

Abstract
Epilepsy genes deliver critical insights into the molecular control of brain synchronization and are revolutionizing our understanding and treatment of the disease. The epilepsy-associated genome is rapidly expanding, and two powerful complementary approaches, isolation of de novo exome variants in patients and targeted mutagenesis in model systems, account for the steep increase. In sheer number, the tally of genes linked to seizures will likely match that of cancer and exceed it in biological diversity. The proteins act within most intracellular compartments and span the molecular determinants of firing and wiring in the developing brain. Every facet of neurotransmission, from dendritic spine to exocytotic machinery, is in play, and defects of synaptic inhibition are over-represented. The contributions of somatic mutations and noncoding microRNAs are also being explored. The functional spectrum of established epilepsy genes and the arrival of rapid, precise technologies for genome editing now provide a robust scaffold to prioritize hypothesis-driven discovery and further populate this genetic proto-map. Although each gene identified offers translational potential to stratify patient care, the complexity of individual variation and covert actions of genetic modifiers may confound single-gene solutions for the clinical disorder. In vivo genetic deconstruction of epileptic networks, ex vivo validation of variant profiles in patient-derived induced pluripotent stem cells, in silico variant modeling and modifier gene discovery, now in their earliest stages, will help clarify individual patterns. Because seizures stand at the crossroads of all neuronal synchronization disorders in the developing and aging brain, the neurobiological analysis of epilepsy-associated genes provides an extraordinary gateway to new insights into higher cortical function.
About 25 years have elapsed since the first gene linked to the clinical expression of seizures was recognized1, and the list has now exploded to 500 loci in human and mouse (Mouse Genome Informatics; http://www.informatics.jax.org/humanDisease.shtml). Because the overlap of orthologous genes is small, there is little reason to believe that their number is saturated in either organism or that their utility is fully realized. However, defying the laws of economics, neither is their individual value declining. Although none of the genes directly explains why, with little warning, an electrographic seizure starts, spreads or stops, and only a few pinpoint the precise circuitry involved, each can help provide a molecular diagnosis for the physician and patient, and a deeper understanding of the natural history of the disorder. For the neurobiologist, the gene is a molecular entry point that triggers a logical sequence of questions designed to understand how a given mutation altering membrane excitability or synaptic release creates one or more epilepsy phenotypes, and how developmental plasticity and permissive brain network wiring properties allow dysrhythmic electroencephalographic (EEG) signatures to appear. For neurologists, the genes also force continuous reappraisal of the classifications of the clinical disorder itself2. Individuals with epilepsy and their families are gratified to learn that their mysterious condition can be explained by a singular inborn error, and that this information may ultimately benefit their care. Most importantly, each new gene calls for immediate consideration of whether it offers a critical opportunity worthy of major investment to target new drug development.
Now that individuals with unexplained epilepsy are under increasing genomic scrutiny, powerful bioinformatic techniques3–6 are being applied to filter their variome for candidate monogenic causes of seizures. Early results of these approaches suggest the impending arrival of a golden era of diagnostic and therapeutic opportunity for neurodevelopmental disorders once a list of causal gene variants from an individual can be generated and interpreted in a timely way7,8, while simultaneously acknowledging their current limitations in analyzing noncoding regions and resolving smaller copy number variants. Even larger obstacles remain, however. First, in most cases, estimating the protein damage inflicted by a novel variant requires supportive functional validation, even in genes already known to be causative. Gain or loss of function, sometimes separated by variation in only a single base pair, is an essential distinction for both prognosis and therapy, and high-throughput assays of the functional motifs among alternatively spliced proteins have not yet been devised for most neuronal genes. Second, in the absence of a high-value singular de novo candidate mutation, in silico models and algorithms to interpret the aggregate clinical valence of a complex variant profile composed of multiple genes of minor causative, if not suppressant, effects on relevant cortical microcircuits remain on the distant horizon.
Ironically, the most compelling reason for optimism that the accuracy of human molecular diagnosis of this condition will steadily improve amid this embarrassment of riches is the certainty that many more causative genes for epilepsy remain to be found. In the first large-scale exome sequencing study in epilepsy, only 7% of a clinically stratified patient cohort of 264 patient-parent trios with epileptic encephalopathy could be given a confident genetic diagnosis, and these cases were considered solved because a link between epilepsy and these genes, if not the mutations themselves, had been previously validated9. The other protein-damaging de novo mutations unearthed in novel candidate genes, however plausible, still require functional evidence of pathogenicity for epileptic encephalopathy before meaningful clinical utility is assured. This leaves over 90% of these carefully studied, but clinically heterogeneous, cases still without an individual genetic diagnosis despite the presence of >300 other de novo mutations and substantial inherited gene variation in this group.
In surveying the broadening genetic landscape of epilepsy, this review will focus on the neurogenetic pathways and the next generation of experimental opportunities available for combining genomic discovery with rapid mutagenesis to validate and prioritize emerging targets. Fortuitously, advances in stem cell biology and genetic engineering are keeping pace with the $1,000 genome, and we have reached a point where gene validation and hypothesis-driven functional discovery can be efficiently harnessed. Ultra-rapid, single-base-pair editing technologies can now be applied to create and validate human gene defects in mouse models10,11 and patient-derived stem cells12,13. Rather than wait for the expected influx of human variants, we can preemptively explore still-uncharted mechanisms of epileptogenesis to fill in the gaps of a biological scaffold that converges on impaired inhibition in brain networks. This discovery engine is well oiled and fueled by ongoing human sequencing and conventional mutagenesis (Fig. 1). If targeted scaffold genes were also selected based on drugability or intersection with other neurological disease phenotypes, their utility might be even more quickly leveraged. This realization means that the molecular neurobiology of each candidate gene is an invaluable guide to prioritization, because a denser functional network interactome will speed the positioning of novel patient-derived variants into a mechanistic epilepsy pathway and refine the disease map for better diagnostic utility.
Figure 1.
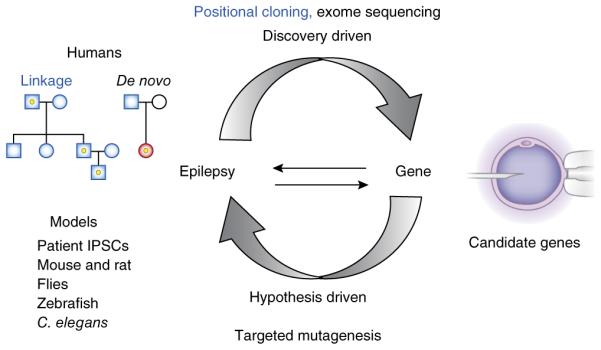
Inherited gene variants are discovered in human epilepsy pedigrees and de novo variants are detected in proband-parental trios. These require physiological verification in orthologous models to demonstrate epileptic phenotypes. Pathway-driven candidate genes are mutagenized and validated in models for rediscovery in human exomes. Each revolution of this discovery cycle results in a validated diagnostic gene and an experimental biological test system to search for therapy. IPSC, induced pluripotent stem cell.
The genetic roots: first-, second- and third-wave gene discovery
The early gene discoveries create a robust initial template to populate with inborn biochemical errors. The first human Mendelian genes appeared in the analysis of pedigrees of syndromic epilepsies, disorders where seizures are a notable, but inconsistent, element of a complex disease phenotype affecting multiple tissues, beginning with myoclonic epilepsy with ragged red fibers, a mitochondrial disorder affecting brain and skeletal muscle1. Dozens of subsequent examples involving membrane proteins and intermediary metabolism in tissues ranging from cochlear hair cells to pancreas, thyroid, adrenal gland, kidney, heart and skin are now found in the Online Mendelian Inheritance in Man database. The second wave arrived shortly thereafter, based on mapped loci in human pedigrees with generalized epilepsy syndromes, where seizures form the major if not sole clinical presentation, subcategorized by seizure type, EEG pattern, age of onset and other salient features, which arise as a monogenic and typically dominant trait. A missense mutation in the gene encoding the neuronal nicotinic acetylcholine receptor-α 4 subunit was the first in this category14. Members of the voltage- and ligand-gated membrane ion channel and transporter gene superfamily, first identified in the Drosophila hyperexcitability mutant shaker15 and followed by many more identified in mouse mutants, now define the predominant mechanistic pathway for momentary epileptiform instability in neural circuits—namely, the instantaneous control of intrinsic membrane excitability and synaptic transmission. These functionally validated genes, further supported by evidence of de novo origin16, have rapidly solidified the use of genetic testing in the pediatric epilepsy clinic. Recent expert reviews17–20 describe the best-studied clinical phenotypes and pathophysiology of specific individual genes.
Among these clinically defined syndromes, as in all neurological disease, evidence for substantial genetic heterogeneity quickly arose, and it became necessary to claim merely that “a gene,” rather than “the gene,” for each of these syndromes had been found. Indeed, few seizure syndromes classified by the International League Against Epilepsy (ILAE) arise from a solitary gene (for example, 135 entries for epileptic encephalopathy are listed in the Online Mendelian Inheritance in Man database). Each of the genes for a named syndrome shows its own partially overlapping regional brain distribution with independent patterns of developmental expression in specific cell types, and distinct mutations create many possible alterations in protein isoforms and function, adding to the potential clinical spectrum of each gene. The ILAE classification therefore portrays only an idealized, consensus clinical picture; for now, genotype and phenotype classifications are viewed differently by clinicians and basic researchers. One major effect of the emerging genetic heterogeneity of epilepsy syndromes has been to force a careful reexamination of both the clinical attributes and the most appropriate target for gene-directed therapy when multiple potentially causative mechanisms coexist, and the field is currently undecided on the prognostic precision, which may be age-dependent, for many of the genes. Clinicians point out that the disease in most individuals with sporadic epilepsy older than 12 years of age does not fit into any electroclinical syndrome, leaving these cases in a large diagnostic no-man’s-land21.
Understandably, this category of epilepsy raises its own critical deterministic issues, and the spectrum of temporal onsets during childhood and adolescence may be one of its more mysterious attributes. There is also substantial puzzlement when genes with disparate anatomical expression patterns within neuronal networks lead to a shared epilepsy syndrome. Equally problematic is the occurrence of multiple distinct clinical phenotypes apparently arising from mutation of a single epilepsy-associated gene. For example, mutations in KCNQ2, a gene that was originally linked to a familial benign neonatal epilepsy syndrome22, are now identified in catastrophic lifelong epileptic encephalopathy23,24. Recent evidence suggests the more severe clinical form may be due to a dominant-negative mechanism25. Sodium channel β1-subunit missense mutations (SCN1B) were originally described in individuals with the readily treated generalized epilepsy with febrile seizures syndrome26; however, mutation of this regulatory subunit, along with some missense mutations and essentially all de novo truncations in the partner SCN1A pore-forming subunit, can result in a severe pharmacoresistant epileptic encephalopathy (Dravet syndrome)27. Mutations in the sodium-gated potassium channel KCNT1 (refs. 28–30) and glucose transporter GLUT1 (ref. 31) are found in a particularly broad spectrum of phenotypes. The basis for the dramatic pleiotropic role of these genes, and whether the phenotype is a product of a specific mutated allele, epigenetic and posttranscriptional changes, epistatic variation, or the age of each individual, is a goal of continuing genotype-phenotype correlation studies for each gene. Rapid mutagenesis brings some tractability to understanding the major categories of function controlled by each gene; ultimately human sequence databases with phenotypic curation, such as ClinVar32, will point to common variants to prioritize the analysis. In general, all of these attributes, including still-unknown environmental factors, contribute to the final phenotype.
A third wave of genes achieving prominence includes those found in syndromic epilepsies composed of purely neurological but seemingly unrelated comorbidities, such autism33,34, familial Alzheimer’s disease35,36, cognitive disorders9,37, dyskinesias38, migraine39 and ataxias40,41, illustrating how a seizure disorder can appear alongside or in the wake of another neurological deficit during its neurodegenerative trajectory. The genetic overlap with autism is particularly notable42. In general, as circuits fail in the brain, they may progress through a stage where an unbalanced inhibitory deficit predominates, leading to network dysrhythmia and seizures. These genes bridge the traditionally narrow perspective of epilepsy as a static hyperexcitability disorder with the broader view of synchronization disorders that mark different stages of brain disease arising from errors of cell proliferation43, cell migration44 and synaptogenesis on the one hand, and inflammation, cell death and homeostatic repair on the other.
Genes linking seizures, central autonomic disorders and premature mortality underlying sudden unexpected death in epilepsy (SUDEP) are vivid examples of hypothesis- and pathway-driven gene discovery in epilepsy. Beginning with the finding that SCN5A, a sodium channel gene associated with cardiac long QT syndrome (LQTS) linked to sudden death, is expressed in the amygdala, the limbic nucleus regulating cardiac representation in the forebrain45, mutations in the human gene encoding the KCNQ1 potassium channel (the most common cause of human LQTS and sudden death) were evaluated in mouse models and shown to replicate the SUDEP phenotype46, opening the door to many LQT genes as candidates for shared brain and cardiac arrhythmia. KCNA1 encodes a second, non-LQTS potassium channel linked to brainstem and vagal nerve-driven cardiac arrhythmias, severe seizures and early mortality47. Sentrin/SUMO protease protein 2 (SENP2), a gene linked to SUMOylation and membrane trafficking of these potassium ion channels in heart and brain, is a new member of this category48. Interestingly, molecular autopsy of human SUDEP cases reveals that mutations of these genes can also form complex risk profiles, opening an opportunity for early prognosis and potential for intervention in this life-threatening condition49.
Defining major and branch pathways: lessons from cancer
The many failed genome-wide association studies seeking to identify variants of genes enriched in the epilepsy population, despite technical limitations50, have nevertheless succeeded in producing aggregate support for the portrayal of the genetic landscape of epilepsy as a broadly heterogeneous group of disorders largely composed of individuals with rare (<1%) and private mutations drawn from an expansive list of single genes. By this definition, no gene is a common cause of epilepsy, and no common epilepsy syndrome is caused by only a single gene, contributing to the reason why even the best known loci for generalized epilepsies escape or barely reach that level of significance. With so many disparate biochemical functions represented, we need a more detailed map of how to envision minor genetic convergence on what ultimately must be a widely shared biological substrate for neuronal network instability.
Cancer genomics may offer some instructive analogies for genetic pathway building in epilepsy. Neoplasia is a defect contained within single cells that continuously divide due to uncontrolled cell growth and, to avoid destruction, is abetted by a permissive immune system. Inherited epilepsy represents a network defect in the development of eurhythmic electrical signaling among an ensemble of neurons and their dynamic stabilization by intrinsic and extrinsic inhibitory processes. Both diseases arise from germline and somatic mutations, but in very different proportions. In cancer, there are relatively few defined cancer genes in the germ line51,52, whereas the majority of tumors arise sporadically from de novo mutation in distinctive cell types. When exome sequencing is carried out in these tumors and compared with genomic DNA, nearly all mutations found (current census ~522 genes; COSMIC Database, http://cancer.sanger.ac.uk/cancergenome/projects/census/) are somatic53. In epilepsy, the reverse is true; although somatic mutation in neurons leading to epilepsy has long been considered rare simply due to lack of evidence, this mechanism is receiving new attention. For example, structural imaging of bilateral brain malformations helped identify neuronal migration defects underlying distinctive phenotypes such as double cortex and perinodular heterotopias, leading to isolation of their germline mutations54. In contrast to diffuse bilateral defects, somatic mutation is an attractive mechanism for focal cerebral dysplasias, and recent analysis has revealed somatic mutations in these and related genes (DCX and PAFAH1B1 (LIS1)) in cases of subcortical band heterotopia and polymicrogyria55, as well as somatic mutation in the MTOR pathway genes PIK3CA, AKT3 and MTOR in one form of hemimegancephaly56. Technological advances allowing increased imaging resolution to identify what were formerly considered microscopic structural lesions signal the opportunity to isolate other somatic gene mutations from surgical tissue samples when a genomic mutation is not found in blood55,57. Even without imaging findings, seizures in restricted cortical foci could still arise from somatic mutation of intrinsic excitability genes that do not alter cell migration and patterning. Similar to tumor exome profiling, exomic analysis of resected epileptic foci for somatic variants may reveal further examples of pathogenic mosaicism, and represents an interesting new direction for gene discovery. While little is known regarding the genetic complexity of single neurons in the brain vis-à-vis their immediate neighbors58, strategies are emerging to better understand single cell identity in the brain59,60.
Variation in the cancer genome occurs in genes encoding nuclear enzymes, intracellular signal transduction, and cell surface/secreted growth factors in three main pathways: DNA damage, mismatch repair, and the fidelity of replicative machinery. Among 522 tumor genes, some 140 are considered to be ‘drivers’ and the remainder, in some cases many thousands, ‘passengers’ with minor individual contributions52,61. In epilepsy, mutations of hundreds of genes with driver or monogenic status are drawn chiefly from cellular pathways including membrane excitability, neurotransmission, and neuronal migration, but perhaps two-thirds of them have not yet been assigned to these categories. Nevertheless it is likely that a significant fraction of the epilepsy genome will be determined to exert a predominant impact on inhibitory processes. In a major form of childhood epilepsy (absence epilepsy), essentially all known genes are ion channels involved in phasic and tonic inhibition in the thalamocortical circuit62.
If defective inhibition has emerged as a central point of convergence, the number of pathways to disinhibition in epilepsy, as told by its genes, is far more intricate. Not only are interneuron cell number, migration, maturation and synaptogenesis key to effective inhibition, but all mechanisms that contribute to inhibitory neurotransmitter synthesis, vesicular packaging, release and reuptake, postsynaptic receptor activation and the intracellular chloride gradient are included, along with metabolic substrates required for normal firing and synaptic plasticity (Fig. 2). In epilepsy, defects in synaptic inhibition that begin as a germline, cell-autonomous defect may engender significant activity-driven molecular63 and cellular64 remodeling of the affected networks throughout development, which may homeostatically reinforce or weaken their role. Within each of these genetically regulated steps, there is ample evidence to explain the cellular diversity65, developmental ontogeny66 and plasticity67 of inhibitory defects underlying epilepsy phenotypes.
Figure 2.

A significant monogenic driver pathway for epilepsy can be constructed from current gene evidence implicating mutations in genes that impair every aspect of synaptic inhibitory transmission from early development through maturation of adult GABA neurotransmission. Only a partial listing of known genes is shown, and many others remain to be functionally assigned to their target location and impact on excitation and inhibition in epileptic microcircuits.
Much work is necessary to clarify the roles of the still functionally unassigned genes and position them in the overall biology of synaptic inhibition, whether expressed in interneurons, excitatory cells or even astrocytes. Interestingly, within the larger gene list are oncogenes directly linked to epilepsy phenotypes (PTEN, TSC1, TSC2, CDKL5, RBFOX1, WWOX). Like cancer, epilepsy also can be unleashed by autoimmune dysregulation, both in the presence and absence of neoplasia (paraneoplastic and non-paraneoplastic autoimmune epilepsies). Targets of the inflammatory proepileptic autoantibodies overlap with those genetically linked to epilepsy (LGI1, GABAA receptor, K+ channels, NMDA receptors, Ca2+ channels)68.
In cancer research, characterizing tumors by their genomic mutation burden is the current standard of analysis, and their contribution is assessed numerically by how frequently they are found, a purely bioinformatic measure of causality. Although the primary defect begins in a single cell, clonal expansion in a tumor diversifies the mutation pattern, and tumors that respond initially to one treatment may become resistant as their mutational profile evolves. Pharmacoresistance in cancer may therefore depend on mutational complexity, a fundamental principle that may also apply to epilepsy. Although the most potent cancer gene pathways (Wnt, transforming growth factor-β, RTK/RAS/P13K and p53) are few in number61, the distribution tail of all genes mutated in tumors is long and characteristic of a given subtype51. These mutations have an unknown contribution to the underlying process, but fall somewhere along the causative pathways; thus the pathogenicity and aggressiveness of a tumor is likely not the result of a single driver gene, but the extent and nature of genetic disruption in secondary branches. Defining these branches for epilepsy will facilitate mapping future epilepsy-associated genes and building genetic profiles of pharmacosensitivity.
Intracellular, extracellular and circuit complexity
Prospecting for new candidate genes begins with an exploration of the relevant protein interactome, but linking them in a GO/KEGG biological pathway is challenging when they are not expressed at the same time in the same cell, and it is usually unclear at what point disease genes interact anatomically and biochemically during development. Therefore, solving an individual case of genetic epilepsy requires confronting the reality of the three major forms of complexity in the disease: syndrome heterogeneity, downstream complexity and epistatic complexity. A beginning step for the first of these, to understand how two genes cause or modify the same phenotype, is to localize their temporal and spatial overlap within a common circuit. However, the analysis of single-gene mouse mutants with epilepsy provides clear examples of the challenge of inferring epileptic circuitry from expression of the mutated gene alone, especially in neurons which are designed for activity-dependent changes in their transcriptome (Box 1).
Box 1. Four caveats to localizing epileptic circuitry by expression patterns of mutated genes.
Widespread mutated gene transcripts mask selective functional failure in specific cells and networks94
Selective vulnerability is determined not by the presence of defective proteins, but by the absence of homologous subunits with overlapping function75
Mutation triggers maladaptive downstream remodeling, leading to EEG phenotype95
• Elevated mutated gene dose leads to ectopic expression in epileptic brain96
A subsequent strategy to localize and define the impact of a variant on disinhibition is to genetically deconstruct epileptic microcircuits using conditional genetic engineering, starting by, for example, selectively expressing cell type-specific mutations impairing neurotransmitter release in the neocortex. Using this innovative strategy, selective ablation of P/Q type calcium ion channels encoded by Cacna1a from cortical GABAergic interneurons containing parvalbumin and somatostatin reveals distinct EEG seizure patterns when these circuit breakers are switched off alone or in combination with excitatory neurons69, demonstrating the balancing contribution of each subtype. This strategy has also been used to explore the developmental timing of longer-range inputs on the maturation of forebrain synchronization, for example, those from cerebellum to the thalamus70,71. Conditional modification of GABAergic interneurons versus excitatory cells adds further evidence supporting the overall model of epilepsy as a disruption of cortical excitatory/inhibitory imbalance. Selective elimination of Scn1a from inhibitory interneurons produces a seizure phenotype in a mouse model of Dravet syndrome that is more severe than that produced by global deletion72. When the gene is eliminated from only excitatory cells, seizures are not present, suggesting that excitation counterbalances the disinhibition. A similar analysis in mouse models of Rett syndrome showed seizures when Mecp2 was removed from interneurons73; in this model, when the gene is eliminated from only excitatory cells, seizures are still present, but there is a secondary impairment of GABAergic inhibition74. Together, these conditional expression studies show that cell type-specific levels of expression of the mutated gene throughout brain circuitry have a major role in seizure expressivity, pointing to the potential for strong modulatory influences of microRNAs and other determinants of patterned gene transcription levels among neuronal networks.
Downstream gene complexity is also exemplified by mutation of a single gene that alters multiple proteins. For example, when Cacnβ4, a single cytosolic regulatory subunit of a voltage-gated calcium ion channel that promiscuously heterodimerizes with multiple, pore-forming α-subunits, is lost, other homologous family members (β1–3) with alternative functional effects are reshuffled, modulating multiple distinct calcium currents in unpredictable patterns75. Another example is when a locus exerts editorial control over transcription and splice patterns of dozens of genes. Dysregulation of microRNAs with hundreds of gene targets has been identified in epilepsy tissue, and their targets may be related to lesion progression76. The fact that seizures themselves dynamically regulate microRNAs creates additional complexity but may offer therapeutic opportunities77. Mutations affecting DNA and RNA binding proteins also pose a major challenge to understanding how the network has been remodeled78,79. For example, epilepsy caused by loss of NOVA2, an RNA binding protein with control over the splice forms of 34 synaptic proteins, many of which are known to cause epilepsy, is an excellent example of this group80 (Fig. 3).
Figure 3.

Two extreme examples of single and multigenic biological complexity in epileptic brain. Left, epileptic phenotype in mouse mutant with a solitary Nova2 knockout arises from alterations in splice patterns of a cassette of 34 synaptic proteins78 (reproduced with permission from ref. 78). Many of these targets are known monogenic causes of epilepsy. Right, extensive multigenic complexity in the ion channel variant profile (channotype) of individuals with sporadic epilepsy is similar to that found in unaffected cases. Many affected individuals show multiple non-synonymous single nucleotide variants in human epilepsy genes (hEP), and clinical status depends on the pattern, not absolute load, of variants. In silico modeling of combinatorial effects of multiple ion channel variants in a single model neuron demonstrates how profiles produce a spectrum of single cell firing patterns81 (reproduced from ref. 81 with permission from Elsevier).
Epistatic complexity, in which combinatorial effects of multiple independent gene mutations shape the phenotype, has been explored in human by parallel sequence profiling of ion channel subunit genes, the largest single group of causal genes for epilepsy81. Over 400 ion channel genes are known, and multiple deleterious variants in this family colocalize in cellular compartments where they share precise control over membrane excitability and can finely tune the oscillatory behavior of any circuit. Their contribution to cellular firing patterns during development is cell-type specific and activity-dependent67,82. The fact that two causative gene mutations can mask each other’s phenotype by counteracting their opposing effects on transmitter release is strong proof of principle for their ability to act as potent comodifiers83. This Janus-like function of deleterious epileptogenic channel mutations playing the dual parts of causative gene and modifier can explain their presence in multiple clinical phenotypes, including neurologically unaffected cohorts81 and, given their prevalence in epilepsy, could also be a source of pharmacoresistance. Because each ion channel contributes widely differing amounts of depolarizing or hyperpolarizing current, it is also easy to appreciate why the absolute number of mutated genes present in an individual (channel gene mutation load) is far less informative than the specific pattern and localization of aberrant membrane conductances. Exploring this dimensionality in CNS microcircuits will greatly benefit from advances in in silico analysis of single neurons and small, defined invertebrate networks84,85. Assigning phenotypic causation to any single channel variant will become more complex as exome variant profiles from other disease categories are aggregated and become more accessible in ClinVar and other databases. When deleterious mutations are detected in unaffected individuals, the meaning of ‘causation’ becomes significantly less clear.
Suppressor gene discovery
Defining modifier alleles in neural hyperexcitability pathways was the early province of Drosophila genetic research86, and is now an essential direction for future epilepsy neurogenetics and drug discovery. There has been a small but steady push to uncover suppressor genes using classical mapping87–89 and combinatorial approaches83,90 in experimental models. Now that changes in these genes can be routinely detected in human exome studies, incorporating them in a diagnostic variant profile analysis will be critical to accurately define risk. The size and structure of this category is unknown. Some modifiers are obvious and may be highly dependent on a specific causative gene, for example, heteromeric subunits of a voltage- or ligand gated ion channel. Others may be more generic; for example, genetic deletion of Mapt (formerly Tau), encoding a microtubule-associated protein that colocalizes in axons with two proepileptic ion channels (encoded by Kcna1 and Scn1a), suppresses seizures and sudden unexpected death (SUDEP) in both of these mouse models91,92.
Acknowledging the presence of modifiers has long been the central caveat for functional analysis of ion channel variants in heterologous cells93. Cells derived from induced pluripotent stem cells will have an extremely important role in clarifying the native behavior of ion channel variants by studying membrane currents in context amidst a patient’s own genomic background. Comparisons of these currents in cells from probands and unaffected parental trios can help functionally validate candidate de novo mutations. In pioneering studies, Dravet syndrome mutations in neurons and cardiomyocytes are being analyzed in individuals to directly verify the effects of their mutations on the biophysical behavior of the channel and to guide therapy13.
Epilogue
Given the potential scientific and clinical return from each new gene, there is a high priority to hasten their discovery, study their singular and collective neurobiological impact, and decipher the genetic script of epilepsy. By analogy with the history of linguistics, we are leaving the stone age of proto-writing, when language was characterized by single ideographic glyphs etched into rocks, and entering the era of smelting copper and alloying it with tin, when writing merged with grammar, encoding ideas so they could be replicated and their meaning reconstructed. This is the bronze age of epilepsy neurogenetics, when single genes are no longer interrogated one at a time, but cast with dozens of interactors and reassembled to define neural network behavior. The convergence of epilepsy and many other neurodevelopmental disorders on synaptic inhibition suggests that assembling scaffolds of genes that regulate interneuron phenotypes, and defining permissive and protective combinations of variants, will be of broad significance to help predict risk and accelerate the discovery of preventative therapies for these devastating disorders.
ACKNOWLEDGMENTS
Supported by US National Institutes of Health NS29709, NINDS Center for SUDEP Research (NS090340) and The Blue Bird Circle Foundation.
Footnotes
COMPETING FINANCIAL INTERESTS The author declares no competing financial interests.
Reference
- 1.Shoffner JM, et al. Myoclonic epilepsy and ragged-red fiber disease (MERRF) is associated with a mitochondrial DNA tRNA(Lys) mutation. Cell. 1990;61:931–937. doi: 10.1016/0092-8674(90)90059-n. [DOI] [PubMed] [Google Scholar]
- 2.Berg AT, et al. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 3.MacArthur DG, et al. Guidelines for investigating causality of sequence variants in human disease. Nature. 2014;508:469–476. doi: 10.1038/nature13127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petrovski S, et al. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013;9:e1003709. doi: 10.1371/journal.pgen.1003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knecht C, Krawczak M. Molecular genetic epidemiology of human diseases: from patterns to predictions. Hum. Genet. 2014;133:425–430. doi: 10.1007/s00439-013-1396-y. [DOI] [PubMed] [Google Scholar]
- 6.Katsonis P, et al. Single nucleotide variations: biological impact and theoretical interpretation. Protein Sci. 2014;23:1650–1666. doi: 10.1002/pro.2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Veltman JA, Brunner HG. De novo mutations in human genetic disease. Nat. Rev. Genet. 2012;13:565–575. doi: 10.1038/nrg3241. [DOI] [PubMed] [Google Scholar]
- 8.Hoischen A, Krumm N, Eichler EE. Prioritization of neurodevelopmental disease genes by discovery of new mutations. Nat. Neurosci. 2014;17:764–772. doi: 10.1038/nn.3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epi4K Consortium et al. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Wang H, Jaenisch R. Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc. 2014;9:1956–1968. doi: 10.1038/nprot.2014.134. [DOI] [PubMed] [Google Scholar]
- 11.Yin H, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32:551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Velasco I, et al. Generation of neurons from somatic cells of healthy individuals and neurological patients through induced pluripotency or direct conversion. Stem Cells. 2014;32:2811–2817. doi: 10.1002/stem.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Y, et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol. 2013;74:128–139. doi: 10.1002/ana.23897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Steinlein OK, et al. A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 1995;11:201–203. doi: 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- 15.Tempel BL, et al. Sequence of a probable potassium channel component encoded at Shaker locus of Drosophila. Science. 1987;237:770–775. doi: 10.1126/science.2441471. [DOI] [PubMed] [Google Scholar]
- 16.De Jonghe P. Molecular genetics of Dravet syndrome. Dev. Med. Child Neurol. 2011;53(suppl. 2):7–10. doi: 10.1111/j.1469-8749.2011.03965.x. [DOI] [PubMed] [Google Scholar]
- 17.Hildebrand MS, et al. Recent advances in the molecular genetics of epilepsy. J. Med. Genet. 2013;50:271–279. doi: 10.1136/jmedgenet-2012-101448. [DOI] [PubMed] [Google Scholar]
- 18.Hirose S, et al. SCN1A testing for epilepsy: application in clinical practice. Epilepsia. 2013;54:946–952. doi: 10.1111/epi.12168. [DOI] [PubMed] [Google Scholar]
- 19.Maheshwari A, Noebels JL. Steinlein OK, editor. Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. in Genetics of Epilepsy. Prog. Brain Res. 2014;213:223–252. doi: 10.1016/B978-0-444-63326-2.00012-0. [DOI] [PubMed] [Google Scholar]
- 20.Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th. Oxford Univ. Press; 2012. [PubMed] [Google Scholar]
- 21.French JA. ILAE classification redux: ready for prime time? Epilepsy Curr. 2014;14:84–85. doi: 10.5698/1535-7597-14.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh NA, et al. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat. Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 23.Cooper EC. Potassium channels (including KCNQ) and epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th. Bethesda, Maryland, USA; National Center for Biotechnology Information: 2012. [PubMed] [Google Scholar]
- 24.Claes LR, et al. De novo KCNQ2 mutations in patients with benign neonatal seizures. Neurology. 2004;63:2155–2158. doi: 10.1212/01.wnl.0000145629.94338.89. [DOI] [PubMed] [Google Scholar]
- 25.Orhan G, et al. Dominant-negative effects of KCNQ2 mutations are associated with epileptic encephalopathy. Ann. Neurol. 2014;75:382–394. doi: 10.1002/ana.24080. [DOI] [PubMed] [Google Scholar]
- 26.Wallace RH, et al. Febrile seizures and generalized epilepsy associated with a mutation in the Na+-channel β1 subunit gene SCN1B. Nat. Genet. 1998;19:366–370. doi: 10.1038/1252. [DOI] [PubMed] [Google Scholar]
- 27.Brunklaus A, Zuberi SM. Dravet syndrome–from epileptic encephalopathy to channelopathy. Epilepsia. 2014;55:979–984. doi: 10.1111/epi.12652. [DOI] [PubMed] [Google Scholar]
- 28.Kim GE, Kaczmarek LK. Emerging role of the KCNT1 Slack channel in intellectual disability. Front. Cell. Neurosci. 2014;8:209. doi: 10.3389/fncel.2014.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barcia G, et al. De novo gain-of-function KCNT1 channel mutations cause malignant migrating partial seizures of infancy. Nat. Genet. 2012;44:1255–1259. doi: 10.1038/ng.2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heron SE, et al. Missense mutations in the sodium-gated potassium channel gene KCNT1 cause severe autosomal dominant nocturnal frontal lobe epilepsy. Nat. Genet. 2012;44:1188–1190. doi: 10.1038/ng.2440. [DOI] [PubMed] [Google Scholar]
- 31.Pearson TS, et al. Phenotypic spectrum of glucose transporter type 1 deficiency syndrome (Glut1 DS) Curr. Neurol. Neurosci. Rep. 2013;13:342. doi: 10.1007/s11910-013-0342-7. [DOI] [PubMed] [Google Scholar]
- 32.Landrum MJ, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 2014;42:D980–D985. doi: 10.1093/nar/gkt1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peñagarikano O, et al. Absence of CNTNAP2 leads to epilepsy, neuronal migration abnormalities, and core autism-related deficits. Cell. 2011;147:235–246. doi: 10.1016/j.cell.2011.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ozkan ED, et al. Reduced cognition in Syngap1 mutants is caused by isolated damage within developing forebrain excitatory neurons. Neuron. 2014;82:1317–1333. doi: 10.1016/j.neuron.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Palop JJ, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron. 2007;55:697–711. doi: 10.1016/j.neuron.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lopera F, et al. Clinical features of early-onset Alzheimer disease in a large kindred with an E280A presenilin-1 mutation. J. Am. Med. Assoc. 1997;277:793–799. [PubMed] [Google Scholar]
- 37.Zhu PJ, et al. Suppression of PKR promotes network excitability and enhanced cognition by interferon-γ-mediated disinhibition. Cell. 2011;147:1384–1396. doi: 10.1016/j.cell.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heron SE, et al. PRRT2 mutations cause benign familial infantile epilepsy and infantile convulsions with choreoathetosis syndrome. Am. J. Hum. Genet. 2012;90:152–160. doi: 10.1016/j.ajhg.2011.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinzen EL, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol. 2014;13:503–514. doi: 10.1016/S1474-4422(14)70011-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ortolano S, et al. Loss of GABAergic cortical neurons underlies the neuropathology of Lafora disease. Mol. Brain. 2014;7:7. doi: 10.1186/1756-6606-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuura T, et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000;26:191–194. doi: 10.1038/79911. [DOI] [PubMed] [Google Scholar]
- 42.Betancur C. Etiological heterogeneity in autism spectrum disorders: more than 100 genetic and genomic disorders and still counting. Brain Res. 2011;1380:42–77. doi: 10.1016/j.brainres.2010.11.078. [DOI] [PubMed] [Google Scholar]
- 43.Liu M, et al. Loss of BETA2/NeuroD leads to malformation of the dentate gyrus and epilepsy. Proc. Natl. Acad. Sci. USA. 2000;97:865–870. doi: 10.1073/pnas.97.2.865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ross ME, Walsh CA. Human brain malformations and their lessons for neuronal migration. Annu. Rev. Neurosci. 2001;24:1041–1070. doi: 10.1146/annurev.neuro.24.1.1041. [DOI] [PubMed] [Google Scholar]
- 45.Hartmann HA, et al. Selective localization of cardiac SCN5A sodium channels in limbic regions of rat brain. Nat. Neurosci. 1999;2:593–595. doi: 10.1038/10147. [DOI] [PubMed] [Google Scholar]
- 46.Goldman AM, et al. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci. Transl. Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glasscock E, et al. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J. Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qi Y, et al. Hyper-SUMOylation of the Kv7 potassium channel diminishes the M-current leading to seizures and sudden death. Neuron. 2014;83:1159–1171. doi: 10.1016/j.neuron.2014.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klassen TL, et al. High-resolution molecular genomic autopsy reveals complex sudden unexpected death in epilepsy risk profile. Epilepsia. 2014;55:e6–e12. doi: 10.1111/epi.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heinzen EL, et al. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am. J. Hum. Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wheeler DA, Wang L. From human genome to cancer genome: the first decade. Genome Res. 2013;23:1054–1062. doi: 10.1101/gr.157602.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vogelstein B, et al. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Alexandrov LB, Stratton MR. Mutational signatures: the patterns of somatic mutations hidden in cancer genomes. Curr. Opin. Genet. Dev. 2014;24:52–60. doi: 10.1016/j.gde.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hu WF, Chahrour MH, Walsh CA. The diverse genetic landscape of neurodevelopmental disorders. Annu. Rev. Genomics Hum. Genet. 2014;15:195–213. doi: 10.1146/annurev-genom-090413-025600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jamuar SS, et al. Somatic mutations in cerebral cortical malformations. N. Engl. J. Med. 2014;371:733–743. doi: 10.1056/NEJMoa1314432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JH, et al. De novo somatic mutations in components of the PI3K–AKT3-mTOR pathway cause hemimegalencephaly. Nat. Genet. 2012;44:941–945. doi: 10.1038/ng.2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cai X, et al. Single-cell, genome-wide sequencing identifies clonal somatic copy-number variation in the human brain. Cell Reports. 2014;8:1280–1289. doi: 10.1016/j.celrep.2014.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fishell G, Heintz N. The neuron identity problem: form meets function. Neuron. 2013;80:602–612. doi: 10.1016/j.neuron.2013.10.035. [DOI] [PubMed] [Google Scholar]
- 59.Gerfen CR, Paletzki R, Heintz N. GENSAT BAC cre-recombinase driver lines to study the functional organization of cerebral cortical and basal ganglia circuits. Neuron. 2013;80:1368–1383. doi: 10.1016/j.neuron.2013.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eberwine J, et al. The promise of single-cell sequencing. Nat. Methods. 2014;11:25–27. doi: 10.1038/nmeth.2769. [DOI] [PubMed] [Google Scholar]
- 61.Stephens PJ, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–404. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maheshwari A, Noebels JL. Monogenic models of absence epilepsy: windows into the complex balance between inhibition and excitation in thalamocortical microcircuits. Prog. Brain Res. 2014;213:223–252. doi: 10.1016/B978-0-444-63326-2.00012-0. [DOI] [PubMed] [Google Scholar]
- 63.Brooks-Kayal AR, Russek SJ. Regulation of GABAA receptor gene expression and epilepsy. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s Basic Mechanisms of the Epilepsies. 4th. National Center for Biotechnology Information; Bethesda, Maryland, USA: 2012. [PubMed] [Google Scholar]
- 64.Houser CR. Do structural changes in GABA neurons give rise to the epileptic state? Adv. Exp. Med. Biol. 2014;813:151–160. doi: 10.1007/978-94-017-8914-1_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petilla Interneuron Nomenclature Group et al. Petilla terminology: nomenclature of features of GABAergic interneurons of the cerebral cortex. Nat. Rev. Neurosci. 2008;9:557–568. doi: 10.1038/nrn2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kepecs A, Fishell G. Interneuron cell types are fit to function. Nature. 2014;505:318–326. doi: 10.1038/nature12983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Okaty BW, et al. Transcriptional and electrophysiological maturation of neocortical fast-spiking GABAergic interneurons. J. Neurosci. 2009;29:7040–7052. doi: 10.1523/JNEUROSCI.0105-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lancaster E, Dalmau J. Neuronal autoantigens–pathogenesis, associated disorders and antibody testing. Nat. Rev. Neurosci. 2012;8:380–390. doi: 10.1038/nrneurol.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rossignol E, et al. CaV 2.1 ablation in cortical interneurons selectively impairs fast-spiking basket cells and causes generalized seizures. Ann. Neurol. 2013;74:209–222. doi: 10.1002/ana.23913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mark MD, et al. Delayed postnatal loss of P/Q-type calcium channels recapitulates the absence epilepsy, dyskinesia, and ataxia phenotypes of genomic Cacna1a mutations. J. Neurosci. 2011;31:4311–4326. doi: 10.1523/JNEUROSCI.5342-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maejima T, et al. Postnatal loss of P/Q-type channels confined to rhombic-lip-derived neurons alters synaptic transmission at the parallel fiber to purkinje cell synapse and replicates genomic Cacna1a mutation phenotype of ataxia and seizures in mice. J. Neurosci. 2013;33:5162–5174. doi: 10.1523/JNEUROSCI.5442-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ogiwara I, et al. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum. Mol. Genet. 2013;22:4784–4804. doi: 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chao HT, et al. Dysfunction in GABA signalling mediates autism-like stereotypies and Rett syndrome phenotypes. Nature. 2010;468:263–269. doi: 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W, et al. Loss of MeCP2 from forebrain excitatory neurons leads to cortical hyperexcitation and seizures. J. Neurosci. 2014;34:2754–2763. doi: 10.1523/JNEUROSCI.4900-12.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Burgess DL, et al. β subunit reshuffling modifies N- and P/Q-type Ca2+ channel subunit compositions in lethargic mouse brain. Mol. Cell. Neurosci. 1999;13:293–311. doi: 10.1006/mcne.1999.0748. [DOI] [PubMed] [Google Scholar]
- 76.Zucchini S, et al. Identification of miRNAs differentially expressed in human epilepsy with or without granule cell pathology. PLoS ONE. 2014;9:e105521. doi: 10.1371/journal.pone.0105521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Henshall DC. MicroRNA and epilepsy: profiling, functions and potential clinical applications. Curr. Opin. Neurol. 2014;27:199–205. doi: 10.1097/WCO.0000000000000079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ule J, et al. Nova regulates brain-specific splicing to shape the synapse. Nat. Genet. 2005;37:844–852. doi: 10.1038/ng1610. [DOI] [PubMed] [Google Scholar]
- 79.St Laurent G, et al. Genome-wide analysis of A-to-I RNA editing by single-molecule sequencing in Drosophila. Nat. Struct. Mol. Biol. 2013;20:1333–1339. doi: 10.1038/nsmb.2675. [DOI] [PubMed] [Google Scholar]
- 80.Eom T, et al. NOVA-dependent regulation of cryptic NMD exons controls synaptic protein levels after seizure. Elife. 2013;2:e00178. doi: 10.7554/eLife.00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Klassen T, et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Toledo-Rodriguez M, et al. Correlation maps allow neuronal electrical properties to be predicted from single-cell gene expression profiles in rat neocortex. Cereb. Cortex. 2004;14:1310–1327. doi: 10.1093/cercor/bhh092. [DOI] [PubMed] [Google Scholar]
- 83.Glasscock E, et al. Masking epilepsy by combining two epilepsy genes. Nat. Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 84.Marder E. Neuromodulation of neuronal circuits: back to the future. Neuron. 2012;76:1–11. doi: 10.1016/j.neuron.2012.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- 86.Ganetzky B, Wu CF. Drosophila mutants with opposing effects on nerve excitability: genetic and spatial interactions in repetitive firing. J. Neurophysiol. 1982;47:501–514. doi: 10.1152/jn.1982.47.3.501. [DOI] [PubMed] [Google Scholar]
- 87.Frankel WN, et al. Unraveling genetic modifiers in the gria4 mouse model of absence epilepsy. PLoS Genet. 2014;10:e1004454. doi: 10.1371/journal.pgen.1004454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Meisler MH, O’Brien JE, Sharkey LM. Sodium channel gene family: epilepsy mutations, gene interactions and modifier effects. J. Physiol. (Lond.) 2010;588:1841–1848. doi: 10.1113/jphysiol.2010.188482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Miller AR, et al. Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav. 2014;13:163–172. doi: 10.1111/gbb.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Howlett IC, et al. Drosophila as a model for intractable epilepsy: gilgamesh suppresses seizures in para(bss1) heterozygote flies. G3 (Bethesda) 2013;3:1399–1407. doi: 10.1534/g3.113.006130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holth JK, et al. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J. Neurosci. 2013;33:1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gheyara AL, et al. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014;76:443–456. doi: 10.1002/ana.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tomaselli GF. Cardiac Ito, KCNE2, and Brugada syndrome: promiscuous subunit interactions, or what happens in HEK cells stays in HEK cells? Heart Rhythm. 2010;7:206–207. doi: 10.1016/j.hrthm.2009.10.029. [DOI] [PubMed] [Google Scholar]
- 94.Yu FH, et al. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 95.Zhang Y, et al. Mutations in high-voltage-activated calcium channel genes stimulate low-voltage-activated currents in mouse thalamic relay neurons. J. Neurosci. 2002;22:6362–6371. doi: 10.1523/JNEUROSCI.22-15-06362.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sutherland ML, et al. Overexpression of a Shaker-type potassium channel in mammalian central nervous system dysregulates native potassium channel gene expression. Proc. Natl. Acad. Sci. USA. 1999;96:2451–2455. doi: 10.1073/pnas.96.5.2451. [DOI] [PMC free article] [PubMed] [Google Scholar]