nM (FK228)) (Figure 4).
Figure 4.
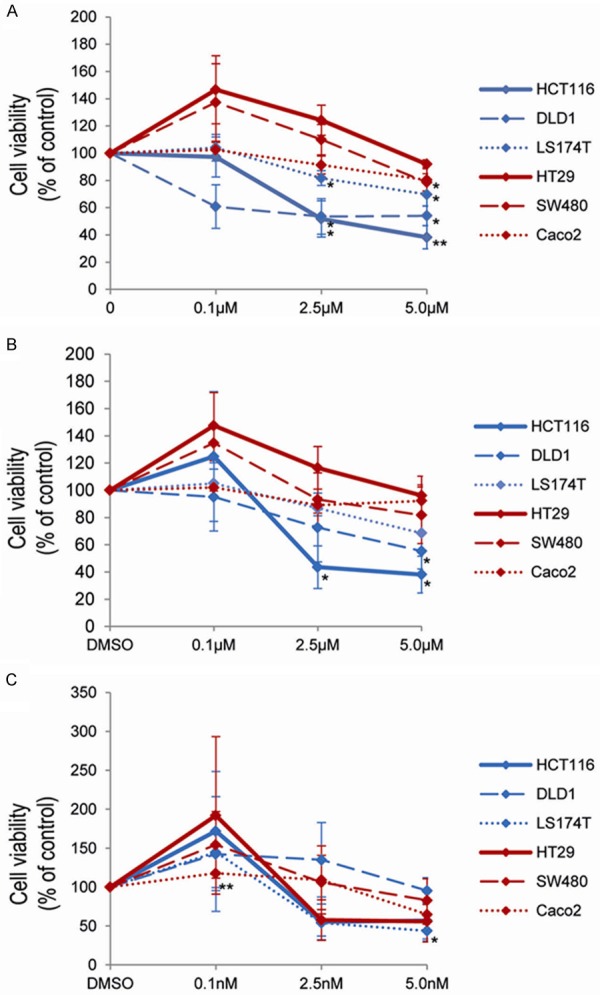
HDACi treatment decreases cell viability in CRC cell lines. Cell viability was analyzed 72 h post inhibitor addition by MTS assays for the broad HDACi SAHA (A) and for two specific HDACi with MS-275 (B) and FK228 (C). Diagrams show the mean +/- SEM of three independent experiments, each performed in technical triplicates. P-values are provided as *: 0.05-0.01 and **: ≤ 0.01-0.001.
The broad HDACi SAHA (Figure 4A) showed loss of cell viability with >50% primarily in DLD1 (already at 0.1 µM), HCT116 (2.5 µM) and almost to 50% in LS174T (5 µM) cells. In contrast, Caco2, HT29 and SW480 cells were mainly unresponsive to SAHA.
A similar effect was seen for the specific HDACi MS-275 (Figure 4B), leading to a reduction of >50% cell viability in HCT116 (2.5 µM) and to almost 50% in LS174T (5 µM) and DLD1 (5 µM) cells. Again, Caco2, HT29 and SW480 cells remained largely unaffected.
In contrast, the most specific HDACi tested - FK228 - actually displayed a broader profile of cell responses (Figure 4C): In fact, here cell viability was reduced to >50% in HCT116 (2.5 nM), LS147T (2.5 nM), HT29 (2.5 nM) and almost to 50% in Caco2 (5 nM).
Interestingly, in HT29 and SW480 cells there was a notable increase of cell viability for all three HDACi given at low doses, whereas HCT116, DLD1 and LS174T cells showed this increase in cell viability only at low doses of MS-275 and FK228 (Figure 4A-C).
Thus, the responses to broad and specific HDACi are not entirely related to HDAC expression and activity pattern of the individual cell lines. Moreover, microsatellite-instable CRC cell lines (HCT116, DLD1, LS174T) appear to be more sensitive to HDACi (SAHA, MS-275) as compared to microsatellite-stable CRC cell lines.
The sensitivity of CRC cell lines to HDACi is not mirrored by HDAC “by-pass” activity or specific p21 expression and DNA damage response patterns
In view of the different efficacy of HDACi in the six CRC cell lines, the response to HDACi was next evaluated regarding reduction of HDAC activity and associated histone acetylation as well as induction of p21 expression and DNA damage response. For this, the HDACi doses reaching >50% loss of cell viability or the maximum tested doses were used (Table 2).
As depicted in Figure 5A, all six cell lines - irrespective of either loss or maintained cell viability - showed marked reduction (>50%) of HDAC activity for all three tested inhibitors at the given dose. This was significant for all HDACi in Caco2 cells, for FK228 in SW480 and LS174T cells and for MS-275 and FK228 in HCT116 cells. In addition, HT29, DLD1, SW480 and LS174T cells showed marked reduction of HDAC activity for SAHA and MS275.
Figure 5.
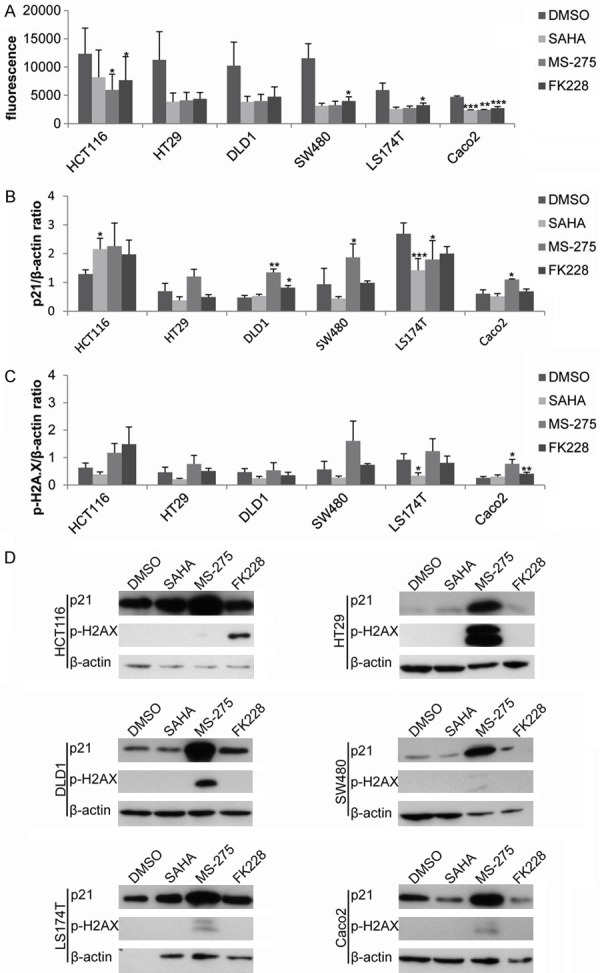
Cellular responses induced by HDACi. HDAC activity was measured 24 h post HDACi addition (A). Immunoblotting with subsequent quantification was performed for induction of p21 expression (B) and DNA damage by γH2A-X (C). One representative immunoblot is shown for both p21 and γH2A-X (D). Diagrams show the mean +/- SEM of three independent experiments. P-values are provided as *: 0.05-0.01, **: ≤ 0.01-0.001 and ***: ≤ 0.001.
In addition, patterns of p21 protein expression, which are considered to be up-regulated during HDACi responses [31], did not reflect the patterns of cell viability responses in most CRC cell lines (Figure 5B, 5D). Thereby, p21 expression was for example increased by MS-275 in HT29, DLD1, SW480 and Caco2 cells, however at these doses only DLD1, but not HT29, SW480 or Caco2 cells showed a loss of cell viability. Vice versa, there was a slight reduction of p21 protein expression for all three HDACi in LS174T cells, which lost cell viability at these inhibitor doses. Only in HCT116 cells, an induction of p21 expression was observed in parallel to and matching cell viability responses to all three HDACi.
Finally, DNA damage responses - as measured by induction of γH2A-X levels - were induced predominantly by MS-275 in HT29, SW480 and Caco2 as well as by FK228 in HCT116 cells (Figure 5C, 5D). However, again increased levels of γH2A-X levels did not match cell viability responses.
Thus, together these data imply that despite efficient inhibition of HDAC activity and in part induction of p21 and DNA damage response by specific HDACi (MS-275, FK228), CRC cell lines appear to act by so far unknown and rather individual cellular mechanisms to HDACi.
Discussion
Colorectal carcinomas (CRC) comprise different histological and morphological sub-entities, which reflect diverse mechanisms of development, progression and therapy responses. Thus, chromosomal instable (CIN), microsatellite instable (MIN) and CpG Island Methylator Phenotype (CIMP) CRCs currently define the molecular classes of CRC [32,33]. Besides DNA methylation, also histone modifications may play an important role in development CRCs. For example butyrate - an HDAC inhibitor - levels were associated with high fibre diet to reduced risk of CRC carcinogenesis [31,34,35]. Moreover, histone modifications may also be a valuable starting point for broadening CRC treatment options [36] - either directly by inhibitors to histone modifiers or indirectly by defining epigenetic sub-classes of CRCs for prognostication and associated adjuvant chemo-/radiotherapy. Previous studies have addressed single histone marks or histone modifier expression in CRC tissue specimens [37,38] or examined response to selected histone modifier inhibitors in a single CRC cell line [39].
The present study is however the first, which comprehensively examining 1) active and repressive histone marks as well as histone deacetylases (HDAC) in human CRC tissue specimens, and 2) the response of six CRC cell lines to both broad and specific HDAC inhibitors (HDACi). Out data point to a complex pattern of activating and repressive histone marks in most (>75%) CRCs. The “net” effect on gene regulation of these patterns of histone marks however requires detailed further investigation, such as by chromatin immunoprecipitation/sequencing or correlation to RNA transcriptional profiles. Nevertheless, we found that the active histone mark H3K9ac was significantly associated with higher pT stages, whilst the repressive mark H3K9me3 decreased at higher pT stages. This underlines the known mutually exclusive presence of these two histone marks [40] and suggests the involvement of these histone modifications in driving a more aggressive tumor behavior. The high levels of H3K9ac may - at least in part - be generated by altered (reduced) levels of HDAC [41], which may be also induced by HDACi.
Similarly to other previous studies [42], HDAC class I expression was frequent in our cohort of CRCs and was higher in tumor compared to non-dysplastic colonic mucosa (HDAC2) and correlated to lymph node metastasis (HDAC1). However, despite using the same IRS quantification as a previous study [10], we detected differences of HDAC2 expression between non-dysplastic colonic mucosa and tumor cells. This is most likely due to the fact that in our study entire tissue specimen sections in contrast to tissue microarrays [10] were examined. In addition, our detailed analyses of entire tissue specimen sections furthermore revealed intratumoral heterogeneity for HDAC1 expression, even within the same tumor gland. Indeed, it was HDAC1 expression that was linked to lymph node metastasis, possibly indicating that some high (or low) HDAC1 expressing tumor cells are involved in dissemination to lymph nodes [43,44].
Irrespective of this, the presence of HDAC1, HDAC2 and HDAC3 expression in two-thirds of CRC patients in our and other studies [10,42] clearly calls for HDAC being a potential therapeutic target. In fact, new HDACi substances were previously studied mainly in selected CRC cell lines, such as I-7ab in HCT116 [45], SAHA in Colo320HSR cells [46], MPT0G030 in HT29 cells [47]. Since we observed partial heterogeneous HDAC expression and since treatment responses are most likely also defined by the specific molecular sub-class of CRCs, we here for the first time measured treatment responses of six CRC cell lines to broad and specific HDACi. Indeed, it was shown before that HDACi with similar specificities may exert different cellular responses [48], depending on the molecular background of the cells studied. In our case, we examined three CIN- and three MIN-type CRC cell lines, which reflected the HDAC expression patterns observed in situ. Despite carrying distinct mutations in key CRC associated genes, such as KRAS, BRAF and/or TP53, there was a clear association of HDACi efficacy with the two molecular sub-classes of CRC: Whilst both CIN- and MIN-type cell lines showed similar effects on HDAC activity, only MIN-type CRC cells responded to HDACi with a robust reduction of cell viability. Known alterations induced by HDACi treatment, such as cell cycle arrest (p21 restoration) and DNA damage induction (phosphorylation of γH2A-X), were induced by especially MS-275 in several CRC cell lines and by FK228 in HCT116. However, this cellular response varied between cell lines, indicating their heterogeneous molecular characteristics [24]. In fact, the cell cycle dynamic control is guided via TP53 and p21, so that mutated TP53 may influence p21 levels upon HDACi treatment: The TP53 mutated cell lines (HT29, DLD1, SW480, Caco2) showed lower p21 levels as compared to wildtype TP53 cell lines (HCT116, LS174T). Upon HDACi treatment, especially MS-275, the TP53 mutated cell lines then indeed showed a marked increase of p21 levels, whereas TP53 wildtype cell lines responded with varying p21 levels. Indeed, a recent study showed that isogenic HCT116 cells with induced TP53 mutation also exhibited a reduced p21 induction post MS-275 treatment compared to HCT116 TP53 wildtype cells [49].
Still, our data suggest that the specific HDACi MS-275 was more effective than FK228 or the broad HDACi SAHA in the six CRC cell lines, which was paralleled by observable increased in histone acetylation levels by MS-275 exclusively. This may indicate that from the three investigated HDAC HDAC3 expression may play a prominent role, since only MS-275, but not FK228 targets HDAC3 [17] or that the class of benzamides are to be favored. Indeed, low/high HDAC3 expression in CRC cell lines (HT29, HCT116) were linked to strong/weak p21 induction post the HDACi butyrate [31]. Clearly further studies need to clarify the precise role of HDAC3 expression with sensitivity or resistance to different types of HDACi.
In summary, this study revealed specific histone marks and histone modifiers to be frequently present in CRCs, to be linked to more aggressive tumor stages at which adjuvant therapy is necessary, and to define histone-related sub-classes of CRCs, with inter- and intra-tumoral (intra-gland) heterogeneity of HDAC. That these histone modifications may indeed be targeted by treatment with predominantly a specific HDACi (MS-275) was demonstrated as proof of principle in vitro using six CRC cell lines. Thus, the present study provides a step forward in the translation of epigenetic alterations to a clinico-pathological setting.
Acknowledgements
The authors acknowledge the excellent laboratory work of Bianca Riedel and Nicola Bittermann. The authors thank Prof. Dr. A. Hecht for providing CRC cell lines LS174T and SW480. This study has been funded by the DKTK Molecular Diagnostics Program as well as in part the Mushett Family Foundation (grants to SL, MW) and the DFG SFB992 (grant to SL).
Disclosure of conflict of interest
None.
References
- 1.Bosman FT, Carneiro F, Hruban RH, Theise ND. WHO Classification of Tumours of the Digestive System. 4 ed. Vol 3. Lyon, France: IAR; 2010. pp. 131–180. [Google Scholar]
- 2.Rosty C, Young JP, Walsh MD, Clendenning M, Walters RJ, Pearson S, Pavluk E, Nagler B, Pakenas D, Jass JR, Jenkins MA, Win AK, Southey MC, Parry S, Hopper JL, Giles GG, Williamson E, English DR, Buchanan DD. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol. 2013;26:825–34. doi: 10.1038/modpathol.2012.240. [DOI] [PubMed] [Google Scholar]
- 3.Douillard JY, Oliner KS, Siena S, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocákova I, Ruff P, Błasińska-Morawiec M, Šmakal M, Canon JL, Rother M, Williams R, Rong A, Wiezorek J, Sidhu R, Patterson SD. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med. 2013;369:1023–34. doi: 10.1056/NEJMoa1305275. [DOI] [PubMed] [Google Scholar]
- 4.Heinemann V, von Weikersthal LF, Decker T, Kiani A, Vehling-Kaiser U, Al-Batran SE, Heintges T, Lerchenmüller C, Kahl C, Seipelt G, Kullmann F, Stauch M, Scheithauer W, Hielscher J, Scholz M, Müller S, Link H, Niederle N, Rost A, Höffkes HG, Moehler M, Lindig RU, Modest DP, Rossius L, Kirchner T, Jung A, Stintzing S. FOLFIRI plus cetuximab versus FOLFIRI plus bevacizumab as first-line treatment for patients with metastatic colorectal cancer (FIRE-3): a randomised, open-label, phase 3 trial. Lancet Oncol. 2014;15:1065–75. doi: 10.1016/S1470-2045(14)70330-4. [DOI] [PubMed] [Google Scholar]
- 5.Van Engeland M, Derks S, Smits KM, Meijer GA, Herman JG. Colorectal cancer epigenetics: Complex simplicity. J. Clin. Oncol. 2011;29:1382–91. doi: 10.1200/JCO.2010.28.2319. [DOI] [PubMed] [Google Scholar]
- 6.Zoratto F, Rossi L, Verrico M, Papa A, Basso E, Zullo A, Tomao L, Romiti A, Lo Russo G, Tomao S. Focus on genetic and epigenetic events of colorectal cancer pathogenesis: Implications for molecular diagnosis. Tumor Biology. 2014:6195–206. doi: 10.1007/s13277-014-1845-9. [DOI] [PubMed] [Google Scholar]
- 7.Ogino S, Goel A. Molecular classification and correlates in colorectal cancer. J Mol Diagn. 2008;10:13–27. doi: 10.2353/jmoldx.2008.070082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Funkhouser WK Jr, Lubin IM, Monzon FA, Zehnbauer BA, Evans JP, Ogino S, Nowak JA. Relevance, Pathogenesis, and testing algorithm for mismatch repair-defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn. 2012;14:91–103. doi: 10.1016/j.jmoldx.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 9.Seppälä TT, Böhm JP, Friman M, Lahtinen L, Väyrynen VM, Liipo TK, Ristimäki AP, Kairaluoma MV, Kellokumpu IH, Kuopio TH, Mecklin JP. Combination of microsatellite instability and BRAF mutation status for subtyping colorectal cancer. Br J Cancer. 2015:1966–75. doi: 10.1038/bjc.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weichert W, Röske A, Niesporek S, Noske A, Buckendahl AC, Dietel M, Gekeler V, Boehm M, Beckers T, Denkert C. Class I histone deacetylase expression has independent prognostic impact in human colorectal cancer: specific role of class I histone deacetylases in vitro and in vivo. Clin Cancer Res. 2008;14:1669–77. doi: 10.1158/1078-0432.CCR-07-0990. [DOI] [PubMed] [Google Scholar]
- 11.Nakazawa T, Kondo T, Ma D, Niu D, Mochizuki K, Kawasaki T, Yamane T, Iino H, Fujii H, Katoh R. Global histone modification of histone H3 in colorectal cancer and its precursor lesions. Hum Pathol. 2012;43:834–42. doi: 10.1016/j.humpath.2011.07.009. [DOI] [PubMed] [Google Scholar]
- 12.Ishihama K, Yamakawa M, Semba S, Takeda H, Kawata S, Kimura S, Kimura W. Expression of HDAC1 and CBP/p300 in human colorectal carcinomas. J Clin Pathol. 2007;60:1205–10. doi: 10.1136/jcp.2005.029165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Spurling CC, Godman CA, Noonan EJ, Rasmussen TP, Rosenberg DW, Giardina C. HDAC3 overexpression and colon cancer cell proliferation and differentiation. Mol Carcinog. 2008;47:137–47. doi: 10.1002/mc.20373. [DOI] [PubMed] [Google Scholar]
- 14.Nosho K, Shima K, Irahara N, Kure S, Firestein R, Baba Y, Toyoda S, Chen L, Hazra A, Giovannucci EL, Fuchs CS, Ogino S. SIRT1 histone deacetylase expression is associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2009;22:922–32. doi: 10.1038/modpathol.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marks PA, Xu WS. Histone deacetylase inhibitors: Potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JH, Choy ML, Ngo L, Foster SS, Marks PA. Histone deacetylase inhibitor induces DNA damage, which normal but not transformed cells can repair. Proc Natl Acad Sci U S A. 2010;107:14639–44. doi: 10.1073/pnas.1008522107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 18.Gissot M, Kim K. How epigenomics contributes to the understanding of gene regulation in Toxoplasma gondii. J Eukaryot Microbiol. 2008;55:476–80. doi: 10.1111/j.1550-7408.2008.00366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Munshi A, Shafi G, Aliya N, Jyothy A. Histone modifications dictate specific biological readouts. J Genet Genomics. 2009;36:75–88. doi: 10.1016/S1673-8527(08)60094-6. [DOI] [PubMed] [Google Scholar]
- 20.Hehlgans S, Storch K, Lange I, Cordes N. The novel HDAC inhibitor NDACI054 sensitizes human cancer cells to radiotherapy. Radiother Oncol. 2013;109:126–32. doi: 10.1016/j.radonc.2013.08.023. [DOI] [PubMed] [Google Scholar]
- 21.Kim JC, Shin ES, Kim CW, Roh SA, Cho DH, Na YS, Kim TW, Kim MB, Hyun YL, Ro S, Kim SY, Kim YS. In vitro evaluation of histone deacetylase inhibitors as combination agents for colorectal cancer. Anticancer Res. 2009;29:3027–34. [PubMed] [Google Scholar]
- 22.Flis S, Gnyszka A, Flis K, Spławiński J. MS275 enhances cytotoxicity induced by 5-fluorouracil in the colorectal cancer cells. Eur J Pharmacol. 2010;627:26–32. doi: 10.1016/j.ejphar.2009.10.033. [DOI] [PubMed] [Google Scholar]
- 23.Morelli MP, Tentler JJ, Kulikowski GN, Tan AC, Bradshaw-Pierce EL, Pitts TM, Brown AM, Nallapareddy S, Arcaroli JJ, Serkova NJ, Hidalgo M, Ciardiello F, Eckhardt SG. Preclinical activity of the rational combination of selumetinib (AZD6244) in combination with vorinostat in KRAS-mutant colorectal cancer models. Clin Cancer Res. 2012;18:1051–62. doi: 10.1158/1078-0432.CCR-11-1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ahmed D, Eide PW, Eilertsen IA, Danielsen SA, Eknæs M, Hektoen M, Lind GE, Lothe RA. Epigenetic and genetic features of 24 colon cancer cell lines. Oncogenesis. 2013;2:e71. doi: 10.1038/oncsis.2013.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sobin LH, Gospodarowicz MK, Wittekind C. Clinical Oncology. 7th ed. Wilmington: Wiley-Blackwell; 2009. TNM classification of malignant tumours; pp. 94–99. [Google Scholar]
- 26.Parc Y, Gueroult S, Mourra N, Serfaty L, Fléjou JF, Tiret E, Parc R. Prognostic significance of microsatellite instability determined by immunohistochemical staining of MSH2 and MLH1 in sporadic T3N0M0 colon cancer. Gut. 2004;53:371–5. doi: 10.1136/gut.2003.019190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shia J. Immunohistochemistry versus microsatellite instability testing for screening colorectal cancer patients at risk for hereditary nonpolyposis colorectal cancer syndrome Part I. The utility of immunohistochemistry. J Mol Diagn. 2008;10:293–300. doi: 10.2353/jmoldx.2008.080031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ahrens TD, Timme S, Hoeppner J, Ostendorp J, Hembach S, Follo M, Hopt UT, Werner M, Busch H, Boerries M, Lassmann S. Selective inhibition of esophageal cancer cells by combination of HDAC inhibitors and Azacytidine. Epigenetics. 2015;10:431–45. doi: 10.1080/15592294.2015.1039216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herz C, Schlürmann F, Batarello D, Fichter CD, Schöpflin A, Münch C, Hauschke D, Werner M, Lassmann S. Occurrence of Aurora A positive multipolar mitoses in distinct molecular classes of colorectal carcinomas and effect of Aurora A inhibition. Mol Carcinog. 2012;51:696–710. doi: 10.1002/mc.20823. [DOI] [PubMed] [Google Scholar]
- 30.Fichter CD, Timme S, Braun JA, Gudernatsch V, Schöpflin A, Bogatyreva L, Geddert H, Faller G, Klimstra D, Tang L, Hauschke D, Werner M, Lassmann S. EGFR, HER2 and HER3 dimerization patterns guide targeted inhibition in two histotypes of esophageal cancer. Int J Cancer. 2014;135:1517–30. doi: 10.1002/ijc.28771. [DOI] [PubMed] [Google Scholar]
- 31.Archer SY, Meng S, Shei A, Hodin RA. p21WAF1 is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc Natl Acad Sci U S A. 1998;95:6791–6. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jass JR. Classification of colorectal cancer based on correlation of clinical, morphological and molecular features. Histopathology. 2007;50:113–30. doi: 10.1111/j.1365-2559.2006.02549.x. [DOI] [PubMed] [Google Scholar]
- 33.Sanford M, Bertagnolli M. Molecular Basis of Colorectal Cancer. N Engl J Med. 2009;361:2449–60. doi: 10.1056/NEJMra0804588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenetics. 2012;4:4. doi: 10.1186/1868-7083-4-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Burkitt DP, Walker AR, Painter NS. Effect of dietary fibre on stools and the transit-times, and its role in the causation of disease. Lancet. 1972;2:1408–12. doi: 10.1016/s0140-6736(72)92974-1. [DOI] [PubMed] [Google Scholar]
- 36.Takeshima H, Wakabayashi M, Hattori N, Yamashita S, Ushijima T. Identification of coexistence of DNA methylation and H3K27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis. 2015;36:192–201. doi: 10.1093/carcin/bgu238. [DOI] [PubMed] [Google Scholar]
- 37.Saijo K, Imamura J, Narita K, Oda A, Shimodaira H, Katoh T, Ishioka C. Biochemical, biological and structural properties of romidepsin (FK228) and its analogs as novel HDAC/PI3K dual inhibitors. Cancer Sci. 2015;106:208–15. doi: 10.1111/cas.12585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karczmarski J, Rubel T, Paziewska A, Mikula M, Bujko M, Kober P, Dadlez M, Ostrowski J. Histone H3 lysine 27 acetylation is altered in colon cancer. Clin Proteomics. 2014;11:24. doi: 10.1186/1559-0275-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lee HY, Tsai AC, Chen MC, Shen PJ, Cheng YC, Kuo CC, Pan SL, Liu YM, Liu JF, Yeh TK, Wang JC, Chang CY, Chang JY, Liou JP. Azaindolylsulfonamides, with a more selective inhibitory effect on histone deacetylase 6 activity, exhibit antitumor activity in colorectal cancer HCT116 cells. J Med Chem. 2014;57:4009–22. doi: 10.1021/jm401899x. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Zang C, Rosenfeld JA, Schones DE, Barski A, Cuddapah S, Cui K, Roh TY, Peng W, Zhang MQ, Zhao K. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weinberger L, Voichek Y, Tirosh I, Hornung G, Amit I, Barkai N. Expression Noise and Acetylation Profiles Distinguish HDAC Functions. Mol Cell. 2012;47:193–202. doi: 10.1016/j.molcel.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mariadason JM. HDACs and HDAC inhibitors in colon cancer. Epigenetics. 2008;3:28–37. doi: 10.4161/epi.3.1.5736. [DOI] [PubMed] [Google Scholar]
- 43.Park SY, Jun JA, Jeong KJ, Heo HJ, Sohn JS, Lee HY, Park CG, Kang J. Histone deacetylases 1, 6 and 8 are critical for invasion in breast cancer. Oncol Rep. 2011;25:1677–81. doi: 10.3892/or.2011.1236. [DOI] [PubMed] [Google Scholar]
- 44.Kim NH, Kim SN, Kim YK. Involvement of HDAC1 in E-cadherin expression in prostate cancer cells; its implication for cell motility and invasion. Biochem Biophys Res Commun. 2011;404:915–21. doi: 10.1016/j.bbrc.2010.12.081. [DOI] [PubMed] [Google Scholar]
- 45.Yang L, Liang Q, Shen K, Ma L, An N, Deng W, Fei Z, Liu J. A novel class I histone deacetylase inhibitor, I-7ab, induces apoptosis and arrests cell cycle progression in human colorectal cancer cells. Biomed Pharmacother. 2015;71:70–8. doi: 10.1016/j.biopha.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 46.Jin JS, Tsao TY, Sun PC, Yu CP, Tzao C. SAHA inhibits the growth of colon tumors by decreasing histone deacetylase and the expression of cyclin D1 and survivin. Pathol Oncol Res. 2012;18:713–20. doi: 10.1007/s12253-012-9499-7. [DOI] [PubMed] [Google Scholar]
- 47.Wang LT, Liou JP, Li YH, Liu YM, Pan SL, Teng CM. A novel class I HDAC inhibitor, MPT0G030, induces cell apoptosis and differentiation in human colorectal cancer cells via HDAC1/PKCδ and E-cadherin. Oncotarget. 2014;5:5651–62. doi: 10.18632/oncotarget.2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hu E, Dul E, Sung CM, Chen Z, Kirkpatrick R, Zhang GF, Johanson K, Liu R, Lago A, Hofmann G, Macarron R, de los Frailes M, Perez P, Krawiec J, Winkler J, Jaye M. Identification of novel isoform-selective inhibitors within class I histone deacetylases. J Pharmacol Exp Ther. 2003;307:720–8. doi: 10.1124/jpet.103.055541. [DOI] [PubMed] [Google Scholar]
- 49.Sonnemann J, Marx C, Becker S, Wittig S, Palani CD, Krämer OH, Beck JF. P53-dependent and p53-independent anticancer effects of different histone deacetylase inhibitors. BJC. 2014;110:656–67. doi: 10.1038/bjc.2013.742. [DOI] [PMC free article] [PubMed] [Google Scholar]