

Abstract
Chlamydia trachomatis is auxotrophic for a variety of essential metabolites. Inhibitors that interrupt host cell catabolism may inhibit chlamydial growth and reveal Chlamydia metabolite requirements. We used the known indoleamine-2,3-dioxygenase (IDO)-inhibitor 4-phenyl imidazole (4-PI) to reverse Interferon (IFN)-γ-induced chlamydial growth inhibition. However, at elevated inhibitor concentrations chlamydial growth was arrested even in the absence of IFN-γ. Since 4-PI is known to interfere with cholesterol metabolism, the effect of cholesterol add-back was tested. Chlamydia growth was restored in the presence of cholesterol in serum-containing, but not serum-free medium suggesting that cholesterol and other serum components are required for growth recovery. When serum factors were tested, either cholesteryl linoleate or the combination of cholesterol and linoleic acid restored chlamydial growth. However, growth was not restored when either cholesterol or linoleic acid were added alone, suggesting that the production of cholesteryl esters from cholesterol and fatty acids was affected by 4-PI treatment. In eukaryotic cells, the enzyme Acyl-CoA:cholesterol acyltransferase (ACAT) catalyzes the production of cholesteryl esters. When HeLa cells were treated with the ACAT-specific inhibitor 4-hydroxycinnamicacid amide C. trachomatis growth was interrupted, but was restored by the addition of cholesteryl linoleate, suggesting that ACAT activity is necessary for intracellular Chlamydia growth.
Keywords: cholesteryl esters, sterol o-acyltransferase
This manuscript shows that cholesterol esterification by the important eukaryotic enzyme Acyl-CoA:cholesterol acyltransferase (ACAT) is necessary for intracellular Chlamydia growth.
Graphical Abstract Figure.
This manuscript shows that cholesterol esterification by the important eukaryotic enzyme Acyl-CoA:cholesterol acyltransferase (ACAT) is necessary for intracellular Chlamydia growth.
INTRODUCTION
The obligate intracellular bacterial pathogen Chlamydia trachomatis is responsible for ocular or genital infections in humans. The C. trachomatis serovars D–K are the world-wide leading cause of bacterial sexually transmitted diseases with more than 1.4 million newly reported cases in the USA alone in 2012 (STD surveillance report 2012, CDC: http://www.cdc.gov/std/stats12/default.htm, 13 April, date last accessed)
The unique biphasic developmental cycle with two morphologically distinguishable forms is characteristic for Chlamydia. The extracellular elementary bodies (EBs) exhibit limited metabolic activity and are the infectious form of Chlamydia. Once EBs invade the host cell they are found in a membrane-bound compartment called the inclusion. Here, the EBs transform into metabolically active, but non-infectious reticulate bodies (RBs). After multiple rounds of replication RBs convert back to the infectious EBs, which are released either by cell lysis or by extrusion and can invade new eukaryotic cells (Hybiske and Stephens 2007).
The Chlamydia genome is small (∼1 Mbp) compared to most other bacteria and reflects the adaptation to an obligate intracellular life style with an auxotrophy for many metabolites (Stephens et al. 1998; Kalman et al. 1999; Read et al. 2000; Read et al. 2003; Horn et al. 2004; Azuma et al. 2006; Thomson et al. 2008) that are obtained from the eukaryotic host during infection. One important group of metabolites obtained from the host cell is lipids. Although Chlamydia produce some lipids on their own (phosphatidylethanolamine, phosphatidylglycerol and phosphatidylserine), others (phosphatidylcholine, phosphatidylinositol, sphingomyelin, cardiolipin and cholesterol), are transported from various sources within the host cell into the inclusion and are acquired by RBs (reviewed in Elwell and Engel 2012). For example, it is known that exogenous lipids may be obtained via receptor‐mediated uptake (e.g. low-density lipoprotein (LDL), CD36 scavenger receptor), which are re-routed to the inclusion (Kalayoglu et al. 1999). Other eukaryotic de novo synthesized lipids are acquired by Chlamydia by manipulating exocytic pathways and re-routing lipid-containing vesicles to the inclusion. Beatty (2006) demonstrated that multivesicular bodies are re‐directed from the host cytoplasm to the inclusion. In addition, a Brefeldin A‐independent transport of Golgi particles has also been reported (Wylie, Hatch and McClarty 1997; Heuer et al. 2009). Lipid droplets can be found in the lumen of inclusions (Kumar, Cocchiaro and Valdivia 2006; Cocchiaro et al. 2008), which are transported to the inclusion in association with peroxisomes (Boncompain et al. 2014). The lipid transporter ABCA1 and the lipid acceptor protein ApoA1 also are recruited to the inclusion (Cox et al. 2012).
In human cell lines Interferon (IFN)-γ-induced persistence in Chlamydia is mainly a result of the depletion of tryptophan due to the induction of the tryptophan de-cyclizing, heme-containing enzyme indoleamine-2,3-dioxygenase (IDO) (Byrne, Lehmann and Landry 1986). To study IDO-independent effects of IFN-γ on Chlamydia growth, we used the IDO-inhibitor 4-phenyl imidazole (4-PI), but at higher concentrations the inhibitor blocked chlamydial growth independent of IFN-γ treatment. 4-PI is known to target enzymes other than IDO, including members of the cytochrome P450 family (McLean et al. 2002). In addition, derivatives of 4-PI inhibit the eukaryotic enzyme Acyl:CoA: cholesterol o-acetyltransferase and these chemical inhibitors are used as treatment for hypercholesterolemia (Kimura et al. 1993). We found that chlamydial growth inhibition by 4-PI was reversed by cholesterol addition in serum-containing, but not in serum-free medium. We identified phosphocholine and cholesteryl linoleate, as essential components in serum for Chlamydia growth recovery. Interestingly, cholesterol and linoleic acid reversed the inhibitory effect of 4-PI only when added together to the medium. When we inhibited the enzyme that catalyzes cholesterol esterification in eukaryotic cells, Acyl-CoA:cholesterol acyltransferase (ACAT), with the specific ACAT-inhibitor 4‐hydroxycinnamicacid amide (4-HCAA), we observed growth reduction of three different C. trachomatis strains representing each C. trachomatis biovar. Taken together, our data demonstrate that C. trachomatis requires cholesterol esterification by eukaryotic ACAT before it is available for chlamydial growth.
MATERIALS AND METHODS
Chemicals
IFN-γ was purchased from R&D Systems (Minneapolis, MN) and the ACAT-inhibitor 4-HCAA (chemical name: N-[3-(4-hydroxyphenyl)-1-oxo-2-propenyl]-L-phenylalanine, methyl ester) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Unless stated otherwise all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO). All lipids were dissolved at a concentration of 1 mg ml−1 in medium containing 200 μg ml−1 methyl-β-cyclodextrin.
Cell culture and bacterial strains
The human epithelial cell line HeLa 229 (American Type Culture Collection, Manassas, VA) was cultured in Dulbecco's modified Eagle medium (DMEM, Lonza, Walkersville, MD) supplemented with 10% fetal bovine serum, 0.01 mg ml−1 gentamicin (Gibco, Grand Island, NY), 1 mM sodium pyruvate, 2 mM L-glutamine, 10 mM Hepes and 0.055 mM β-mercaptoethanol at 37°C with 5% CO2 in a humidified atmosphere. For some experiments, the cell culture medium was used without the addition of FBS. Two different media were used in the course of this work: DMEM (full) refers to serum (FBS) containing; and DMEM (-serum) for FBS-free medium. Chlamydia trachomatis serovar D/UW-3Cx and C. trachomatis serovar A/HAR-13 were routinely grown in HeLa cells at 37°C, 7% CO2 and stocks were purified as previously described (Schachter and Wyrick 1994). Chlamydia trachomatis serovar L2/434/Bu was kindly provided by R. Belland (UTHSC Memphis, TN).
Inhibitor treatment and C. trachomatis infection of HeLa cells
HeLa cells were seeded at a density of 3.75 × 104 cells per well in 48 well plates 48 h prior to infection. Twenty four hours before infection, the medium was removed and replaced with DMEM containing 50 ng ml−1 IFN-γ or inhibitor solution. Unless stated otherwise experiments were done with inhibitor concentrations of 1 mM 4-PI or 200 μM 4-HCAA. At the day of infection, the medium was transferred to a tube (maintained medium), the cells were washed in HBSS (Gibco, Grand Island, NY) and incubated with 1 × DEAE-Dextran in HBSS for 10 min at 37°C. DEAE-Dextran was removed, the cells were washed in HBSS, and infected with C. trachomatis diluted in sucrose-phosphate-glutamic acid buffer (SPG, 0.22 M sucrose, 3.8 mM KH2PO4, 10 mM Na2HPO4, 5 mM L-glutamic acid; pH7.4). Cells were infected with an MOI of 0.5 for immunofluorescence or an MOI of 1 for titration experiments, respectively. Infected cells were centrifuged at 1200 rpm for 1 h at 30°C and incubated for 30 min at 37°C. The inoculum was removed (start of infection), and the maintained medium was returned. The infected cells were further incubated at 37°C with 7% CO2 until either fixed in methanol for immunofluorescence or harvested for titration.
Immunofluorescence microscopy and EB titration
For fixation the cells were washed in PBS at the indicated times post infection (p.i.), and fixed in methanol for 10 min at RT. Chlamydia trachomatis was identified by immunofluorescence microscopy using a C. trachomatis specific staining kit (Argene, North Massapequa, NY) as described by the manufacturer's protocol. HeLa cells were counterstained with 2 μg ml−1 Evans blue. The stained cells were examined with a Nikon Eclipse TE200-U fluorescence microscope using a 200× magnification. Images were obtained with a Retiga 1300 cooled 12-bit camera (QImaging, Surrey, Canada) using the IPLab software version 3.9.2 (Becton, Dickinson and Company, Franklin Lakes, NJ). For determination of inclusion forming units (IFU) cells were harvested at indicated time points, sonicated, pelleted by centrifugation at 14 000 rpm at 4°C for 20 min and resuspended in SPG buffer. Fresh HeLa cells were infected with dilutions as described above. After 24 h, the cells were fixed in methanol and stained with Giemsa stain for IFU determination as described in (Conn et al. 1960).
Statistics
All experiments were done with three biologically independent replicate samples and experimental data is represented as mean ± standard deviation (SD). All statistic calculations were done in Excel using a one-tailed paired Student's t-test to compare two groups of data or ANOVA to compare more than two groups. Only P values ≤ 0.05 were considered significant.
RESULTS
The IDO-inhibitor 4-PI, but not L-1-methyl tryptophan, blocks chlamydial growth
The original intent of the study was to study IDO-independent effects of IFN-γ induced persistence. We therefore treated HeLa cells with IFN-γ with or without the addition of IDO inhibitors at concentrations of 10 nM, 100 nM and 1 μM (1-MT); or 100 μM and 1 mM (4-PI), and infected the cells 24 h post-treatment with C. trachomatis serovar D as described in the section ‘Material and Methods’. Treatment with IFN-γ resulted in growth inhibition of C. trachomatis D with reduced and abnormal inclusion size (as seen in Fig. 1) consistent with persistent Chlamydia morphology (Beatty, Byrne and Morrison 1993). Cotreatment with 1-MT at 1 μM and 4-PI at 100 μM led to large inclusion with normal morphology suggesting that addition of the inhibitors blocked the effect of IDO. Interestingly, growth of the chlamydiae was significantly reduced at higher concentration of 4-PI (1 mM) and showed smaller inclusions compared to untreated cells. Growth reduction was observed with or without IFN-γ demonstrating an IFN-γ and IDO independent growth inhibition in the presence of this inhibitor.
Figure 1.
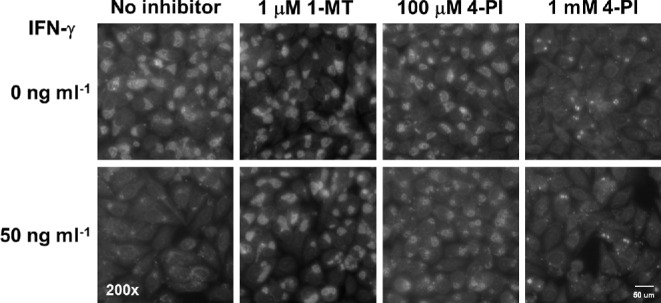
The IDO-inhibitor 4-PI inhibits C. trachomatis growth independent of IFN-γ. Cell were left untreated (upper panel) or pre-treated with 50 ng ml−1 IFN-γ treated (lower panel) and incubated for 24 h in the absence or presence of either 1 μM L-1-methyl tryptophan (1-MT), 100 μM or 1 mM 4-Phenyl imidazole (4-PI). The cells were infected after 24 h with an MOI of 1 and were fixed in methanol after 24 h p.i. The fixed cells were stained with an FITC-conjugated antibody against C. trachomatis and counterstained with Evans blue. Representative images (200× magnification fluorescence) are shown.
Inhibition of 4-PI reduces chlamydial growth during the metabolically active phase
To identify at which stage during the chlamydial developmental cycle chlamydiae are affected by 4‐PI, we added 4-PI at different time points during the infection and varied the length of treatment time. We first analyzed if 4-PI affected chlamydial attachment or invasion. For some samples, we pre-treated the cells for 24 h before the infection and either removed 4-PI before or after the infection. For another set of samples the inhibitor was only present during the time of infection. We infected the cells, fixed the cells at 24 h p.i., and stained for C. trachomatis. As Fig. 2A shows, only cells pre-treated with 4-PI in the continuous presence of the inhibitor after infection showed inhibited growth. We next tested if 4-PI inhibited the chlamydiae at later stages of the developmental cycle. We pre-treated cells for 24 h with 4-PI, or added the inhibitor after 0, 12 or 24 h p.i. and harvested EBs after 48 h p.i. Fig. 2B shows that the amount of recovered EBs was only reduced when the inhibitor was added at 12 h p.i. or earlier. Treatment after 24 h p.i. showed similar IFU production as did infections of untreated cells. It is known that C. trachomatis converts from EBs to RBs at around 12 h p.i. (Miyairi et al. 2006) and that many chlamydiae convert back to EBs by 24 h p.i. These data indicate that the 4-PI induced growth inhibition affects Chlamydia during the metabolically active phase of RB development.
Figure 2.
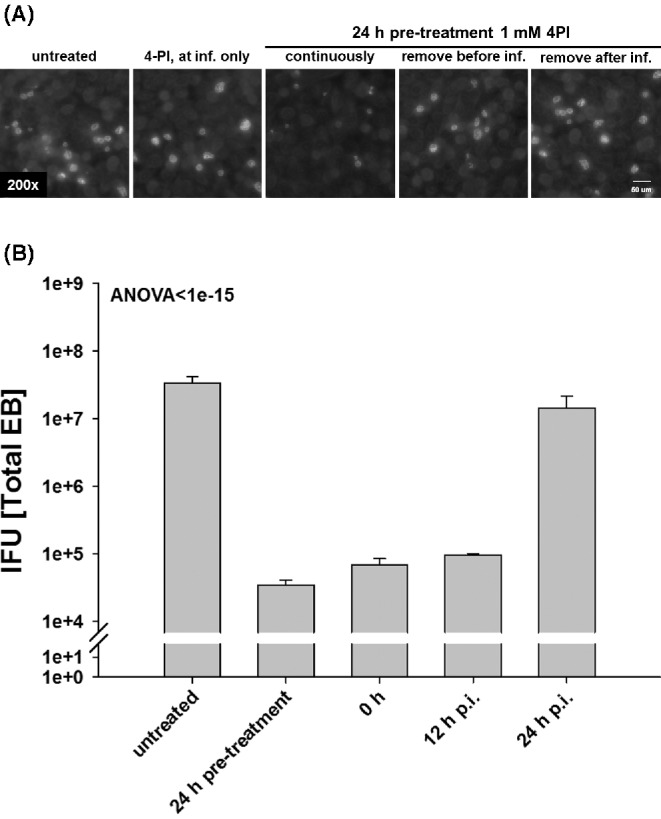
4-PI inhibits C. trachomatis growth when added before EB to RB transition. HeLa cells were treated at different time points prior to or after infection with C. trachomatis D. (A) HeLa cells were left untreated, pre-treated for 24 h with 1 mM 4-PI, inhibitor added only during or directly after infection. The cells were infected with C. trachomatis with an MOI of 0.5 and were methanol fixed 48 h p.i. Chlamydia trachomatis was stained with an FITC-conjugated antibody to C. trachomatis and cells counterstained with Evans blue. Representative images (200× magnification fluorescence) are shown. (B) HeLa cells were left untreated, pre-treated for 24 h with 1 mM 4-PI, or the inhibitor added directly after infection (0 h), 12 h or 24 h p.i. The cells were infected with an MOI of 1 and harvested after 48 h p.i. Fresh HeLa cells were infected with dilutions to titrate the C. trachomatis IFU. All titrations are the mean of three biologically independent replicate samples and error bars represent SD.
4-PI affects cholesterol-dependent metabolic pathways during chlamydial infections
It is known that 4-PI and its derivatives affect enzymes that are part of the catabolism of cholesterol and other sterols. To test if cholesterol depletion is a limiting factor for chlamydial growth after 4-PI treatment, we first inhibited chlamydial growth with 4-PI. The medium was then removed from the cells after 24 h p.i. and either 25 μg ml−1 cholesterol was added directly to inhibitor containing medium or replaced with fresh cell culture medium without inhibitor. We harvested the cells 24 h after rescue and determined the recovered EBs by IFU measurement. The addition of cholesterol to the inhibitor-containing medium partially restored chlamydial growth as shown in Fig. 3. These results demonstrated that cholesterol depletion is one limiting factor for chlamydial growth inhibition during 4‐PI treatment.
Figure 3.
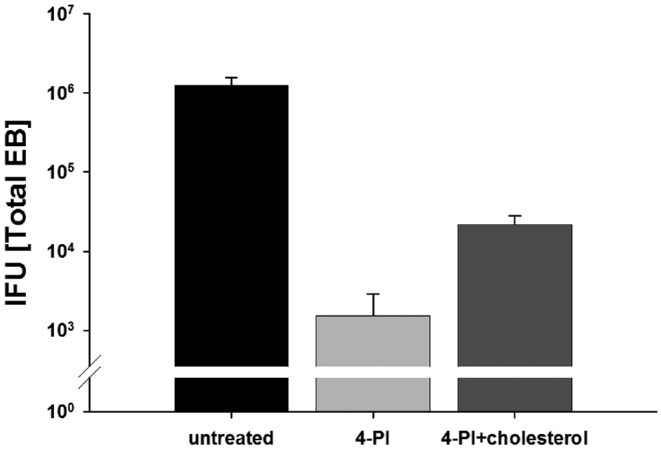
4-PI induced growth inhibition can be restored after removing the inhibitor or adding exogenous cholesterol. Chlamydia trachomatis infected cells with or without 1 mM 4-PI were incubated for 24 h and the medium exchanged with inhibitor, with inhibitor and 25 μg ml−1 cholesterol, or medium without inhibitor and further incubated for additional 24 h. Cells were harvested 48 h p.i. and HeLa cells fresh infected with dilutions of the harvested samples to titrate the C. trachomatis IFU. All titrations are the mean of three biologically independent replicate samples. Error bars represent SD.
Fetal bovine serum contains components that are required for Chlamydia recovery after 4-PI inhibition
To further characterize the nature of the 4-PI inhibition and to identify a potential enzyme target for the inhibitor, we used serum-free medium for better defined growth conditions. Surprisingly, chlamydial growth did not resume after 4-PI induced inhibition for infected cells cultured in serum-free medium, while the addition of cholesterol in complete medium resulted in an increase of infectious EBs (data not shown). To test if serum was required for cholesterol mediated recovery, we pre-treated cells with 4-PI in complete or serum-free medium. For recovery, we either replaced the medium completely or added cholesterol directly to the inhibitor containing medium (Fig. 4). The replaced medium had either no additional supplements or contained cholesterol with final concentrations of FBS at 0%, 5%, 10% or 20% (v/v). When cholesterol in serum-free medium alone was used to recover the growth inhibition, no recovery was observed. However, fresh serum-containing complete medium without the addition of cholesterol also restored chlamydial growth, demonstrating that FBS contains the necessary components to reverse the effect of 4-PI inhibition. In addition, we found that 4-PI induced growth inhibition could be reversed when cholesterol was added to the inhibitor in complete medium in an FBS-dose-dependent manner verifying that FBS contains components that are required to restore chlamydial growth after 4-PI inhibition. Collectively, the data suggest that additional components in serum are necessary for cholesterol dependent rescue.
Figure 4.
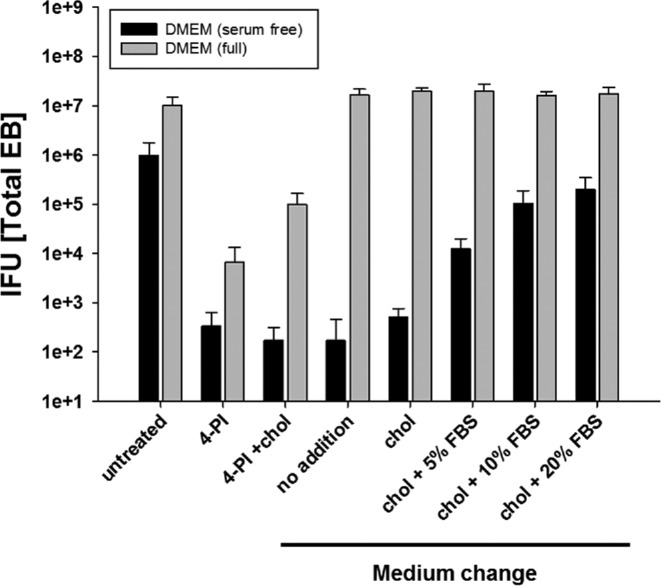
Serum is required to reverse the 4-PI-induced C. trachomatis growth inhibition. HeLa cells were left untreated or pre-treated for 24 h with 1 mM 4-PI in serum-free or serum-containing complete medium. The cells were infected with C. trachomatis with an MOI of 1 and after 24 h p.i. cells either maintained in medium or the medium replaced. Cholesterol without or with different concentrations of FBS in the medium was added as supplement. Cells were harvested 48 h p.i. and fresh HeLa cells infected with dilutions of the harvested samples to titrate the C. trachomatis IFU. All titrations are the mean of three biologically independent replicate samples and error bars represent SD.
Vitamins and lipids in serum contribute to chlamydial recovery after 4-PI inhibition
FBS contains many different possible components such as proteins, hormones, salts, lipids and vitamins. We concentrated on nutrition groups known to be required by Chlamydia (salts, lipids, vitamins) although we are aware that other components in serum may be also required for recovery after 4-PI induced growth inhibition. We first used pools of the tested supplements. For the pool of salts, we used CaCl2, FeCl2, FeCl3, CuSO4 and MgCl2 at final concentration of 25 μg ml−1 for each individual salt. For vitamins, we used a commercially available mix of all vitamins required by eukaryotic cells (RPMI 1640 100 × vitamins, Sigma). The lipids tested were a mix of phosphocholine, cholesterol, linoleic acid, cholesteryl linoleate and glyceride trilinoleate at a final concentration of 25 μg ml−1. These additions represent lipids typically found in serum. HeLa cells were pre-treated with 4-PI in serum-free medium and the medium replaced after 24 h p.i. with fresh serum-free medium containing the different nutrition pools or medium with 10% FBS final concentration. Fig. 5A shows that FBS, vitamins and lipids, but not the mix of salts, reversed the 4-PI induced inhibition. We focused on the groups of lipids and repeated the experiment using single lipids. The main source of lipids in serum is from LDL. The LDL lipids break down as 10% triacylglycerols, 11% free cholesterol, 29% phospholipids and 50% cholesteryl esters (from Christy 2014): http://lipidlibrary.aocs.org/Lipids/lipoprot/index.htm, 13 April 2015, date last accessed). When we added the single lipids to serum-free medium (Fig. 5B), only phosphocholine and cholesteryl linoleate, but not cholesterol, linoleic acid or glyceride trilinoleate resulted in recovery of chlamydial growth. However, the combination of cholesterol and linoleic acid also reversed the effect of 4-PI indicating that this esterified cholesterol compound may be required for the rescue of Chlamydia after 4-PI induced growth inhibition.
Figure 5.
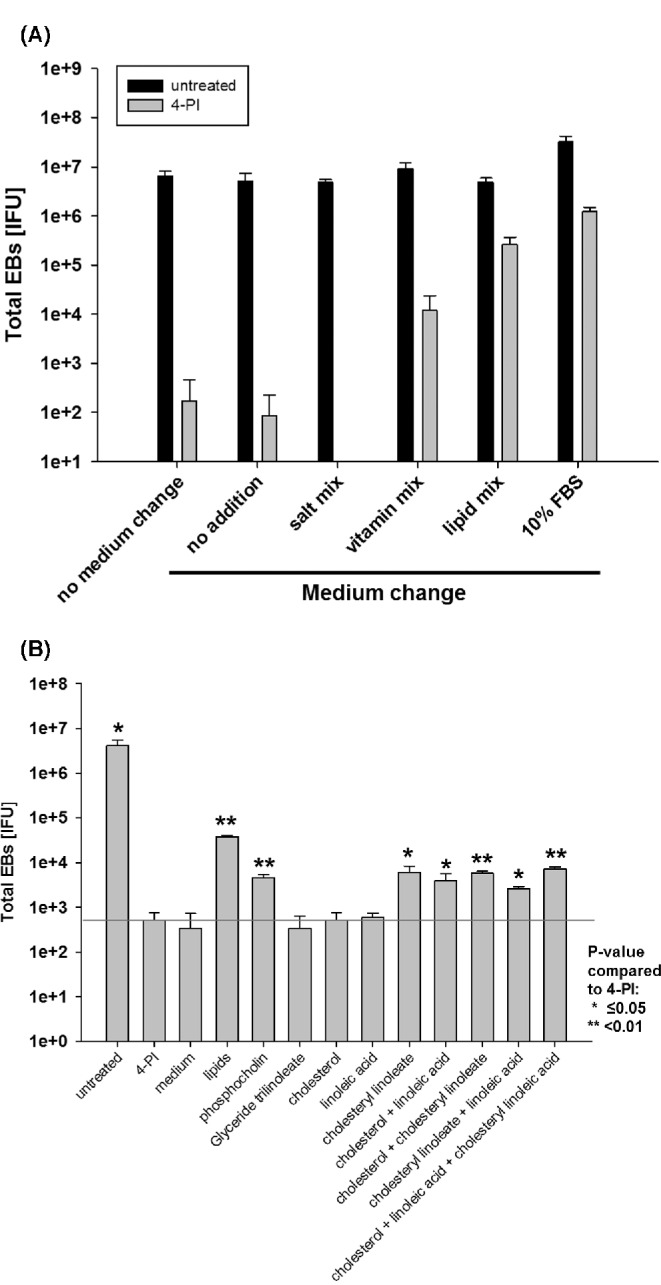
Lipids in serum are the main contributing components during recovery of C. trachomatis after 4-PI growth inhibition. HeLa cells were left untreated or pre-treated with 1 mM 4-PI in serum-free medium and infected with an MOI of 1. (A) After 24 h, the medium was replaced with fresh medium with or without supplements of cholesterol, or pools of vitamins, salts or lipids, respectively. (B) After 24 h the medium was replaced with fresh medium with or without supplements of glyceride trilinoleate, phosphocholine, cholesterol, linoleic acid, cholesteryl linoleate or combinations of the latter three. Cells were harvested 48 h p.i. and HeLa cells fresh infected with dilutions the harvested samples to titrate the C. trachomatis IFU. All titrations are the mean of three biologically independent replicate samples and error bars represent SD.
Chlamydial growth is reduced by an ACAT-specific inhibitor in a dose-dependent manner and is restored by cholesteryl linoleate
In eukaryotic cells, the main enzyme that catalyzes the production of cholesteryl esters from cholesterol and fatty acids is ACAT also called sterol o-acyltransferase. The previous result suggests that ACAT is a possible target of 4-PI and that cholesteryl ester production may be required for the transport of cholesterol into Chlamydia. To test if ACAT activity is necessary for chlamydial growth, we used the specific ACAT-inhibitor 4-HCAA (Lee et al. 2004).
We first tested if the inhibitor affected HeLa cell growth by doing a dose response of 4-HCAA on uninfected cells and incubated for 24 h p.i. We found that ACAT-inhibitor concentrations ≥250 μM reduced HeLa cell growth (data not shown). Therefore, only 4-HCAA concentrations of 100, 150 and 200 μM were used for further analysis, which did not affect growth of the HeLa cells even after 48 h of treatment (upper panel in Fig. 6A). HeLa cells were infected, fixed after 24 h p.i. and stained for IFA as described above. Inclusion size was measured as an indicator for growth. Images were randomly taken and the inclusion size calculated by ImageJ. The average size of 150 chlamydial inclusions with inhibitor was plotted against untreated cells. Fig. 6A shows IFA of untreated or 4-HCAA treated cells in uninfected or infected cells. Fig. 6B illustrates the graph of the average size of 150 chlamydial inclusions. As seen in the graph, the inclusion size is reduced in a dose-dependent manner with increasing amounts of 4-HCAA. These data suggest that ACAT activity is required for chlamydial growth in eukaryotic cells.
Figure 6.
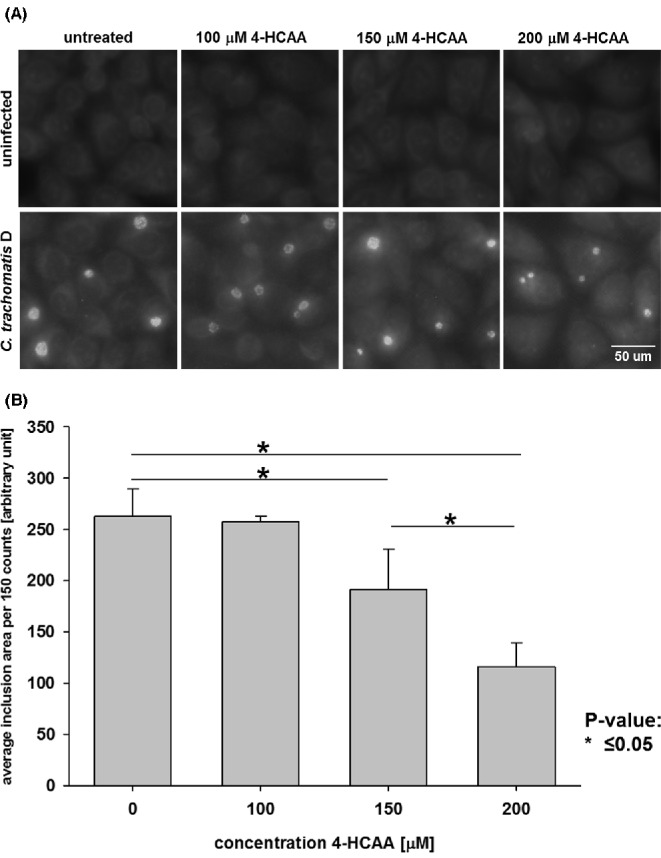
Chlamydial growth reduction by the specific ACAT inhibitor is concentration dependent. HeLa cells were left untreated or pre-treated for 24 h p.i. with ACAT-inhibitor concentrations between 100–200 μM. Cells were infected with an MOI of 0.5 and fixed after 24 h p.i. Chlamydia trachomatis was stained with an FITC-conjugated antibody to C. trachomatis and cells counterstained with Evans blue. (A) Representative images of C. trachomatis inclusions are shown (200× magnification) (B) Images were randomly taken and the area size in the green channel determined with ImageJ. The graph shows the mean of 150 different inclusion areas and error bars represent SD.
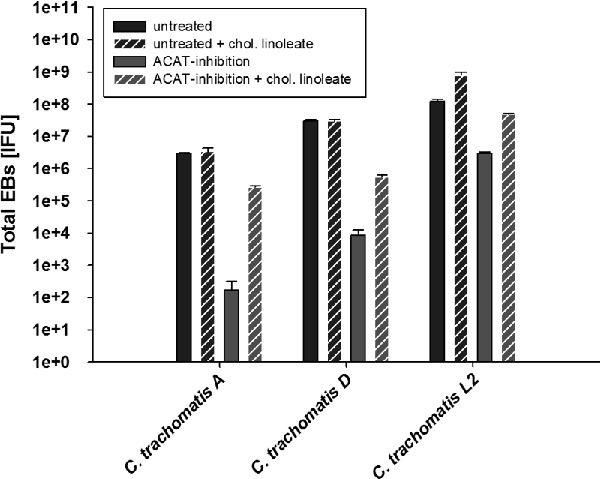
We then treated HeLa cells for 24 h with either 4-PI, 4-HCAA or left the cells untreated in serum-free medium. HeLa cells were infected with C. trachomatis for EB titration and incubated for 24 h p.i. To test if C. trachomatis D growth inhibition was reversed, we removed the inhibitor-containing medium and added fresh medium with 25 μg ml−1 cholesteryl linoleate to some samples for rescue. The cells were harvested after an additional 24 h for titration of the total amount of infectious EBs in the samples. Fig. 7 shows the total amount of EBs during inhibition and recovery in the presence of cholesteryl linoleate compared to untreated samples. The graph illustrates that each inhibitor reduced the amount of IFUs significantly and that growth can be partially reversed by adding cholesteryl linoleate to the medium, although the inhibitory effect of 4-PI was more dramatic compared to 4-HCAA. It is possible that ACAT is only one potential target of 4-PI and other, not yet identified, enzymes are also inhibited during 4-PI treatment. A previous publication demonstrated in in vitro assays that even at a concentration of 200 μM ACAT-specific inhibitor still retained some residual activity (Lee et al. 2004). Therefore, it is possible that the produced amount of cholesteryl esters in the cells even with low ACAT activity is sufficient for minimal Chlamydia growth. To test if other C. trachomatis strains are similarly affected by ACAT inhibition, we repeated the experiment with 4-HCAA using the ocular C. trachomatis serovar A strain and the lymphogranuloma venereum C. trachomatis serovar L2 strain. As seen in Fig. 8, both strains showed statistically significant reduced amounts of EBs recovered after ACAT inhibition. However, while the total amount of recovered EBs for C. trachomatis serovar A and serovar D were reduced 3–4 logs compared to untreated cells, Chlamydia trachomatis serovar L2 showed 1 log reduction compared to the untreated sample, likely due to the faster growth of serovar L2 strain that can ‘out-compete’ the cell for the lipid.
Figure 7.
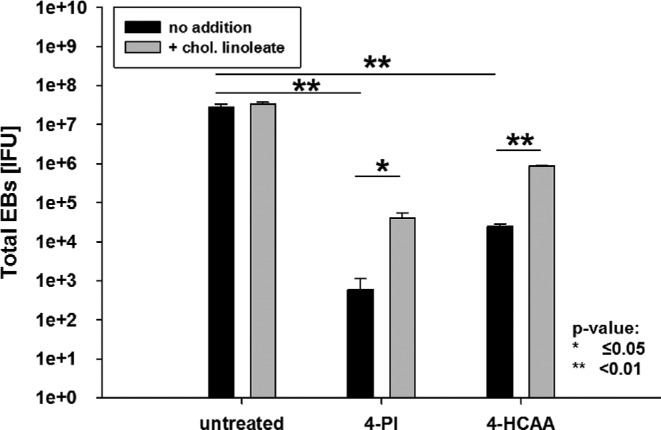
Chlamydial growth is reduced by the specific ACAT inhibitor and can be restored with cholesteryl linoleate. HeLa cells were left untreated or pre-treated with either 1 mM 4-PI or 200 μM ACAT inhibitor. HeLa cells were infected with an MOI of 1 and after 24 h p.i. the inhibitor containing medium replace with 25 μg ml−1 cholesteryl linoleate. The cells were harvested after 48 h p.i. and fresh HeLa cells were infected with dilutions of harvested samples to titrate the C. trachomatis IFU. All titrations are the mean of three biologically independent replicate samples and error bars represent SD.
Figure 8.
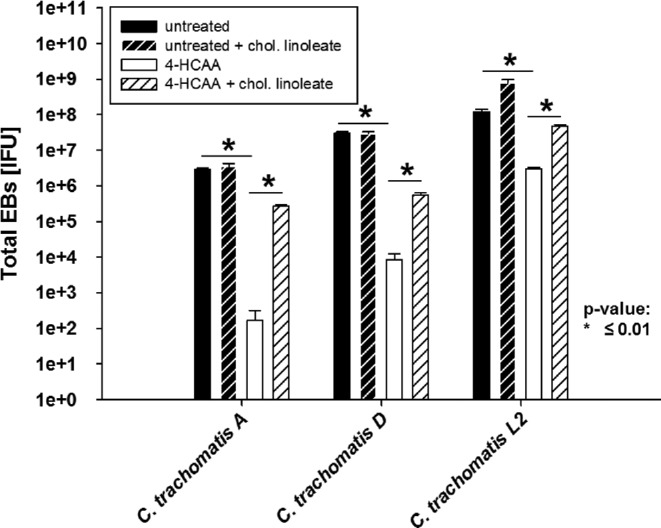
Growth of C. trachomatis serovar A and L2 is affected by ACAT inhibition in a manner similar to C. trachomatis D. HeLa cells were left untreated or pre-treated with 200 μM 4-HCAA. HeLa cells were infected with an MOI of 1 using C. trachomatis serovars A, D and L2. After 24 h p.i. the inhibitor containing medium was replaced with 25 μg ml−1 cholesteryl linoleate. The cells were harvested after 48 h p.i. and fresh HeLa cells were infected with dilutions of harvested samples to titrate the C. trachomatis IFU. All titrations are the mean of three biologically independent replicate samples and error bars represent SD.
DISCUSSION
Chlamydia has a minimal genome reflecting extreme auxotrophy and dependence on host-cell derived metabolites. Lipids, including phospholipids, cholesterol and cholesteryl esters, are among the most imported eukaryotic metabolites by Chlamydia. Despite the current knowledge of many pathways by which Chlamydia transport lipids to the inclusion (Wylie, Hatch and McClarty 1997; Beatty 2006; Kumar, Cocchiaro and Valdivia 2006; Cocchiaro et al. 2008; Heuer et al. 2009; Cox et al. 2012; Boncompain et al. 2014), many steps of the lipids transport and uptake processes during the chlamydial developmental cycle are not fully understood.
We used the IDO-inhibitor 4-PI to investigate tryptophan depletion independent effects on C. trachomatis, and coincidentally found that 4-PI inhibited chlamydial growth even in the absence of IFN-γ suggesting that the activity of enzymes other than IDO were responsible for the inhibitory effect. Most of the known targets of 4-PI and its derivatives bind to enzymes catabolizing either cholesterol or other steroids. Chlamydiae were able to recover after 4-PI induced growth inhibition once exogenous cholesterol was added indicating that cholesterol metabolism was affected. We found that in serum-free medium phosphocholine and cholesteryl linoleate restored Chlamydia growth, while cholesterol, linoleic acid and glyceride trilinoleate did not reverse the inhibition. Interestingly, cholesterol and linoleic acid in combination also reversed the effect of 4-PI suggesting that the production of cholesteryl linoleate in the host cells is blocked by 4-PI. The enzyme in eukaryotic cells that is largely responsible for cholesteryl ester synthesis is ACAT and derivatives of 4-PI were used to target the eukaryotic enzyme previously (Kimura et al. 1993). We found a concentration-dependent growth reduction of C. trachomatis when using an ACAT-specific inhibitor, which we could reverse by the addition of cholesteryl esters.
There are two known paralogs of ACAT in eukaryotic cells called ACAT-1 and ACAT-2. In eukaryotic cells, ACAT-1 catalyzes the formation of cholesteryl esters, the main transport and storage form of cholesterol, while ACAT-2 is also involved in phospholipid synthesis. ACAT is essential in eukaryotic cells for the homoeostasis of cholesterol since high concentrations of cholesterol are toxic for eukaryotic cells. Excess amounts of cholesterol are esterified with fatty acids by ACAT and either stored in lipid droplets (Guo et al. 2009) or shuttled to the ABCA1 exporter where the lipids are transported to extracellular high-density lipoprotein (HDL) (Dove et al. 2005).
In our experiments, the addition of the phospholipid phosphocholine (PC) to serum-free medium also restored chlamydial growth after inhibition by 4-PI. Although it is not known if 4‐PI or derivatives can also inhibit ACAT-2 and prevent esterification of phospholipids, it is possible that, similar to cholesterol, esterification of phospholipids with fatty acids is necessary before they can be transported into the inclusion. Both PC and CEs are transported by ABCA1 and Cox et al. (2012) showed recently that the transporter ABCA1 and its lipid-binding-associated protein ApoA1 are recruited to the inclusion. It is further known that ACAT in the ER is required for lipid droplet formation (Guo et al. 2009). The expression of ACAT-1 is upregulated during C. pneumoniae infection of macrophages and is required for foam cell formation in macrophages (He et al. 2009). Additionally, a previous publication demonstrated that C. psittaci 6BC infected MK-2 cells contained increased amounts of cholesteryl esters compared to uninfected cells (Makino et al. 1970), but the requirement of ACAT activity by Chlamydia was not known. ACAT is found in many tissues and cell types that are infected by Chlamydia such as macrophages and epithelial cells (Sakashita et al. 2000).
Our current work demonstrates that ACAT-dependent cholesterol esterification is important for Chlamydia and it is likely that cholesteryl esterification is an essential step before cholesterol is imported into the inclusion (illustrated in Fig. 9). The advantage of transporting cholesteryl esters into chlamydiae is that two necessary lipids, cholesterol and fatty acids, are transported in a one-step process rather than being transported separately. In chlamydiae, the cholesteryl esters can be hydrolyzed and, if required, the lipids further modified. We recently published that the C. trachomatis ct149 gene product has esterase activity and hydrolyzes cholesteryl linoleate (Peters et al. 2012). This protein is therefore a possible candidate for a lipase that hydrolyzes imported eukaryotic lipids. Future research will focus on additional enzymatic activities of CT149 and other potential candidate enzymes in Chlamydia that can hydrolyze esters and serve as nutrient for Chlamydia.
Figure 9.

Model of ACAT influence on C. trachomatis growth. A model of the influence of ACAT activity on intracellular chlamydial growth. The ER localized eukaryotic enzyme catalyzes the esterification of cholesterol in the lumen of the smooth ER and the produced cholesteryl esters are transported to lipid droplets. Lipids in the lipid droplets are then accessible for Chlamydia and imported into the chlamydiae. Here, chlamydial enzymes such as CT149 can break down the esters and utilize cholesterol and the free fatty acid.
Acknowledgments
We would like to thank Bob Belland and Yasser AbdelRahman for providing us C. trachomatis L2/434/Bu.
FUNDING
This work was supported by the National Institutes of Health [grant number AI 19782] to [GIB].
Conflict of interest. None declared.
REFERENCES
- Azuma Y, Hirakawa H, Yamashita A, et al. Genome sequence of the cat pathogen, Chlamydophila felis. DNA Res. 2006;13:15–23. doi: 10.1093/dnares/dsi027. [DOI] [PubMed] [Google Scholar]
- Beatty WL. Trafficking from CD63-positive late endocytic multivesicular bodies is essential for intracellular development of Chlamydia trachomatis. J Cell Sci. 2006;119:350–9. doi: 10.1242/jcs.02733. [DOI] [PubMed] [Google Scholar]
- Beatty WL, Byrne GI, Morrison RP. Morphologic and antigenic characterization of interferon gamma-mediated persistent Chlamydia trachomatis infection in vitro. P Natl Acad Sci USA. 1993;90:3998–4002. doi: 10.1073/pnas.90.9.3998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boncompain G, Muller C, Meas-Yedid V, et al. The intracellular bacteria Chlamydia hijack peroxisomes and utilize their enzymatic capacity to produce bacteria-specific phospholipids. PLoS One. 2014;9:e86196. doi: 10.1371/journal.pone.0086196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne GI, Lehmann LK, Landry GJ. Induction of tryptophan catabolism is the mechanism for gamma-interferon-mediated inhibition of intracellular Chlamydia psittaci replication in T24 cells. Infect Immun. 1986;53:347–51. doi: 10.1128/iai.53.2.347-351.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy WW. Plasma Lipoproteins: Composition, Structure And Biochemistry. 2014. http://lipidlibrary.aocs.org/Lipids/lipoprot/index.htm. [Google Scholar]
- Cocchiaro JL, Kumar Y, Fischer ER, et al. Cytoplasmic lipid droplets are translocated into the lumen of the Chlamydia trachomatis parasitophorous vacuole. P Natl Acad Sci USA. 2008;105:9379–84. doi: 10.1073/pnas.0712241105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn HJ, Darrow MA, Emmel VM, et al. Staining Procedures Used by the Biological Stain Commission. Baltimore, MD:: The Williams and Wilkins Company; 1960. [Google Scholar]
- Cox JV, Naher N, Abdelrahman YM, et al. Host HDL biogenesis machinery is recruited to the inclusion of Chlamydia trachomatis-infected cells and regulates chlamydial growth. Cell Microbiol. 2012;14:1497–512. doi: 10.1111/j.1462-5822.2012.01823.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove DE, Su YR, Zhang W, et al. ACAT1 deficiency disrupts cholesterol efflux and alters cellular morphology in macrophages. Arterioscl Thromb Vas. 2005;25:128–34. doi: 10.1161/01.ATV.0000148323.94021.e5. [DOI] [PubMed] [Google Scholar]
- Elwell CA, Engel JN. Lipid acquisition by intracellular Chlamydiae. Cell Microbiol. 2012;14:1010–8. doi: 10.1111/j.1462-5822.2012.01794.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Cordes KR, Farese RV, Jr, et al. Lipid droplets at a glance. J Cell Sci. 2009;122:749–52. doi: 10.1242/jcs.037630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He P, Mei C, Cheng B, et al. Chlamydia pneumoniae induces macrophage-derived foam cell formation by up-regulating acyl-coenzyme A: cholesterol acyltransferase 1. Microbes Infect. 2009;11:157–63. doi: 10.1016/j.micinf.2008.11.001. [DOI] [PubMed] [Google Scholar]
- Heuer D, Lipinski A Rejman, Machuy N, et al. Chlamydia causes fragmentation of the Golgi compartment to ensure reproduction. Nature. 2009;457:731–5. doi: 10.1038/nature07578. [DOI] [PubMed] [Google Scholar]
- Horn M, Collingro A, Schmitz-Esser S, et al. Illuminating the evolutionary history of chlamydiae. Science. 2004;304:728–30. doi: 10.1126/science.1096330. [DOI] [PubMed] [Google Scholar]
- Hybiske K, Stephens RS. Mechanisms of host cell exit by the intracellular bacterium Chlamydia. P Natl Acad Sci USA. 2007;104:11430–5. doi: 10.1073/pnas.0703218104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalayoglu MV, Miranpuri GS, Golenbock DT, et al. Characterization of low-density lipoprotein uptake by murine macrophages exposed to Chlamydia pneumoniae. Microbes Infect. 1999;1:409–18. doi: 10.1016/s1286-4579(99)80044-6. [DOI] [PubMed] [Google Scholar]
- Kalman S, Mitchell W, Marathe R, et al. Comparative genomes of Chlamydia pneumoniae and C. trachomatis. Nat Genet. 1999;21:385–9. doi: 10.1038/7716. [DOI] [PubMed] [Google Scholar]
- Kimura T, Watanabe N, Matsui M, et al. Structure-activity relationship of a series of phenylureas linked to 4-phenylimidazole. Novel potent inhibitors of acyl-CoA:cholesterol O-acyltransferase with antiatherosclerotic activity. 2. J Med Chem. 1993;36:1641–53. doi: 10.1021/jm00063a014. [DOI] [PubMed] [Google Scholar]
- Kumar Y, Cocchiaro J, Valdivia RH. The obligate intracellular pathogen Chlamydia trachomatis targets host lipid droplets. Curr Biol. 2006;16:1646–51. doi: 10.1016/j.cub.2006.06.060. [DOI] [PubMed] [Google Scholar]
- Lee S, Han JM, Kim H, et al. Synthesis of cinnamic acid derivatives and their inhibitory effects on LDL-oxidation, acyl-CoA:cholesterol acyltransferase-1 and -2 activity, and decrease of HDL-particle size. Bioorg Med Chem Lett. 2004;14:4677–81. doi: 10.1016/j.bmcl.2004.06.101. [DOI] [PubMed] [Google Scholar]
- McLean KJ, Marshall KR, Richmond A, et al. Azole antifungals are potent inhibitors of cytochrome P450 mono-oxygenases and bacterial growth in mycobacteria and streptomycetes. Microbiology. 2002;148:2937–49. doi: 10.1099/00221287-148-10-2937. [DOI] [PubMed] [Google Scholar]
- Makino S, Jenkin HM, Yu HM, et al. Lipid composition of Chlamydia psittaci grown in monkey kidney cells in defined medium. J Bacteriol. 1970;103:62–70. doi: 10.1128/jb.103.1.62-70.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyairi I, Mahdi OS, Ouellette SP, et al. Different growth rates of Chlamydia trachomatis biovars reflect pathotype. J Infect Dis. 2006;194:350–7. doi: 10.1086/505432. [DOI] [PubMed] [Google Scholar]
- Peters J, Onguri V, Nishimoto SK, et al. The Chlamydia trachomatis CT149 protein exhibits esterase activity in vitro and catalyzes cholesteryl ester hydrolysis when expressed in HeLa cells. Microbes Infect. 2012;14:1196–204. doi: 10.1016/j.micinf.2012.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Brunham RC, Shen C, et al. Genome sequences of Chlamydia trachomatis MoPn and Chlamydia pneumoniae AR39. Nucleic Acids Res. 2000;28:1397–406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read TD, Myers GS, Brunham RC, et al. Genome sequence of Chlamydophila caviae (Chlamydia psittaci GPIC): examining the role of niche-specific genes in the evolution of the Chlamydiaceae. Nucleic Acids Res. 2003;31:2134–47. doi: 10.1093/nar/gkg321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakashita N, Miyazaki A, Takeya M, et al. Localization of human acyl-coenzyme A: cholesterol acyltransferase-1 (ACAT-1) in macrophages and in various tissues. Am J Pathol. 2000;156:227–36. doi: 10.1016/S0002-9440(10)64723-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachter J, Wyrick PB. Culture and isolation of Chlamydia trachomatis. Method Enzymol. 1994;236:377–90. doi: 10.1016/0076-6879(94)36028-6. [DOI] [PubMed] [Google Scholar]
- Stephens RS, Kalman S, Lammel C, et al. Genome sequence of an obligate intracellular pathogen of humans: Chlamydia trachomatis. Science. 1998;282:754–9. doi: 10.1126/science.282.5389.754. [DOI] [PubMed] [Google Scholar]
- Thomson NR, Holden MT, Carder C, et al. Chlamydia trachomatis: genome sequence analysis of lymphogranuloma venereum isolates. Genome Res. 2008;18:161–71. doi: 10.1101/gr.7020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wylie JL, Hatch GM, McClarty G. Host cell phospholipids are trafficked to and then modified by Chlamydia trachomatis. J Bacteriol. 1997;179:7233–42. doi: 10.1128/jb.179.23.7233-7242.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]