

Abstract
Cardiorespiratory collapse after a seizure is the leading cause of sudden unexpected death in epilepsy (SUDEP) in young persons, but why only certain individuals are at risk is unknown. To identify a mechanism for this lethal cardiorespiratory failure, we examined whether genes linked to increased SUDEP risk lower the threshold for spreading depolarization (SD), a self-propagating depolarizing wave that silences neuronal networks. Mice carrying mutations in Kv1.1 potassium channels (−/−) and Scn1a sodium ion channels (+/R1407X) phenocopy many aspects of human SUDEP. In mutant, but not wild-type mice, seizures initiated by topical application of 4-aminopyridine to the cortex led to a slow, negative DC potential shift recorded in the dorsal medulla, a brainstem region that controls cardiorespiratory pacemaking. This irreversible event slowly depolarized cells and inactivated synaptic activity, producing cardiorespiratory arrest. Local initiation of SD in this region by potassium chloride microinjection also elicited electroencephalographic suppression, apnea, bradycardia, and asystole, similar to the events seen in monitored human SUDEP. In vitro study of brainstem slices confirmed that mutant mice had a lower threshold for SD elicited by metabolic substrate depletion and that immature mice were at greater risk than adults. Deletion of the gene encoding tau, which prolongs life in these mutants, also restored the normal SD threshold in Kv1.1-mutant mouse brainstem. Thus, brainstem SD may be a critical threshold event linking seizures and SUDEP.
Introduction
Sudden unexpected death in epilepsy (SUDEP) is the leading cause of mortality in individuals with seizure disorders (1). Among neurological disorders, SUDEP is second only to stroke in the number of potential life years lost (2). One major class of causative genes expressed in the heart and brain (3–5) and two epidemiological SUDEP risk factors (younger age and high incidence of pharmacoresistant seizures) have been identified, but the mechanisms leading to sudden autonomic collapse at the moment of death remain to be elucidated. Effective prediction or intervention is currently unavailable.
Most individuals with epilepsy experience tachycardia and only transient hypoxia during a seizure as a result of central respiratory depression or tonic spasm of the diaphragm. Rarely, paradoxical slowing of heart rate or even prolonged cardiac asystoles are recorded, but both are reversible during or within moments after the seizure (6). One-third of unwitnessed SUDEP instances lack evidence of a recent seizure (7). However, more than 10% of patients with chronic refractory temporal lobe epilepsy eventually fail to self-resuscitate after a seizure, with variable patterns of electroencephalographic (EEG) and cardiorespiratory shutdown at the moment of death (8–10). Studies in humans (11, 12) and in mouse models (2, 5) suggest the presence of a rare genetic dysregulation of a centrally mediated autonomic function in individuals at risk for SUDEP, arising from intrinsic membrane hyperexcitability or reduced synaptic inhibition in pathways that slow the heart rate or alter gasp reflexes. However, the basis of the irreversibility is unclear, and why, especially in a person at high genetic risk, the first seizure is not also the last is unknown.
Spreading depolarization (SD) is a pathological, self-regenerating wave of depolarization in neurons and glia that is associated with excess glutamate release and extracellular potassium elevation. A variety of factors regulate the onset and propagation of the slow (2 to 6 mm/min) wave, which contributes to transient human neurological deficits during cerebral ischemia, trauma, and migraine (13). SD has been studied in the neocortex, hippocampus, and brainstem, where it produces profound reversible or irreversible loss of neural activity (14, 15). Although SD can be evoked experimentally by high potassium or tetanic neuronal stimulation, it can also arise spontaneously during limited energy substrate availability (hypoxia and ischemia) or hyperthermia. Each of these conditions may also provoke seizures. Despite this shared vulnerability, seizure and SD have nonoverlapping age- and region-dependent thresholds (16–18). We reasoned that a high threshold for SD normally acts as a protective barrier against dysregulation of the central autonomic output initiated by the seizure itself, preventing a fatal outcome. Therefore, we investigated whether the threshold for brainstem SD is lower in genetic SUDEP mouse models and explored the possibility that such a decreased threshold could mediate central autonomic death.
Results
Brainstem SD coincides with terminal cardiorespiratory dysregulation during seizures
Kv1.1 channels conduct a critical potassium current in neurons that prevents hyperexcitability, and mice lacking the gene recapitulate critical SUDEP phenotypes, including frequent generalized seizures, autonomic instability, and premature death at a young age (4, 19). We investigated the excitability in the dorsal medulla during seizures triggered in the cortex of these anesthetized juvenile mice [postnatal day 18 (P18) to P25], an age when roughly 50% of Kv1.1 mice die suddenly. We generated recurrent cortical seizures by topical focal application of 4-aminopyridine (4AP) within a small cranial window over the somatosensory cortex (Fig. 1, A and B) of wild-type and Kv1.1−/− [knockout (KO)] animals. Application of 4AP onto Kv1.1 KO cortex triggered seizures with postictal suppression of cortical EEG, ictal and peri-ictal apnea, transient bradycardia, and asystoles (Fig. 1 and figs. S1 and S2). At least one episode of reversible apneic bradycardia/asystole was observed in 84% (21 of 24) of Kv1.1 KO mice tested. We performed DC recordings in the dorsal medulla of 18 of these mice, and 50% (9 of 18) showed sudden brainstem SD coincident with autonomic failure, which was uniformly and rapidly fatal (Fig. 1C). In all nine mice, fatal autonomic failure and cortical EEG suppression coincided with a slow DC potential shift (mean amplitude, −17.8 ± 7.4 mV) in the dorsal medulla (Fig. 1, D and E), linking this brainstem event to fatal autonomic collapse. In contrast, cortical seizures had minimal cardiorespiratory effects in wild-type mice (Fig. 2, A to D), with 100% postictal survival (n = 15; Fig. 1C). Brainstem SD was not detected in these mice (Fig. 2A).
Fig. 1. Premorbid cardiorespiratory dysregulation and brainstem SD in Kv1.1 mutant associated with cortical seizures in vivo.
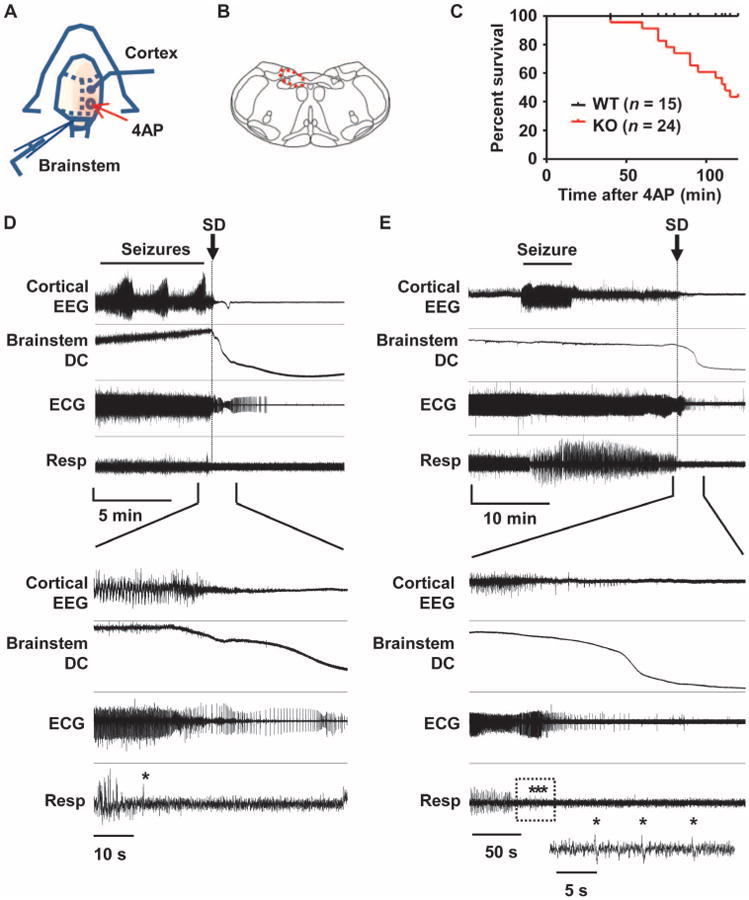
(A) Diagram of experimental setup for application of 4AP and recording of EEG and brainstem DC potentials in spontaneously breathing urethane-anesthetized juvenile mice (P18 to P25). (B) Illustration of brainstem recording area (red circle). (C) Time until death in Kv1.1 wild-type (WT) and KO mice after focal 4AP application. (D and E) Representative traces of premorbid sequence of the cortical EEG, brainstem DC current, ECG, and respiration in two Kv1.1 KO mice. Expanded traces shown in the lower half of the panels illustrate the temporal association between loss of cortical EEG activity, brainstem SD, and development of cardiorespiratory arrhythmias. Asterisk, gasping. (D) Immediate postictal EEG flattening tightly coupled to onset of cardiorespiratory dysregulation and brainstem SD. Vertical scale: cortical EEG, 0.35 mV; brainstem DC, 5 mV; ECG, 0.22 mV; respiration, arbitrary units. (E) Delayed cortical suppression and cardiorespiratory shutdown >10 min after final intense seizure activity. The respiratory trace in the box is further expanded and shown in the inset. Vertical scale: cortical EEG, 0.31 mV; brainstem DC, 18 mV; ECG, 0.43 mV; respiration, arbitrary units.
Fig. 2. Cardiorespiratory dysregulation and brainstem SD in WT mice associated with cortical seizures in vivo.
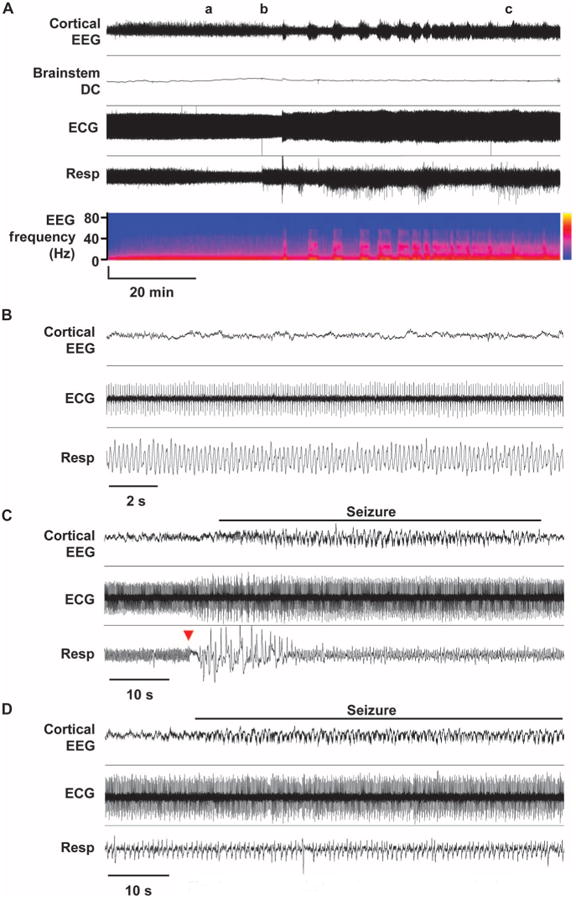
(A to D) Representative traces of premorbid sequence of the cortical EEG, brainstem DC current, ECG, and respiration in one WT mouse (Kv1.1+/+) with expanded traces from time points a, b, and c shown in (B) to (D). In the EEG power spectrum, vertical axis shows EEG frequency band, and the color map indicates power. (B) Interictal cardiorespiratory tone. No deleterious cardiorespiratory rhythms were observed during the interictal period. (C) Ictal apnea and recovery. Cortical seizure triggered a very brief respiratory arrest (red arrow). (D) Breathing during seizure. During sustained seizure, breathing rate slowed but remained regular.
There was a variable latency (1 to 3 min) between the onset of arrhythmias and apneas, which occurred during the seizure, and detection of SD after the end of the seizure (Fig. 3A), which may have been in part due to microscopic differences in the placement of the brainstem recording electrode and in the patterns of SD propagation. The time to complete cardiac arrest after the onset of irrecoverable sinus bradycardia was ∼3 min and was usually preceded by loss of cortical EEG activity and apneas, although variations in this sequence were observed, as in monitored human cases (8).
Fig. 3. Characterization of premorbid seizures and autonomic deregulation in Kv1.1 KO mice.
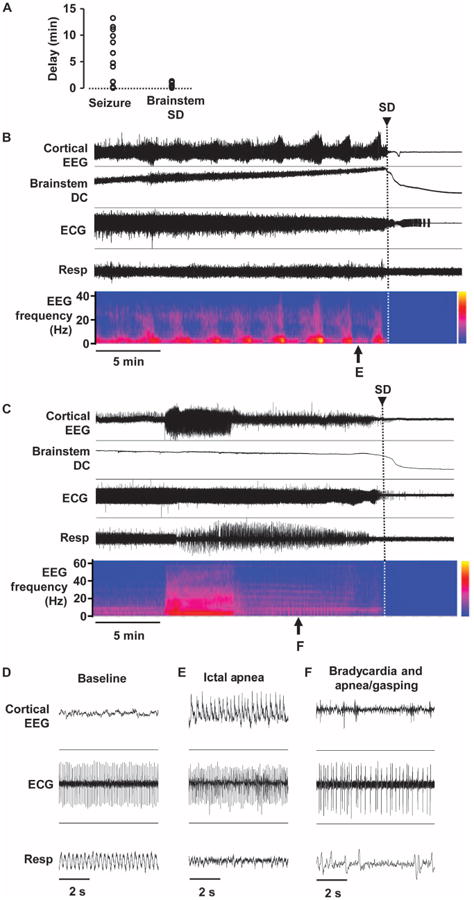
(A) Time delay to the onset of lethal cardiorespiratory failure from the end of intense seizure (left) or the beginning of brainstem SD (right). (B to F) Representative traces showing development of cortical seizures and terminal cardiorespiratory deregulation. Expanded traces are shown in (D) to (F). (B) Recurring high-amplitude seizures until brainstem SD and death. Regular cardiorespiratory tone during baseline (D) and failure during seizure (E). (C) An intense cortical seizure triggering cardiorespiratory deregulation (F), leading to brainstem SD and death.
We noted a variable amount of time (up to ∼13 min) between the end of a cortical seizure and the onset of the lethal cardiorespiratory depression, whereas the brainstem SD was followed closely by cardiorespiratory arrest and death in KO mice (Fig. 3A). Figure 3 (B and C) shows a detailed analysis of the two recordings from the Kv1.1 KO mice shown in Fig. 1. In Fig. 3B, brainstem SD occurred 17 s after the termination of a brief seizure and coincided with postictal cortical EEG suppression and cardiorespiratory depression. Full cardiac arrest occurred 230 s later. The power spectra of the recurrent cortical seizures before death were similar and did not clearly predict the onset of brainstem SD. In another case (Fig. 3C), brainstem SD appeared over 11 min after the seizure. In every case, however, the normal respiration patterns seen before the seizure (Fig. 3D) were severely disrupted during (Fig. 3E) and after (Fig. 3F) the seizure, and audible agonal breathing (that is, inspiratory stridor) was frequently detected. Ventilatory compromise produced a severe drop in blood oxygen saturation in two mice, falling from >90% to 38 and 36%.
Similar experiments were conducted in Scn1a R1407X mice, a second SUDEP mouse model (20), containing a knock-in (Ki) truncation mutation that causes Dravet Syndrome in humans, an epileptic encephalopathy with a highly elevated risk for postictal EEG suppression, cardiac instability, and SUDEP (21). In six juvenile mutant mice (P20 to P30), the evoked seizures resulted in bradycardia and apneas, and four mice died of sudden cardiorespiratory arrest coinciding with the onset of brainstem DC shifts (Fig. 4, A and B).
Fig. 4. Characterization of premorbid seizures and autonomic deregulation in the Scn1a (+/R1407X) mouse.
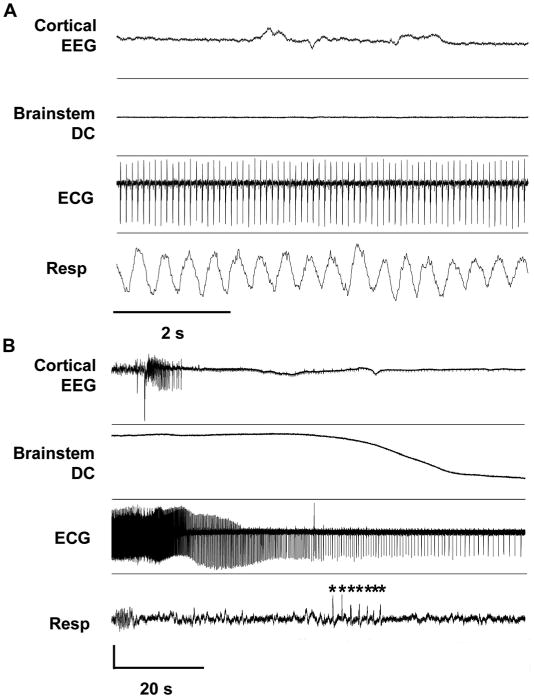
(A) Stable baseline cardiorespiratory tone. (B) Degenerative cardiorespiratory response during the onset of brainstem SD. Sudden onsets of agonal breathing, cardiac arrhythmias, and cortical EEG suppression preceded the onset of negative DC potential shift in brainstem DC trace. Asterisk, gasping. Vertical scale: cortical EEG, 1.1 mV; brainstem DC, 7.5 mV; ECG, 0.22 mV; respiration, arbitrary units.
These in vivo results implicate hypoxia, a critical determinant of SD threshold, in the demise of Kv1.1 KO animals, as reported for humans (1, 22). By inactivating the nucleus tractus solitarius (NTS) and dorsal vagal motor nuclei, which are critical central pathways supporting tissue oxygenation, SD in this region may produce further hypoxia, leading to a vicious spiral progressing to terminal cardiorespiratory arrest.
Locally evoked brainstem SD triggers cardiorespiratory arrest and cortical EEG suppression
To separate the contribution of brainstem SD–associated cortical and cardiorespiratory collapse from other potential causes of cortical seizure–driven bradycardia and hypoxia, we initiated local brainstem SDs in the absence of cortical 4AP-induced seizures in juvenile mice (P20 to P30). In 50% of the trials in wild-type animals, a pulse of potassium chloride (KCl) delivered by micropipette generated a slow DC brainstem potential shift (Fig. 5A). Unexpectedly, the brainstem SD induced brief suppression of cortical EEG amplitude in both wild-type and Kv1.1 KO animals, similar to that seen after cortical seizures in mice and in monitored SUDEP cases (8). The brainstem-driven EEG suppression in the absence of cortical seizure was nevertheless accompanied by transient bradycardia and apnea in all Kv1.1 KO mice (Fig. 5A) and, less reliably, in wild-type mice. A cortical negative DC potential shift was absent during this period, suggesting that the depressed EEG amplitude was not a result of intrinsic SD propagation in the cortex. Sinus bradycardia was more severe (P = 0.03; Fig.5B) and lasted longer (P = 0.011; Fig. 5C) in Kv1.1 KO mice than in control animals (Fig. 5C) and always outlasted the brainstem DC potential shift. On the other hand, there was no genotype difference in the durations of EEG suppression (Fig. 5D) or apnea (Fig. 5E). A short duration of brainstem SD was critical for recovery; prolonged SD (>10 min), triggered by KCl microinjection total in wild-type mice, produced persistent bradycardia, apnea, and death, as in the cortical seizure model (Fig. 5F). Therefore, isolated brainstem SD can cause the neurophysiological and cardiorespiratory hallmarks of imminent death, even in the absence of a cortical seizure.
Fig. 5. Locally evoked brainstem SD triggers cortical suppression and cardiorespiratory collapse.
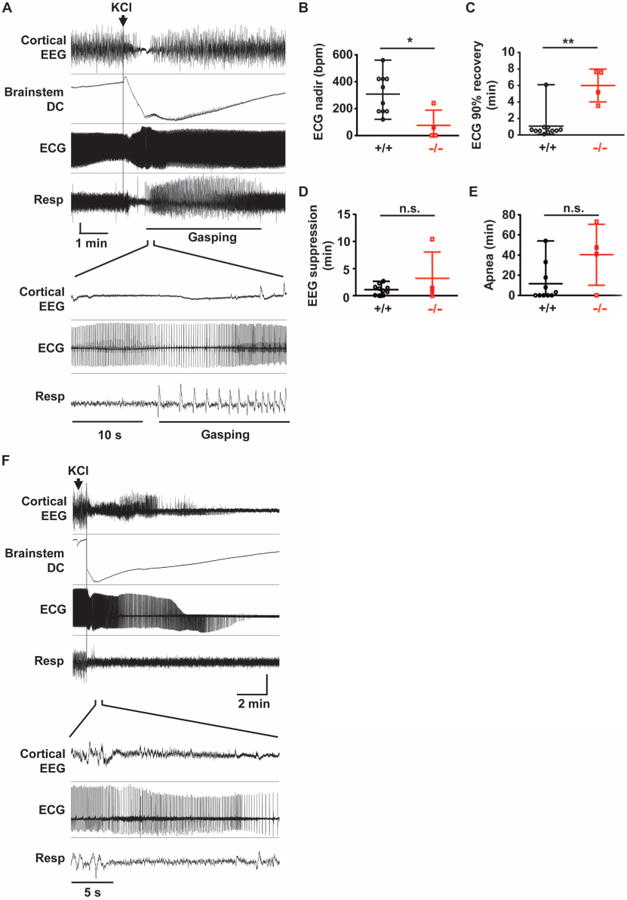
Local brainstem SD initiated by KCl microinjection into the dorsal medulla of anesthetized spontaneously breathing mouse. (A) Representative cortical and cardiorespiratory responses in juvenile WT mouse. Brainstem SD transiently suppressed cortical EEG, ECG, and spontaneous respiration. The expanded trace shown below illustrates periodic cortical EEG suppression, bradycardia, and apnea. Gasping was observed during the recovery from apnea. Vertical scale: cortical EEG, 0.15 mV; brainstem DC, 4.5 mV; ECG, 0.2 mV; respiration, arbitrary unit. (B to E) Quantification of central and peripheral consequences of brainstem SD. Duration of bradycardia was longer and the peak heart rate decrease (analyzed every 1-s bin) was lower in Kv1.1 KO mice. There was no difference in the duration of cortical EEG suppression and the duration of apnea. WT, n = 10; Kv1.1 KO, n = 4. *P < 0.05, **P < 0.01; n.s., not significant. We note that 63% (7 of 11) of Kv1.1 KO mice died after SD and were excluded from the analyses. (F) Microinjection of KCl triggered prolonged DC potential shift, followed by death. Vertical scale: cortical EEG, 0.25 mV; brainstem DC, 10 mV; ECG, 0.18 mV; respiration, arbitrary unit.
SUDEP mutations lower brainstem SD threshold in vitro
We used an in vitro brainstem slice preparation to test whether Kv1.1 channel deficiency lowered the threshold for intrinsic brainstem SD under conditions of constant metabolic substrate depletion. Coronal brainstem slices from juvenile mice at the age of peak incidence of premature death in Kv1.1 KO mice (P18 to P25) (4) were deprived of oxygen and glucose (OGD), and SD was visualized by changes in intrinsic optical signal (IOS). Exposure to OGD solution reliably generated an IOS wave of cellular depolarization in dorsomedial nuclei (Fig. 6A and movie S1), the same area in which we had detected SD in vivo. In this slice orientation, SD arose spontaneously and reproducibly in the lateral NTS and spread medially to invade vagal and hypoglossal nuclei, the microcircuits involved in vital cardiorespiratory reflexes. Voltage-clamp recordings revealed a slow inward current (Fig. 6B) and suppression of excitatory transmission from the visceral afferent axons (that is, tractus solitarius) onto NTS neurons upon arrival of the IOS peak (Fig. 6C). The propagation pattern and speed of SD were indistinguishable between juvenile wild-type and Kv1.1 KO slices (wild-type: 0.60 ± 0.21 mm/min, KO: 0.55 ± 0.13 mm/min, n = 9 each, P > 0.5). However, SD onset was significantly earlier in mutant slices (Fig. 6D), indicating a lower intrinsic SD threshold during substrate deprivation in Kv1.1 KO than in wild-type brainstem. The difference became even more obvious when the glucose concentration was elevated to 5 mM [50% of normal artificial cerebrospinal fluid (ACSF)]. Under this condition, SD was delayed or prevented in wild-type slices, whereas SD was still reliably generated in KO slices (Fig. 6D). A further increase in glucose concentration to 10 mM completely prevented hypoxic SD in all Kv1.1 wild-type and KO tissues (Kv1.1 wild type and KO, n = 9 each).
Fig. 6. SD threshold is influenced by age, ion channel deficiency, and tau deletion in brainstem slices.
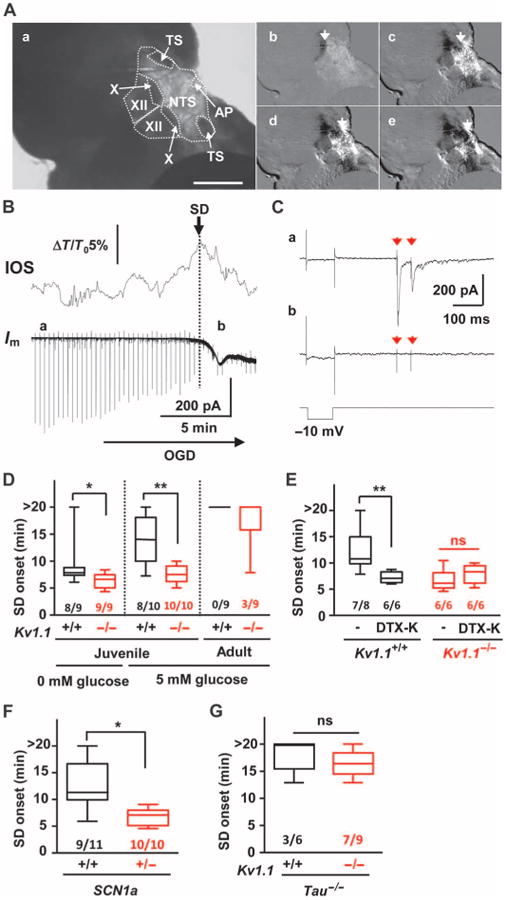
SD was triggered in coronal medulla slices by OGD and monitored by IOS. (A) Raw image (a) and montage of video frames (b to e, see movie S1). TS, tractus solitarius; AP, area postrema; X, dorsal motor nucleus of vagus; XII, hypoglossal nucleus. Scale bar, 500 µm (B) IOS and whole-cell recording of NTS neurons showing compromised afferent input after SD onset. Vertical lines in the whole-cell current trace are test responses of membrane resistance to a train of excitatory postsynaptic currents (EPSCs) triggered every 20 s. Im, whole-cell current; ΔT/T0, relative transparency change. (C) Expanded traces of (a) and (b) from (B). (a) stimulation artifacts (red arrows) followed by EPSCs at baseline; (b) EPSC absent during SD. (D) SD onset was significantly faster in slices obtained from juvenile Kv1.1 KO (red). Adult slices showed a marked increase in SD threshold and no genotype difference. (E) The Kv1.1 channel inhibitor DTX-K (50 nM) reduced latency to SD onset in WT (black) but not in Kv1.1 KO slices (red). (F) Scn1a-deficient mice also showed lowered SD threshold (red). (G) Compound Kv1.1/tau deletion (red) abolished low SD threshold in Kv1.1 KO slices. The numbers indicate numbers of slices with SD out of total slices tested. *P < 0.05, **P < 0.01. n.s., not significant.
We next examined the SD threshold for adult Kv1.1 mutants (age > 90 days) that had survived the early SUDEP susceptibility period. Consistent with previous studies (23, 24), brainstem slices of older animals were highly resistant to hypoxia-induced SD. In adult Kv1.1 KO mice, SD was detected in only 33% (3 of 9) of slices and never (0 of 9) in wild-type slices (Fig. 6D; P = 0.2, Fisher's exact probability test). Because genetic Kv1.1 deletion may alter brain development, we examined the direct role of Kv1.1 channels on the OGD-SD threshold in adult wild-type mice by acute pharmacologic inhibition of Kv1.1 currents, which are typically generated through heteromeric Kv1.1/Kv1.2 channels. Exposure of slices to the Kv1.1/Kv1.2 specific inhibitor dendrotoxin-K (DTX-K; 50 nM) significantly decreased the latency to SD onset in wild-type slices compared to that observed in Kv1.1 KO slices, whereas the toxin was without additional effect in KO slices (Fig. 6E). These results indicate that loss of the Kv1.1 channel is sufficient to lower the brainstem SD threshold, consistent with its effect in the cerebellar cortex (25). We also found that bath application of the N-methyl-d-aspartate receptor antagonist MK801 (30 µM), which inhibits SD in other models (13), also inhibited SD in Kv1.1 KO brainstem (SD onset: control, 5.7 ± 2.8 min; MK801, 16.3 ± 2.6 min; P < 0.001, n = 6 each). Together, these in vitro findings confirm that the SUDEP risk factors identified in vivo (Kv1.1 deficiency and younger age) are intrinsic to Kv1.1 KO neuronal networks and increase the vulnerability of brainstem dorsal nuclei to hypoxic SD generation.
To determine whether Kv1.1 KO mice are unique among SUDEP models in exhibiting a decreased SD threshold, we examined brainstem slices from juvenile Scn1a R1407X mice, a mouse model of Dravet Syndrome (20). Pathogenic mutations in this syndrome elevate network excitability by impairing sodium conductance and by network disinhibition (16, 26, 27). We found that the threshold for brainstem SD was also significantly lowered in this mouse loss-of-function model (Fig. 6F), whereas SD propagation rate was unchanged (wild-type: 0.46 ± 0.17 mm/min, heterozygote: 0.62 ± 0.21 mm/min, P > 0.1, n = 9 and 10, respectively). We also tested whether a known genetic modifier of life span in SUDEP mice influenced SD threshold. Premature death in both Kv1.1 KO and Scn1a Ki mutant mice is largely rescued by ablation of the gene MapT that encodes the axonal microtubule-associated protein tau (28, 29). Consistent with its striking prolongation of survival, tau deletion normalized the latency to onset of brainstem SD in Kv1.1 KO slices (Fig. 6G). Thus, Kv1.1/tau double KO (Kv1.1−/−, tau−/−) mice showed a raised brainstem SD threshold, and onset latency was delayed to that seen in control slices (Kv1.1+/+, tau−/−).
Discussion
Our data point to an active intermediary role of SD in the postictal cardiorespiratory collapse that leads to sudden mortality in epilepsy. This conclusion rests on several lines of evidence in two genetically validated mouse models of human SUDEP and suggests an approach for therapeutic intervention in this life-threatening condition.
First, we demonstrated that a deleterious positive feedback relationship occurs when SD silences brainstem dorsal nuclei whose function is to ensure oxygenation of the nervous system; depolarizing blockade of these cells prevents normal autoresuscitation. This central shutdown of the cardiorespiratory centers may explain the failure of efforts to revive epilepsy patients by cardioresuscitation after cardiac arrest. Second, we found a lowered brainstem threshold to SD in two validated SUDEP models generated by decreased function in the genes Kcna1 and Scn1a. These genes alter excitability through distinct membrane and synaptic mechanisms, reinforcing the generality of our findings. Loss of Kv1.1 current has a powerful effect on both excitatory and inhibitory neurons (19, 30), whereas Scna1a channel deficiency preferentially decreases the excitability of interneurons in the forebrain (20, 26). Nevertheless, both mouse models exhibited a heightened sensitivity to SD. Human SCN1A mutations are also linked to familial hemiplegic migraine type 3 (FHM3) (31), and two other genes for FHM phenotypes also lower thresholds for SD, seizures, and premature death in mouse models. A gain-of-function mutation, S218L, in CACNA1A (FHM1) encoding P/Q-type calcium channels lowers SD threshold, and 20% of FHM patients carrying this mutation develop seizures during attacks (13). Mice bearing the S218L Cacna1a mutation show low SD threshold, seizures, and premature death (32), whereas a loss-of-function Cacna1a mutation raises SD threshold (33) and prolongs survival in Kv1.1 KO mutants (34). Humans and mice with mutations in the Na+, K+-ATPase genes ATP1A2 and ATP1A3 cause hemiplegic migraine. Atp1a2 mutations (FHM2) lower SD threshold (35), and Atp1A3 mutations cause seizures and premature death in mice (36) and seizures in 33% of patients with variable autonomic and respiratory disturbances (37). Finally, we and others have recently shown that the loss-of-function modifier gene MapT (tau) prevents seizures and prolongs longevity in Kv1.1 and Scn1a mutants (28, 29).
Here, light anesthesia was required for stable and noninjurious DC recordings in the small mouse brainstem during seizures. Urethane anesthesia conserves cardiovascular reflexes (38) and creates a physiological status similar to sleep (39), which may be relevant given the frequent occurrence of SUDEP during sleep (8). Because SD can be readily triggered by a mechanical pinprick, motor movements prevented us from recording unprovoked brainstem DC responses during spontaneous convulsive seizures in freely moving animals. This technical challenge must be overcome to directly monitor spontaneous brainstem death in unanesthetized animal models.
Our study identifies functional mutations in genes regulating SD threshold as a significant contributor to the risk of SUDEP. Like those for cardiac long QT syndrome (LQTS), the list of pathogenic SD mutations will continue to grow and serve as candidate targets for individualized therapeutic strategies. Computer simulations indicate that SD threshold, latency, and duration are under the coordinate control of membrane conductances regulated by multiple voltage- and ligand-gated channels, supporting an oligogenic model underlying cardiorespiratory instability and SUDEP (40, 41). Given the complexity of ion channel gene variation contributing to epilepsy itself (12), an integrated biological and in silico analysis of variant function will be essential to refine the prognostic value of epistatic variant profiles in evaluating personal risk for SUDEP.
Materials and Methods
Study design
In vivo studies were first conducted in a set of Kv1.1 KO animals to examine cardiorespiratory abnormalities as well as brainstem SD events. Later experiments alternated between littermate wild-type control mice to characterize contribution of the genotype. The in vivo experiments could not be blinded to the genotype of the animal because the genotype was apparent by the behavior of the mutant animal during the surgical preparation. We did not perform an a priori power analysis for in vivo analyses of physiological variables. All post hoc quantitative analysis of digitized data was performed identically using objective measurement algorithms and maintained in chronological order on the laboratory computer.
Prospective quantitative comparison was conducted for the in vitro analyses of SD onset (Fig. 3). Experiments were designed to test (i) at least two brainstem slices from each mouse to evaluate experimental error and (ii) at least four mice to evaluate between-animal differences by using nonparametric analysis (see below). Variability within or between animals was similar and small in Kv1.1 KO preparations, and thus SD onset of each slice was used as individual data sample.
Animals
All experimental protocols were approved by Institutional Animal Care and Use Committee of Baylor College of Medicine (BCM). Kv1.1 wild-type (+/+) and KO (−/−) were obtained from heterozygous (Kv1.1+/−/C57BL6×C3HeB/FeJ hybrid) breeding colonies established at BCM. Scn1a R1407X Ki mice (19) were obtained by crossing wild-type and heterozygous mice, maintained in C57BL6/SV129 hybrid (75 to 88% C57BL6) background. Kv1.1/tau double KO mice were generated as described previously (27). Retrospectively, ∼55 and ∼15% of Kv1.1 KO and Scn1a (R1407X) heterozygotes, respectively, were found dead before P30. Our study therefore used juvenile (P16 to 25) and adult mice (P >90). Both male and female mice were used and were not randomized. Genotypes of Kcna1 and Scn1a mutant alleles were determined by polymerase chain reaction methods (4, 19). Genotype could be readily identified on the basis of their behavior and phenotype, and thus experiments were not blinded to the experimenter. All mice were bred at BCM and housed at 22°C with a 12-hour light cycle with ad libitum access to food and water.
In vivo electrophysiology
Mice were anesthetized with urethane (1.0 to 1.5 g/kg, intraperitoneally) and mounted on a stereotaxic frame. Body temperature was maintained at 37°C with a thermistor feedback-controlled thermal blanket. Electrocardiogram (ECG) was recorded with subcutaneous electrodes over the thorax. Respiration was measured by temperature fluctuations detected with a thermocouple placed at the mouth after occlusion of the nares. Cortical EEG was recorded with a silver/silver chloride ball electrode placed on the thinned skull and held with superglue. Seizures were evoked by application of a pledget soaked with 4AP (100 mM, ∼10 ml) on the cortical surface accessed through a small cranial window. Glass electrodes (1 to 2 megohms) were inserted in the dorsal brainstem (∼500-µm caudal from obex, ∼500-µm depth) through an opening in the occipital bone using a micromanipulator visually guided by a surgical microscope. The meninges overlying the brainstem are thin and easily penetrated by the glass electrode. After 1 hour of postsurgical recovery, oxygen saturation (SpO2) and blood glucose levels were tested using MouseOX (Starr Life Sciences) and FreeStyle glucose oxidase strips (Abbott), respectively. Recordings were made for 2 hours after 4AP application. Brainstem SD was evoked by KCl microinjection with Picosplitzer II (Parker) in the superficial layer (∼500 µm from the surface, pipette tip diameter ∼10 µm, 1 M KCl, 10 to 100 ms, 30 psi, ∼10 nl volume) in the caudal region of the fourth ventricular floor containing the NTS (<0.5 mm from the obex) while monitoring with the glass micropipette electrode placed superficially (<1 mm below the surface and >1 mm distant from the injection site). Prolonged SD was triggered by a repetitive train of pressure pulses (0.1 to 0.2 Hz, total ∼1 s duration). Microinjection >1.5 mm rostral to the NTS or ventral to the hypoglossal nucleus (>2 mm below the surface) did not evoke SD (n = 3 each). Death was not associated with the presence (9 of 18) or absence (5 of 7) of the brainstem recording electrode (P = 0.7, Fisher's exact test). The anesthetized mouse typically had a heart rate of ∼600 beats per minute and a respiration rate of ∼80 breaths per minute before the recordings. Mice with abnormal heart rate and respiratory rate, SpO2 less than 95%, or blood glucose <100 mg/dl were euthanized. Cortical and brainstem electrical activities were amplified with Axoclamp-2A (Molecular Devices), and ECG and respiration signals were amplified with a Model 410 amplifier (Brownlee Precision). All electrical data were digitized with Digidata 1334 and analyzed with pClamp9.2 software. Here, SD was defined as a negative electrical potential shift >−4 mV/min in the DC recording.
After recording, brains were extracted and kept in 4% paraformaldehyde at 4°C for >48 hours and were transferred in 30% sucrose. Brains were frozen and cut into 100-µm sections in a cryostat. Sections were directly mounted on glass slides, and the electrode position was confirmed on the basis of a slit created by electrode insertion.
Slice electrophysiology
Coronal slices (300-µm thickness) were prepared from forebrains containing dorsal hippocampus and brainstem covering the NTS. Slices obtained from ±500 µm from the obex showed similar SD onsets, and thus the slices within this anatomical range were used for the analyses. Briefly, mice were deeply anesthetized with Avertin (250 mg/kg, intraperitoneally) and decapitated. Brains were extracted in cold dissection solution (220 mM sucrose, 10 mM glucose, 6 mM magnesium sulfate, 3 mM KCl, 25 mM sodium bicarbonate, 1.25 mM sodium monophosphate, 2 mM calcium chloride, and 0.4 mM ascorbic acid equilibrated with 95% O2/5% CO2), trimmed, and glued on the cutting stage. Brain slices were prepared on a vibratome and incubated in ACSF (126 mM sodium chloride, 10 mM glucose, 1 mM magnesium sulfate, 3 mM KCl, 25 mM sodium bicarbonate, 1.25 mM sodium monophosphate, 2 mM calcium chloride, and 0.4 mM ascorbic acid equilibrated with 95% O2/5% CO2) at 35°C for 1 hour. After recovery, the slices were kept in ACSF at room temperature until used for experiments.
For electrophysiological recording, the slices were transferred to a submerged chamber (RC27, Warner Instruments) and were superfused with oxygenated ACSF at 2 ml/min. Slices were visualized with an upright microscope (Axioskop, Zeiss; BX51, Olympus) with a charge-coupled device (CCD) camera (C2400, Hamamatsu). Whole-cell recordings were obtained from visually identified NTS neurons in the translucent region of the dorsal medulla. Patch pipettes were filled with intracellular solution [130 mM cesium gluconate, 1 mM magnesium chloride, 5 mM sodium chloride, 10 mM 1,2-bis(2-aminophenoxy) ethane-N,N,N′,N′-tetraacetic acid (BAPTA), 10 mM Hepes, 5 mM lid-ocaine N-ethyl chloride (QX314), 2 mM adenosine triphosphate (ATP), 0.3 mM guanosine triphosphate, and pH adjusted to 7.2 with cesium hydroxide]. Signals were amplified (Multiclamp 200B), digitized by Digidata 1322A, and analyzed with pClamp9.2 software (Molecular Devices). Pairs of EPSCs were evoked by a bipolar electrode placed on the adjacent tractus solitarius controlled by an IsoFlex and Master 8 (100 (µs, ∼0.2 mA, 100-ms interval). Recorded Kv1.1 wild-type and KO NTS neurons had similar input resistances (Kv1.1 wild type: 1.84 ± 1.2 gigohms, n = 8, and KO: 1.99 ± 0.65 gigohms, n = 7) and capacitances (Kv1.1 wild type: 10.5 ± 3.1 pF, n = 8, and KO: 10.6 ± 4.1 pF, n = 7). Series resistances were between 15 and 20 megohms, with compensation of 50 to 70%. SD was generated by OGD. We used the 5 mM glucose level for comparative analyses, except where stated. Recordings were made for 20 min after OGD exposure. SD was detected by a band of increased light transmission (that is, IOS) traveling across the brain tissue using the CCD camera. A sequence of images was acquired at 1 Hz and was analyzed by ImageJ software.
At least four mice were used for each comparison to evaluate variances from animals. All experiments were interleaved between slices obtained from the test genotype or treated with drugs.
Reagents
The Kv1.1/Kv1.2 inhibitor DTX-K was purchased from Alomone Labs. All other chemicals were obtained from Sigma-Aldrich.
Statistics
All data are presented as means ± SD in text and/or box-and-whisker graph with minimum to maximum data range bars. Data were compared by nonparametric Mann-Whitney U test, unless otherwise mentioned. P < 0.05 was considered significant. In experiments of SD onsets, experiments with latency to SD onset >20 min were considered as “SD blocked” and were treated as the highest rank value in the rank test. Sample size was determined on the basis of pilot experiments. Statistical power was not calculated.
Supplementary Material
Fig. S1. Peri-ictal autonomic abnormality in Kv1.1 KO mouse #1.
Fig. S2. Peri-ictal autonomic abnormality in Kv1.1 KO mouse #2.
Table S1. Raw data
Movie S1. OGD-SD in brainstem slice
Acknowledgments
We thank K. Yamakawa and J. Ziburkas for sharing SCN1A R1407X Ki mice, and E. Roberson for the gift of tau KO mice. Funding: This work was supported by a Postdoctoral Fellowship award (14POST20130031) from the American Heart Association (I.A.), NINDS NS029709 and NS090340 (J.L.N.), and the Blue Bird Circle Foundation.
Footnotes
Author contributions: I.A. and J.L.N. conceived and designed the experiments and wrote the manuscript. I.A. conducted the experiments and analyzed the data.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All raw data are available online in table S1.
References and Notes
- 1.Massey CA, Sowers LP, Dlouhy BJ, Richerson GB. Mechanisms of sudden unexpected death in epilepsy: The pathway to prevention. Nat Rev Neurol. 2014;10:271–282. doi: 10.1038/nrneurol.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thurman DJ, Hesdorffer DC, French JA. Sudden unexpected death in epilepsy: Assessing the public health burden. Epilepsia. 2014;55:1479–1485. doi: 10.1111/epi.12666. [DOI] [PubMed] [Google Scholar]
- 3.Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glasscock E, Yoo JW, Chen TT, Klassen TL, Noebels JL. Kv1.1 potassium channel deficiency reveals brain-driven cardiac dysfunction as a candidate mechanism for sudden unexplained death in epilepsy. J Neurosci. 2010;30:5167–5175. doi: 10.1523/JNEUROSCI.5591-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest. 2013;123:1798–1808. doi: 10.1172/JCI66220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cole AJ, Eskandar E, Mela T, Noebels JL, Gonzales RG, McGuone D. Case records of the Massachusetts General Hospital. Case 18-2013—A 32-year-old woman with recurrent episodes of altered consciousness. N Engl J Med. 2013;368:2304–2312. doi: 10.1056/NEJMcpc1215969. [DOI] [PubMed] [Google Scholar]
- 7.Stecker EC, Reinier K, Uy-Evanado A, Teodorescu C, Chugh H, Gunson K, Jui J, Chugh SS. Relationship between seizure episode and sudden cardiac arrest in patients with epilepsy: A community-based study. Circ Arrhythm Electrophysiol. 2013;6:912–916. doi: 10.1161/CIRCEP.113.000544. [DOI] [PubMed] [Google Scholar]
- 8.Ryvlin P, Nashef L, Lhatoo SD, Bateman LM, Bird J, Bleasel A, Boon P, Crespel A, Dworetzky BA, Høgenhaven H, Lerche H, Maillard L, Malter MP, Marchal C, Murthy JM, Nitsche M, Pataraia E, Rabben T, Rheims S, Sadzot B, Schulze-Bonhage A, Seyal M, So EL, Spitz M, Szucs A, Tan M, Tao JX, Tomson T. Incidence and mechanisms of cardiorespiratory arrests in epilepsy monitoring units (MORTEMUS): A retrospective study. Lancet Neurol. 2013;12:966–977. doi: 10.1016/S1474-4422(13)70214-X. [DOI] [PubMed] [Google Scholar]
- 9.Shorvon S, Tomson T. Sudden unexpected death in epilepsy. Lancet. 2011;378:2028–2038. doi: 10.1016/S0140-6736(11)60176-1. [DOI] [PubMed] [Google Scholar]
- 10.Jeppesen J, Fuglsang-Frederiksen A, Brugada R, Pedersen B, Rubboli G, Johansen P, Beniczky S. Heart rate variability analysis indicates preictal parasympathetic overdrive preceding seizure-induced cardiac dysrhythmias leading to sudden unexpected death in a patient with epilepsy. Epilepsia. 2014;55:e67–e71. doi: 10.1111/epi.12614. [DOI] [PubMed] [Google Scholar]
- 11.Sowers LP, Massey CA, Gehlbach BK, Granner MA, Richerson GB. Sudden unexpected death in epilepsy: Fatal post-ictal respiratory and arousal mechanisms. Respir Physiol Neurobiol. 2013;189:315–323. doi: 10.1016/j.resp.2013.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klassen T, Davis C, Goldman A, Burgess D, Chen T, Wheeler D, McPherson J, Bourguin T, Lewis L, Villasana D, Morgan M, Muzny D, Gibbs R, Noebels J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pietrobon D, Moskowitz MA. Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nat Rev Neurosci. 2014;15:379–393. doi: 10.1038/nrn3770. [DOI] [PubMed] [Google Scholar]
- 14.Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab. 2011;31:17–35. doi: 10.1038/jcbfm.2010.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Richter F, Bauer R, Lehmenkühler A, Schaible HG. The relationship between sudden severe hypoxia and ischemia-associated spreading depolarization in adult rat brainstem in vivo. Exp Neurol. 2010;224:146–154. doi: 10.1016/j.expneurol.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Rogawski MA. In: Jasper's Basic Mechanisms of the Epilepsies. Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. U.S. National Center for Biotechnology Information; Bethesda, MD: 2012. [PubMed] [Google Scholar]
- 17.Mody I, Lambert JD, Heinemann U. Low extracellular magnesium induces epileptiform activity and spreading depression in rat hippocampal slices. J Neurophysiol. 1987;57:869–888. doi: 10.1152/jn.1987.57.3.869. [DOI] [PubMed] [Google Scholar]
- 18.Hablitz JJ, Heinemann U. Alterations in the microenvironment during spreading depression associated with epileptiform activity in the immature neocortex. Brain Res Dev Brain Res. 1989;46:243–252. doi: 10.1016/0165-3806(89)90288-5. [DOI] [PubMed] [Google Scholar]
- 19.Smart SL, Lopantsev V, Zhang CL, Robbins CA, Wang H, Chiu SY, Schwartzkroin PA, Messing A, Tempel BL. Deletion of the KV1.1 potassium channel causes epilepsy in mice. Neuron. 1998;20:809–819. doi: 10.1016/s0896-6273(00)81018-1. [DOI] [PubMed] [Google Scholar]
- 20.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K. Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: A circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci. 2007;27:5903–5914. doi: 10.1523/JNEUROSCI.5270-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim SH, Nordli DR, Jr, Berg AT, Koh S, Laux L. Ictal ontogeny in Dravet syndrome. Clin Neurophysiol. 2014;126:446–455. doi: 10.1016/j.clinph.2014.06.024. [DOI] [PubMed] [Google Scholar]
- 22.Bateman LM, Li CS, Lin TC, Seyal M. Serotonin reuptake inhibitors are associated with reduced severity of ictal hypoxemia in medically refractory partial epilepsy. Epilepsia. 2010;51:2211–2214. doi: 10.1111/j.1528-1167.2010.02594.x. [DOI] [PubMed] [Google Scholar]
- 23.Funke F, Kron M, Dutschmann M, Müller M. Infant brain stem is prone to the generation of spreading depression during severe hypoxia. J Neurophysiol. 2009;101:2395–2410. doi: 10.1152/jn.91260.2008. [DOI] [PubMed] [Google Scholar]
- 24.Richter F, Rupprecht S, Lehmenkühler A, Schaible HG. Spreading depression can be elicited in brain stem of immature but not adult rats. J Neurophysiol. 2003;90:2163–2170. doi: 10.1152/jn.00388.2003. [DOI] [PubMed] [Google Scholar]
- 25.Chen G, Gao W, Reinert KC, Popa LS, Hendrix CM, Ross ME, Ebner TJ. Involvement of kv1 potassium channels in spreading acidification and depression in the cerebellar cortex. J Neurophysiol. 2005;94:1287–1298. doi: 10.1152/jn.00224.2005. [DOI] [PubMed] [Google Scholar]
- 26.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci. 2006;9:1142–1149. doi: 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- 27.Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, Yamakawa K. Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet. 2013;22:4784–4804. doi: 10.1093/hmg/ddt331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holth JK, Bomben VC, Reed JG, Inoue T, Younkin L, Younkin SG, Pautler RG, Botas J, Noebels JL. Tau loss attenuates neuronal network hyperexcitability in mouse and Drosophila genetic models of epilepsy. J Neurosci. 2013;33:1651–1659. doi: 10.1523/JNEUROSCI.3191-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gheyara AL, Ponnusamy R, Djukic B, Craft RJ, Ho K, Guo W, Finucane MM, Sanchez PE, Mucke L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann Neurol. 2014;76:443–456. doi: 10.1002/ana.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goldberg EM, Clark BD, Zagha E, Nahmani M, Erisir A, Rudy B. K+ channels at the axon initial segment dampen near-threshold excitability of neocortical fast-spiking GABAergic interneurons. Neuron. 2008;58:387–400. doi: 10.1016/j.neuron.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cestèle S, Labate A, Rusconi R, Tarantino P, Mumoli L, Franceschetti S, Annesi G, Mantegazza M, Gambardella A. Divergent effects of the T1174S SCN1A mutation associated with seizures and hemiplegic migraine. Epilepsia. 2013;54:927–935. doi: 10.1111/epi.12123. [DOI] [PubMed] [Google Scholar]
- 32.van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD. High cortical spreading depression susceptibility and migraine-associated symptoms in Cav2.1 S218L mice. Ann Neurol. 2010;67:85–98. doi: 10.1002/ana.21815. [DOI] [PubMed] [Google Scholar]
- 33.Ayata C, Shimizu-Sasamata M, Lo EH, Noebels JL, Moskowitz MA. Impaired neurotransmitter release and elevated threshold for cortical spreading depression in mice with mutations in the α1A subunit of P/Q type calcium channels. Neuroscience. 2000;95:639–645. doi: 10.1016/s0306-4522(99)00446-7. [DOI] [PubMed] [Google Scholar]
- 34.Glasscock E, Qian J, Yoo JW, Noebels JL. Masking epilepsy by combining two epilepsy genes. Nat Neurosci. 2007;10:1554–1558. doi: 10.1038/nn1999. [DOI] [PubMed] [Google Scholar]
- 35.Leo L, Gherardini L, Barone V, De Fusco M, Pietrobon D, Pizzorusso T, Casari G. Increased susceptibility to cortical spreading depression in the mouse model of familial hemiplegic migraine type 2. PLOS Genet. 2011;7:e1002129. doi: 10.1371/journal.pgen.1002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clapcote SJ, Duffy S, Xie G, Kirshenbaum G, Bechard AR, Schack VR, Petersen J, Sinai L, Saab BJ, Lerch JP, Minassian BA, Ackerley CA, Sled JG, Cortez MA, Henderson JT, Vilsen B, Roder JC. Mutation I810N in the α3 isoform of Na+,K+-ATPase causes impairments in the sodium pump and hyperexcitability in the CNS. Proc Natl Acad Sci U S A. 2009;106:14085–14090. doi: 10.1073/pnas.0904817106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rosewich H, Thiele H, Ohlenbusch A, Maschke U, Altmüller J, Frommolt P, Zirn B, Ebinger F, Siemes H, Nürnberg P, Brockmann K, Gärtner J. Heterozygous de-novo mutations in ATP1A3 in patients with alternating hemiplegia of childhood: A whole-exome sequencing gene-identification study. Lancet Neurol. 2012;11:764–773. doi: 10.1016/S1474-4422(12)70182-5. [DOI] [PubMed] [Google Scholar]
- 38.Kasparov S, Paton JF. Changes in baroreceptor vagal reflex performance in the developing rat. Pflugers Arch. 1997;434:438–444. doi: 10.1007/s004240050418. [DOI] [PubMed] [Google Scholar]
- 39.Pagliardini S, Gosgnach S, Dickson CT. Spontaneous sleep-like brain state alternations and breathing characteristics in urethane anesthetized mice. PLOS One. 2013;8:e70411. doi: 10.1371/journal.pone.0070411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kager H, Wadman WJ, Somjen GG. Conditions for the triggering of spreading depression studied with computer simulations. J Neurophysiol. 2002;88:2700–2712. doi: 10.1152/jn.00237.2002. [DOI] [PubMed] [Google Scholar]
- 41.Wei Y, Ullah G, Schiff SJ. Unification of neuronal spikes, seizures, and spreading depression. J Neurosci. 2014;34:11733–11743. doi: 10.1523/JNEUROSCI.0516-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Peri-ictal autonomic abnormality in Kv1.1 KO mouse #1.
Fig. S2. Peri-ictal autonomic abnormality in Kv1.1 KO mouse #2.
Table S1. Raw data
Movie S1. OGD-SD in brainstem slice